Zomacton 4 Mg
sp.zn.sukls231159/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1 NÁZEV PŘÍPRAVKU ZOMACTON 4 mg
Prášek a rozpouštědlo pro injekční roztok
2 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Somatropinum* ...............................................................4 mg
(1,3 mg/ml nebo 3,3 mg/ml po naředění)
* Připravený rekombinantní DNA technologií produkovaný Escherichia coli
Celkové množství v lahvičce zahrnuje přebytek, aby bylo možné odebrat předepsané množství.
Úplný seznam pomocných látek viz bod 6.1.
3 LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok
Zomacton je bílý až špinavě bílý lyofilizovaný prášek. Rozpouštědlo je čirý bezbarvý roztok
4 KLINICKÉ ÚDAJE
4. 1 Terapeutické indikace
Zomacton je určen k dlouhodobé léčbě dětí:
a) s poruchou růstu v důsledku neodpovídajícího výdeje růstového hormonu
b) u dívek s Turnerovým syndromem.
U dospělých je určen k léčbě nedostatku růstového hormonu:
a) při nedostatku zjištěném již v dětství. V tomto případě by měl být pacient znovu vyšetřen a deficit růstového hormonu by měl být potvrzen dvěma různými dynamickými stimulačními testy.
b) při nedostatku růstového hormonu vzniklého v dospělosti při známém hypotalamo-hypofyzárním onemocnění, kde je prokázán deficit dalšího, s onemocněním souvisejícího hormonu, kromě prolaktinu. Deficit by měl být prokázán dvěma různými dynamickými stimulačními testy, které by měly být prováděny až po zahájení adekvátní substituce v ostatních deficitních osách.
4. 2 Dávkování a způsob podání
Dávkování
Léčba přípravkem Zomacton může probíhat pouze pod dohledem kvalifikovaného lékaře, se zkušenostmi v léčbě pacientů s deficitem růstového hormonu.
Dávkování a harmonogram podávání přípravku Zomacton by měl být pro každého pacienta individuální.
Doba léčby, obvykle období několika let, bude záviset na maximálním dosažitelném léčebném úspěchu.
Dávkování u dětí:
Všeobecně se doporučuje dávka 0,17 - 0,23 mg/kg tělesné hmotnosti (odpovídající 4,9 - 6,9 mg/m
plochy tělesného povrchu) na týden, rozdělená na 6 - 7 podkožních injekcí (to odpovídá denní injekční
2
aplikaci 0,02 - 0,03 mg/kg tělesné hmotnosti nebo 0,7 - 1,0 mg/m plochy tělesného povrchu).
2
Celková týdenní dávka nemá přesáhnout 0,27 mg/kg tělesné hmotnosti nebo 8 mg/m plochy tělesného povrchu, tj. každodenní injekční aplikaci do 0,04 mg/kg hmotnosti.
Turnerův syndrom
2
Při léčbě Turnerova syndromu se obvykle podává dávka 0,33 mg/kg těl. hm. (to odpovídá 9,86 mg/m plochy tělesného povrchu) týdně, rozdělená do 6 - 7 denních podkožních injekcí (to odpovídá denní injekční aplikaci 0,05 mg/kg tělesné hmotnosti nebo 1,40-1,63 mg/m2 plochy tělesného povrchu).
Dávkování u dospělých:
Počáteční dávka by měla být co nejnižší. Doporučovaná počáteční dávka je 0,042 mg/kg/týden (odpovídá 1,26 mg/ m2 plochy tělesného povrchu/týden). Tuto dávku je možné postupně zvyšovat, neměla by však být překročena dávka 0,084 mg/kg/týden (to odpovídá 2,25 mg / m2 plochy tělesného povrchu/týden).
Vodítkem pro stanovení optimální dávky by měl být případný výskyt nežádoucích účinků a hladina IGF-1 v krevním séru (růstový faktor podobný inzulínu). Pacient by měl užívat nejnižší účinnou dávku, která se obvykle snižuje s věkem.
Způsob podání
Subkutánní aplikace růstového hormonu může vést k úbytku nebo nárůstu tukové tkáně v místě vpichu. Proto je nutno místo aplikace injekcí měnit.
4. 3 Kontraindikace
Hypersensitivita na léčivou látku nebo kteroukoli pomocnou látku uvedenou v bodě 6.1.
Přípravek Zomacton se nesmí podávat novorozencům a nedonošeným dětem, protože rozpouštědlo obsahuje benzylalkohol.
Somatropin se nesmí používat v případě prokázání nádorové aktivity. Intrakraniální nádory musí být inaktivní a před zahájením léčby růstovým hormonem musí být protinádorová léčba ukončena. Léčba by měla být ukončena v případě průkazného růstu nádoru.
Somatropin se nesmí používat k urychlení růstu u dětí s uzavřenými epifýzami.
Somatropinem nesmí být léčeni pacienti s akutním závažným onemocněním způsobeným komplikacemi po otevřených operacích srdce a břicha, vícečetnými poraněními při nehodě, pacienti s akutním respiračním selháním nebo podobnými stavy.
U dětí s chronickým selháním ledvin je třeba léčbu somatropinem přerušit v době transplantace ledvin.
4. 4 Zvláštní upozornění a opatření pro použití
Zomacton může způsobit toxickou reakci a anafylaktickou reakci u kojenců a dětí do 3 let věku, protože rozpouštědlo obsahuje benzyl alkohol._Přípravek Zomacton se nesmí podávat novorozencům a nedonošeným dětem.
Zomacton není indikován k dlouhodobé léčbě pediatrických pacientů s nedostatečným vzrůstem, který je podmíněn geneticky potvrzeným syndromem Prader - Williho, pokud u nich nebyla stanovena diagnóza nedostatku růstového hormonu. Byla publikována sdělení o výskytu spánkové apnoe a náhlého úmrtí po zahájení terapie růstovým hormonem u pediatrických pacientů se syndromem Prader - Williho, kteří měli jeden nebo více následujících rizikových faktorů: extrémní obezita, anamnéza obstrukce horních dýchacích cest nebo spánkové apnoe, nebo nezjištěné respirační infekce.
Byly hlášeny vzácné případy nitrolební hypertenze. V případě silné nebo opakované bolesti hlavy, problémů s viděním a nauzey/zvracení se doporučuje provést vyšetření očního pozadí k posouzení možného edému papily. Pokud je potvrzena přítomnost edému papily, je nutno zvážit možnost onemocnění nezhoubnou nitrolební hypertenzí, a pokud se potvrdí, je třeba léčení růstovým hormonem ukončit (viz též bod 4.8). V současné době není dostatek údajů, které by napomohly v klinickém rozhodování u pacientů, u nichž intrakraniální hypertenze ustoupila. Pokud se léčba růstovým hormonem obnoví, je třeba pečlivě monitorovat symptomy intrakraniální hypertenze.
Leukémie byla hlášena u malého počtu pacientů s deficitem růstového hormonu léčených somatropinem, právě tak jako u neléčených pacientů. Nejsou však žádné důkazy o tom, že by se u pacientů, kteří dostávají růstový hormon, zvyšovala incidence leukémie, pokud nemají jiné predisponující faktory (vzniku leukémie).
Je možné, podobně jako u všech přípravků obsahujících somatropin, že u malého procenta pacientů dojde k rozvoji protilátek proti somatropinu. Vazebná schopnost těchto protilátek je malá, a tyto protilátky nemají žádný vliv na rychlost růstu. U všech pacientů, kteří neodpovídají na terapii, by mělo být provedeno vyšetření na protilátky proti somatropinu.
Růstový hormon urychluje extratyroidální konverzi T4 na T3 a může odhalit latentní hypotyreózu. Z tohoto důvodu je třeba u všech pacientů monitorovat funkci štítné žlázy. U pacientů s hypopituitarismem je při probíhající terapii somatropinem nutno provádět pečlivé monitorování standardní substituční terapie.
Vzhledem k tomu, že somatropin může snížit citlivost na insulin, měli by pacienti být monitorováni, aby byly zjištěny případné známky glukózové intolerance. U pacientů s diabetes mellitus může být po zahájení terapie přípravkem obsahujícím somatropin nutné upravit dávku insulinu. U pacientů s diabetem nebo glukózovou intolerancí by mělo být v průběhu terapie somatropinem prováděno pečlivé monitorování. Zomacton by měl být používán se zvýšenou opatrností také u pacientů s rodinnou anamnézou predisponující k tomuto onemocnění (diabetes mellitus).
U pacientů se sekundárním deficitem růstového hormonu při intrakraniální lézi se doporučují časté kontroly stavu z hlediska progrese nebo recidivy základního onemocnění. Pokud dojde k progresi nebo recidivě léze, je nutno terapii přípravkem Zomacton přerušit.
U pacientů léčených v minulosti na maligní onemocnění se doporučuje sledovat znaky a příznaky relapsu maligního onemocnění.
U některých dětí se během rychlého růstu může objevit skolióza. V průběhy léčby by měly být monitorovány příznaky skoliózy.
U pacientů s endokrinními poruchami může dojít častěji k posunu epifýzy stehenní kosti. Pacienti léčení přípravkem Zomacton , kteří začnou kulhat nebo mají bolesti v kyčli nebo v koleni, by měli být vyšetřeni lékařem.
Účinnost léčby růstovým hormonem na uzdravení byla studována ve dvou placebem kontrolovaných studiích zahrnujících 522 kriticky nemocných dospělých pacientů postižených komplikacemi po otevřených srdečních operacích, břišních operacích, vícenásobných zraněních při nehodách nebo po akutním respiračním selhání.
V porovnání s pacienty užívajícími placebo byla úmrtnost vyšší (42 % oproti 19 %) u pacientů léčených růstovými hormony (v dávkách 5,3 až 8 mg/den). Na základě těchto údajů by takto postižení pacienti neměli být růstovými hormony léčeni. Jelikož nejsou k dispozici informace o léčbě akutně kriticky nemocných pacientů náhradou růstového hormonu, musí se za dané situace prospěšnost souvislé léčby poměřovat s potenciálními riziky.
U všech pacientů, u kterých dojde ke vzniku jiných nebo podobných akutního kritického stavu se musí poměřit možný přínos léčby růstovým hormonem s potenciálními riziky.
Maximální doporučená denní dávka by neměla být překročena (viz bod 4.2).
4. 5 Interakce s jinými léčivými přípravky a jiné formy interakce
Současné léčení glukokortikoidy inhibuje účinky přípravků obsahujících somatropin spočívající v podpoře růstu. U pacientů s nedostatkem ACTH je třeba provést pečlivou úpravu substituční terapie glukokortikoidy, aby se předešlo jakémukoliv inhibičnímu účinku na růstový hormon.
Vysoké dávky androgenů, estrogenů nebo anabolických steroidů mohou urychlit zrání kostí a potlačovat stimulaci růstu.
Protože lidský růstový hormon může vyvolat stav inzulínové rezistence, je třeba v případě nutnosti přizpůsobit dávky inzulínu u pacientů, kteří zároveň dostávají Zomacton.
Údaje z interakční studie provedené u dospělých osob s deficitem růstového hormonu ukazují, že aplikace somatropinu může významně zvyšovat clearance sloučenin metabolizovaných cytochromem P450 3A4 (např. pohlavních hormonů, steroidů, kortikosteroidů, antikonvulziv, a cyklosporinu), což má za následek nižší hladinu těchto sloučenin v plazmě. Klinický význam tohoto zjištění není znám.
4. 6 Těhotenství a kojení
Reprodukční studie přípravků obsahujících somatropin provedené u zvířat neprokázaly zvýšené riziko nežádoucích účinků na embryo nebo plod.
Nejsou k dispozici žádné údaje o použití somatropinu v průběhu březosti u zvířat. (Viz bod 5.3 Předklinické údaje vztahující se k bezpečnosti)
Přípravky obsahující somatropin proto nejsou doporučeny v průběhu těhotenství a u fertilních žen, které nepoužívají účinnou kontracepční metodu.
U kojících žen nebyly provedeny žádné klinické studie přípravků obsahujících somatotropin. Není známo, zda je somatropin vylučován do lidského mateřského mléka. Při podávání přípravků obsahujících somatropin kojícím ženám je proto třeba postupovat opatrně.
4. 7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek nemá žádný nebo zanedbatelný vliv na schopnost řízení motorových vozidel a obsluhy strojů.
4. 8 Nežádoucí účinky
Subkutánní aplikace růstového hormonu může vést ke ztrátě tukové tkáně nebo k jejímu růstu v místě vpichu. Ve vzácných případech se u pacientů v místě vpichu objevila bolest a svědivá vyrážka.
Somatropin vede ke zvýšení tvorby protilátek přibližně u 1 % pacientů. Vazebná schopnost těchto protilátek je nízká a s jejich tvorbou nesouvisí žádné klinické změny.
|
Vyjadřování frekvence podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>1/10 000 až <1/1000) |
Velmi vzácné (<1/10 000) |
|
Poruchy krve a lymfatického systému | |||||
|
Srdeční poruchy |
Tachykardie, hypertenze u dospělých |
Hypertenze u dětí | |||
|
Poruchy ucha a labyrintu |
|
Endokrinní poruchy |
Hypotyreóza | ||||
|
Poruchy oka |
Papiloedém, diplopie | ||||
|
Gastrointestinání poruchy |
Zvracení, bolest břicha, nadýmání, nauzea | ||||
|
Celkové poruchy a reakce v místě aplikace |
Otok a periferní otok u dospělých |
Otok a periferní otok u dětí, reakce v místě vpichu, astenie |
Slabost, atrofie v místě vpichu, krvácení v místě vpichu, zatvrdnutí, hypertrofie v místě vpichu | ||
|
Poruchy imunitního systému |
Tvorba protilátek | ||||
|
Vyšetření |
Abnormální hodnoty při vyšetření funkce ledvin | ||||
|
Poruchy metabolismu a výživy |
Střední hyperglykemie u dospělých |
Poruchy glukózové tolerance u dětí |
Hypoglykémie, hyperfosfatemie |
Diabetes mellitus typu II | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie a myalgie u dospělých |
Artralgie a myalgie u dětí Ztuhlost dolních končetin a paží |
Svalová atrofie, bolest kostí, syndrom karpálního tunelu, ztuhlost končetin a paží u dětí | ||
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) |
Maligní nádor, nádor |
Leukémie | |||
|
Poruchy nervového systému |
Bolest hlavy a parestézie u dospělých |
Bolest hlavy, hypertonie, nespavost u dospělých |
Somnolence, nystagmus |
Neuropatie, zvýšený intrakraniální tlak, nespavost u dětí, parestézie u dětí | |
|
Poruchy nervového systému |
Poruchy osobnosti | ||||
|
Poruchy ledvin a močových cest |
Močová inkontinence, hematurie, polyurie, častější močení/polakisurie, abnormalita moče | ||||
|
Poruchy reprodukčního |
Výtok z genitálií, gynekomastie u |
Gynekomastie u dětí |
|
systému a prsu |
dospělých | ||||
|
Poruchy kůže a podkožní tkáně |
Lipodystrofie, atrofie kůže, exfoliativní kopřivka, hirsutismus, hypertrofie kůže |
Protilátky proti somatropinu: protein somatropin může vyvolat tvorbu protilátek. V souvislosti s tímto přípravkem byla u určitého procenta léčené populace zjištěna přítomnost těchto protilátek. Jejich vazebná schopnost a jejich titry jsou však většinou nízké a nevedou k žádným klinickým projevům. Vyšetření protilátek proti somatropinu by mělo být provedeno v případě chybějící odpovědi na terapii somatotropinem.
Leukémie: u dětí s nedostatkem růstového hormonu, z nichž některé byly léčeny somatotropinem v rámci postmarketingové studie, byly popsány případy leukémie (velmi vzácné). Nejsou však žádné důkazy o zvýšení rizika leukémie v nepřítomnosti predisponujících faktorů.
U dětí léčených růstovým hormonem byl hlášen výskyt skluzu horní epifýzy stehenní kosti a juvenilní osteochondrózy hlavice kosti stehenní [Leggovy-Calvéovy-Perthesovy choroby]. Skluz horní epifýzy stehenní kosti se vyskytuje častěji při endokrinních poruchách a juvenilní osteochondróza hlavice kosti stehenní je častější při nízkém vzrůstu. Není však známo, zda tato dvě onemocnění jsou nebo nejsou častější v průběhu léčby somatropinem. K podezření na jejich přítomnost by měl vést nepříjemný pocit nebo bolest v kyčli a/nebo koleni.
Další nežádoucí účinky je možno považovat za skupinový účinek (tzv. class effect), jako například hyperglykemie v důsledku snížené citlivosti na insulin, snížená koncentrace volného tyroxinu a možný rozvoj nezhoubné nitrolební hypertenze.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek 4.9 Předávkování
Doporučená dávka přípravku Zomacton by neměla být překračována.
Ačkoli nejsou žádné zprávy o předávkování přípravkem Zomacton, akutní předávkování může vést k počáteční hypoglykémii s následnou hyperglykémií.
Účinky dlouhodobého, opakovaného užívání přípravku Zomacton v dávkách přesahujících doporučené nejsou známy. Nicméně je možné, že takové užívání může vyvolat podobné příznaky jako při nadbytku lidského růstového hormonu (např. akromegalie).
5 FARMAKOLOGICKÉ VLASTNOSTI
5. 1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hormony, somatotropin a analogy ATC kód: H01AC01
Růstový hormon Zomacton je identický s lidským hypofyzámím růstovým hormonem (pit-hGH) v aminokyselinové sekvenci, délce řetězce (191 aminokyselin) a farmakokinetickém profilu. Lze u něj očekávat stejné farmakologické účinky jako u endogenního hormonu.
Kosterní systém:
Růstový hormon působí u člověka celkový proporcionální růst dlouhých kostí. Po exogenním podávání přípravku Zomacton byl prokázán zvýšený lineární růst u dětí s potvrzeným nedostatkem pit-hGH. Měřitelný výškový přírůstek po podávání přípravku Zomacton je důsledkem působení na epifyzární štěrbiny dlouhých kostí. U dětí, které mají nedostatek odpovídajícího množství pit-hGH, působí Zomacton urychlení růstu a zvýšení koncentrací IGF-1 (inzulinový růstový faktor/somatomedin - C), které jsou podobné koncentracím zjištěným po terapii pit-hGH. Také dochází k zvýšení koncentrace alkalické fosfatázy v krevním séru.
Ostatní orgány a tkáně:
V důsledku působení růstového hormonu dochází k růstu ostatních tkání úměrně celkovému zvýšení tělesné hmotnosti. Změny zahrnují: zvýšený růst pojivových tkání, kůže a vaziva; zvětšení kosterních svalů se zvětšením počtu a velikosti buněk; růst brzlíku; zvětšení jater se zvýšenou buněčnou proliferací; mírné zvětšení gonád, nadledvin a štítné žlázy.
Disproporcionální růst kůže a plochých kostí a urychlení pohlavního dospívání nebyly v souvislosti se substituční terapií růstovým hormonem hlášeny.
Metabolismus bílkovin, uhlohydrátů a lipidů:
Růstový hormon vyvolává retenci dusíku a zvyšuje transport aminokyselin do tkání. Oba procesy zesilují syntézu bílkovin. Využití uhlohydrátů a lipogenese jsou růstovým hormonem potlačeny. Ve velkých dávkách nebo při absenci inzulínu působí růstový hormon jako diabetogenní agens.
Metabolismus minerálních látek:
V důsledku léčby růstovým hormonem dochází k retenci sodíku, draslíku a fosforu. Zvýšení ztrát vápníku ledvinami je kompenzováno zvýšením jeho absorpce ve střevech. Koncentrace vápníku v séru se u pacientů léčených přípravkem Zomacton nebo pit-hGH výrazně nemění. Po podávání přípravku Zomacton a pit-hGH bylo pozorováno zvýšení koncentrace anorganických fosfátů v séru. Hromadění těchto nerostných látek signalizuje zvýšení potřeby během tkáňové syntézy.
5. 2 Farmakokinetické vlastnosti
Farmakokinetický profil je obdobný jako u přirozeného hormonu.
Po aplikaci dávky 0,1 mg/kg těl. hmotnosti osmi zdravým jedincům byla maximální hladina v plazmě v hodnotě asi 64 ng/ml naměřena 6 hodin po podání.
5. 3 Předklinické údaje vztahující se k bezpečnosti
Toxicita jednotlivé dávky:
Studie o toxicitě jednotlivé dávky byly prováděny na potkanech (intramuskulární aplikace 10 mg/kg), psech a opicích (intramuskulární dávka 5 mg/kg , což odpovídá 50 - 100 násobku terapeutické dávky u člověka). U žádného z těchto druhů nebyly zjištěny důkazy toxicity v souvislosti s léčebným přípravkem.
Toxicita opakované dávky:
Žádné známky toxicity nebyly pozorovány ve studii na potkanech, kde byly zvířatům podávány dávky 1,10 mg/kg/den po dobu 30 dnů a 0,37 mg/kg/den po dobu 90 dnů.
Reprodukční toxikologie, mutagenní a kancerogenní potenciál:
Geneticky získaný růstový hormon je identický s endogenním lidským hypofyzárním růstovým hormonem. Má stejné biologické vlastnosti a je obvykle podáván ve fyziologických dávkách. Proto nebylo považováno za nezbytné provádět tyto toxikologické studie v plném rozsahu. Nežádoucí vliv na reprodukční orgány a na průběh těhotenství a laktace je nepravděpodobný a také není třeba předpokládat žádné kancerogenní působení. Mutagenní studie neprokázaly žádné mutagenní účinky.
6 FARMACEUTICKÉ ÚDAJE
6. 1 Seznam pomocných látek
Rozpouštědlo obsahuje benzylalkohol 9 mg/ml
Prášek
Mannitol.
Rozpouštědlo
Chlorid sodný, voda na injekci, benzylalkohol jako konzervační činidlo.
6. 2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky
6. 3 Doba použitelnosti
3 roky
Po naředění se může roztok uchovávat v chladničce (2°C - 8°C) maximálně 14 dní.
Injekční lahvičky uchovávejte ve vzpřímené poloze.
6. 4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8 °C). Lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem. Chraňte před mrazem.
Podmínky uchovávání naředěného léčivého přípravku viz bod 6.3 6. 5 Druh obalu a velikost balení
a) lahvička s práškem - lahvička z bezbarvého skla a pryžovou zátkou a hliníkovým uzávěrem s odtrhovacím plastovým kroužkem.
b) Rozpouštědlo - ampule z bezbarvého skla
Bezjehlový aplikátor ZomaJet 2 Vision nebo jehlový aplikátor Ferring Pen nejsou součástí balení. Velikost balení:
1 lahvička s práškem a 1 ampule rozpouštědla 5 lahviček s práškem a 5 ampulí rozpouštědla 10 lahviček s práškem a 10 ampulí rozpouštědla
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Před použitím se Zomacton lyofilizovaný prášek musí rozpustit příslušným rozpouštědlem.
Roztok může být připraven ve dvou různých koncentracích: 3,3 mg/ml (pro použití aplikátorem ZomaJet 2 Vision, Ferring Pen nebo pro běžné injekční stříkačky) nebo 1,3 mg/ml (pouze pro injekční stříkačky)
Roztok o koncentraci 3,3 mg/ml lze připravit odebráním 1,3 ml roztoku z ampule s rozpouštědlem (balení 4 mg). K odebrání je třeba použít vhodně dělenou stříkačku. Toto množství rozpouštědla se vstříkne do lahvičky se suchým práškem Zomacton.
K získání roztoku o koncentraci 1,3 mg/ml se do lahvičky se suchým práškem přidá 3,2 ml roztoku z ampule s rozpouštědlem.
Aby se zabránilo zpěnění roztoku, musí proud rozpouštědla směřovat proti stěně lahvičky. Prášek je potom třeba rozpouštět mírnými rotačními pohyby, dokud obsah není úplně rozpuštěn a dokud nevznikne čirý bezbarvý roztok. Protože je Zomacton protein, nedoporučuje se třepání nebo prudké míchání. Pokud je po zamíchání roztok kalný nebo obsahuje částečky hmoty, nesmí být použit. V případě, že se obsah zakalí po ochlazení, může se lahvička nechat ohřát na pokojovou teplotu. Pokud zůstává kalný, vyhoďte nádobku i s jejím obsahem.
Podrobné informace pro rozředění jsou uvedeny v příbalové informace pro uživatele.
Aplikace:
Požadovaná dávka Zomactonu se podává bezjehlovým aplikátorem ZomaJet 2 Vision, jehlovým
aplikátorem Ferring Pen nebo běžnou injekční stříkačkou, které nejsou součástí balení.
Všeobecný popis rozředění a aplikaci pomocí bezjehlového aplikátoru ZomaJet 2 Vision je uveden
níže. Rozředění se má provést v souladu s pravidly správné praxe pro použití zejména co se týká
asepse.
1. Důkladně si umyjte ruce.
2. Odstraňte ochranný uzávěr z lahvičky.
3. Očistěte vrchní část lahvičky aseptickým roztokem, aby se zabránilo kontaminaci obsahu lahvičky.
4. Namiřte hrot adaptéru do středu gumové zátky a pevně zatlačte adaptér směrem dolů, až pokud nezapadne na lékovku. Během připojování adaptéru na lahvičku opatrně otáčejte adaptérem, aby hrot adaptéru snadněji propíchl zátku lahvičky.
5. Pro odstranění ochranného krytu z adaptéru jednoduše podržte lahvičku a adaptér v jedné ruce a rovnou vytáhněte víčko. Uložte kryt pro pozdější uchovávání.
Přesný návod k použití aplikátoru ZomaJet 2 Vision je uveden v brožuře, která je k němu přiložena.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
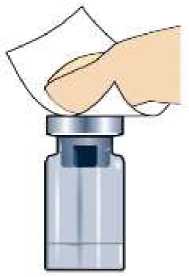
Očistěte gumovou zátku lahvičky alkoholem, aby se zbavila nečist a změkčil se gumový materiál zátky před připojením adaptéru.
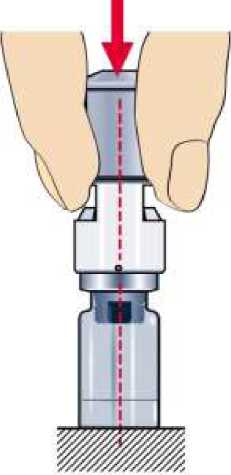
Namiřte hrot adaptéru do středu gumové zátky a pevně zatlačte adaptér směrem dolů, až pokud nezapadne na lékovku. Během připojování adaptéru na lahvičku opatrně otáčejte adaptérem, aby hrot adaptéru snadněji propíchl zátku lahvičky.

Pro odstranění ochranného krytu z adaptéru jednoduše podržte lahvičku a adaptér v jedné ruce a rovnou vytáhněte víčko. Uložte kryt pro pozdější uchovávání.

7 DRŽITEL ROZHODNUTÍ O REGISTRACI
FERRING Pharmaceuticals CZ s.r.o.
K Rybníku 475
252 42 Jesenice u Prahy
Česká republika
8 REGISTRAČNÍ ČÍSLO
56/607/96-C
9 DATUM PRVNÍ REGISTRACE/ PRODLOUŽENÍ REGISTRACE
9. 10. 1996 /13.11.2013
10 DATUM REVIZE TEXTU
11.3.2016
10/10