Fibclot 1,5 G
sp. zn. sukls10610/2015
Souhrn údajů o přípravku
'V Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Fibclot 1,5 g, prášek a rozpouštědlo pro injekční/infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Fibrinogenum humanum
Jedna injekční lahvička přípravku Fibclot obsahuje nominálně fibrinogenum humanum 1,5 g.
Po rekonstituci ve 100 ml rozpouštědla (voda na injekci) obsahuje přípravek Fibclot nominálně fibrinogenum humanum 15 mg/ml.
Účinnost je stanovena v souladu s monografií Evropského lékopisu pro lidský fibrinogen (fibrinogenum humanum).
Vyrobeno z plazmy lidských dárců.
Pomocné látky se známým účinkem:
Jedna injekční lahvička přípravku obsahuje maximálně 69 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok. Bílý nebo světle žlutý prášek v injekční lahvičce.
4 KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
Léčba a perioperační profylaxe krvácení u pacientů s vrozenou hypofibrinogenemií nebo afibrinogenemií se sklonem ke krvácení.
4.2. Dávkování a způsob podání
Léčba se má zahajovat pod dohledem lékaře se zkušenostmi s léčbou poruch koagulace.
Dávkování
Dávkování a doba trvání substituční terapie závisí na závažnosti poruchy, místě a rozsahu krvácení a na klinickém stavu pacienta.
Je třeba stanovit (funkční) hladinu fibrinogenu, aby bylo možné vypočítat individuální dávkování. Dávka a četnost podání se musí stanovit u každého pacienta individuálně na základě pravidelného měření hladiny plazmatického fibrinogenu, neustálého sledování klinického stavu pacienta a jiných použitých substitučních terapií.
Normální hladina plazmatického fibrinogenu se pohybuje v rozmezí 1,5 - 4,5 g/l. U vrozené hypofibrinogenemie nebo afibrinogenemie představuje kritickou hladinu plazmatického fibrinogenu, pod kterou může dojít ke krvácením, přibližně hodnota 0,5 - 1,0 g/l.
V případě velké chirurgické intervence je nezbytné přesné sledování substituční terapie pomocí koagulačních testů.
Léčba krvácení a profylaxe u pacientů s vrozenou hypofibrinogenemií nebo afibrinogenemií a se známým sklonem ke krvácení.
K léčbě nechirurgických krvácivých příhod se doporučuje zvýšit hladinu fibrinogenu na 1 g/l a udržet fibrinogen na této hladině, dokud není hemostáza pod kontrolou, a na hladině vyšší než 0,5 g/l, dokud nedojde k úplnému zhojení.
K prevenci nadměrného krvácení během chirurgických zákroků se doporučuje profylaktická léčba na zvýšení hladiny fibrinogenu na 1 g/l a udržování fibrinogenu na této hladině, dokud není hemostáza pod kontrolou, a na hladině vyšší než 0,5 g/l, dokud nedojde k úplnému zhojení.
V případě chirurgického zákroku nebo léčby nechirurgického krvácení se dávka vypočítá takto:
dávka (g) = (cílová hladina (g/l) - výchozí hladina (g/l)) x 0,043 x tělesná hmotnost (kg). přičemž 0,043 odpovídá 1/recovery ((g/l)/(g/kg)).
V případě naléhavé situace, kdy výchozí hladina fibrinogenu není známa, je doporučená počáteční dávka 0,05 g na kg tělesné hmotnosti podávaná intravenózně.
Následné dávkování (dávky a četnost injekcí) se upraví podle klinického stavu pacienta a laboratorních výsledků.
Biologický poločas fibrinogenu je 3-4 dny. Při absenci spotřeby tak není opakovaná léčba lidským fibrinogenem obvykle nutná. Vzhledem k tomu, že v případě opakovaného podání za účelem profylaxe dochází k nahromadění přípravku, se má dávka a četnost podávání stanovit na základě terapeutických cílů lékaře pro daného pacienta.
Pediatrická populace
V současnosti dostupné údaje jsou uvedeny v bodě 4.8 a 5.1, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování u dětí.
Způsob podání
Intravenózní podání infuzí nebo injekcí.
Přípravek Fibclot se má podávat pomalou intravenózní infuzí. Maximální rychlost 4 ml/min.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.2 a 6.6.
4.3. Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4. Zvláštní upozornění a opatření pro použití
Tromboembolické příhody
Jsou-li pacienti léčeni lidským fibrinogenem, existuje riziko trombózy, zejména při vysoké dávce nebo po opakovaném podávání dávek. Pacienti, kterým je podáván lidský fibrinogen, musí být pečlivě sledováni s ohledem na známky a příznaky trombózy.
U pacientů s ischemickou chorobou srdeční nebo infarktem myokardu v anamnéze, u pacientů s onemocněním jater, u peri a pooperačních pacientů, u novorozenců, u pacientů vystavených riziku tromboembolických příhod nebo diseminované intravaskulární koagulace se musí potenciální přínos léčby lidským plazmatickým fibrinogenem zvážit oproti riziku tromboembolických komplikací. Je nutná opatrnost a pečlivé sledování pacienta.
Alergické reakce nebo reakce anafylaktického typu
Objeví-li se alergické reakce nebo reakce anafylaktického typu, musí se podávání injekce/infuze okamžitě ukončit. V případě anafylaktického šoku je třeba použít standardní lékařský postup léčby šoku.
Přenosná agens
Standardní opatření pro prevenci infekcí způsobených použitím léčivých přípravků připravených z lidské krve nebo plazmy zahrnují výběr dárců, screening jednotlivých a směsných vzorků plazmy na přítomnost specifických infekčních markerů a zavedení účinných výrobních postupů k inaktivaci/odstranění virů. Přes všechna tato opatření nelze při podávání léčivých přípravků vyrobených z lidské krve nebo plazmy zcela vyloučit možnost přenosu infekčních agens. To platí i pro jakékoli neznámé nebo nově vznikající viry či jiné patogeny.
Přijatá opatření se považují za účinná vůči obaleným virům, jako je virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV), a vůči neobalenému viru hepatitidy A (HAV). Přijatá opatření mohou mít omezený užitek vůči neobaleným virům, jako je parvovirus B19. Infekce způsobená parvovirem B19 může být závažná u těhotných žen (infekce plodu) a u jedinců s imunodeficiencí nebo zvýšenou erytropoézou (např. hemolytická anemie).
U pacientů, kterým je pravidelně/opakovaně podáván lidský fibrinogen získaný z plazmy je třeba zvážit vhodnou vakcinaci (proti hepatitidě A a B).
Důrazně se doporučuje zaznamenat při každém podání přípravku Fibclot pacientovi název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Imunogenita
V případě substituční terapie koagulačními faktory u jiných vrozených deficitů byly pozorovány protilátkové reakce, avšak v současnosti nejsou k dispozici žádné údaje v souvislosti s fibrinogenem.
Hladina sodíku
Přípravek obsahuje maximálně 3 mmol (nebo 69 mg) sodíku/injekční lahvičku. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Pediatrická populace
Pro pediatrickou populaci platí stejná upozornění a opatření.
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou známy žádné interakce přípravků obsahujících lidský fibrinogen s jinými léčivými přípravky.
4.6. Fertilita, těhotenství a kojení
Bezpečnost použití přípravků obsahujících lidský plazmatický fibrinogen u člověka v těhotenství a během kojení nebyla prokázána v kontrolovaných klinických studiích.
Klinické zkušenosti s přípravky obsahujícími fibrinogen při léčbě porodních komplikací naznačují, že se nemusí očekávat žádné škodlivé účinky na průběh těhotenství nebo na zdraví plodu či novorozence.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Přípravek Fibclot nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8. Nežádoucí účinky Tabulkový seznam nežádoucích účinků
Nežádoucí účinky uvedené v tabulce níže byly hlášeny u 35 pacientů s vrozeným deficitem fibrinogenu zařazených do dvou klinických intervenčních studií a jedné neintervenční postmarketingové studie bezpečnosti. V průběhu těchto studií bylo hlášeno 36 nežádoucích účinků u 13/35 (37,1 %) pacientů, kterým bylo podáno celkem 572 infuzí přípravku Fibclot.
Nejvýznamnější účinky jsou popsány podle klasifikace MedDRA (třída orgánových systémů a preferovaný termín). Četnosti byly určeny na jednu infuzi podle těchto pravidel: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10 000 až <1/1000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). V rámci každé skupiny četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Standardní třída orgánových systémů dle MedDRA |
Nežádoucí účinky |
Četnost na jednu infuzi (N = 572) |
|
Poruchy imunitního systému |
Alergické reakce/reakce anafylaktického typu (včetně anafylaktického šoku, bledosti, zvracení, kašle, sníženého krevního tlaku, třesavky, kopřivky) |
Méně časté |
|
Poruchy nervového systému |
Bolest hlavy Závrať |
Časté Méně časté |
|
Poruchy ucha a labyrintu |
Tinitus |
Méně časté |
|
Cévní poruchy |
Tromboembolické příhody (včetně hluboké žilní trombózy, povrchové tromboflebitidy) (viz bod 4.4) |
Méně časté |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
Erytematózní vyrážka |
Méně časté |
|
Erytém |
Méně časté | |
|
Podráždění kůže |
Méně časté | |
|
Noční pocení |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pocit horka |
Méně časté |
Informace o bezpečnosti vzhledem k přenosným agens viz bod 4.4.
Celkový bezpečnostní profil u pacientů léčených přípravkem Fibclot v jiných klinických situacích vyžadujících terapii fibrinogenem se neliší.
Pediatrická populace:
Z 35 pacientů zařazených do analýzy bezpečnosti u vrozeného deficitu fibrinogenu bylo 14 pacientů ve věku méně než 18 let, z toho 10 pacientů bylo mladších 12 let a 3 byli mladší 6 let.
Celkový bezpečnostní profil se mezi dospělými a pediatrickými pacienty neliší.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: http://www.sukl.cz/nahlasit-nezadouci-ucinek
4.9. Předávkování
Aby se zabránilo předávkování, je indikována pravidelná kontrola hladiny fibrinogenu v plazmě během léčby (viz bod 4.2).
V případě předávkování se zvyšuje riziko rozvoje tromboembolických komplikací.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostatika, lidský fibrinogen ATC kód: B02BB01
Lidský fibrinogen (koagulační faktor I) se v přítomnosti trombinu, aktivovaného koagulačního faktoru XIII (FXIIIa) a vápníkových iontů mění na stabilní a elastickou trojrozměrnou fibrinovou hemostatickou sraženinu.
Podání lidského fibrinogenu vede ke zvýšení hladiny plazmatického fibrinogenu a může dočasně napravit koagulační poruchu u pacientů s deficitem fibrinogenu.
V klinické farmakologické studii (dávka přípravku Fibclot 0,06 g/kg) bylo dosaženo normalizace souhrnných koagulačních testů (např. aktivovaný parciální tromboplastinový čas a protrombinový čas) při hladině fibrinogenu 0,5 g/l a vyšší a tato normalizace trvala alespoň 3 dny.
V rámci všech studií u vrozeného deficitu fibrinogenu byl přípravek Fibclot podán při:
- 100 nechirurgických krvácivých příhodách u 18 pacientů (včetně 17 velkých příhod u 9 pacientů),
- 38 chirurgických zákrocích u 15 pacientů (včetně 10 velkých zákroků u 7 pacientů).
Většinu (92,8 %) příhod (128/138) bylo možné zvládnout pomocí jediné dávky asi 3 g přípravku Fibclot, odpovídající střední dávce na infuzi 0,050 g/kg.
V postmarketingové studii bylo 9 pacientů léčeno v rámci dlouhodobé profylaxe alespoň po dobu 12 měsíců střední dávkou 0,059 g/kg jednou týdně.
Pediatrická populace
Přípravek Fibclot byl v klinických studiích podáván 14 pacientům ve věku méně než 18 let. Střední dávka na jednu infuzi byla 0,059 g/kg k léčbě 26 nechirurgických krvácivých příhod nebo k prevenci nadměrného krvácení během 14 chirurgických zákroků.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Fibclot u pacientů mladších 12 let v souladu s Plánem pediatrického výzkumu (PIP) (informace o použití u dětí viz bod 4.2).
5.2. Farmakokinetické vlastnosti
Biologický poločas fibrinogenu v plazmě je 3-4 dny.
Přípravek se podává intravenózně a je okamžitě dostupný v plazmě v koncentraci odpovídající podané dávce.
V klinické farmakologické studii bylo hodnoceno 14 pacientů po dobu 14 dnů. Po infuzi dávky 0,06 g/kg přípravku Fibclot, bylo maximální koncentrace fibrinogenu dosaženo během 1 hodiny a poté následoval pomalý pokles, přičemž kritické hladiny plazmatického fibrinogenu 0,5 g/l bylo dosaženo za přibližně 3 až 4 dny.
|
Farmakokinetické |
i.v. infuze jedné dávky přípravku Fibclot |
|
parametry |
(geometrický průměr (geometrický CV%)) |
|
Cmax (g/l) |
1,4 (24,4) |
|
t1/2 (h) |
69,3 (21,8) |
|
AUCq-„ (g.h/l) |
114 (23,4) |
|
MRT (h) |
95,6 (20,7) |
|
Cl (ml/h/kg) |
0,53 (22,0) |
|
Vss (ml/kg) |
50,7 (16,7) |
|
IR ((g/l)/(g/kg)) |
23,5 (23,2) |
|
R (%) |
93,6 (20,9) |
Cmax = maximální koncentrace (aktivita) t1/2 = konečný eliminační poločas AUC = plocha pod křivkou
MRT = průměrná doba setrvání přípravku v těle (mean residence time)
Cl = clearance
Vss = distribuční objem v ustáleném stavu IR = přírůstek uzdravení (incremental recovery)
R = in vivo recovery CV = variační koeficient
Pediatrická populace
Nejsou k dispozici žádné farmakokinetické údaje u pediatrických pacientů ve věku < 12 let.
5.3. Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po jedné dávce a po opakovaném podávání a rovněž trombogenicity neodhalily žádné zvláštní riziko pro člověka.
Vzhledem k povaze přípravku nebyly provedeny studie karcinogenity. Studie reprodukce na zvířatech nebyly provedeny, neboť fibrinogen je normální složkou lidského těla.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
Prášek: Arginin-hydrochlorid
Isoleucin
Lysin-hydrochlorid
Glycin
Dihydrát natrium-citrátu Rozpouštědlo: Voda na injekci.
6.2. Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
K intravenóznímu podání rekonstituovaného roztoku při pokojové teplotě se doporučuje použít standardní infuzní set.
6.3. Doba použitelnosti
3 roky.
Chemická a fyzikální stabilita přípravku byla prokázána po dobu 24 hodin při teplotě 25 °C. Z mikrobiologického hlediska se má přípravek použít okamžitě.
6.4. Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Chraňte před mrazem.
Uchovávejte v původním vnějším obalu, aby byl přípravek chráněn před světlem a vlhkostí. Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5. Druh obalu a obsah balení
Jedno balení obsahuje:
- Prášek (1,5 g lidského fibrinogenu) v injekční lahvičce z bezbarvého skla třídy I uzavřené silikonizovanou brombutylovou zátkou, hliníkovým uzávěrem a plastovým víčkem.
- Rozpouštědlo (100 ml vody na injekci) v injekční lahvičce ze skla třídy II uzavřené brombutylovou zátkou, hliníkovým uzávěrem a plastovým víčkem.
- Převodní systém vybavený sterilním filtračním vzduchovým otvorem.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním Rekonstituce:
Řiďte se aktuálními pokyny pro aseptický postup.
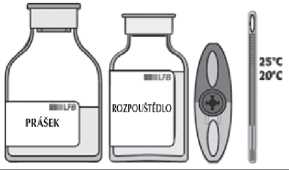
V případě potřeby zahřejte obě injekční lahvičky (s práškem a rozpouštědlem) na pokojovou teplotu.
Sejměte ochranné víčko z injekční lahvičky s rozpouštědlem a z injekční lahvičky s práškem.
Dezinfikujte povrch obou zátek.
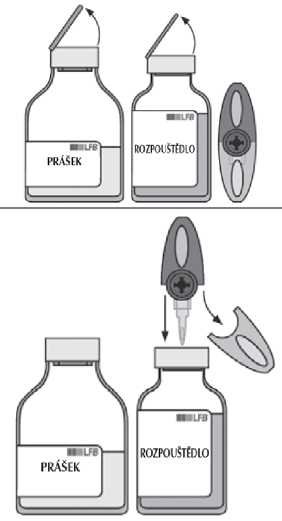
Sejměte průsvitný ochranný kryt z převodního systému a obnažený ostrý hrot zaveďte za současného otáčení zcela skrz střed zátky injekční lahvičky s rozpouštědlem.
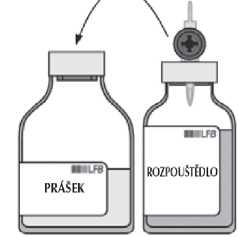
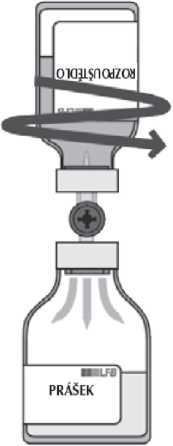
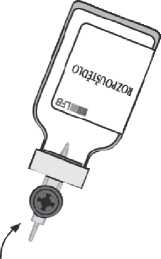

Sejměte šedý ochranný kryt z druhého konce převodního systému. Otočte lahvičku s rozpouštědlem a rychle protlačte volný konec ostrého hrotu skrz střed zátky injekční lahvičky s práškem, aby rozpouštědlo mohlo přetéci do prášku.
Dbejte na to, aby byl hrot neustále ponořen v rozpouštědle, aby se zabránilo předčasnému uvolnění podtlaku.
Během přetékání směrujte krouživým horizontálním pohybem proud rozpouštědla po celém povrchu prášku a podél stěny injekční lahvičky. Ujistěte se, že veškeré rozpouštědlo přeteklo. Podtlak se na konci převádění automaticky uvolní sterilním vzduchem, pronikajícím vzduchovým otvorem převodního systému.
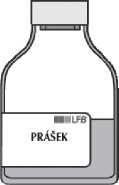
Oddělte prázdnou injekční lahvičku (od rozpouštědla) s převodním systémem.
Několik minut lahvičkou s přípravkem jemně kružte, abyste zabránili vzniku pěny, dokud se prášek zcela nerozpustí.
Rekonstituovaný přípravek se musí před podáním vizuálně zkontrolovat, zda neobsahuje částice. Rekonstituovaný roztok má být téměř bezbarvý, lehce opalizující. Nepoužívejte roztoky, které jsou zakalené nebo obsahují usazeniny.
Podání:
Přípravek Fibclot má být podáván pouze intravenózně, v jedné dávce, ihned po rekonstituci, rychlostí do 4 ml/min.
Pokud se rekonstituovaný roztok ihned nepodá, nemá doba uchovávání přesáhnout 24 hodin při pokojové teplotě (maximálně 25 °C).
Doporučuje se použít infuzní set s nesterilizujícím 15pm filtrem.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Laboratoire Fran^ais du Fractionnement et des Biotechnologies
3 Avenue des Tropiques ZA de Courtaboeuf 91940 Les Ulis Francie
Tel.: + 33 (0)1 69 82 70 10 Fax: + 33 (0)1 69 82 19 03
8. REGISTRAČNÍ ČÍSLO(A)
75/071/16-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 4.5.2016
10. DATUM REVIZE TEXTU
4.5.2016
9