Betafact 50 Iu/Ml
sp.zn.sukls117327/2013
Příbalová informace: informace pro uživatele BETAFACT 50 IU/ml, prášek a rozpouštědlo pro injekční roztok Lidský koagulační faktor IX
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek BETAFACT a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek BETAFACT používat
3. Jak se přípravek BETAFACT používá
4. Možné nežádoucí účinky
5. Jak přípravek BETAFACT uchovávat
6. Obsah balení a další informace
1. Co je přípravek BETAFACT a k čemu se používá
BETAFACT je lék patřící do skupiny antihemoragik . Léčivou látkou je lidský koagulační faktor IX, tedy protein, který je běžně přítomen v lidském těle. Úkolem tohoto proteinu je zajistit normální srážlivost (koagulaci) krve a zabránit příliš dlouhému trvání krvácení.
Přípravek BETAFACT se používá ke kompenzaci nedostatku koagulačního faktoru IX a tím tedy k prevenci a léčbě krvácení (hemoragie) u pacientů s hemofilií B.
Hemofilie B je dědičné onemocnění charakterizované nedostatkem proteinu zvaného koagulační faktor IX. Tento nedostatek způsobuje poruchy srážlivosti.
2. Čemu musíte věnovat pozornost, než začnete přípravek BETAFACT používat Nepoužívejte přípravek BETAFACT
■ jestliže jste alergický(á) na léčivou látku (faktor IX) nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6. „Obsah balení a další informace“),
■ jestliže jste alergický(á) na heparin nebo jeho deriváty,
■ jestliže u Vás v minulosti došlo k poklesu počtu krevních destiček v důsledku užívání léku obsahujícího heparin,
■ jestliže Vám Váš lékař sdělil, že jste alergický(á) na heparin, kontaktujte jej před použitím tohoto léku.
Zvláštní opatrnosti při použití přípravku BETAFACT je zapotřebí
Je nutné, aby Váš lékař zhodnotil možné přínosy léčby přípravkem BETAFACT
Vzhledem k riziku abnormální tvorby sraženin v krvi (tromboembolické komplikace)
■ u pacientů se známkami rozpadu krevních sraženin (fibrinolýza),
■ u pacientů vykazujících tvorbu většího množství sraženin v cirkulující krvi (diseminovaná intravaskulární koagulace),
■ u novorozenců,
■ jestliže jste právě podstoupil(a) operaci,
■ jestliže máte abnormálně vysokou srážlivost krve,
■ jestliže trpíte onemocněním jater.
Váš lékař Vám provede krevní testy, aby bylo možné co nejdříve odhalit známky těchto komplikací. Riziko alergických reakcí
Vzhledem k riziku alergií (viz bod 4. „Možné nežádoucí účinky“) během podávání faktoru IX musí být první injekce přípravku BETAFACT podány pod lékařským dohledem, aby bylo možné v případě potřeby ihned poskytnout protialergickou léčbu.
Váš lékař Vás bude informovat o varovných známkách alergické reakce (viz bod 4. „Možné nežádoucí účinky“). Pokud se kterýkoli z těchto účinků objeví, ihned přerušte léčbu a neprodleně informujte lékaře, který zahájí vhodnou léčbu v závislosti na typu a závažnosti reakce.
Po opakované léčbě přípravkem BETAFACT může Váš imunitní systém reagovat na faktor IX vytvořením inhibitorů (protilátky proti faktoru IX). Přítomnost těchto protilátek může snížit účinnost léčby. Váš lékař Vám musí pravidelně provádět krevní testy, aby mohl kontrolovat přítomnost těchto inhibitorů a měřit jejich množství.
Byla prokázána spojitost mezi přítomností inhibitorů faktoru IX a výskytem alergických reakcí. Proto:
■ Jestliže se u Vás po použití faktoru IX objeví alergické reakce, je nutné provést testy na přítomnost inhibitorů.
■ Jestliže jsou zjištěny inhibitory faktoru IX, existuje vyšší riziko rozvoje závažné alergické reakce během injekčního podání faktoru IX.
BETAFACT obsahuje stopy lidských proteinů jiných než faktor IX. Tyto proteiny by také mohly hrát roli ve výskytu alergické reakce.
Přípravek BETAFACT se vyrábí z lidské plazmy (tekutá složka krve).
Při přípravě léků z lidské krve nebo plazmy se přijímají určitá opatření, která mají zamezit přenosu infekcí na pacienty. Tato opatření zahrnují:
• pečlivý výběr dárců krve a plazmy prostřednictvím lékařského pohovoru, aby se zajistilo vyloučení osob, které představují riziko přenosu infekcí,
• testování jednotlivých odběrů krve a plazmatických poolů na přítomnost známek virových infekcí,
• zařazení kroků do zpracování krve nebo plazmy, které jsou schopny inaktivovat nebo odstranit viry.
Navzdory těmto opatřením nelze při podávání léčivých přípravků připravených z lidské krve nebo plazmy možnost přenosu infekce zcela vyloučit. Totéž platí pro neznámé nebo vznikající viry a jiné typy infekcí.
Přijatá opatření u přípravku BETAFACT se považují za účinná u tzv. obalených virů, jako jsou virus lidské imunitní nedostatečnosti (virus HIV nebo AIDS), virus hepatitidy B a virus hepatitidy C.
Přijatá opatření mohou mít omezenou účinnost proti určitým neobaleným virům, jako jsou virus hepatitidy A a parvovirus B19. Infekce parvovirem B19 může být závažná u těhotných žen (infekce plodu) a u jedinců postižených se sníženou imunitou nebo určitými typy anémie (např. srpkovitá anémie nebo hemolytický anémie).
Váš lékař Vám může doporučit očkování proti hepatitidě A a B, pokud jsou Vám pravidelně/opakovaně podávány přípravky s faktorem IX odvozeným z lidské plazmy.
Rovněž se důrazně doporučuje při každém podání dávky přípravku BETAFACT zaznamenat název a číslo šarže přípravku, aby bylo možné dohledat čísla použitých šarží přípravku.
Děti
Uvedená upozornění a opatření se vztahují jak na dospělé, tak na děti.
Další léčivé přípravky a BETAFACT
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně léků, které jsou dostupné bez lékařského předpisu.
Dosud nebyly pozorovány žádné interakce mezi přípravkem BETAFACT a jinými léčivými přípravky.
Těhotenství a kojení
Použití přípravku BETAFACT nebylo u těhotných nebo kojících žen hodnoceno.
Jestliže jste těhotná nebo kojíte, musí Váš lékař zhodnotit přínosy léčby přípravkem BETAFACT. Potenciální přínos této léčby je nutné porovnat s možnými riziky.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Nic nenasvědčuje tomu, že faktor IX má vliv na schopnost řídit dopravní prostředky nebo obsluhovat stroje.
BETAFACT obsahuje sodík
Tento léčivý přípravek obsahuje přibližně 2,6 mg sodíku na 1 ml přípravku (13 mg v 5ml injekční lahvičce, 26 mg v 10ml injekční lahvičce, 52 mg ve 20ml injekční lahvičce). Tuto skutečnost je nutné brát v úvahu, jestliže jste na dietě s omezeným nebo zcela zakázaným příjmem soli.
BETAFACT obsahuje heparin
Tento léčivý přípravek může způsobovat alergické reakce a pokles počtu krvinek, což může mít vliv na srážlivost krve.
3. Jak se přípravek BETAFACT používá
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Dávka
Váš lékař Vám určí vhodnou dávku přípravku BETAFACT.
Vhodná dávka a četnost podávání závisí na následujících faktorech:
■ Vaše tělesná hmotnost,
■ závažnost vaší hemofilie,
■ místo a rozsah krvácení,
■ Váš zdravotní stav,
■ a v určitých případech také operační výkon, který se chystáte podstoupit (např. chirurgický zákrok, extrakce (vytržení) zubu atd.).
Váš lékař Vám doporučí, abyste během léčby podstoupil(a) krevní testy, aby bylo možné kontrolovat:
■ hladiny faktoru IX,
■ přítomnost inhibitorů faktoru IX.
Na základě výsledků těchto testů může Váš lékař rozhodnout o úpravě dávky a četnosti podávání injekcí.
Vhodná dávka je vyjádřena v mezinárodních jednotkách (IU).
Četnost podání
Váš lékař Vám upraví četnost injekcí podle závažnosti Vašeho krvácení a účinnosti léčby.
Tabulka popisující četnost injekcí a dobu trvání léčby v různých situacích je uvedena na konci této příbalové informace v bodě určeném pro zdravotnické pracovníky.
Způsob a cesta podání
Tento lék se podává do žíly ve formě infuze.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Jestliže jste použil(a) více přípravku BETAFACT, než jste měl(a)
Ihned kontaktujte svého lékaře nebo lékárníka.
U lidského koagulačního faktoru IX však nebyl zaznamenán žádný případ předávkování.
Jestliže jste zapomněl(a) použít přípravek BETAFACT
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento léčivý přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Riziko alergických reakcí
• Reakce přecitlivělosti nebo alergické reakce se mohou vyskytnout vzácně. V některých případech tyto reakce přešly v závažnou alergickou reakci.
• Alergické reakce se mohou vyskytnout společně s rozvojem inhibitoru faktoru IX a mohou ovlivnit funkci ledvin (viz také bod 2. „Riziko alergických reakcí“).
Varovné signály alergických reakcí jsou:
• otok tváře a krku,
• pocity pálení a brnění v místě aplikace injekce,
• zimnice,
• zarudnutí,
• svědění a vyrážka,
• nízký krevní tlak,
• extrémní únava (letargie),
• pocit na zvracení (nauzea), zvracení,
• neklid,
• zrychlená srdeční frekvence,
• tíseň na hrudi,
• mravenčení,
• sípání (astmatického charakteru).
Pokud nastane kterýkoliv z těchto účinků, okamžitě léčbu ukončete a upozorněte lékaře, aby zahájil vhodnou léčbu v závislosti na typu a závažnosti reakce._
V klinických studiích přípravku Betafact byly přímo pozorovány následující nežádoucí účinky a mohou se objevit vzácně (mohou ovlivnit až 1 z 1 000 injekcí):
• reakce přecitlivělosti a alergické reakce (viz také bod 2 a 4 „Riziko alergických reakcí“),
• bolest hlavy,
• svědění,
• alergický otok,
• pocit na zvracení (nauzea),
• reakce na injekci (nevolnost, bolest na hrudi),
• reakce v místě vpichu.
Během postmarketingového období používání byly u přípravku BETAFACT hlášeny 2 případy aktivitu neutralizujících protilátek (inhibitorů) u jednoho dříve neléčeného pacienta a u jednoho dříve léčeného pacienta.
Následující nežádoucí účinky nebyly pozorovány v klinických studiích přípravku Betafact, ale byly pozorovány u pacientů užívajících stejnou skupinu léčiv jako Betafact:
Krevní sraženiny
Krevní sraženiny se mohou vyskytnout při používání přípravků faktoru IX s nízkou čistotou. Mohou:
• blokovat dodávku krve a kyslíku do srdce a způsobit srdeční příhodu.
• blokovat dodávku krve a kyslíku do plic a vést k závažnému zdravotnímu stavu zvanému plicní embolie,
• způsobit sraženinu v žíle (žilní trombóza),
• způsobit krevní sraženiny uvnitř cév v celém těle (diseminovaná intravaskulární koagulace). Betafact je faktor IX vysoké čistoty a vzácně je spojován s tímto typem účinku.
Rozvoj inhibitorů
• U pacientů užívajících přípravky s faktorem IX se mohou rozvinout protilátky proti faktoru IX (tzv. inhibitory - viz bod 2. „Riziko alergických reakcí“).
• Tyto inhibitory nebyly pozorovány v klinických studiích prováděných s přípravkem Betafact u 11 dříve neléčených pacientů.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek BETAFACT uchovávat
Uchovávejte tento léčivý přípravek mimo dohled a dosah dětí.
Nepoužívejte tento léčivý přípravek po uplynutí doby použitelnosti uvedené na štítku injekční lahvičky a na krabičce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek by měl být po rekonstituci okamžitě použit.
Pro účely ambulantního použití může být před otevřením přípravek z chladničky vyjmut, bez náhrady, maximálně na dobu 6 měsíců a uchováván při teplotě do 25 °C.
Na vnější obal by se mělo zapsat datum, kdy se léčivý přípravek vyjme, a nová doba použitelnosti. Tato nová doba použitelnosti by nikdy neměla překročit dobu původně uvedenou na vnější krabičce. Pokud nebyl léčivý přípravek použit před novou dobou použitelnosti, měl by se zlikvidovat.
Nepoužívejte tento přípravek, pokud si všimnete, že je roztok zakalený nebo obsahuje usazeniny.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte likvidovat přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek BETAFACT obsahuje
Léčivou látkou je Factor IX coagulationis sang. humani cryod. (lidský koagulační faktor IX lyofilizovaný) v koncentraci 50 IU/ml po rekonstituci. Po rekonstituci 5, 10 nebo 20 ml vody na injekci jedna injekční lahvička obsahuje 250 IU/5 ml, 500 IU/10 ml, respektive 1000 IU/20 ml lidského koagulačního faktoru IX.
Specifická aktivita přípravku BETAFACT je přibližně 110 IU/mg celkového proteinu.
Dalšími složkami jsou:
chlorid sodný, sodná sůl heparinu, lysin-hydrochlorid, arginin, dihydrát natrium-citrátu a voda na injekci (viz bod 2. „Čemu musíte věnovat pozornost, než začnete přípravek BETAFACT používat“)
Jak přípravek BETAFACT vypadá a co obsahuje toto balení
Přípravek BETAFACT se dodává ve formě prášku pro přípravu injekčního roztoku s rozpouštědlem ve skleněných injekčních lahvičkách, společně s převodní soupravou a filtrační jehlou. Přípravek BETAFACT je dostupný v následujících baleních: 250 IU/5 ml, 500 IU/10 ml a 1000 IU/20 ml.
Držitel rozhodnutí o registraci
LFB-BIOMEDICAMENTS
3, avenue des Tropiques BP 305 - Les Ulis 91958 Courtaboeuf Cedex FRANCIE
Tento léčivý přípravek je v členských státech EHP registrován pod těmito názvy:
Rakousko: BETAFACT 50 I.E./ml Česká republika: BETAFACT 50 IU/ml Francie: BETAFACT 50 UI/ml Německo: BETAFACT 250 I.E./ 500 I.E./1000 I.E.
Řecko: BETAFACT 50 IU/ml Maďarsko: BETAFACT 50 NE/ml Polsko: BETAFACT 250 IU/500 IU/1000 IU Portugalsko: BETAFACT 50 UI/ml
Rumunsko: BETAFACT 50 UI/ml, pulbere si solvent pentru solutie injectabila
Slovenská republika: BETAFACT 50 IU/ml
Španělsko: BETAFACT 50 IU/ml
Nizozemsko: BETAFACT 50 I.E/ml
Velká Británie: BETAFACT 50 IU/ml
Tato příbalová informace byla naposledy revidována 14.5.2014.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Vhodná dávka je vyjádřena v mezinárodních jednotkách (IU) a vypočítá se podle následujícího vzorce:
počet jednotek, které mají být podány = tělesná hmotnost (kg) x cílový nárůst hladin faktoru IX (%) (IU/dl) x 0,93
Dávkování
U všech níže uvedených krvácivých příhod by neměla aktivita faktoru IX klesnout v daném období pod uvedenou hladinu. Tuto tabulku lze použít pro určení účinného dávkování u uvedených krvácivých příhod a chirurgických zákroků:
|
Intenzita krvácení / typ chirurgického zákroku |
Cílová hladina faktoru IX v plazmě (%) (IU/dl) |
Frekvence injekcí (hodiny) / doba trvání léčby (dny) |
|
Krvácení: | ||
|
Časná hemartróza, krvácení do svalů nebo dutiny ústní. |
20 až 40 |
Opakujte každých 24 hodin nejméně po dobu 1 dne, dokud neodezní bolest nebo se nezastaví krvácení. |
|
Rozsáhlá hemartróza, krvácení do svalů nebo hematom. |
30 až 60 |
Opakujte injekce každých 24 hodin po dobu 3-4 dnů nebo déle, dokud se neobnoví plný rozsah pohybů a neodezní bolest. |
|
Život ohrožující krvácení. |
60 až 100 |
Opakujte injekce každých 8 až 24 hodin, dokud není krvácení pod kontrolou. |
|
Chirurgický zákrok: | ||
|
Menší zákrok včetně extrakce zubu |
30 až 60 |
Opakujte každých 24 hodin nejméně po dobu 1 dne, dokud se nezastaví krvácení. |
|
Větší zákrok |
80 až 100 (před operací a po operaci) |
Opakujte injekce každých 8 až 24 hodin, dokud se nedosáhne adekvátního zhojení rány, a poté pokračujte v léčbě nejméně dalších 7 dní, abyste udrželi aktivitu faktoru IX mezi 30 % a 60 % (IU/dl). |
Za určitých okolností mohou být potřebná větší než vypočtená množství přípravku BETAFACT, především v případě počáteční dávky.
V průběhu léčby se jako vodítko k určení požadované dávky a frekvence opakování infuzí doporučuje sledování hladin faktoru IX. Především v případě větších chirurgických zákroků je přesné sledování substituční léčby pomocí koagulační analýzy (aktivity faktoru IX v plazmě) nezbytné. Odpovědi jednotlivých pacientů na faktor IX se mohou lišit různými poločasy a zotavením.
Pro dlouhodobou profylaxi u pacientů se závažnou hemofilií B by se měly podávat dávky od 20 do 40 IU přípravku BETAFACT na kilogram tělesné hmotnosti každé 3 až 4 dny.
V určitých případech, především u mladších pacientů, může být nutné zvýšení dávky nebo zkrácení intervalu mezi dvěma injekcemi.
U pacientů je nutné sledovat vývoj inhibitorů faktoru IX. Pokud není dosaženo očekávaných hladin aktivity faktoru IX v plazmě, nebo pokud se po příslušné dávce krvácení nezastaví, je nutné provést test na přítomnost inhibitoru faktoru IX. Pokud je hladina přítomného inhibitoru nižší než 10 Bethesda jednotek (BU) na 1 ml, může podání dalšího lidského koagulačního faktoru IX inhibitor zneutralizovat. U pacientů s titry inhibitoru přesahujícími 10 BU nebo s vysokou anamnestickou odpovědí je nutné zvážit použití (aktivovaného) koncentrátu protrombinového komplexu (aPCC) nebo aktivovaného faktoru VII (FVIIa). Takovou léčbu by měl vést lékař se zkušeností s péčí o pacienty s hemofilií.
V klinických studiích bylo 11 dětí mladších 6 let léčeno přípravkem betafact a jejich dávkování bylo podobné dávkám podávaným dospělým pacientům.
Rekonstituce:
Rekonstituci přípravku proveďte vodou na injekci podle níže uvedených pokynů. Postupujte podle aktuálních předpisů pro aseptickou proceduru.



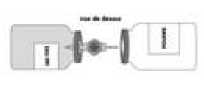


• Pokud je to nutné, zahřejte obě injekční lahvičky (prášek a rozpouštědlo) na pokojovou teplotu.
• Odstraňte z injekční lahvičky s rozpouštědlem (vodou na injekci) a z injekční lahvičky s práškem ochranné víčko.
• Dezinfikujte povrch obou zátek.
• Odstraňte průhledný ochranný kryt z převodní soupravy a odkrytou jehlu za jejího současného otáčení kompletně vpíchněte středem zátky do injekční lahvičky s rozpouštědlem.
• Odstraňte druhý ochranný kryt z druhého konce převodní soupravy.
• Udržujte obě injekční lahvičky v horizontální poloze (hrot s průduchem směřuj te nahoru) a rychle vpíchněte volný konec převodní jehly do středu
zátky injekční lahvičky s práškem.
• Ujistěte se, že je jehla neustále ponořená v rozpouštědle, aby nedošlo k předčasnému uvolnění vakua.
• Ihned umístěte soupravu do vertikální polohy, přičemž držte injekční lahvičku s rozpouštědlem přímo nad injekční lahvičkou s práškem, aby mohlo rozpouštědlo přejít do prášku.
• Během převodu se snažte směrovat proud rozpouštědla po celém povrchu prášku. Ujistěte se, že došlo k převodu veškerého rozpouštědla.
• Na konci převodu se vakuum automaticky uvolní (sterilní vzduch).
• Odpojte prázdnou injekční lahvičku (od rozpouštědla) od převodní
soupravy.
• Aby se zamezilo tvorbě pěny, je nutné plnou injekční lahvičkou několik minut lehce kroužit do úplného rozpuštění prášku.
K rozpouštění prášku většinou dochází okamžitě a do 5 minut by měl být kompletně rozpuštěn. Roztok musí být čirý.
Nepoužívejte roztok, který je zakalený nebo obsahuje usazeniny.
Nemíchejte přípravek s jinými léčivými přípravky.
Rekonstituovaný přípravek dále neřeďte.
Podání:
Natáhněte přípravek do sterilní injekční stříkačky prostřednictvím dodané filtrační jehly.
Odpojte jehlu od injekční stříkačky.
Připojte injekční stříkačku k intravenózní jehle, vytlačte z injekční stříkačky vzduch, vydezinfikujte pokožku nad žílou a vpíchněte jehlu.
Aplikaci injekcí provádějte intravenózně v jedné dávce ihned po rekonstituci a s maximální rychlostí podávání 4 ml/min.
Uchovávání po rekonstituci:
Z důvodu zachování sterility má být přípravek použit okamžitě. Nicméně, chemická a fyzikální stabilita po rekonstituci před použitím byla prokázána na dobu 3 hodin při 25 °C.
12/12