Berinert 1500 Iu
sp. zn. sukls222991/2013
Souhrn údajů o přípravku
1. NÁZEV PŘÍPRAVKU
Berinert 500 IU
Prášek a rozpouštědlo pro injekční/infuzní roztok.
Berinert 1500 IU
Prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Léčivá látka: antiesterasum-C1 (humanum)
Berinert 500 IU obsahuje 500 IU v jedné injekční lahvičce.
Berinert 1500 IU obsahuje 1500 IU v jedné injekční lahvičce.
Účinnost inhibitoru C1-esterázy je vyjádřená v mezinárodních jednotkách (IU), které se vztahují k aktuálnímu WHO standardu pro přípravky s inhibitorem C1-esterázy.
Berinert 500 IU obsahuje 50 IU/ml inhibitoru C1-esterázy po rekonstituci s 10 ml vody na injekci. Berinert 1500 IU obsahuje 500 IU/ml inhibitoru C1-esterázy po rekonstituci s 3 ml vody na injekci.
Celkový obsah bílkovin v 500 IU roztoku po rekonstituci je 6,5 mg/ml.
Celkový obsah bílkovin v 1500 IU roztoku po rekonstituci je 65 mg/ml.
Pomocné látky se známým účinkem:
100 ml roztoku obsahuje až 486 mg sodíku (přibližně 21 mmol).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Berinert 500 IU:
Prášek a rozpouštědlo pro injekční/infuzní roztok. Berinert 1500 IU:
Prášek a rozpouštědlo pro injekční roztok.
Bílý prášek
4. KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
Dědičný angioedém typu I a II (DAE)
Léčba a předoperační prevence akutních příhod.
4.2. Dávkování a způsob podání
Léčba má být započata pod dohledem lékaře, který má zkušenosti s léčbou deficitu inhibitoru C1-esterázy.
Dávkování
Dospělí
Léčba akutních záchvatů angioedému:
20 IU na kg tělesné hmotnosti (20 IU/kg těl. hm.).
Předoperační prevence záchvatů angioedému:
1000 IU méně než 6 hodin před lékařským, stomatologickým nebo chirurgickým zákrokem.
Pediatrická populace
Léčba akutních záchvatů angioedému:
20 IU na kilogram tělesné hmotnosti (20 IU / kg těl.hm.).
Předoperační prevence záchvatů angioedému:
15 až 30 IU na kilogram tělesné hmotnosti (15-30 IU / kg těl.hm.) méně než 6 hodin před lékařským, stomatologickým nebo chirurgickým zákrokem. Dávka má být zvolena s ohledem na klinické okolnosti (např. typ zákroku a závažnosti onemocnění).
Způsob podání
Berinert se rekonstituuje podle návodu uvedeného v bodě 6.6. Roztok po rekonstituci pro
- Berinert 500 IU musí být bezbarvý a čirý,
- Berinert 1500 IU musí být bezbarvý a čirý až slabě opalescentní.
Roztok se podává pomalu i.v. injekcí. Berinert 500 IU se může také podávat jako infuze (4 ml/min.).
4.3. Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4. Zvláštní upozornění a opatření pro použití
U pacientů se známou tendencí ke vzniku alergií mají být profylakticky podávána antihistaminika a kortikosteroidy.
Pokud se vyskytne alergická nebo anafylaktická reakce, podávání přípravku Berinert musí být okamžitě zastaveno (např. přerušením injekce/infuze) a zahájena vhodná léčba. Léčebná opatření závisí na typu a závažnosti nežádoucího účinku. V případě šoku je třeba se řídit standardními lékařskými postupy pro léčbu šoku.
Pacienti s laryngeálním edémem vyžadují obzvláště pozorné sledování a léčebnou pohotovost pro naléhavé stavy.
Nepovolené použití nebo léčba „capillary-leak“ syndromu (CLS) pomocí přípravku Berinert se nedoporučuje (viz také bod 4.8 Nežádoucí účinky).
Berinert obsahuje až 486 mg sodíku (přibližně 21 mmol) na 100 ml roztoku. Toto by měli vzít v úvahu pacienti, kteří jsou na kontrolované sodíkové dietě.
Domácí léčba a autoaplikace
K dispozici jsou omezené údaje o použití tohoto léčivého přípravku v domácí léčbě nebo autoaplikace. Potenciální rizika spojená s domácí léčbou, jsou spojená s vlastním podáním, stejně jako zvládnutí nežádoucích účinků, zejména hypersenzitivity. Rozhodnutí o využití domácí léčby u každého jednotlivého pacienta má být učiněno ošetřujícím lékařem, který má zajistit příslušné školení a v pravidelných intervalech prověřovat používání injekční techniky.
Virová bezpečnost
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a zařazení efektivních výrobních postupů k inaktivaci/eliminaci virů. Přes všechna tato opatření při podávání léčivých přípravků vyráběných z lidské krve nebo plazmy, nelze zcela vyloučit možnost přenosu infekčních agens. To se týká také neznámých nebo nově vznikajících virů a jiných patogenů.
Přijatá opatření jsou považována za účinná proti obaleným virům jako je HIV, HBV a HCV a u neobalených virů HAV a parvovirus B19.
Doporučuje se zvážit vhodné očkování (hepatitida A a B) u těch pacientů, kteří pravidelně/opakovaně dostávají přípravky vyrobené z lidské plazmy.
Důrazně se doporučuje, aby vždy při každém podání přípravku Berinert pacientovi byl zaznamenán název a číslo šarže použitého přípravku k zajištění možnosti dohledání spojení mezi pacientem a šarží přípravku.
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Neprováděly se žádné studie interakcí.
4.6. Fertilita, těhotenství a kojení Těhotenství
K dispozici je omezené množství údajů, které neukazují zvýšené riziko plynoucí z použití přípravku Berinert u těhotných žen. Léčivá látka přípravku Berinert je fyziologickou složkou lidské plazmy. Proto nebyly prováděny žádné studie reprodukční nebo vývojové toxicity na zvířatech a nejsou očekávány žádné nežádoucí účinky na fertilitu, prenatální a postnatální vývoj u lidí. Z těchto důvodů má být Berinert podáván těhotným ženám pouze pokud jsou indikace zcela jasné.
Kojení
Není známé, jestli se Berinert vylučuje do mateřského mléka, ale vzhledem k jeho vysoké molekulární hmotnosti se přenos přípravku Berinert do mateřského mléka jeví jako nepravděpodobný. Nicméně kojení u žen trpících dědičným angioedémem je sporné. Musí se zvážit, zda přerušit kojení nebo přerušit léčbu přípravkem Berinert s ohledem na prospěch kojení pro dítě a prospěch léčby pro matku.
Fertilita
Léčivá látka přípravku Berinert je fyziologickou složkou lidské plazmy. Proto nebyly provedeny žádné studie reprodukce a vývojové toxicity na zvířatech a neočekávají se žádné nežádoucí účinky na fertilitu, pre- a postnatální vývoj u člověka.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Berinert nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8. Nežádoucí účinky
Následující nežádoucí účinky jsou založeny na posmarketingových zkušenostech a na údajích z odborné literatury. Byly použity následující standardní kategorie četnosti:
> 1/10
Velmi časté Časté
Méně časté Vzácné Velmi vzácné
> 1/100 až < 1/10
> 1/1000 až < 1/100
> 1/10000 až < 1/1000
< 1/10000 (včetně hlášených jednotlivých případů)
Nežádoucí účinky při podávání přípravku Berinert jsou vzácné.
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
|
Cévní poruchy |
Vývoj trombózy* | ||||
|
Celkové poruchy a reakce v místě aplikace |
Zvýšená teplota, reakce v místě aplikace | ||||
|
Poruchy imunitního systému |
Alergické nebo anafylaktické reakce (např. tachykardie, hyper-nebo hypotenze, zarudnutí vyrážka, dušnost, bolest hlavy, závratě, nauzea |
Šok |
* Při pokusném podání vysokých dávek přípravku Berinert na profylaxi nebo léčbu „capillary-leak“ syndromu (CLS) před, během nebo po operaci srdce s mimotělním krevním oběhem (nepovolená indikace a dávka), v jednotlivých případech s následkem smrti.
Informace o bezpečnosti s ohledem na přenos infekčních agens viz bod 4.4
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9. Předávkování
Nebyl hlášen žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva úžívaná u hereditárního angioedému, C1 inhibitor, získaný z plazmy
ATC kód: B06AC01
Inhibitor C1-esterázy je plazmatický glykoprotein s molekulovou hmotností 105 kDa a s obsahem uhlohydrátové složky 40 %. Jeho koncentrace v lidské plazmě je přibližně 240 mg/l. Kromě lidské plazmy je inhibitor C1-esterázy obsažen také v placentě, jatemích buňkách, monocytech a trombocytech.
Inhibitor C1-esterázy patří do systému inhibitorů serinových proteáz (serpin) lidské plazmy spolu s jinými proteiny jako je antitrombin III, alfa-2-antiplazmin, a alfa-1-antitrypsin a další.
Za fyziologických podmínek inhibitor C1-esterázy blokuje klasickou cestu systému komplementu tím, že inaktivuje enzymatické aktivní složky C1s a C1r. Aktivní enzym vytváří komplex s inhibitorem ve stechiometrickém poměru 1:1.
Inhibitor C1-esterázy představuje také nejdůležitější inhibitor kontaktní aktivace koagulace tím, že inhibuje faktor XIIa a jeho fragmenty. Navíc slouží, kromě alfa-2-makroglobulinu, jako hlavní inhibitor plazmatického kalikreinu.
Léčebný efekt přípravku Berinert u dědičného angioedému spočívá v substituci chybějící aktivity inhibitoru C1-esterázy.
5.2. Farmakokinetické vlastnosti
Přípravek se podává intravenózně a je okamžitě dostupný v plazmě v koncentraci odpovídající podané dávce.
Farmakokinetické vlastnosti přípravku Berinert byly sledovány ve dvou studiích.
Klinická studie fáze I provedená u 15 zdravých dospělých osob poskytla farmakokinetické (PK) údaje, které byly použity k posouzení relativní biologické dostupnosti přípravků Berinert 1500 IU a Berinert 500 IU. Byla prokázána srovnatelná biologická dostupnost pro obě prezentace přípravku Berinert. Pro C1-INH antigen byly koncentrace Cmax a AUC0_last odchylky geometrického průměru (90% CIs) 1,02 (0,99-1,04) a 1,02 (0,99-1,05). Biologický poločas byl odhadnut u subjektů pomocí non-kompartmentové PK analýzy. Průměrný biologický poločas přípravku Berinert 1500 IU byl 87,7 hodiny a přípravku Berinert 500 IU 91,4 hodiny.
Farmakokinetické vlastnosti byly sledovány u pacientů s dědičným angioedémem (34 pacientů ve věku > 18 let, 6 pacientů <18 let). Z těchto pacientů bylo 15 pacientů na profylaktické léčbě (s častými/závažnými ataky) a 25 pacientům s méně častými/mírnými ataky se přípravek podával v případě potřeby. Tyto údaje se získávaly v období mezi ataky.
Střední in vivo recovery (IVR) byl 86,7 % (rozsah: 54,0 - 254,1 %). IVR u dětí byl nepatrně vyšší (98,2 %, rozsah: 69,2 - 106,8 %) než u dospělých (82,5 %, rozsah: 54,0 - 254,1 %). Pacienti se závažnými ataky měli vyšší IVR (101,4 %) v porovnání s pacienty s mírnými ataky (75,8 %, rozsah: 57,2 - 195,9 %).
Střední zvýšení aktivity bylo 2,3 %/ IU/kg tělesné hmotnosti (rozsah: 1,4 - 6,9 %/IU/kg tělesné hm.). Nebyl pozorován žádný významný rozdíl mezi dospělými a dětmi. Pacienti se závažnými ataky vykazovali mírně vyšší zvýšení aktivity než pacienti s mírnými ataky (2,9, rozsah: 1,4 - 6,9 oproti 2,1, rozsah: 1,5 - 5,1 %/IU/kg těl. hm.).
Maximální koncentrace aktivity inhibitoru C1-esterázy v plazmě byla dosažena 0,8 hodiny po podání přípravku Berinert bez významných rozdílů mezi jednotlivými skupinami pacientů.
Střední biologický poločas byl 36,1 hodiny. Tento poločas byl o něco kratší u dětí než u dospělých (32,9 oproti 36,1 hodiny) a u pacientů se závažnými ataky než u pacientů s mírnými ataky (30,9 oproti 37,0).
5.3. Předklinické údaje vztahující se k bezpečnosti
Léčivou látkou přípravku Berinert je inhibitor C1-esterázy. Je vyrobený z lidské plazmy a působí jako endogenní složka plazmy. Při jednorázovém podání přípravku Berinert potkanům a myším a při opakovaném podávání potkanům se neprokázala toxicita.
Předklinické studie s opakovaně aplikovanými dávkami na vyšetření kancerogenity a reprodukční toxicity nemohly být provedeny na konvenčních živočišných modelech vzhledem ke vzniku protilátek, které vznikají po podání heterologní lidské bílkoviny.
In vitro Ouchterlonyho test a in vivo PCA model na morčatech neprokázaly existenci nově vzniklých antigenních determinantů u pasterizovaného přípravku Berinert.
Testy in-vivo na trombogenicitu byly provedeny u králíků s dávkami přípravku Berinert až 800 IU/kg. Testy neprokázaly žádné pro-trombotické riziko spojené s i.v. podáním přípravku Berinert až do dávky 800 IU/kg.
Studie lokální snášenlivosti na králících prokázaly, že přípravek Berinert byl po intravenózní, subkutánní, intraarteriální a intramuskulární aplikaci klinicky, lokálně a histologicky dobře tolerován.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
Prášek:
Glycin
Chlorid sodný Dihydrát natrium-citrátu
Rozpouštědlo:
Voda na injekci
6.2. Inkompatibility
Vzhledem k absenci studií kompatibility se tento léčivý přípravek nesmí mísit s jinými léčivými přípravky a rozpouštědly v injekční stříkačce/infuzní soupravě.
6.3. Doba použitelnosti
Berinert 500 IU: 30 měsíců Berinert 1500 IU: 36 měsíců
Fyzikálně-chemická stabilita po rekonstituci byla prokázána po dobu 48 hodin při pokojové teplotě (max. 25 °C). Z mikrobiologického hlediska, vzhledem k tomu, že Berinert neobsahuje žádné konzervační látky, se má přípravek po rekonstituci podat okamžitě. Pokud se přípravek nepodá okamžitě, jeho uchovávání nemá překročit 8 hodin při pokojové teplotě.
Rekonstituovaný přípravek musí být uchováván pouze v injekční lahvičce.
6.4. Zvláštní opatření pro uchovávání
Berinert 500 IU: Uchovávejte při teplotě do 25 °C.
Berinert 1500 IU: Uchovávejte při teplotě do 30 °C.
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5. Druh obalu a obsah balení
Berinert 500 IU: Prášek (500 IU) v injekční lahvičce (sklo třídy II) se zátkou
(brombutylová pryž), víčkem (hliník) a odklápěcím krytem (plast).
10 ml rozpouštědla v injekční lahvičce (sklo třídy I) se zátkou (chlorbutylová pryž), víčkem (hliník) a odklápěcím krytem (plast).
Berinert 1500 IU: Prášek (1500 IU) v injekční lahvičce (sklo třídy I) se zátkou
(brombutylová pryž), víčkem (hliník) a odklápěcím krytem (plast).
3 ml rozpouštědla v injekční lahvičce (sklo třídy I) se zátkou (chlorbutylová pryž), víčkem (hliník) a odklápěcím krytem (plast).
Aplikační souprava:
1 přepouštěcí adaptér s filtrem 20/20, 1 jednorázová injekční stříkačka (Berinert 500 IU: 10 ml, Berinert 1500 IU: 5 ml), 1 venepunkční set, 2 tampony s alkoholem, 1 náplast.
Velikost balení: 1.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Způsob podání
Všeobecné pokyny:
- Roztok pro přípravek Berinert 500 IU má být bezbarvý a čirý.
- Roztok pro přípravek Berinert 1500 IU má být bezbarvý a čirý až slabě opalescentní
- Po filtraci/nasátí (viz níže) má být rekonstituovaný přípravek před podáním zkontrolován vizuálně, nesmí obsahovat částice a musí být bezbarvý.
- Nepoužívejte roztoky, které jsou zakalené nebo obsahují částice.
- Rekonstituce a nasátí musí být provedeno za aseptických podmínek. Použijte injekční stříkačku dodávanou s přípravkem.
Rekonstituce
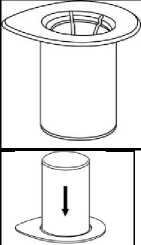
Zahřejte rozpouštědlo na pokojovou teplotu. Sejměte vyklápěcí víčka z lahviček s práškem i s rozpouštědlem, očistěte pryžové zátky antiseptickým roztokem a nechte je oschnout než otevřete balení Mix2Vial.
1

2
1. Otevřete balení Mix2Vial tím, že vyklopíte víčko. Nevytahujte Mix2Vial z blistru!
2. Postavte injekční lahvičku s rozpouštědlem na rovný a čistý povrch, držte ji pevně. Uchopte Mix2Vial společně s blistrem a zatlačte hrot modrého konce adaptéru rovně dolů skrz pryžovou zátku injekční lahvičky s rozpouštědlem.
3
4

5
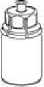

6
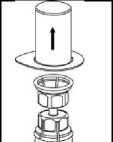
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle vzhůru. Přesvědčte se, že jste vytáhli jen blistrový obal a ne soupravu Mix2Vial.
4. Postavte injekční lahvičku s přípravkem na
rovný a pevný povrch. Obraťte injekční lahvičku s rozpouštědlem spolu s nasazenou soupravou
Mix2Vial a zatlačte hrot průhledného konce
adaptéru přímo dolů skrz pryžovou zátku injekční lahvičky s přípravkem. Rozpouštědlo se samo automaticky nasaje do injekční lahvičky
s přípravkem.
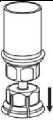
5. Uchopte jednou rukou tu část soupravy
Mix2Vial, kde je injekční lahvička s přípravkem a druhou rukou tu část, kde je injekční lahvička od rozpouštědla a odšroubujte je od sebe opatrně na dvě části. Odstraňte injekční lahvičku od rozpouštědla s připojeným modrým adaptérem
soupravy Mix2Vial.
6. Jemně otáčejte injekční lahvičkou s přípravkem s připojeným průhledným adaptérem, dokud se prášek zcela nerozpustí. Netřepejte s ní.
1
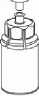
7
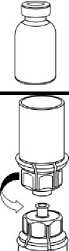
7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Použijte injekční stříkačku dodávanou s přípravkem. Zatímco je injekční lahvička s přípravkem dnem dolů, spojte injekční stříkačku s koncovkou Luer Lock soupravy Mix2Vial a vstříkněte vzduch do injekční lahvičky s přípravkem.
Natáhnutí a aplikace

8
8. Zatímco držíte píst injekční stříkačky stlačený, obraťte celý systém dnem vzhůru. Pomalým vytahováním pístu natáhněte roztok do injekční stříkačky.
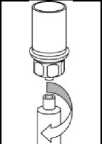
9
9. Po natažení roztoku do injekční stříkačky uchopte pevně válec injekční stříkačky (píst stále směřuje dolů) a odpojte průhledný adaptér soupravy Mix2Vial od injekční stříkačky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH Emil-von-Behring-Strasse 76 35041 Marburg Německo
8. REGISTRAČNÍ ČÍSLO(A)
Berinert 500 IU: 16/5 92/09-C Berinert 1500 IU: 16/267/15-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Berinert 500 IU:
Datum první registrace: 29.7.2009
Datum posledního prodloužení registrace: 23.10.2013
Berinert 1500 IU:
Datum první registrace: 27.5.2015
10.DATUM REVIZE TEXTU
27.5.2015
9