Antithrombin Iii Nf Baxter
zastaralé informace, vyhledat novějšíPříloha č. 2 ke sdělení sp.zn. sukls116308/2012
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
ANTITHROMBIN III NF BAXTER, prášek pro přípravu infuzního roztoku s rozpouštědlem
2. SLOŽENÍ KVALITATIVNÍ A KVANTITATIVNÍ
1 ml rozpuštěného přípravku obsahuje antithrombinum III 50 IU
proteinům 20-50mg
ANTITHROMBIN III NF BAXTER je dodáván jako prášek pro přípravu infuzního roztoku, obsahující nominálně 500 IU (1000 IU) antithrombinu z lidské plazmy v jedné injekční lahvičce.
Po rekonstituci v 10 ml (20 ml) vody na injekci obsahuje přípravek přibližně 50 IU/ml (500 IU/10 ml, 1000 IU/20 ml) antithrombinu z lidské plazmy.
Síla (IU) se stanovuje pomocí chromogenního testu dle Evropského lékopisu. Specifická aktivita přípravku ANTITHROMBIN III NF BAXTER je minimálně 3 IU AT/mg plazmatického proteinu.
Tento léčivý přípravek obsahuje přibližně 3,77 mg sodíku/ml.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro přípravu infuzního roztoku s rozpouštědlem. Popis přípravku: a) bílý až nažloutlý lyofilizát
b) bezbarvá tekutina (voda na injekci)
4. KLINICKÉ ÚDAJE
4.1 Indikace
Nedostatek antithrombinu může být vrozený nebo získaný v rámci různých klinických poruch.
Získaný nedostatek antithrombinu může být způsoben buď zvýšenou spotřebou nebo ztrátou proteinu či poruchou syntézy antithrombinu.
Podání přípravku ANTITHROMBIN III NF BAXTER k profylaxi a léčbě trombotických a tromboembolických poruch je indikováno u pacientů s aktivitou antithrombinu v plazmě nižší než 70% normálu. Infuze antithrombinu mohou být potřebné zejména v následujících klinických situacích:
- chirurgické výkony nebo těhotenství a porod u pacientů s kongenitálním deficitem antithrombinu;
neadekvátní nebo chybějící odpověď na heparin;
- existence nebo riziko diseminované intravaskulární koagulace (např. při polytraumatu, septických komplikacích, šoku, preeklampsii a jiných stavech spojených s akutní konsumpční koagulopatií);
existence nebo riziko trombózy u pacientů s nefrotickým syndromem nebo zánětlivým střevním onemocněním;
- chirurgický výkon nebo krvácení u pacientů se závažným selháním jater, především jsou-li pacienti léčeni koncentráty koagulačních faktorů.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře se zkušenostmi s léčbou pacientů s nedostatkem antithrombinu.
Dávkování
Při vrozeném nedostatku by mělo být dávkování přizpůsobeno potřebám každého pacienta a měla by se zvážit rodinná anamnéza s ohledem na tromboembolické příhody, aktuální klinické rizikové faktory a výsledky laboratorních vyšetření.
Dávkování a délka trvání substituční terapie u získaného nedostatku závisí na plazmatické hladině antithrombinu, přítomnosti známek zvýšeného obratu, základním onemocnění a závažnosti klinického stavu. Množství, jež má být podáno, a frekvence podání by měly být vždy založeny na klinické účinnosti a laboratorním hodnocení v jednotlivém případě.
Počet podaných jednotek antithrombinu se vyjadřuje v mezinárodních jednotkách (IU), jež se vztahují k současnému standardu WHO pro antithrombin. Aktivita antithrombinu v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro antithrombin v plazmě).
Jedna mezinárodní jednotka (IU) aktivity antithrombinu je ekvivalentní množství antithrombinu v jednom mililitru normální lidské plazmy. Výpočet požadované dávky antithrombinu je založen na empirickém zjištění, že 1 mezinárodní jednotka (IU) antithrombinu na kilogram tělesné hmotnosti zvyšuje aktivitu antithrombinu v plazmě zhruba o 2 %.
Úvodní dávka se stanoví pomocí následujícího vzorce:
Požadovaný počet jednotek = tělesná hmotnost (kg) x (cílová hladina - skutečná aktivita antithrombinu [%]) x 0,5
Úvodní cílová aktivita antithrombinu závisí na klinickém stavu. Je-li stanovena indikace pro substituci antithrombinu, mělo by být dávkování dostatečné, aby byla dosažena cílová aktivita antithrombinu a aby byla udržena účinná hladina. Dávkování by mělo být stanoveno a monitorováno na základě laboratorních testů aktivity antithrombinu, které by měly být prováděny do stabilizace pacienta minimálně dvakrát denně, poté jednou denně, nejlépe bezprostředně před následující infuzí. Úprava dávkování by měla zohlednit jak známky zvýšeného obratu antithrombinu podle laboratorních hodnot, tak i klinický průběh. Aktivita antithrombinu by měla být po dobu trvání léčby udržována nad 80 %, pokud by klinické údaje nesvědčily pro jinou účinnou hladinu.
Obvyklá počáteční dávka při vrozeném nedostatku bývá 30-50 IU/kg.
Poté by měly být dávkování a frekvence i doba trvání léčby upraveny podle biologických údajů a klinické situace.
Pediatrická populace
Není k dispozici dostatek údajů k tomu, aby mohlo být doporučeno podávání přípravku ANTITHROMBIN III NF BAXTER dětem do 6 let.
Způsob podání
Rekonstituujte přípravek podle pokynů v bodu 6.6. Přípravek by měl být podáván intravenózně. Maximální rychlost podání je 5 ml/min.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku.
Heparinem vyvolaná trombocytopenie v anamnéze.
4.4 Zvláštní upozornění a opatření pro použití
Stejně jako u jiných intravenózních proteinových přípravků jsou možné hypersenzitivní reakce alergického typu. Pacienti musí být po celou dobu infuze pečlivě monitorováni a všechny příznaky během infuze se musí pečlivě sledovat. Pacienti by měli být seznámeni s prvními známkami hypersenzitivních reakcí, k nimž patří vyrážka, generalizovaná kopřivka, tíže na hrudi, sípání, hypotenze a anafylaxe. Objeví-li se po podání tyto symptomy, měli by kontaktovat svého lékaře.
V případě šoku je třeba zahájit standardní léčbu.
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a zařazení účinných výrobních kroků, při nichž jsou inaktivovány/odstraněny viry. Přesto nemůže být při podávání léčiv vyráběných z lidské krve nebo plazmy zcela vyloučena možnost přenosu infekce. To platí i pro jakékoli neznámé nebo vznikající viry a jiné patogeny.
Přijatá opatření jsou považována za účinná u tzv. obalených virů, například virů HIV, HBV a HCV, a u neobaleného viru HAV. Omezený účinek mohou mít tato opatření u neobalených virů jako je parvovirus B19. Infekce parvovirem B19 může být závažná u těhotných žen (infekce fetu) a u jedinců s imunodeficitem nebo zvýšenou erytropoézou (např. hemolytická anemie).
U pacientů, kteří pravidelně/opakovaně dostávají přípravky antithrombinu z lidské plazmy, je třeba zvážit vhodnou vakcinaci (hepatitida A a B).
Při každé aplikaci přípravku ANTITHROMBIN III NF BAXTER doporučujeme zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Klinické a biologické sledování při souběžném podávání antithrombinu a heparinu:
- aby se upravilo dávkování heparinu a aby se zamezilo nadměrné hypokoagulabilitě, mají být
pravidelně a v krátkých intervalech prováděny kontroly antikoagulace (APPT a tam, kde je to vhodné, aktivita anti-FXa), především v prvních minutách a hodinách po zahájení používání antithrombinu.
- denní měření hladin antithrombinu, aby se upravila individuální dávka s ohledem na riziko
snížení hladin antithrombinu delší léčbou nefrakcionovaným heparinem.
Tento přípravek obsahuje přibližně 3,77 mg sodíku/ml. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Pediatrická populace
Bezpečnost a účinnost používání přípravku ANTITHROMBIN III NF BAXTER u dětských pacientů nebyla v klinických studiích společností Baxter hodnocena.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Heparin: substituce antithrombinu během podávání heparinu v léčebné dávce zvyšuje riziko krvácení. Účinek antithrombinu je heparinem značně zvýšen. Poločas antithrombinu se může podstatně snížit souběžnou léčbou heparinem z důvodu zrychleného obratu antithrombinu. Proto musí být souběžné podávání heparinu a antithrombinu pacientovi se zvýšeným rizikem krvácení klinicky a biologicky monitorováno.
4.6 Fertilita, těhotenství a kojení
Zkušenosti ohledně bezpečnosti přípravků lidského antithrombinu během těhotenství jsou omezené. Bezpečnost podávání přípravku ANTITHROMBIN III NF BAXTER těhotným nebo kojícím ženám nebyla v kontrolovaných klinických studiích ověřena. ANTITHROMBIN III NF BAXTER by měl být těhotným nebo kojícím ženám s nedostatkem antithrombinu podáván pouze tehdy, je-li jednoznačně indikován, přičemž je třeba vzít v úvahu, že těhotenství s sebou u těchto pacientek přináší zvýšené riziko tromboembolických příhod.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly pozorovány účinky na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Občas byly pozorovány hypersenzitivní nebo alergické reakce (k nimž může patřit angioedém, pálení a píchání v místě infuze, mrazení, zčervenání, generalizovaná kopřivka, bolest hlavy, vyrážka, hypotenze, letargie, nauzea, neklid, tachykardie, tíže na prsou, brnění, zvracení či sípání), které mohou v některých případech vyústit v závažnou anafylaxi (včetně šoku).
Ve vzácných případech byla pozorována horečka.
Vzácně se může objevit heparinem vyvolaná a protilátkami zprostředkovaná trombocytopenie (typu II). Může být zaznamenán počet trombocytů nižší než 100 000/pl nebo pokles počtu trombocytů o 50%.
Během postmarketingových zkušeností s přípravkem ANTITHROMBIN III NF BAXTER byly hlášeny následující nežádoucí účinky, seřazené podle tříd orgánových systémů MedDRa (TOS), dále podle upřednostňovaných termínů v pořadí závažnosti, pokud je to možné:
Frekvence byly hodnoceny podle následující konvence: velmi časté (> 1/10), časté (> 1/100 to < 1/10), méně časté (> 1/1000 to < 1/100), vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
|
Frekvence výskytu nežádoucích účinků (NÚ) | ||
|
MedDRA Třídy orgánových systémů (TOS) |
Upřednostňovaný termín MedDRA |
NÚ kategorie frekvence |
|
Poruchy nervového systému |
Není známo | |
|
Cévní poruchy |
Není známo | |
Informace o virové bezpečnosti viz bod 4.4.
4.9 Předávkování
Nebyly zaznamenány příznaky předávkování antithrombinem.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antitrombotika: skupina heparinu. ATC kód: B01AB02.
Antithrombin, glykoprotein o molekulové hmotnosti 58 kD, se 432 aminokyselinami, patří ke skupině serpinů (inhibitorů serinové protézy). Je jedním z nejdůležitějších přirozených inhibitorů krevní koagulace. Nejsilněji inhibovanými faktory jsou thrombin a faktor Xa, ale také faktory kontaktní aktivace, vnitřního systému a komplex faktoru VIIa/ tkáňového faktoru. Aktivita antithrombinu je značně posilována heparinem a antikoagulační účinky heparinu závisí na přítomnosti antithrombinu.
Antithrombin obsahuje dvě funkčně důležité domény. První obsahuje reakční centrum a poskytuje místo štěpení pro proteinázy jako je thrombin, což je nutná podmínka pro vytvoření stabilního komplexu proteináza-inhibitor. Druhou je doména, která váže glykosaminoglykan a odpovídá za interakci s heparinem a příbuznými látkami, což urychluje inhibici thrombinu. Komplexy inhibitoru a koagulačního enzymu jsou odbourávány retikuloendoteliárním systémem.
Aktivita antithrombinu u dospělých se pohybuje v rozmezí 80-120 % a hladiny u novorozenců jsou zhruba 40-60 %.
5.2 Farmakokinetické vlastnosti
Farmakokinetické studie antithrombinu prokázaly průměrný biologický poločas přibližně 3 dny. Souběžnou léčbou heparinem se může poločas snížit na zhruba 1,5 dne. Poločas může klesnout na hodiny u klinických stavů spojených s vysokou spotřebou.
5.3 Předklinické údaje vztahující se k bezpečnosti
Antithrombin je normální složkou lidské plazmy. Testování toxicity po jednorázovém podání na zvířecích modelech má pro podmínky u pacientů jen malý význam. Testování toxicity po opakovaném podávání u zvířat je neproveditelné vzhledem k tvorbě protilátek proti cizímu (lidskému) proteinu. Nebyla zaznamenána souvislost antithrombinu s embryo-fetální toxicitou, onkogenním nebo mutagenním potenciálem.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam všech pomocných látek
Prášek:
glukóza
chlorid sodný
dihydrát citronanu sodného
trometamol
Rozpouštědlo: voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Lyofilizovaný přípravek má dobu použitelnosti 3 roky.
Po rekonstituci má být přípravek ANTITHROMBIN III NF BAXTER použit okamžitě, protože neobsahuje žádné konzervační látky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C).
Uchovávejte mimo dosah a dohled dětí.
6.5 Druh obalu a velikost balení
2 bezbarvé skleněné lahvičky s pryžovou zátkou, kovový uzávěr, plastikový kryt, set k rozpuštění a aplikaci, krabička.
Velikost balení:
1x 500 IU + 10 ml solvens 1x 1000 IU + 20 ml solvens
Každé balení také obsahuje:
- 1 převodní jehlu
- 1 filtrační jehlu
- 1 zavzdušňovací jehlu
- 1 jehlu k jednorázovému použití
- 1 infuzní set
6.6 Návod k použití přípravku, zacházení s ním a k jeho likvidaci
Přípravek ANTITHROMBIN III NF BAXTER má být rekonstituován bezprostředně před použitím a podáván pouze pomocí přiloženého infuzního setu. Během celého procesu rekonstituce je třeba používat aseptickou techniku. Roztok má být ihned použit (přípravek neobsahuje konzervační látky). Rekonstituovaný přípravek má být před podáním vizuálně zkontrolován s ohledem na přítomnost částic či změnu zabarvení. Roztok je čirý nebo lehce opalescentní. Zakalené roztoky nebo roztoky obsahující usazeniny nepoužívejte.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Rekonstituce prášku:
1. Zahřejte neotevřenou lahvičku s rozpouštědlem (voda na injekci) na pokojovou teplotu (max.
37oC).
2. Odstraňte ochranná víčka z lahviček s koncentrátem a rozpouštědlem (obr. A) a dezinfikujte pryžové zátky obou lahviček.
3. Sejměte ochranný kryt z jednoho konce přiložené převodní jehly pootočením a povytažením (obr. B). Zapíchněte tento konec jehly skrze pryžovou zátku do lahvičky s rozpouštědlem (obr. C).
4. Sejměte ochranný kryt z druhého konce převodní jehly tak, abyste se přitom nedotkli odkrytého konce.
5. Obraťte lahvičku s rozpouštědlem nad lahvičku s koncentrátem a propíchněte převodní jehlou pryžovou zátku lahvičky s koncentrátem (obr. D). Rozpouštědlo přejde pomocí vakua do lahvičky s koncentrátem.
6. Oddělte obě lahvičky vyjmutím jehly z lahvičky s koncentrátem (obr. E). Lehce protřepejte nebo promíchejte, abyste urychlili rozpouštění.
7. Po úplném rozpuštění koncentrátu vpíchněte do lahvičky přiloženou zavzdušňovací jehlu (obr. F), čímž odstraníte případně vzniklou pěnu. Zavzdušňovací jehlu vyjměte.
Podání:
8. Pootočením a povytažením sejměte ochranný kryt z přiložené filtrační jehly a nasaďte ji na sterilní jednorázovou stříkačku. Natáhněte roztok do stříkačky (obr. G).
9. Odpojte filtrační jehlu od stříkačky a roztok aplikujte pomalu intravenózně (max. rychlost 5 ml/min) pomocí přiložené jehly k jednorázovému použití (nebo pomocí přiloženého infuzního setu).
Pokud se neprovádí filtrace během rekonstituce, je třeba použít infuzní set s odpovídajícím filtrem (max. rychlost infuze 5 ml/min).
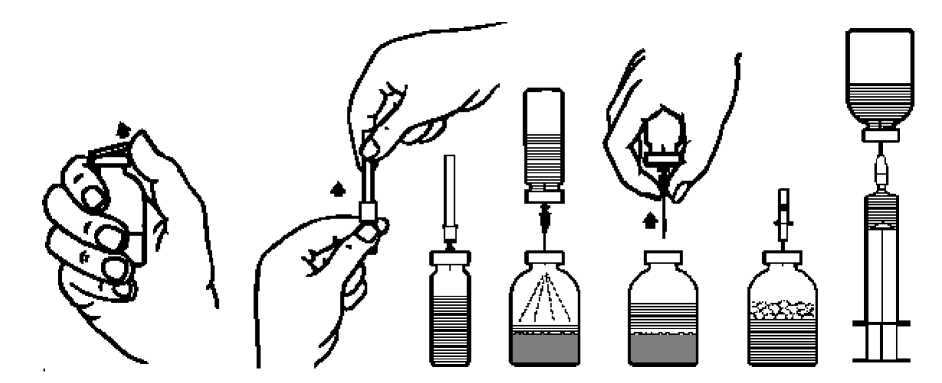
obr. A obr. B
obr. C obr. D obr. E
obr. F obr. G
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň, Rakousko
8. REGISTRAČNÍ ČÍSLO
16/144/89-C
9. DATUM REGISTRACE/DATUM PRODLOUŽENÍ REGISTRACE
18.5.1994 / 20.5. 2009
10. DATUM POSLEDNÍ REVIZE TEXTU
6.6.2012
Stránka 7 z 7