Atrovent N
sp.zn. sukls218984/2015 a sp.zn. sukls175903/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU Atrovent N
roztok k inhalaci v tlakovém obalu
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna odměřená dávka obsahuje ipratropii bromidům 0,020 mg (ve formě ipratropii bromidum monohydricum 0,021 mg).
Pomocná látka se známým účinkem: jedna odměřená dávka obsahuje až 8,415 mg bezvodého ethanolu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA Roztok k inhalaci v tlakovém obalu
Popis přípravku: čirý, bezbarvý nebo téměř bezbarvý roztok s charakteristickou vůní po ethanolu, bez viditelných částic.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Atrovent N je bronchodilatační přípravek a je určen k udržovací léčbě bronchospasmu spojeného s chronickou obstrukční plicní nemocí, včetně chronické bronchitidy, emfyzému a astmatu.
4.2 Dávkování a způsob podání
Dávkování
Dávkování musí být přizpůsobeno individuální potřebě pacienta a pacient musí být během léčby sledován lékařem. Nedoporučuje se překračovat doporučenou denní dávku ani při akutním ani při chronickém podávání.
Jestliže léčba nevede k významnému zlepšení nebo pokud se stav pacienta zhoršuje, je třeba vyhledat lékařskou pomoc a změnit způsob léčby. Pacient by měl být poučen, že v případě akutní a rychle se zhoršující dyspnoe musí být okamžitě vyhledána lékařská pomoc.
Doporučeny jsou následující dávky:
Udržovací léčba:
Dospělí a děti od 6 let:
2 odměřené dávky (vdechy) 4x denně
V případě potřeby vyšších dávek může být počet dávek zvýšen, maximální denní dávka 12 vdechů by však neměla být překročena.
Při akutním zhoršení chronické obstrukční plicní nemoci (CHOPN) se doporučuje použít přípravek Atrovent ve formě roztoku k inhalaci (roztoku k rozprašování pomocí nebulizačního zařízení).
Protože neexistuje dostatek údajů o podávání přípravku dětem, měl by být u této skupiny Atrovent N roztok k inhalaci v tlakovém obalu podáván pouze na doporučení lékaře a pod dohledem dospělé osoby.
Způsob použití
Čtěte pečlivě následující pokyny, aby bylo zajištěno správné podání přípravku.
Správný způsob použití inhalátoru je předpokladem úspěšné léčby.
Před prvním použitím stiskněte dvakrát dávkovací ventil.
Před každým použitím dodržujte následující postup:
1. Odstraňte ochranný kryt.
2. Zhluboka vydechněte.
3. Inhalátor držte v poloze zobrazené na obrázku č. 1, ústní nástavec sevřete rty. Dno tlakového obalu (kovové tlakové nádobky) a šipka na ní směřuje vzhůru.
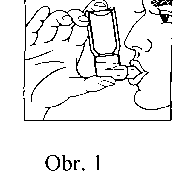
4. Zhluboka se nadechněte a na začátku tohoto nádechu stiskněte rázně dávkovací ventil, který uvolní jednu odměřenou dávku. Zadržte dech na několik sekund, vyjměte ústní nástavec z úst a vydechněte. Při podání druhé dávky, pokud je to potřebné, postupujte stejným způsobe m.
5. Po použití nasaďte zpět ochranný kryt.
6. Pokud nebyl přípravek použit 3 dny, je třeba před aplikací 1x stisknout dávkovací ventil.
Kovová tlaková nádobka je neprůhledná, nelze tedy sledovat hladinu přípravku a zrakem zjistit, kdy je spotřebován. Přípravek obsahuje 200 dávek léku. Poté, co byly aplikovány, může kovová tlaková nádobka stále ještě obsahovat určité množství tekutiny. Je však nutno užít nové balení přípravku, protože při dalších aplikacích by dávka inhalovaného léku nemusela být správná.
Množství zbývajícího léku lze ověřit následovně:
Protřepáním kovové tlakové nádobky můžete zjistit, zda ještě obsahuje tekutinu. Nebo vyjměte kovovou tlakovou nádobku s roztokem z umělohmotného ústního nástavce a vložte ji do větší nádoby naplněné vodou. Obsah roztoku v kovové tlakové nádobce lze odhadnout podle pozice, kterou tato nádobka ve vodní lázni zaujme (viz obr. 2).
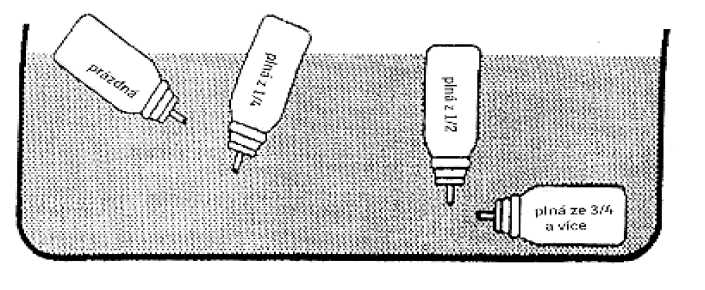
Obr. 2
Čištění ústního nástavce inhalátoru provádějte nejméně jednou týdně. Je důležité udržovat ústní nástavec inhalátoru v čistotě, aby nedošlo k poruše funkce spreje ucpáním nečistotami.
Při čištění nejprve sejměte ochranný kryt a vyjměte kovovou tlakovou nádobku z inhalátoru. Proplachujte teplou vodou ústní nástavec inhalátoru tak dlouho, dokud nejsou viditelné žádné nečistoty.
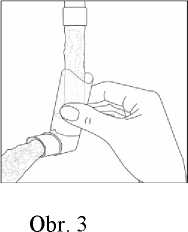
Po vyčištění ústní nástavec inhalátoru vytřepejte a nechte volně uschnout na vzduchu. Při sušení nepoužívejte teplo.
Jakmile ústní nástavec inhalátoru vyschne, složte ochranný kryt a kovovou tlakovou nádobku opět dohromady.
Obr. 4
Varování-. Umělohmotný ústní nástavec inhalátoru byl vyroben pouze pro použití s roztokem k inhalaci v tlakovém obalu Atrovent N tak, aby vždy byla zajištěna inhalace optimálního množství přípravku. Ústní nástavec inhalátoru proto nesmí být používán s jinými roztoky k inhalaci a roztok k inhalaci v tlakovém obalu Atrovent N nesmí být používán s jiným ústním nástavcem, než který je dodáván spolu s přípravkem.
4.3 Kontraindikace
Hypersenzitivita na atropin, j eho deriváty (jako je léčivá látka ipratropium-bromid) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Po podání přípravku Atrovent N se mohou ve vzácných případech vyskytnout okamžité reakce přecitlivělosti jako kopřivka, angioedém, kožní vyrážka, bronchospasmus, orofaryngeální edém a anafylaktické reakce.
Paradoxní bronchospazmus
Stejně jako jiné inhalační léky může Atrovent N vyvolat paradoxní bronchospazmus, který může vést až k ohrožení života. Pokud se paradoxní bronchospazmus objeví, je nutno léčbu přípravkem Atrovent N okamžitě přerušit a nahradit ji léčbou jinou.
Oční komplikace
Přípravek Atrovent N musí být podáván se zvýšenou opatrností u pacientů s dispozicí ke vzniku glaukomu s úzkým úhlem.
Při vniknutí aerosolové formy ipratropium-bromidu samotného nebo v kombinaci s beta2-agonisty do očí jsou ojediněle zmiňovány oční komplikace (např. mydriáza, zvýšený nitrooční tlak, glaukom s úzkým úhlem, bolestivost očí).
Bolesti očí nebo oční problémy, rozostřené vidění, vizuální haló nebo duhové vidění spojené se zčervenáním očí, které je způsobeno překrvením spojivek, a otok rohovky, mohou být příznaky akutního glaukomu s úzkým úhlem. Objeví-li se jakákoli kombinace uvedených příznaků, je nutno zahájit léčbu kapkami s miotickým účinkem a vyhledat pomoc očního lékaře.
Pacienti musí být poučeni o správném způsobu podávání přípravku Atrovent N, roztok k inhalaci v tlakovém obalu.
Je třeba dbát na to, aby se přípravek nezasáhl oči. Vzhledem k tomu že se roztok k inhalaci v tlakovém obalu aplikuje pomocí náustku a je kontrolován ručně, je riziko omezené.
Vliv na ledviny a močové cesty
Přípravek Atrovent N je třeba podávat opatrně u pacientů s již existující obstrukcí vývodných cest močových (například při hyperplázii prostaty nebo při obstrukci v oblasti hrdla močového měchýře).
Poruchy gastrointestinální motility
Pacienti s cystickou fibrózou mohou být náchylnější k poruchám gastrointestinální motility.
Tento přípravek obsahuje malé množství alkoholu, méně než 10 mg v jedné dávce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Současné dlouhodobé inhalační podávání přípravku Atrovent N spolu s jinými anticholinergiky nebylo studováno. Z tohoto důvodu se současné dlouhodobé podávání přípravku Atrovent N s jinými naticholinergiky nedoporučuje.
Beta-agonisté a xantinové deriváty mohou zesilovat bronchodilatační účinek.
4.6 Fertilita, těhotenství a kojení
Bezpečnost podávání přípravku Atrovent N v průběhu těhotenství nebyla stanovena. Při potvrzeném nebo předpokládaném těhotenství musí být zvážen prospěch léčby pro matku oproti případným rizikům pro nenarozené dítě. Neklinické studie neprokázaly embryotoxicitu nebo teratogenitu při inhalačním nebo nosním podávání dávek, které výrazně převyšovaly doporučené dávky pro člověka.
Kojení
Není známo, zda je ipratropium-bromid vylučován do mateřského mléka. Je ale nepravděpodobné, že by zvláště při inhalačním podání dosáhl ipratropium-bromid takové koncentrace v mateřském mléce, která by ohrožovala kojence. Při užívání přípravku kojícími matkami je třeba zvýšené opatrnosti.
Fertilita
Klinické údaje vztahující se k plodnosti nejsou pro ipratropium-bromid k dispozici. Neklinické studie prováděné s léčivou látkou ipratropium-bromid neprokázaly nežádoucí účinky na plodnost (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie vlivu přípravku Atrovent N na schopnost řídit a obsluhovat stroje. Nicméně pacienti musí být poučeni, že se mohou vyskytnout nežádoucí účinky jako závrať, poruchy akomodace, mydriáza a rozostřené vidění během léčby přípravkem Atrovent N. Proto je nutná při řízení auta nebo obsluze strojů opatrnost.
Mnohé z uvedených nežádoucích účinků patří k vlastnostem anticholinergik. Tak jako při každé inhalační terapii, také u přípravku Atrovent N se mohou objevit příznaky lokálního podráždění. Nežádoucí účinky byly zjištěny z údajů získaných v klinických studiích a během poregistračního sledování přípravku (farmakovigilance).
Mezi nejčastějšími nežádoucími účinky hlášenými v klinických studiích byly bolesti hlavy, podráždění v krku, kašel, sucho v ústech, poruchy gastrointestinální motility (včetně zácpy, průjmu a zvracení), nevolnost a závratě.
Nežádoucí účinky byly rozděleny podle výskytu za použití následující MedDRA konvence:
velmi časté (> 1/10); časté (> 1/100, < 1/10); méně časté (> 1/1000, < 1/100);
vzácné (> 1/10 000, < 1/1000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze
určit).
Poruchy imunitního systému
Méně časté: hypersenzitivita, anafylaktické reakce
Poruchy nervového systému Časté: bolest hlavy, závrať
Poruchy oka
Méně časté: rozostřené vidění, mydriáza, zvýšení nitroočního tlaku, glaukom, bolest očí, vizuální haló, překrvení spojivek, otok rohovky Vzácné: poruchy akomodace
Srdeční poruchy
Méně časté: palpitace, supraventrikulární tachykardie
Vzácné: fibrilace síní, zrychlená srdeční frekvence
Respirační, hrudní a mediastinální poruchy Časté: podráždění v krku, kašel
Méně časté: bronchospasmus, paradoxní bronchospasmus, laryngospasmus, otok hltanu, sucho v krku
Gastrointestinální poruchy
Časté: sucho v ústech, nauzea, poruchy gastrointestinální motility
Méně časté: průjem, zácpa, zvracení, stomatitida, otok úst
Poruchy kůže a podkožní tkáně
Méně časté: vyrážka, svědění, angioedém
Vzácné: kopřivka
Poruchy ledvin a močových cest Méně časté: retence moči
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
Specifické příznaky předávkování nebyly popsány. Vzhledem ke značné terapeutické šíři a topickému podávání přípravku Atrovent N nelze očekávat závažné anticholinergní příznaky. Mohou se objevovat mírné systémové anticholinergní účinky jako sucho v ústech, poruchy vidění (akomodace) a zrychlená srdeční frekvence.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Anticholinergika.
ATC kód: R03BB01
Ipratropium-bromid je kvartérní amoniová sloučenina s anticholinergními (parasympatolytickými) vlastnostmi. Z neklinických studií lze soudit, že inhibuje vagem zprostředkované reflexy tím, že antagonizuje účinek acetylcholinu - transmiteru, který je uvolňován vagovým nervem. Anticholinergní látky zabraňují zvýšení nitrobuněčné koncentrace Ca2+ , ke kterému dochází po interakci acetylcholinu s muskarinovým receptorem na hladké svalovině bronchů. Uvolnění Ca2+ je zprostředkováno sekundárním přenašečem, který se skládá z IP3 (inositol triphosphate) a DAG (diacylglycerol).
Bronchodilatace po inhalaci přípravku Atrovent N je vyvolána místním (nikoli systémovým) a místně specifickým působením na plíce.
Neklinické a klinické studie nenaznačují škodlivý účinek přípravku Atrovent N na tvorbu sekretu ve sliznici dýchacích cest, mukociliární clearance nebo výměnu dýchacích plynů.
Klinické studie
Ve studiích trvajících až tři měsíce u dospělých pacientů s astmatem nebo s CHOPN a u dětí s astmatem byla prokázána rovnocenná terapeutická účinnost bezfreonové a freonové formy přípravku.
V kontrolovaných 90denních studiích zahrnujících pacienty s bronchospasmem provázejícím CHOPN (chronická bronchitida s emfyzémem) bylo prokázáno významné zlepšení plicních funkcí během
15 minut od podání. Vrcholu účinku bylo dosaženo za 1 - 2 hodiny a u většiny pacientů přetrvával až 4 - 6 hodin.
V kontrolovaných 90denních studiích u pacientů s bronchospasmem provázejícím astma bylo prokázáno významné zlepšení plicních funkcí (FEV1 vzrostlo o 15 % a více) u 51 % pacientů.
5.2 Farmakokinetické vlastnosti
Absorpce
Terapeutický účinek přípravku Atrovent N je dán místním působením v dýchacích cestách. Z tohoto důvodu není časový průběh bronchodilatace a systémové farmakokinetiky souběžný.
Po inhalaci se v plicích obvykle ukládá 10-30 % inhalované dávky přípravku v závislosti na jeho formě a inhalační technice. Větší část dávky je spolknuta a dostává se do gastrointestinálního traktu.
Část dávky, která se ukládá v plicích, se dostává rychle do oběhu (během minut).
Kumulativní renální exkrece (0-24 hod) mateřské sloučeniny je přibližně 46% intravenózně podané dávky, což je pod 1% perorální dávky a přibližně 3 až 13% inhalační dávky. Na základě těchto údajů se celková systémová biologická dostupnost perorální a inhalační dávky ipratropium-bromidu odhaduje na 2% a 7% až 28%. Proto spolknuté množství ipratropium-bromidu nemůže přispívat k systémové expozici.
Distribuce
Kinetické parametry popisující dispozice ipratropium-bromidu byly vypočteny z plazmatických koncentrací po intravenózním podání.
Byl pozorován rychlý dvoufázový pokles plazmatických koncentrací. Zdánlivý distribuční objem (Vd) v rovnovážném stavu je 176 l (přibližně 2,4 l/kg). Léčivá látka se minimálně (méně než 20%) váže na plazmatické proteiny. Neklinické údaje svědčí o tom, že kvartérní ipratropium bromid nepřestupuje transplacentárně a neproniká hematoencefalickou bariérou.
Biotransformace
Po nitrožilním podání se přibližně 60 % dávky metabolizuje, hlavní část pravděpodobně v játrech oxidací.
Známé metabolity, které vznikají hydrolýzou, dehydratací nebo eliminací hydroxymethylové skupiny kyseliny tropové, vykazují velmi malou nebo žádnou afinitu k muskarinovému receptoru a je nutno je považovat za neúčinné.
Eliminace
Poločas terminální eliminační fáze je přibližně 1,6 hodiny.
Průměrná hodnota celkové clearance léčivé látky je 2,3 l/min a renální clearance je 0,9 l/min.
V exkrečně bilančních studiích byla zjištěna kumulativní renální exkrece (6 dní radioaktivně značené látky (mateřské sloučeniny a všech jejích metabolitů) 72,1% po intravenózním podání, 9,3% po perorálním podání a 3,2% po inhalačním podání. Celkové množství radioaktivně značeného přípravku vyloučeného stolicí bylo 6,3% po intravenózním podání, 88,5% po perorálním podání a 69,4% po inhalačním podání.
Radioaktivně značený ipratropium-bromid je po intravenózním podání vylučován především ledvinami. Poločas eliminace radioaktivně značeného přípravku (mateřské sloučeniny a všech jejích metabolitů) je 3,6 hodin.
5.3 Předklinické údaje vztahující se k bezpečnosti
Lokální a systémová snášenlivost ipratropium-bromidu byla komplexně zkoumána u několika druhů zvířat pomocí různých cest podání.
Toxicita po jedné dávce
Studie akutní toxicity po inhalačním, perorálním a intravenózním podání byly prováděny na různých druzích laboratorních zvířat (hlodavci i jiná zvířata).
Po inhalačním podání byla zjištěna minimální letální dávka pro samce morčat 199 mg/kg.
U potkanů nebyla zaznamenána žádná mortalita ani při podávání nejvyšších technicky aplikovatelných dávek (např. 0,05 mg/kg po 4 hodinách podávání nebo 160 vdechů ipratropium-bromidu (0,02 mg/vdech).
Hodnoty LD50 po perorálním podání u myší, potkanů a králíků byly 1585, 1925 a 1920 mg/kg. Hodnoty LD50 po intravenózním podání u myší, potkanů a psů byly 13,6, 15,8 a přibližně 18,2 mg/kg. Klinické příznaky zahrnovaly mydriázu, suchost ústní sliznice, dušnost, třes, křeče a/nebo tachykardii.
Toxicita po opakované dávce
Studie toxicity po opakovaném podávání byly prováděny u potkanů, králíků, psů a opic druhu Makak Rhesus.
Při inhalační aplikaci v průběhu 6 měsíců byla stanovena hladina bez nežádoucích účinků (no observed adverse effect level=NOAEL) v dávce 0,38 mg/kg/den u potkanů, 0,18 mg/kg/den u psů a 0,8 mg/kg/den u opic Makak Rhesus.
U psů byla zaznamenána suchost ústní sliznice a tachykardie. V bronchopulmonálním systému ani v jiných orgánech nebyly shledány histopatologické změny související s podáváním přípravku.
U potkanů byla hladina bez nežádoucích účinků stanovena po 18 měsících perorálního podávání na 0,5 mg/kg/den.
Studie toxicity po opakovaném podávání inhalačních dávek u potkanů po dobu až 6 měsíců a psů po dobu až 3 měsíců s jinými formami přípravku (intranazální forma s alternativním hnacím plynem a s laktózovým práškem) nezaznamenaly žádné další informace o obecném profilu toxicity ipratropium-bromidu.
Intranazální podávání po dobu až 6 měsíců stanovilo úroveň dávky bez účinků (NOAEL)
> 0,20 mg/kg/den u psů a potvrdilo výsledky dřívějších studií s podáváním po dobu až 13 týdnů.
Studie toxicity při podávání opakovaných dávek ipratropum-bromidu prokázaly podobné toxikologické profily bezfreonové a freonové formy přípravku.
Lokální tolerance
Vodný roztok ipratropium-bromidu podávaný inhalačně v jednotlivé dávce (0,05 mg/kg - doba podávání více než 4 hodiny) byl potkany lokálně dobře snášen. Ve studiích toxicity při opakovaném podávání byl ipratropium-bromid lokálně dobře snášen.
Imunogenicita
U morčat nebyla zjištěna akutní anafylaktická ani opožděné anafylaktické kožní reakce.
Genotoxicita a karcinogenecita
Genotoxicita nebyla zjištěna ani in vitro v Amesově testu, ani v testech in vivo (mikronukleární test, dominantní letální test u myší, cytogenetický test na buňkách kostní dřeně čínského křečka).
V dlouhodobých studiích na myších a potkanech nebyly nalezeny tumorogenní nebo kancerogenní účinky.
Reprodukční a vývojová toxicita
Embryotoxicita ipratropium-bromidu, vliv na fertilitu, peri- a postnatální vývoj byly studovány u myší, potkanů a králíků. Vysoké použité perorální dávky (tj .1000 mg/kg/den u potkanů a 125 mg/kg/den u králíků) byly toxické pro matky u obou druhů a embryo a fetotoxické u potkanů, kde byla nižší hmotnost plodu. Malformace související s podáním látky nebyla zjištěna. Nejvyšší technicky aplikovatelná inhalační odměřená dávka aerosolu 1,5 mg/kg/den u potkanů a 1,8 mg/kg/den u králíků nevyvolala žádný nežádoucí účinek na reprodukci.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
norfluran
bezvodá kyselina citronová čištěná voda bezvodý ethanol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Chraňte před mrazem.
Kovová tlaková nádobka obsahuje stlačenou kapalinu. Nevystavujte teplotě vyšší než 50 °C. Nádobka nesmí být otevírána násilím.
6.5 Druh obalu a velikost balení
Kovová tlaková nádobka (tlakový obal) uzavřená dávkovacím ventilem uložená do plastového adaptéru s krytem, papírová krabička Velikost balení: 10 ml (200 odměřených dávek)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Ingelheim am Rhein, Německo
8. REGISTRAČNÍ ČÍSLO
14/064/03-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
19. 2. 2003 / 16. 6. 2010
10. DATUM REVIZE TEXTU
4.5.2016
9/9