Willfact 2000 Iu
sp. zn. sukls53368/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Willfact 2000 IU
Prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Willfact je dodáván ve formě prášku a rozpouštědla pro injekční roztok s nominálním obsahem factor von Willebrand humanus 2000 IU v jedné injekční lahvičce.
Přípravek obsahuje přibližně 100 IU/ml factor von Willebrand humanus po rekonstituci 20 ml vody na injekci.
Před přidáním albuminu činí specifická aktivita přípravku Willfact > 50 IU vWF:RCo/mg proteinu.
Účinnost (IU) von Willebrandova faktoru je měřena podle aktivity ristocetinového kofaktoru (vWF:RCo) ve srovnání s mezinárodní normou pro koncentráty von Willebrandova faktoru.
Obsah lidského faktoru VIII v přípravku Willfact činí < 10 IU/100 IU vWF:RCo. Účinnost (IU) faktoru VIII je stanovena pomocí chromogenního testu podle Evropského lékopisu.
Pomocná látka se známým účinkem:
Tento přípravek obsahuje sodík:
Jedna 20 ml lahvička přípravku Willfact 2000 IU obsahuje 0,6 mmol (13,8 mg) sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý až světle žlutý lyofilizovaný prášek nebo drobivá pevná látka.
Rozpouštědlo je čiré a bezbarvé.
4. KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
Willfact je indikován k prevenci a léčbě hemoragií nebo operačního krvácení při von Willebrandově chorobě v případech, kdy je léčba samotným desmopresinem (DDAVP) neúčinná nebo kontraindikovaná.
Willfact nemá být používán k léčbě hemofilie A.
4.2. Dávkování a způsob podání
Na léčbu von Willebrandovy choroby byl měl dohlížet lékař se zkušenostmi s léčbou hemostatických poruch.
Dávkování
Jedna (1) IU/kg von Willebrandova faktoru zpravidla zvyšuje hladinu vWF:RCo v krevním oběhu
0 0,02 IU/ml (2 %).
Má být dosaženo hladiny vWF:RCo v hodnotě > 0,6 IU/ml (60 %) a hladiny FVIII:C > 0,4 IU/ml (40 %).
Hemostázu nelze zajistit, pokud koagulační aktivita FVIII (FVIII:C) nedosáhne 0,4 IU/ml (40 %). Jedna injekce von Willebrandova faktoru nevyvolá maximální zvýšení FVIII:C dříve než za nejméně 6 - 12 hodin. Hladina FVIII:C se jedním podáním von Willebrandova faktoru ihned neupraví. Nachází-li se pacientova počáteční plazmatická hladina FVIII:C pod touto kritickou mezí, je nutno ve všech situacích vyžadujících rychlou úpravu hemostázy, jako např. při léčbě krvácení, těžkém traumatu nebo bezodkladné operaci, podat s první injekcí von Willebrandova faktoru přípravek obsahující faktor VIII, aby bylo dosaženo hemostatické plazmatické hladiny FVIII:C.
Nicméně, není-li okamžité zvýšení FVIII:C nezbytné, například při plánované operaci, nebo je-li počáteční hladina FVIII:C dostatečná pro zajištění hemostázy, může se lékař rozhodnout podat první injekci vWF bez současné injekce faktoru VIII.
Zahájení léčby:
První dávka přípravku Willfact sestává ze 40 až 80 IU/kg k léčbě krvácení nebo traumatu a podává se spolu s požadovaným množstvím přípravku obsahujícího faktor VIII, vypočteným podle základní plazmatické hladiny FVIII:C pacienta, aby se dosáhlo vhodné plazmatické hladiny FVIII:C, a to bezprostředně před zákrokem nebo co nejdříve po začátku krvácivé epizody nebo těžkém traumatu. V případě operace má být přípravek podán 1 hodinu před zákrokem.
Přípravek Willfact může být nutné podat v počáteční dávce 80 IU/kg, zejména u pacientů s von Willebrandovou chorobou (vWD) typu 3, při které může udržování přiměřených hladin vyžadovat vyšší dávky než u ostatních typů vWD.
Při elektivní operaci má léčba přípravkem Willfact začít 12 - 24 hodin před operací a má být opakována
1 hodinu před zákrokem. V tomto případě není současné podání přípravku obsahujícího faktor VIII nutné, protože endogenní FVIII:C obvykle dosáhne kritické hladiny 0,4 IU/ml (40 %) do doby operace. Nicméně je to nutno u každého pacienta potvrdit.
Následující injekce:
V léčbě příslušnou dávkou přípravku Willfact se má v případě potřeby pokračovat, a to v dávce 40 - 80 IU/kg na den podané v 1 nebo 2 injekcích denně po dobu jednoho až několika dnů. Dávka a délka léčby závisí na klinickém stavu pacienta, druhu a závažnosti krvácení a hladinách jak vWF:RCo, tak FVIII:C.
Dlouhodobá profylaxe
Willfact je možno podávat jako dlouhodobou profylaxi v dávce, která je pro každého pacienta stanovena individuálně. Počet krvácivých epizod je snižován dávkami přípravku Willfact mezi 40 a 60 IU/kg podávanými dvakrát až třikrát týdně.
Pediatrická populace
Pro charakteristiku reakce na použití přípravku Willfact u dětí mladších 6 let neexistují data z klinické studie. Použití přípravku Willfact u dětí mladších 12 let je dokumentováno pouze v jednotlivých případech; použití přípravku Willfact u pacientů, kteří dříve nebyli von Willebrandovým faktorem léčeni, není v klinických studiích dokumentováno.
Způsob podání
Přípravek musí být podáván intravenózně, maximální rychlostí 4 ml/min.
Návod k rekonstituci/naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3. Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4. Zvláštní upozornění a opatření pro použití
U pacientů s aktivním krvácením se v první linii léčby doporučuje současné podání přípravku obsahujícího FVIII s přípravkem obsahujícím von Willebrandův faktor s nízkým obsahem FVIII.
Hypersenzitivita
Podobně jako při jakémkoli intravenózním podání proteinu pocházejícího z plazmy, může i při podání přípravku WILLFACT dojít k hypersenzitivním reakcím alergického typu. Pacienti musí být po celou dobu podání injekce pozorně sledováni a pečlivě pozorováni, zda se u nich neobjeví příznaky hypersenzitivní reakce. Pacienty je nutno poučit o časných příznacích hypersenzitivních reakcí, jako např. vyrážka, generalizovaná urtikárie, svírání na hrudi, dýchavičnost, hypotenze a anafylaxe. Pokud se tyto příznaky vyskytnou, musí být podání okamžitě přerušeno. V případě šoku je nutno zavést standardní protišokovou léčbu.
Přenos infekce
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků připravených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a směsí plazmy na specifické ukazatele infekce a zařazení účinných výrobních postupů k inaktivaci/odstranění virů.
Navzdory tomu nelze při podávání léčivých přípravků připravených z lidské krve nebo plazmy možnost přenosu infekčních zárodků úplně vyloučit. To také platí pro neznámé nebo nově vznikající viry a jiné patogeny.
Přijatá opatření se považují za účinná proti obaleným virům, jako např. virus lidské imunitní nedostatečnosti (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV). Tato opatření mohou mít omezenou účinnost proti neobaleným virům, jako např. virus hepatitidy A a parvovirus B19. Infekce parvovirem B19 může být závažná pro těhotné ženy (fetální infekce) a pro osoby s imunodeficiencí nebo zvýšenou erytropoézou (např. hemolytická anemie).
U pacientů pravidelně dostávajících von Willebrandův faktor vyrobený z lidské plazmy se má zvážit příslušné očkování (proti hepatitidě A a hepatitidě B).
Je důrazně doporučeno, aby při každém podání přípravku Willfact pacientovi byly zaznamenány název a číslo šarže léčivého přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Tromboembolie
Existuje riziko výskytu trombotických příhod, zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Proto musí být rizikoví pacienti sledováni, zda se u nich nevyskytnou časné známky trombózy. Má být zavedena profylaxe žilní tromboembolie v souladu s aktuálními doporučeními.
Ošetřující lékař si musí být vědom toho, že nepřetržitá léčba přípravkem obsahujícím spolu s vWF také FVIII může vést k nadměrnému zvýšení FVIII:C. U pacientů dostávajících přípravky obsahující spolu s von Willebrandovým faktorem také faktor VIII je nutno monitorovat plazmatické hladiny FVIII:C, aby se zabránilo trvalým nadměrným plazmatickým hladinám FVIII:C, které by mohly zvyšovat riziko trombotických příhod.
Imunogenicita
U pacientů s von Willebrandovou chorobou, zejména u pacientů s typem 3, může dojít k tvorbě neutralizujících protilátek (inhibitorů) proti vWF. Nebylo-li dosaženo očekávaných plazmatických hladin aktivity vWF:RCo, nebo nelze-li krvácení přiměřenou dávkou kontrolovat, je nutno provést test ke stanovení přítomnosti inhibitoru vWF. U pacientů s vysokými hladinami inhibitoru nemusí být léčba von Willebrandovým faktorem účinná, a je proto nutno zvážit jiné léčebné alternativy.
Pomocná látka vyžadující opatrnost (obsah sodíku)
Tento léčivý přípravek obsahuje sodík. V případě více než 3300 IU injikovaného přípravku (více než 1 mmol sodíku) je třeba toto vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou známy žádné interakce přípravků obsahujících lidský von Willebrandův faktor s jinými léčivými přípravky.
4.6. Fertilita, těhotenství a kojení
Studie fertility, reprodukce, těhotenství, embryonálního/fetálního vývoje nebo perinatálního a postnatálního vývoje na zvířatech jsou nedostatečné pro vyhodnocení bezpečnosti tohoto přípravku.
Bezpečnost přípravku Willfact během těhotenství a kojení nebyla v kontrolovaných klinických studiích hodnocena.
Willfact se má podávat těhotným a kojícím ženám s nedostatkem von Willebrandova faktoru pouze, pokud je to jasně indikováno.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Nebyly pozorovány žádné účinky na schopnost řídit dopravní prostředky nebo obsluhovat stroje.
4.8. Nežádoucí účinky
Shrnutí bezpečnostního profilu
Přecitlivělost a alergické reakce (které mohou zahrnovat angioedém, pálení a píchání v místě infuze, zimnici, zčervenání, kopřivku po celém těle, bolest hlavy, vyrážku, hypotenzi, letargii, nevolnost, neklid, tachykardii, pocit tíhy na hrudi, brnění, zvracení, sípání) byly pozorovány zřídka, v některých případech se ale mohou vyvinout do závažné anafylaxe (včetně šoku). Vzácně byla pozorována horečka.
Velmi vzácně může u pacientů s von Willebrandovou chorobou, zejména u pacientů s typem 3, dojít k tvorbě neutralizujících protilátek (inhibitorů) proti vWF. Pacienty léčené vWF je nutno pečlivě sledovat, zda se u nich nevytvářejí inhibitory, pomocí příslušného klinického pozorování a laboratorních testů. Výskyt takových inhibitorů se projeví jako nedostatečná klinická odpověď. Tyto protilátky se mohou precipitovat a vyskytují se v těsné spojitosti s anafylaktickými reakcemi. Ve všech takových případech se doporučuje kontaktovat specializované hemofilické centrum.
Pacienti, u kterých se vyskytla anafylaktická reakce, by proto měli být vyšetřeni na přítomnost inhibitoru.
Z důvodu rizika trombotické epizody v určitých rizikových situacích je po úpravě nedostatku von Willebrandova faktoru nutno pacienty sledovat, zda se u nich neobjeví časné známky trombózy nebo diseminované intravaskulární koagulace, a zahájit prevenci tromboembolických komplikací v souladu s aktuálními pokyny.
U pacientů dostávajících přípravky obsahující spolu s vWF také faktor VIII mohou trvalé nadměrné plazmatické hladiny FVIII:C zvýšit riziko trombotických příhod.
Bezpečnostní informace týkající se přenosu infekce jsou uvedeny v bodu 4.4.
Tabulkový přehled nežádoucích účinků
Frekvence výskytu nežádoucích účinků je definována podle následující konvence:
Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů četnost nelze určit).
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky |
Frekvence |
|
Poruchy imunitního systému |
Hypersenzitivita nebo alergické reakce. Ty se mohou v některých případech rozvinout až v závažnou anafylaxi (včetně šoku). |
Méně časté |
|
Psychiatrické poruchy |
Neklid |
Méně časté |
|
Poruchy nervového systému |
Bolest hlavy, mravenčení, letargie |
Méně časté |
|
Srdeční poruchy |
Méně časté | |
|
Cévní poruchy |
Hypotenze, návaly |
Méně časté |
|
Respirační, hrudní a mediastinální poruchy |
Dýchavičnost |
Méně časté |
|
Gastrointestinální poruchy |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
Angioedém, generalizovaná urtikarie, kopřivka |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Pálení a píchání v místě infuze, zimnice, svírání na hrudi |
Méně časté |
|
Vzácné | ||
|
Vyšetření |
Neutralizující protilátky (inhibitory) proti vWF |
Velmi vzácné |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu : Státní ústav pro kontrolu léčiv, Šrobárova 48, 100 41 Praha 10 ; Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek.
4.9. Předávkování
Nebyl hlášen žádný případ předávkování přípravkem Willfact.
V případě výrazného předávkování se mohou vyskytnout tromboembolické příhody.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Koagulační faktory: Von Willebrandův faktor
ATC kód: B02BD10
Willfact působí stejným způsobem jako endogenní von Willebrandův faktor.
Podávání von Willebrandova faktoru umožňuje úpravu hemostatických abnormalit vyskytujících se
u pacientů s nedostatkem von Willebrandova faktoru na dvou úrovních:
• vWF obnovuje adhezi krevních destiček k cévnímu subendotelu v místě poškození cévy (jelikož se váže jak na subendotel cévy, tak na membránu krevních destiček), což zajišťuje primární hemostázu, jak je patrno ze zkrácení doby krvácení. O tomto účinku je známo, že do značné míry závisí na úrovni multimerizace léčivé látky.
• Von Willebrandův faktor způsobuje opožděnou korekci souvisejícího nedostatku faktoru VIII. Při intravenózním podání se von Willebrandův faktor váže na endogenní faktor VIII (který pacienti vytvářejí normálně) a stabilizuje ho, čímž brání jeho rychlé degradaci. Následkem toho se hladina FVIII:C po podání čistého von Willebrandova faktoru (přípravek vWF s nízkým obsahem FVIII) navrátí k normálu jako sekundární účinek po první infuzi. Po podání přípravku vWF s obsahem FVIII:C se hladina FVIII:C navrátí k normálu bezprostředně po první infuzi.
5.2. Farmakokinetické vlastnosti
Farmakokinetická studie s přípravkem Willfact byla provedena u 8 pacientů s von Willebrandovou chorobou typu 3. Prokázala, že co se týče vWF:RCo:
• průměrná hodnota AUC0-<X) je 3444 IU.h/dl po podání jedné dávky 100 IU/kg přípravku Willfact,
• vrcholu plazmatické hladiny je dosaženo mezi 30 minutami a 1 hodinou po injekci,
• průměrná recovery je 2,1 [IU/dl]/[IU/kg] injikovaného preparátu,
• poločas se nachází mezi 8 a 14 hodinami, s průměrnou hodnotou 12 hodin,
• průměrná clearance je 3,0 ml/h/kg.
Normalizace hladiny FVIII je progresivní, variabilní a obvykle k ní dochází mezi 6 a 12 hodinami. Tento účinek přetrvává po dobu 2 až 3 dnů.
Vzestup hladiny FVIII je progresivní a normalizuje se po 6 až 12 hodinách. Hladina FVIII se zvyšuje průměrně o 6 % (IU/dl) za hodinu. Proto se i u pacientů s počáteční hladinou FVIII:C nižší než 5 % (IU/dl) hladina FVIII:C zvýší na přibližně 40 % (IU/dl) 6 hodin po injekci, a tato hladina se udržuje po dobu 24 hodin.
5.3. Předklinické údaje vztahující se k bezpečnosti
Na základě údajů získaných z několika předklinických studií provedených na zvířecích modelech neexistují důkazy o jiných toxických účincích přípravku Willfact, než účincích spojených s imunogenicitou lidských proteinů u laboratorních zvířat. Testování toxicity po opakovaných dávkách je neproveditelné z důvodu tvorby protilátek proti heterolognímu proteinu ve zvířecích modelech.
Předklinické údaje vztahující se k bezpečnosti nenaznačují, že by měl Willfact mutagenní potenciál.
6. FARMACEUTICKÉ ÚDAJE 6.1. Seznam pomocných látek
Prášek: roztok lidského albuminu, arginin-hydrochlorid, glycin, dihydrát natrium-citrátu a dihydrát chloridu vápenatého.
Rozpouštědlo: voda na injekci.
6.2. Inkompatibility
Willfact nesmí být mísen s jinými léčivými přípravky s výjimkou koagulačního faktoru FVIII vyrobeného z plazmy od společnosti LFB-BIOMEDICAMENTS, s kterým byla provedena studie kompatibility. Tento koagulační faktor FVIII však není na trhu ve všech evropských zemích.
Mají být použity pouze schválené polypropylenové injekční sety, protože adsorpce lidského von Willebrandova faktoru k vnitřním povrchům některých injekčních vybavení může mít za následek selhání léčby.
6.3. Doba použitelnosti
3 roky.
Chemická a fyzikální stabilita po otevření přípravku byla prokázána po dobu 24 hodin při teplotě 25 °C.
Z mikrobiologického hlediska má být přípravek okamžitě použit. Není-li použit okamžitě, je doba a podmínky skladování po otevření přípravku před použitím odpovědností uživatele.
6.4. Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25 °C. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem. Chraňte před mrazem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5. Druh obalu a obsah balení
1 balení obsahuje: prášek v injekční lahvičce (sklo třídy I) s brombutylovou zátkou, rozpouštědlo v injekční lahvičce (sklo třídy I) s chlorobutylovou zátkou a přepouštěcí set.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rekonstituce:
Musí být dodržovány aktuálně platné pokyny pro aseptické postupy. Přepouštěcí set se používá pouze k rekonstituci léku, tak jak je popsáno níže. Není určen k podání léku pacientovi.
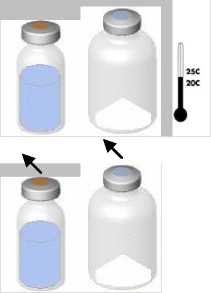
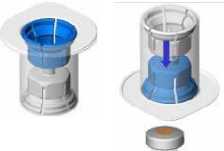
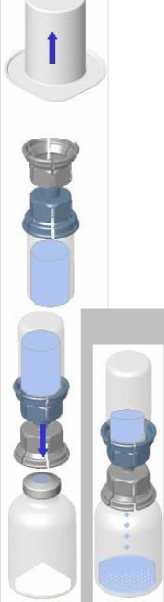
• Zahřejte obě lahvičky (prášek i rozpouštědlo) na teplotu nepřesahující 25 °C.
• Sejměte ochranný uzávěr z lahvičky s rozpouštědlem (voda na injekci) a z lahvičky s práškem.
• Dezinfikujte povrch obou zátek.
Odstraňte víčko ze zařízení Mix2Vial. Bez odstranění zařízení z obalu, k zátce lahvičky s roztokem připevněte modrý konec zařízení Mix2Vial.
• Obal odstraňte a zlikvidujte. Nedotýkejte se nově odhalené části zařízení.
• Spojení zařízení a lahvičkys roztokem otočte dnem vzhůru a připojte k lahvičce s práškem pomocí průhledné části zařízení. Roztok se automaticky přemístí do lahvičky s práškem. Sestavu přidržte a jemně promíchejte, dokud se výrobek zcela nerozpustí.

Roztok by měl být čirý nebo lehce opalescentní, bezbarvý a mírně vazký. Podání:

• Držte lahvičku s vytvořeným produktem ve vertikální pozici a šroubujte sterilní stříkačku na zařízení Mix2Vial. Poté pomalu natáhněte produkt do stříkačky.
• Po přemístění produktu do stříkačky pevně držte stříkačku (pístem směrem dolů), odšroubujte zařízení Mix2Vial a nasaďte intravenózní nebo křídlovou jehlu.
• Vytlačte vzduch ze stříkačky a po dezinfekci povrchu aplikujte do žíly.
• Okamžitě po smíchání jedné dávky pomalu vstřikujte intravenózně při maximální rychlosti 4 ml/minutu.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
LFB-BIOMEDICAMENTS 3, avenue des Tropiques BP 305 - LES ULIS 91958 Courtabreuf Cedex FRANCIE
8. REGISTRAČNÍ ČÍSLO(A) 75/520/15-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 29.10.2015 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
19.11.2015