Gammagard S/D
sp.zn. sukls132128/2016 sukls173503/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
GAMMAGARD S/D
Prášek a rozpouštědlo pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Immunoglobulinum humanum normále ad usum intravenosum (IVIg)
Jeden ml obsahuje:
Immunoglobulinum humanum normale ad usum intravenosum 50 mg nebo 100 mg, přičemž minimálně 90 % proteinu tvoří IgG.
Jedna injekční lahvička obsahuje 5 g nebo 10 g immunoglobulinum humanum normale ad usum intravenosum (IgG). Přípravek se rozpouští ve vodě na injekci na 5% (50 mg/ml) roztok nebo 10% (100 mg/ml) roztok proteinu.
Distribuce podtříd IgG:
IgG: > 56,9 %
IgG2 > 16,0 %
IgGs > 3,3 %
IgG4 > 0,3 %
Maximální obsah imunoglobulinu A (IgA): max. 3 pg/ml v 5% roztoku.
Vyrobeno z plazmy lidských dárců.
Pomocné látky se známým účinkem: sodík (více v bodě 4.4.)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro infuzní roztok.
Přípravek GAMMAGARD S/D je lyofilizovaný, bílý nebo nažloutlý prášek/koláč, bez přítomnosti viditelných cizorodých částic.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Substituční léčba u dospělých a dětí a adolescentů (0-18 let) u:
• Syndromů primárního imunodeficitu (PID) s poruchou tvorby protilátek (viz bod 4.4)
• Hypogamaglobulinémie a rekurentních bakteriálních infekcí u pacientů s chronickou lymfatickou leukemii, u nichž selhala antibiotická profylaxe
• Hypogamaglobulinémie a rekurentních bakteriálních infekcí u pacientů s mnohočetným myelomem ve fázi plateau, kteří nereagovali na imunizaci proti pneumokokům
• Hypogamaglobulinémie u pacientů po alogenní transplantaci hematopoetických kmenových buněk
• Vrozeného AIDS s rekurentními bakteriálními infekcemi.
Nezralé děti s nízkou porodní hmotností
Imunomodulace u dospělých a dětí a adolescentů (0-18 let) u:
• Primární imunitní trombocytopenie (Idiopatická trombocytopenická purpura (ITP)), u pacientů, u nichž je vysoké riziko krvácení, nebo před chirurgickým výkonem k úpravě počtu trombocytů
• Guillain-Barrého syndromu
• Kawasakiho choroby
4.2 Dávkování a způsob podání
Substituční léčba by měla být zahájena a sledována pod dohledem lékaře se zkušenostmi s léčbou imunodeficitů.
Dávkování
Dávka a režim dávkování závisí na indikaci.
Při substituční léčbě je třeba přizpůsobit dávkování individuálně u každého pacienta podle jeho farmakokinetické a klinické odpovědi. Následující režimy dávkování slouží jako vodítko.
Substituční léčba u syndromů primárního imunodeficitu
Režim dávkování by měl být takový, aby nejnižší hladina IgG (měřená před další infuzí) byla minimálně 5-6 g/l. Dosažení rovnováhy trvá tři až šest měsíců od zahájení léčby. Doporučovaná úvodní dávka je 0,4-0,8 g/kg tělesné hmotnosti (TH), po níž následuje dávka minimálně 0,2 g/kg TH podávaná každé tři až čtyři týdny.
Dávka potřebná k dosažení minimálních koncentrací 5-6 g/l před další infuzí se pohybuje mezi 0,20,8 g/kg/TH/měsíc. Po dosažení rovnovážného stavu se interval mezi dávkami pohybuje od tří do čtyř týdnů.
Je třeba měřit hladiny před další infuzí a porovnávat je s incidencí infekcí. Ke snížení počtu infekcí může být zapotřebí zvýšit dávkování s cílem dosáhnout vyšších předinfuzních hladin.
Hypogamaglobulinémie a rekurentní bakteriální infekce u pacientů s chronickou lymfatickou leukémií, u nichž selhala antibiotická profylaxe, hypogamaglobulinémie a rekurentní bakteriální infekce u pacientů s mnohočetným myelomem ve fázi plateau, kteří nereagovali na imunizaci proti pneumokokům, děti s vrozeným AIDS a rekurentními bakteriálními infekcemi.
Doporučená dávka je 0,2-0,4 g/kg TH každé tři až čtyři týdny.
Nezralé děti s nízkou porodní hmotností
K profylaxi pozdního rozvoje infekce u nezralých dětí s nízkou porodní hmotností dostávají novorozenci mladší než 7 dnů 0,5 g/kg TH a stejnou dávku o týden později. Poté následuje celkem 5 infuzí každých 14 dnů nebo do propuštění z nemocnice.
Hypogamaglobulinémie u pacientů po alogenní transplantaci hematopoetických kmenových buněk U léčby infekcí a profylaxe reakce štěpu proti hostiteli se dávky stanovují individuálně.
Doporučená dávka je 0,2-0,4 g/kg TH každé tři až čtyři týdny. Předinfuzní hladiny by měly být udržovány nad 5 g/l.
Primární imunitní trombocytopenie Existují dvě alternativní schémata léčby:
• 0,8-1 g/kg TH podáno v den jedna; tuto dávku lze opakovat jednou během 3 dnů
• 0,4 g/kg podáváno denně po dobu dvou až pěti dnů.
V případě relapsu je možné léčbu zopakovat.
Guillain-Barrého syndrom 0,4 g/kg/den po dobu 5 dnů
Kawasakiho choroba
1,6-2,0 g/kg TH má být podáno v rozdělených dávkách po dobu dvou až pěti dnů nebo 2,0 g/kg jako jedna dávka. Pacientům by měla být souběžně podávána kyselina acetylsalicylová.
Doporučené dávkování je shrnuto v následující tabulce:
|
Indikace |
Dávka |
Frekvence injekcí |
|
Substituční léčba u primárního imunodeficitu |
úvodní dávka: 0,4-0,8 g/kg TH poté: 0,2-0,8 g/kg TH |
každé 3-4 týdny pro zajištění minimální předinfuzní hladiny IgG alespoň 5-6 g/l |
|
Substituční léčba u sekundárního imunodeficitu |
0,2-0,4 g/kg TH |
každé 3-4 týdny pro zajištění minimální předinfuzní hladiny IgG alespoň 5-6 g/l |
|
Vrozený AIDS |
0,2-0,4 g/kg TH |
každé 3-4 týdny |
|
Nezralé děti s nízkou porodní hmotností (novorozenci mladší než 7 dnů) |
0,5 g/kg TH |
dvě injekce s odstupem 1 týdne, poté celkem 5 infuzí každých 14 dnů nebo do doby propuštění z nemocnice |
|
Hypogamaglobulinémie (< 4 g/l) u pacientů po alogenní transplantaci hematopoetických kmenových buněk |
0,2 - 0,4 g/kg TH |
každé 3-4 týdny pro zajištění minimální hladiny IgG nad 5 g/l |
|
- léčba infekcí a profylaxe reakce štěpu proti hostiteli - přetrvávající nedostatečná tvorba protilátek |
0,2 - 0,4 g/kg TH 0,2 - 0,4 g/kg TH |
každý týden ode dne 7 až do 3 měsíců po transplantaci každý měsíc, dokud se hladiny protilátek nevrátí k normálu |
|
Imunomodulace: | ||
|
Primární imunitní trombocytopenie (Idiopatická trombocytopenická purpura) |
0,8-1 g/kg TH nebo 0,4 g/kg TH/d |
v den 1; je-li to možné, opakovat jednou během tří dnů po dobu 2-5 dnů |
|
Guillain-Barrého syndrom |
0,4 g/kg TH/d |
po dobu 5 dnů |
|
Kawasakiho choroba |
1,6-2 g/kg TH nebo 2 g/kg TH |
v rozdělených dávkách po dobu 2-5 dnů v kombinaci s kyselinou acetylsalicylovou v jedné dávce v kombinaci s kyselinou acetylsalicylovou |
Pediatrická populace
Dávkování u dětí a dospívajících (od 0 do18 let) se neliší od dávkování u dospělých, protože dávkování je u každé indikace dáno tělesnou hmotností a upravováno dle klinického přínosu u výše zmíněných stavů.
Způsob podání
K intravenóznímu podání.
Doporučuje se podávat 10% roztok přípravku GAMMAGARD S/D do kubitálních žil. To může u pacienta snížit pravděpodobnost vzniku nepříjemných pocitů v místě vpichu.
Přípravek GAMMAGARD S/D 5% (50 mg/ml) má být podáván intravenózně úvodní rychlostí 0,5 ml/kg TH/hod. Obecně se doporučuje pacientům, kteří začínají s léčbou přípravkem GAMMAGARD S/D nebo jsou převáděni z jiného intravenózního imunoglobulinu na přípravek GAMMAGARD S/D, podat infuzi nejnižší rychlostí a zvýšit rychlost na maximum, pokud tolerovali několik infuzí podávaných střední rychlostí (viz též bod 4.4).
Pokud je dobře snášen, je možno rychlost podání postupně zvyšovat na maximální rychlost 4 ml/kg TH/hod. Pacientům, kteří snášejí infuzi 5% roztoku přípravku GAMMAGARD S/D při rychlosti podání 4 ml/kg TH/hod, je možno infundovat 10% koncentraci při počáteční rychlosti 0,5 ml/kg TH/hod. Pokud se neobjeví žádné nežádoucí účinky, je možno rychlost postupně zvyšovat až na maximální rychlost podání 8 ml/kg TH/hod.
Při přechodu z 5% na 10% roztok by měla být počáteční rychlost podání 10% roztoku nižší, aby byla zachována srovnatelná rychlost podání proteinu IgG.
4.3 Kontraindikace
Hypersenzitivita nebo známá přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 (viz rovněž bod 4.4).
Hypersenzitivita na lidské imunoglobuliny, zvláště u pacientů s protilátkami proti IgA.
Přípravek GAMMAGARD S/D neobsahuje více než 3 mikrogramy IgA v 1 ml 5% roztoku.
Přípravek GAMMAGARD S/D je kontraindikován u pacientů se známou anafylaxí nebo závažnou přecitlivělostí na přípravek GAMMAGARD S/D s méně než 3 mikrogramy/ml IgA v 5% roztoku.
4.4 Zvláštní upozornění a opatření pro použití
Tento léčivý přípravek obsahuje jako pomocnou látku 20 mg glukózy v jednom ml (400 mg/g IgG). Pacient o hmotnosti 70 kg, kterému je podána infúze 1 g/kg přípravku GAMMAGARD S/D, by měl obdržet 28 gramů glukózy (112 kalorií). To je třeba vzít v úvahu v případě latentního diabetu (kde se může objevit přechodná glykosurie), u diabetu nebo u pacientů na dietě s nízkým obsahem cukru. Akutní renální selhání viz níže.
Některé závažné nežádoucí účinky mohou souviset s rychlostí infúze. Je nutno přesně dodržovat doporučenou rychlost infúze, která je uvedena v bodě „4.2 Způsob podání“. Pacienti musí být pečlivě monitorováni a je nutno sledovat, zda se u nich v době infúze neobjeví jakékoli symptomy.
Některé nežádoucí účinky se mohou vyskytovat častěji
- v případě vysoké rychlosti infúze,
- u pacientů s hypo- či agamaglobulinémií s IgA deficitem či bez něj;
- u pacientů s imunodeficitem, kteří dostávají normální lidský imunoglobulin poprvé nebo,
v ojedinělých případech, při změně přípravku normálního lidského imunoglobulinu nebo po uplynutí dlouhé doby od předchozí infúze.
Případným komplikacím je možné často zabránit tím, že:
- pacientům, kteří nejsou citliví na normální lidský imunoglobulin, se podá první dávka přípravku pomalu (0,5 až 1 ml/kg TH/hod);
- zajistí se pečlivé monitorování pacientů a sleduje se, zda se u nich po dobu infúze neobjevují nežádoucí účinky. Týká se to zvláště pacientů, kteří dostávají normální lidský imunoglobulin poprvé, pacientů, kteří jsou převedeni z jiného přípravku intravenózního imunoglobulinu nebo u nichž uplynul dlouhý interval od podání předchozí infúze. Tito pacienti musí být monitorováni během první infúze a jednu hodinu po první infuzi, aby se zachytily možné známky nežádoucího účinku. Všichni ostatní pacienti musí být sledováni minimálně 20 minut po podání;
- zohlední se obsah glukózy (maximální obsah 0,4 g/g IgG) v případě latentního diabetů (kde může dojít k přechodné glykosurii), v případě diabetů nebo u pacientů na dietě s nízkým obsahem cukru.
Podávání intravenózních imunoglobulinů vyžaduje u všech pacientů:
• adekvátní hydrataci před zahájením infuze intravenózního imunoglobulinu
• sledování výdeje moči
• sledování hladin kreatininu v séru
• vyhnout se současnému podávání kličkových diuretik.
Dojde-li k výskytu nežádoucího účinku, je nutné snížit rychlost podávání nebo zastavit infuzi.
Potřebná léčba závisí na povaze a závažnosti nežádoucího účinku. V případě šoku je třeba dodržovat standardní lékařské postupy pro léčbu šoku.
Varování:
Hvpersenzitivita
Pravé hypersenzitivní reakce jsou vzácné. Mohou se vyskytnout ve velmi vzácných případech IgA deficitu s anti-IgA protilátkami.
Vzácně může normální lidský imunoglobulin navodit anafylaktický šok s poklesem krevního tlaku, dokonce i u pacientů, kteří předchozí léčbu normálním lidským imunoglobulinem snášeli dobře. Pacienti s protilátkami proti IgA nebo s deficitem IgA, které jsou součástí základního primárního imunodeficitu, kvůli němuž je indikována léčba IVIg, mohou mít zvýšené riziko anafylaktické reakce. Při použití přípravku GAMMAGARD S/D byla hlášena anafylaxe, přestože obsahuje jen malá množství IgA (viz bod 4.8). Pacienti, u nichž se vyskytla závažná hypersenzitivní reakce, by měli dostat přípravek GAMMAGARD S/D jen pod velmi pečlivým dohledem v prostředí, kde je k dispozici podpůrná péče pro léčbu život ohrožujících reakcí.
Upozornění:
Tromboembolie
Bylo klinicky prokázáno, že existuje souvislost mezi léčbou IVIg (včetně přípravku GAMMAGARD S/D) a trombotickými příhodami, jako je infarkt myokardu, cévní mozková příhoda (včetně mrtvice), plicní embolie a hluboká žilní trombóza. Předpokládá se, že tyto příhody souvisejí s relativním zvýšením viskozity krve, způsobeným vysokým přílivem imunoglobulinu u rizikových pacientů. Zvláštní opatrnosti je zapotřebí při předepisování a infuzi IVIg u obézních pacientů a u pacientů s preexistujícím rizikem vzniku tromboembolických příhod (jako je ateroskleróza v anamnéze, vícečetné kardiovaskulární rizikové faktory, pokročilý věk, porucha srdečního výdeje, zjištěná nebo suspektní hvperviskozita, např. dehydratace nebo paraproteinémie, hyperkoagulační stavy, dlouhodobá imobilizace, obezita, užívání estrogenu, diabetes mellitus, získaná nebo vrozená trombofilie, cévní onemocnění v anamnéze, permanentní cévní katétr, vysoká dávka a rychlá infúze, trombóza nebo tromboembolie v anamnéze).
Před a po podání přípravku zajistěte dostatečnou hydrataci pacienta. Monitorujte známky a příznaky trombózy a stanovte viskozitu krve u pacientů s rizikem hvperviskozitv.
Pacientům s rizikem tromboembolických nežádoucích reakcí by měl být přípravek GAMMAGARD S/D podáván minimální infuzní rychlostí v nejnižší možné dávce.
Renální komplikace
U pacientů léčených IVIg byly hlášeny závažné renální nežádoucí reakce. Může se jednat o akutní selhání ledvin, akutní tubulární nekrózu, proximální tubulární nefropatii a osmotickou nefrózu. Ve většině případů byly identifikovány rizikové faktory, jako jsou preexistující renální insuficience, diabetes mellitus, hypovolémie, nadváha, současné podávání nefrotoxických léčivých přípravků či věk nad 65 let, sepse, hyperviskozita, paraproteinémie.
V případě poruchy funkce ledvin je třeba zvážit přerušení léčby IVIg.
Hlášení o dysfunkci ledvin a akutním selhání ledvin byly spojovány s použitím mnoha registrovaných přípravků IVIg obsahujících různé pomocné látky jako je sacharóza, glukóza a maltóza; z celkového počtu však nepoměrnou většinu představují ty, které obsahují sacharózu jako stabilizátor. U rizikových pacientů lze zvážit použití přípravků IVIg, které neobsahují sacharózu. Přípravek GAMMAGARD S/D neobsahuje sacharózu nebo maltózu.
U pacientů s rizikem akutního renálního selhání by měly být přípravky IVIg podávány minimální infuzní rychlostí v nejnižší možné dávce.
Akutní poškození plic způsobené transfuzí (TRALI)
U pacientů, kterým byly podávány IVIg, byly hlášeny případy nekardiogenního plicního edému (akutní poškození plic způsobené transfuzí, TRALI)
Syndrom aseptické meningitidy (AMS)
Ve spojení s léčbou IVIg (včetně léčby přípravkem GAMMAGARD S/D) byl hlášen syndrom aseptické meningitidy. Přerušení léčby IVIg může vést k remisi AMS během několika dnů. Syndrom obvykle nastupuje během několika hodin až do 2 dnů po léčbě IVIg.
• Vyšetření mozkomíšního moku je často pozitivní, s pleocytózou až několik tisíc buněk v mm3, zejména granulocytů, a zvýšenými hladinami proteinu až na několik set mg/dl.
• AMS se může objevit častěji ve spojení s léčbou vysokými dávkami IVIg (2 g/kg).
Selektivní deficit IgA
Přípravek GAMMAGARD S/D není indikován u pacientů se selektivním deficitem IgA tam, kde je deficit IgA jedinou abnormalitou. Tito pacienti by měli být léčeni jen tehdy, pokud je jejich deficit IgA spojen s imunodeficitem, u kterého je léčba intravenózními imunoglobuliny jasně indikovaná.
Hemolýza
Přípravek GAMMAGARD S/D obsahuje protilátky proti krevním skupinám, které mohou působit hemolyticky a navodit potažení erytrocytů imunoglobulinem in vivo, což může vyvolat pozitivní přímou antiglobulinovou reakci (Coombsův test). Po podání přípravku GAMMAGARD S/D se v důsledku zvýšené sekvestrace erytrocytů může vyvinout pozdní hemolytická reakce. Byla hlášena akutní hemolýza shodující se s intravaskulární hemolýzou. Ke vzniku hemolýzy mohou přispět následující faktory: vysoké dávky (podané jednorázově nebo během několika dnů) a krevní skupiny mimo skupinu 0.
Základní zánětlivé onemocnění v individuálním případě může zvýšit riziko hemolýzy, jeho význam je však nejistý.
Hyperproteinémie
U pacientů léčených IVIg se může objevit hyperproteinémie a zvýšená viskozita séra.
Příjem sodíku
Obsah sodíku v maximální denní dávce může významně ovlivnit doporučený příjem sodíku u pacientů na dietě s nízkým obsahem sodíku. U těchto pacientů je třeba při stanovení příjmu sodíku v dietě spočítat a vzít v úvahu množství sodíku obsaženého v přípravku.
Přípravek GAMMAGARD S/D obsahuje přibližně 0,85% NaCl nebo přibližně 3340 mg sodíku na litr v 5% koncentraci. Pacient hmotnosti 70 kg 1 g/kg (1,4 l) by dostal 4676 mg sodíku.
Přenosná agens
Přípravek GAMMAGARD S/D se vyrábí z lidské plazmy. Standardní opatření k prevenci infekce v souvislosti s použitím léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují výběr dárců, testování jednotlivých odběrů krve a poolů plazmy na specifické ukazatele infekce a zavedení účinných výrobních kroků k inaktivaci / odstranění virů. Přes všechna tato opatření při přípravě léků vyráběných z lidské krve nebo plazmy nelze možnost přenosu infekce zcela vyloučit. To platí i pro jakékoli neznámé nebo nově vznikající viry a jiné patogeny).
Přijatá opatření jsou považována za účinná u obalených virů, jako je virus lidského imunodeficitu (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV), a u neobalených virů hepatitidy A (HAV) a parvoviru B19.
Klinické zkušenosti potvrzují, že k přenosu viru hepatitidy typu A nebo parvoviru B19 pomocí imunoglobulinů nedochází, a předpokládá se, že obsah protilátek významně přispívá k protivirové ochraně.
Při každé aplikaci přípravku GAMMAGARD S/D pacientovi se důrazně doporučuje pečlivě zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Interference s laboratorními testy
• Po aplikaci imunoglobulinu může v krvi pacienta dojít k přechodnému vzestupu pasivně přenesených protilátek, a tím ke vzniku zavádějících pozitivních výsledků v sérologických testech např. hepatitida A, hepatitida B, spalničky a varicela.
Pasivní přenos protilátek proti erytrocytárním antigenům, například A, B, D, může ovlivnit některé sérologické testy na protilátky proti červeným krvinkám, například přímý antiglobulinový test (Coombsův test).
• Podávání přípravku GAMMAGARD S/D může vést k falešně pozitivním výsledkům u testů závislých na detekci beta D-glukanu, které se používají ke stanovení diagnózy mykotických infekcí; tyto falešně pozitivní výsledky mohou přetrvávat týdny po podání infuze přípravku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vakcíny obsahující živé atenuované viry
Podání imunoglobulinu může narušit po dobu minimálně 6 týdnů až 3 měsíců účinnost vakcín, které obsahují živé atenuované viry, např. spalniček, zarděnek, příušnic a planých neštovic. Před očkováním vakcínou, která obsahuje živé atenuované viry, by od podání tohoto přípravku měly uplynout 3 měsíce. V případě vakcíny proti spalničkám může toto narušení přetrvávat až 1 rok. U pacientů očkovaných proti spalničkám by se proto měla zkontrolovat hladina jejich protilátek.
Pediatrická populace
Nebyly provedeny studie interakce s přípravkem GAMMAGARD S/D v pediatrické populaci.
4.6 Fertilita, těhotenství a kojení
Bezpečnost použití přípravku GAMMAGARD S/D v těhotenství u člověka nebyla ověřena v kontrolovaných klinických studiích, proto by měl být u těhotných a kojících žen podáván jen s velkou opatrností. U přípravků IVIg podávaných během těhotenství byl prokázán zvýšený průnik placentou ve třetím trimestru. Klinické zkušenosti s imunoglobuliny nepředpokládají negativní vliv na průběh těhotenství nebo na plod či novorozence.
Kojení
Imunoglobuliny jsou vylučovány do mateřského mléka a mohou přispět k ochraně novorozence před patogeny vstupujícími do organismu přes sliznici.
Fertilita
Klinické zkušenosti s imunoglobuliny nepředpokládají negativní vliv na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nejsou k dispozici informace o účincích přípravku GAMMAGARD S/D na schopnost řídit nebo obsluhovat stroje.
Schopnost řídit a obsluhovat stroje může být ovlivněna některými nežádoucími účinky spojenými s přípravkem GAMMAGARD S/D. Pacienti, kteří mají během léčby nežádoucí reakce, by měli před započetím řízení nebo obsluhy strojů počkat, až tato reakce pomine.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Při intravenózním podání normálních imunoglobulinů se mohou výjimečně objevit nežádoucí účinky jako zimnice, bolesti hlavy, závratě, horečka, zvracení, alergické reakce, nauzea, bolest kloubů, pokles krevního tlaku či mírná bolest v dolní části zad.
Vzácně mohou normální lidské imunoglobuliny způsobit náhlý pokles krevního tlaku a v ojedinělých případech anafylaktický šok, a to i v případech, kdy pacient při předchozím podání žádné známky hypersenzitivity neměl.
Po podání normálních lidských imunoglobulinů byly pozorovány případy aseptické meningitidy a vzácné případy přechodných kožních reakcí. U pacientů byly pozorovány ojedinělé reverzibilní hemolytické reakce, zejména u pacientů krevních skupin A, B a AB. Vzácně se po podání vysokých dávek IVIg může rozvinout hemolytická anémie (viz též bod 4.4).
Bylo pozorováno zvýšení hladin sérového kreatininu a/nebo akutní selhání ledvin.
Velmi vzácně byly pozorovány tromboembolické reakce jako infarkt myokardu, cévní mozková příhoda, plicní embolie a hluboká žilní trombóza.
Klinicky je dokumentována možná souvislost mezi podáním IVIg a sklonem k rozvoji trombotických příhod. Přesná příčina tohoto jevu není známa. U pacientů s anamnézou a predispozicí ke kardiovaskulárním chorobám či trombotickým příhodám je proto třeba postupovat při předepisování a infuzi IVIg opatrně. Analýza hlášení nežádoucích účinků nasvědčuje tomu, že vysoká rychlost infúze může být rizikovým faktorem vzniku obliterujících cévních příhod.
Nežádoucí účinky zjišťující akutní a střednědobou bezpečnost přípravku GAMMAGARD S/D byly shrnuty z pivotní klinické studie s přípravkem GAMMAGARD S/D a ze studie fáze IV. Nežádoucí účinky hlášené z těchto dvou studií a z postmarketingových zkušeností jsou shrnuty a klasifikovány podle tříd orgánových systémů a četností výskytu podle databáze MedDRA v tabulce níže.
Tabulkový přehled nežádoucích účinků
Souhrn níže je řazen podle tříd orgánových systémů MedDRA (TOS a Upřednostňované termíny). Četnost výskytu je vyjádřena za použití následujících kritérií: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Nežádoucí účinky jsou v rámci každé skupiny četností řazeny podle snižující se závažností.
|
Nežádoucí účinky (NÚ) po podání přípravku GAMMAGARD S/D |
D | |
|
Třída orgánového systému MedDRA |
Upřednostňovaný termín MedDRA (Verze 17.0) |
Kategorie četnost výskytu NÚ* |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Aseptická meningitida |
Není známo | |
|
Poruchy krve a lymfatického systému |
Hemolýza, anémie, trombocytopenie, lymfadenopatie. |
Není známo |
|
Poruchy imunitního systému |
Anafylaktická nebo anafylaktoidní reakce, anafylaktický šok, hypersenzitivita |
Není známo |
|
Psychiatrické poruchy |
Úzkost, vzrušení |
Méně časté |
|
Neklid |
Není známo | |
|
Poruchy nervového systému |
Časté | |
|
Letargie |
Méně časté | |
|
Cerebrovaskulární příhoda, přechodná ischemická ataka, křeče, migréna, závrať, parestezie, synkopa, tremor |
Není známo | |
|
Poruchy oka |
Zastřené vidění |
Méně časté |
|
Trombóza retinální žíly, poruchy vidění, bolest očí, fotofobie |
Není známo | |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Infarkt myokardu, cyanóza, tachykardie, bradykardie |
Není známo | |
|
Cévní poruchy |
Zrudnutí |
Časté |
|
Výkyvy krevního tlaku |
Méně časté | |
|
Arteriální trombóza, trombóza duté žíly, hluboká žilní trombóza, tromboflebitida, hypotenze, hypertenze, bledost |
Není známo | |
|
Respirační, hrudní a mediastinální poruchy |
Dyspnoe, epistaxe |
Méně časté |
|
Plicní embolie, plicní edém, hypoxie, bronchospasmus, sípot, hyperventilace, tíseň v krku, kašel |
Není známo | |
|
Gastrointestinální poruchy |
Časté | |
|
Průjem, stomatitida, bolest horní části břicha, abdominální dyskomfort |
Méně časté | |
|
Není známo | ||
|
Poruchy jater a žlučových cest |
Hepatitis (neinfekční hepatitida) |
Není známo |
|
Poruchy kůže a podkožní tkáně |
Kopřivka, pruritus, studený pot, hyperhidróza |
Méně časté |
|
Angioedém, dermatitida, erytém, vyrážka |
Není známo | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest zad, svalová křeč, bolest končetin |
Méně časté |
|
Arthralgie, myalgie |
Není známo | |
|
Poruchy ledvin a močových cest |
Renální selhání |
Není známo |
|
Celkové poruchy a reakce v místě aplikace |
Únava, zimnice, pyrexie |
Časté |
|
Bolest na hrudi, malátnost, bolest, nevolnost na hrudi, pocit abnormality, pocit chladu, pocit horka, onemocnění podobné chřipce, erytém v místě infúze, extravazace v místě infúze, bolest v místě infúze |
Méně časté | |
|
Reakce v místě infúze, astenie, edém |
Není známo | |
|
Vyšetření |
Zvýšení krevního tlaku |
Méně časté |
|
Pozitivní přímý Coombsův test |
Není známo | |
|
Poruchy metabolismu a výživy |
Méně časté | |
*dle procenta infuzí.
Informace o bezpečnosti vzhledem k přenosným agens viz bod 4.4.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Předávkování může vést k přetížení oběhu a hyperviskozitě, zejména u rizikových pacientů, včetně starších pacientů či pacientů se srdeční nebo renální poruchou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunní séra a imunoglobuliny: normální lidský imunoglobulin k intravaskulárnímu podání, ATC kód: J06BA02
Přípravek GAMMAGARD S/D obsahuje převážně funkčně intaktní imunoglobulin G (IgG) se širokým spektrem protilátek proti infekčním agens.
Přípravek GAMMAGARD S/D obsahuje IgG protilátky přítomné v běžné populaci. Obvykle je připraven ze směsné lidské plazmy minimálně od 1 000 dárců. Distribuce podtříd imunoglobulinu G odpovídá distribuci v nativní lidské plazmě. Odpovídající dávky tohoto léčivého přípravku mohou upravit abnormálně nízké hladiny imunoglobulinu G do normálního rozmezí.
Mechanizmus účinku v jiných indikacích než je substituční léčba není zcela objasněn, ale zahrnuje imunomodulační účinky.
5.2 Farmakokinetické vlastnosti
Přípravek GAMMAGARD S/D je po intravenózním podání ihned a úplně biologicky dostupný v oběhu příjemce. Je relativně rychle distribuován mezi plazmou a extravaskulární tekutinou; rovnováhy mezi intravaskulárním a extravaskulárním oddílem je dosaženo přibližně po 3-5 dnech.
Biologický poločas přípravku GAMMAGARD S/D je přibližně 37,7 ± 15 dnů. Tento poločas se může u různých pacientů individuálně lišit, zejména při primárním imunodeficitu.
IgG a IgG-komplexy jsou odbourávány v buňkách retikuloendotelového systému.
5.3 Předklinické údaje vztahující se k bezpečnosti Imunoglobuliny jsou normální složkou lidského těla.
Bezpečnost přípravku GAMMAGARD S/D byla prokázána v řadě neklinických studií. Neklinické údaje z konvenčních farmakologických studií bezpečnosti a toxicity neodhalily žádné zvláštní riziko pro člověka.
Experimentální studie u heterogenních druhů nebyly prováděny, protože klinické zkušenosti neprokázaly karcinogenní potenciál imunoglobulinů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Roztok lidského albuminu (0,06 g/g IgG)
Glycin
Chlorid sodný Monohydrát glukózy
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Přípravek GAMMAGARD S/D nesmí být mísen s jinými léčivými přípravky.
Doporučuje se, aby byl přípravek GAMMAGARD S/D podáván odděleně od jiných léčivých přípravků, které pacient dostává.
6.3 Doba použitelnosti
2 roky
Chemická a fyzikální stabilita rekonstituovaného přípravku GAMMAGARD S/D byla prokázána po dobu 2 hodin při teplotě uchovávání do 25°C. Z mikrobiologického hlediska by měl být přípravek ihned použit. Doba uchovávání připraveného roztoku a podmínky před použitím jsou v odpovědnosti uživatele.
Doba uchovávání může být maximálně 24 hodin při teplotě 2 až 8°C, pokud rekonstituce proběhla za kontrolovaných a validovaných aseptických podmínek. Nejsou-li splněny tyto podmínky, nelze udržet sterilitu rekonstituovaného přípravku.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25°C.
Chraňte před mrazem, lahvička s rozpouštědlem by mohla prasknout.
Uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Nepoužívejte po uplynutí doby použitelnosti.
Uchovávejte mimo dohled a dosah dětí.
Podmínky uchovávání rekonstituovaného přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvičky s práškem a s rozpouštědlem jsou ze skla třídy I, jsou uzavřeny silikonizovanými zátkami z brombutylové pryže, hliníkovým uzávěrem a plastikovým krytem.
Přípravek GAMMAGARD S/D je k dispozici ve velikostech balení 5 g a 10 g.
Každé balení obsahuje rozpouštědlo (96 ml, 192 ml), sterilní převodní zařízení a sterilní infuzní set s filtrem.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Je-li rekonstituce prováděna asepticky mimo sterilní laminární box, měl by být přípravek podán co nejdříve, nejpozději do 2 hodin po rekonstituci. Je-li rekonstituce prováděna asepticky ve sterilním laminárním boxu, může být rekonstituovaný přípravek uchováván chlazený (2-8°C) po dobu až 24 hodin. Nejsou-li splněny tyto podmínky, nelze udržet sterilitu rekonstituovaného přípravku. Částečně spotřebované lahvičky je třeba zlikvidovat.
K úplnému rozpuštění by mělo dojít do 30 minut. Přípravek je třeba před použitím zahřát na pokojovou nebo tělesnou teplotu.
Před rekonstitucí se jedná o bílý až světle žlutý prášek/koláč, bez viditelných cizorodých částic. Rekonstituovaný přípravek je třeba před podáním zkontrolovat s ohledem na přítomnost částic a změnu barvy. Nepoužívejte roztoky, které jsou zakalené nebo obsahují částice.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce - použijte aseptickou techniku
Zahřejte přípravek GAMMAGARD S/D a vodu na injekci (rozpouštědlo) na pokojovou teplotu. Tato teplota se musí udržovat až do úplného rozpuštění přípravku.
A. 5% roztok:
1. Odstraňte ochranné kryty z injekčních lahviček
2. Sejměte ochranný kryt z jednoho konce převodního zařízení. Nedotýkejte se přitom hrotu.
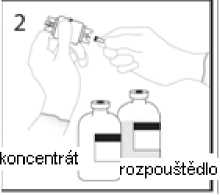
3a. Umístěte injekční lahvičku s rozpouštědlem na vodorovný povrch a propíchněte odkrytým hrotem uzávěr injekční lahvičky s rozpouštědlem v jeho středu.
UPOZORNĚNÍ: Není-li uzávěr injekční lahvičky propíchnut středem, může dojít k vytlačení
uzávěru.
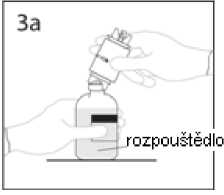
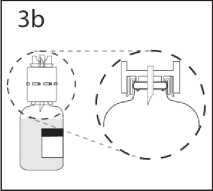
3b. Převodní zařízení pevně zatlačte dolů, aby v něm injekční lahvička pevně držela.
Převodní zařízení s připojenou injekční lahvičkou přidržte a sejměte kryt z druhého hrotu převodního zařízení. Nedotýkejte se přitom hrotu.
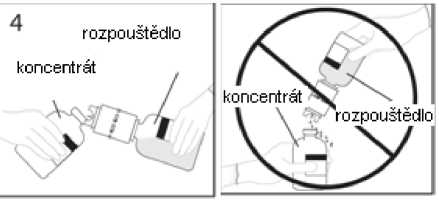
4. Injekční lahvičku s rozpouštědlem a připojenou převodní jehlou přidržte v úhlu k injekční lahvičce s práškem, abyste předešli vylití rozpouštědla.
Poznámka: nedržte injekční lahvičku s rozpouštědlem dnem vzhůru, aby nedošlo k vylití rozpouštědla
5a. Propíchněte injekční lahvičku s práškem středem uzávěru a současně rychle obraťte injekční lahvičku s rozpouštědlem dnem vzhůru, abyste zamezili vylití rozpouštědla. UPOZORNĚNÍ: Není-li uzávěr injekční lahvičky propíchnut středem, může dojít k vytlačení uzávěru a ztrátě vakua.
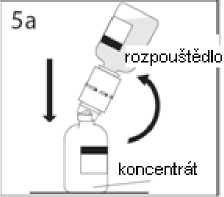
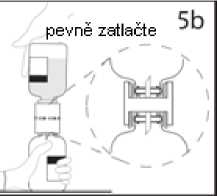
5b. Převodní zařízení pevně zatlačte dolů, aby v něm injekční lahvička pevně držela.
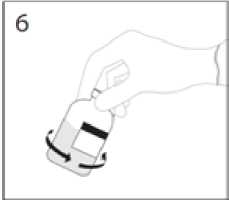
6. Když je přesun rozpouštědla ukončen, vytáhněte převodní zařízení a prázdnou injekční lahvičku s rozpouštědlem z injekční lahvičky s práškem. Injekční lahvičkou s práškem ihned jemně otáčejte, abyste obsah řádně promíchali.
UPOZORNĚNÍ: Netřepejte. Zabraňte zpěnění.
Jehlu, injekční stříkačku a zbytek rozpouštědla po použití zlikvidujte.
B. 10% roztok:
4. Odstraňte ochranné kryty z injekčních lahviček a očistěte uzávěry dezinfekčním roztokem.
5. Pro přípravu 10% roztoku je nutné odstranit polovinu objemu rozpouštědla. Objem rozpouštědla, který je třeba odstranit z injekční lahvičky s rozpouštědlem před napojením převodního zařízení je uveden v tabulce 2. Aseptickým postupem natáhněte nepotřebný objem rozpouštědla pomocí podkožní jehly do injekční stříkačky. Naplněnou injekční stříkačku a jehlu zlikvidujte.
6. S použitím zbývajícího objemu rozpouštědla pokračujte v krocích 2-6, jak jsou popsány v oddílu A.
TABULKA 2
Objem rozpouštědla, který je třeba odstranit
Koncentrace injekční lahvička 5 g injekční lahvička 10 g
5% K rekonstituci 5% roztoku neodstraňujte žádné rozpouštědlo
10% 48 ml 96 ml
Podání - použijte aseptickou techniku.
Postupujte podle návodu na použití infuzního setu, který je součástí každého balení. Pokud použijete jiný infuzní set, ujistěte se, že souprava obsahuje podobný filtr.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Do 30.11.2016:
BAXTER CZECH spol. s.r.o.
Karla Engliše 3201/6 150 00 Praha 5 Česká republika
Od 1.12.2016:
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO
75/152/00-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15.3.2000
Datum posledního prodloužení registrace: 22.12.2015
10. DATUM REVIZE TEXTU
10.8.2016
14/14