Vancomycin Mylan 500 Mg
Sp. zn. sukls211423/2014, sukls211424/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Vancomycin Mylan 500 mg Vancomycin Mylan 1000 mg prášek pro infuzní roztok vancomycini hydrochloridum
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje vancomycini hydrochloridum 500 mg, což odpovídá 500 000 IU.
Po rekonstituci 10 ml vody pro injekci obsahuje 1ml rekonstituovaného roztoku vancomycinum 50 mg.
Jedna injekční lahvička obsahuje vancomycini hydrochloridum 1g, což odpovídá 1 000 000 IU.
Po rekonstituci 20 ml vody pro injekci obsahuje 1ml rekonstituovaného roztoku vancomycinum 50 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro infuzní roztok.
Popis přípravku: bílý až téměř bílý prášek.
pH roztoku po rekonstituci je 2,8 - 4,5.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Intravenózní podání Terapeutické podání
Vancomycin Mylan podávaný intravenózně je indikován k léčbě závažných, potencionálně život ohrožujících infekcí vyvolaných citlivými grampozitivními mikroorganizmy, které nelze léčit jinými účinnými méně toxickými antimikrobiálními přípravky, jako jsou peniciliny a cefalosporiny, nebo jejichž léčba těmito přípravky selhala.
Vankomycin má být vyhrazen jen pro ty případy, u kterých je specificky indikován, s cílem minimalizovat vznik možné rezistence.
Vankomycin je indikován k léčbě následujících závažných infekcí vyvolaných citlivými mikroorganizmy (viz bod. 5.1):
• endokarditida
• infekce kostí (osteomyelitida)
• pneumonie
• infekce měkkých tkání
• bakteriemie způsobená methicilin rezistentními stafylokoky u pacientů s endokarditidou,
pneumonií nebo infekcemi měkkých tkání
V léčbě endokarditidy způsobené Streptococcus viridans nebo S. bovis se vankomycin užívá v kombinaci s aminoglykosidy.
Profylaktická léčba
Vankomycin může být použit v profylaxi perioperačních infekcí způsobených grampozitivní mikroorganizmy, a to zejména při rizikových výkonech jako je kardiovaskulární, hrudní chirurgie, nebo výkony spojené se zaváděním protetických materiálů nebo pomůcek.
Vankomycin má být podáván pacientům s vysokým rizikem infekční endokarditidy (např. s predisponujícím srdečním onemocněním), kteří netolerují beta-laktamová antibiotika, nebo je-li známo, že infekce je způsobena methicilin-rezistentním Staphylococcus aureus (MRSA).
Perorální podání
Vankomycin může být použit perorálně při léčbě pseudomembranózní kolitidy způsobené Clostridium difficile v případě závažné infekce, relapsu nebo selhání jiné léčby.
POZNÁMKA: Intravenózně podaný vancomycin není účinný při léčbě pseudomembranózní kolitidy. Pozornost je třeba věnovat oficiálním doporučením o správném použití antibakteriálních látek.
4.2 Dávkování a způsob podání Intravenózní podání
Nedoporučují se vyšší koncentrace než 5 mg/ml. U vybraných pacientů vyžadujících omezený přívod tekutin, je možné použít koncentraci až 10 mg/ml. Použití vyšších koncentrací může zvýšit riziko vzniku nežádoucích účinků souvisejících s infuzí. (viz bod 6.6)
Infuze má trvat nejméně 60 minut. U dospělých, pokud se použijí dávky nad 500 mg, se doporučuje rychlost infuze maximálně 10 mg/min. Výskyt příhod souvisejících s infuzí závisí jak na koncentraci, tak na rychlosti podání vankomycinu. Dávka a trvání léčby se řídí závažností infekce a její klinickou a bakteriologickou progresí.
Pacienti s normální funkcí ledvin a jater
Dospělí a děti starší 12 let:
Doporučená intravenózní denní dávka je 2000 mg (2g). Podává se rozděleně v dávkách 500 mg každých 6 hodin, anebo 1000 mg každých 12 hodin.
Pro léčbu bakteriální endokarditidy je obecně uznávaný dávkovací režim 1000 mg vankomycinu intravenózně každých 12 hodin po dobu 4 týdnů buď samostatně, nebo v kombinaci s jinými antibiotiky (gentamicin plus rifampicin, gentamicin nebo streptomycin). Enterokoková endokarditida se léčí po dobu 6 týdnů vankomycinem v kombinaci s aminoglykosidy - podle národních standardních postupů.
Perioperační profylaxe: dospělí pacienti dostávají 1000 mg vankomycinu intravenózně (před uvedením do anestezie) a v závislosti na délce a typu operace může být podána dávka 1000 mg vankomycinu i.v. 12 hodin po operaci.
Antibiotická profylaxe má být krátkodobá a omezená na 24hodinové perioperační období. Nemá přesáhnout 48 hodin.
Děti od věku 1 měsíce do 12 let:
Doporučená intravenózní dávka je 10 mg/kg každých 6 hodin nebo 20 mg/kg každých 12 hodin.
Novorozenci a kojenci:
Doporučená úvodní dávka je 15 mg/kg, potom 10 mg/kg každých 12 hodin v průběhu prvního týdne života a potom každých 8 hodin do dosažení věku 1 měsíc. Je doporučována pečlivá kontrola hladin vankomycinu v séru (viz níže).
Starší populace:
Nižší udržovací dávky mohou být nutné vzhledem ke snížení funkce ledvin související s věkem.
Obézní pacienti:
Může být potřebná úprava obvyklého denního dávkování.
Pacienti s poruchou funkce jater
Neexistuje žádný důkaz o tom, že by dávka měla být u pacientů s poruchou funkce jater snížena.
Pacienti s poruchou funkce ledvin
U těchto pacientů musí být dávka upravena a následující nomogram může posloužit jako vodítko. Doporučuje se pečlivé sledování sérové koncentrace vankomycinu (viz níže).
Clearance kreatininu (ml/s)
0.50 1.00 1.50
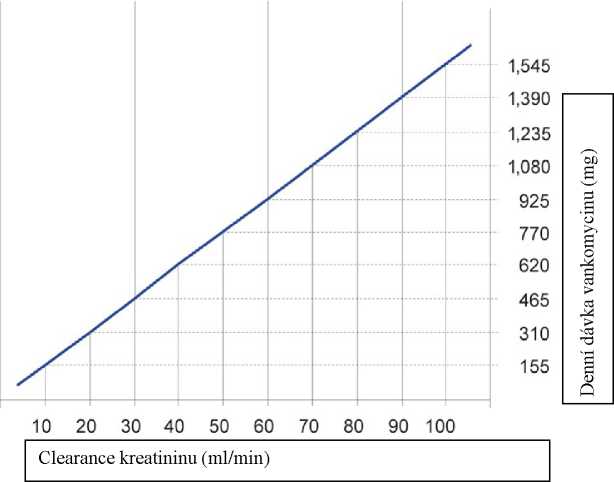
Dávkovací nomogram pro dospělé pacienty s poruchou funkce ledvin
Pokud není clerance kreatininu známa, je možné použít následující výpočet clearance kreatininu na základě věku, pohlaví a hladin sérového kreatininu.
Muži: Tělesná hmotnost [kg] x (140 - věk [v letech])
72 x hladina sérového kreatininu [mg/100 ml]
Ženy: 0,85 x hodnota vypočtená podle vzorce pro muže.
Pokud je to možné, má být vždy určena clearance kreatininu.
U pacientů s mírným nebo středně těžkým renálním selháváním nemá být počáteční dávka nižší než 15 mg/kg. U pacientů se závažným renálním selháváním je vhodnější podávat udržovací dávku mezi 250 mg a 1000 mg v odstupu několika dnů, spíše než podávat nižší denní dávky.
U pacientů s anurií (praktickou absencí renálních funkcí) se podává dávka 15 mg/kg tělesné hmotnosti, dokud není dosaženo terapeutické sérové koncentrace vankomycinu. Udržovací dávky jsou 1,9 mg/kg tělesné hmotnosti/24 hodin.
K usnadnění postupu je možné aplikovat pacientům se závažným postižením funkce ledvin jednu udržovací dávku 250 mg až 1000 mg vždy jednou za několik dní, spíše než každodenně.
Dávkování v případě hemodialýzy:
U pacientů s nefunkčními ledvinami, podstupujících pravidelně hemodialýzu je možné následující dávkování: saturační dávka 1000 mg vankomycinu, udržovací dávka 1000 mg podaná každý sedmý až desátý den.
Pokud se používají při hemodialýze polysulfonové membrány (dialýza s vysokým průtokem), je poločas vankomycinu snížen. U pravidelně hemodialyzovaných pacientů mohou být proto nutné přídatné udržovací dávky.
Monitorování koncentrací vankomycinu v séru:
Sérové koncentrace vankomycinu mají být sledovány od druhého dne léčby bezprostředně před další dávkou a jednu hodinu po infuzi. Terapeutická hladina vankomycinu v krvi jednu hodinu po ukončení infuze má být mezi 30 a 40 mg/l (maximum 50 mg/l), minimální hladina (krátce před další dávkou) pak mezi 5 a 10 mg/l.
Koncentrace vankomycinu v séru má být běžně kontrolována dvakrát nebo třikrát týdně.
Perorální podání
Léčba kolitidy způsobené C. diffcile
Dospělí: Obvyklá denní dávka je 0,5 až 2g rozdělené do 4 dávek (125 mg-500,mg na jednu dávku) po dobu 7 až 10 dnů.
Děti: V terapii dětské pseudomembranózní kolitidy se obvykle podává dávka 40 mg/kg, rozdělená do čtyř dávek. Maximální dávka je 250mg/dávku podávaná po dobu 7 až 10 dní.
Způsob podání:
Pouze k intravenózní infuzi. Není určeno k intramuskulárnímu podání.
Parenterálně se vankomycin může podávat pouze jako pomalá intravenózní infuze (ne více než 10 mg/min - po dobu nejméně 60 min), která je dostatečně zředěná (alespoň 100 ml /500 mg nebo nejméně 200 ml /1000 mg).
Pacienti s omezením přívodu tekutin mohou dostat roztok 500 mg/50 ml nebo 1000 mg/100 ml.
S těmito vyššími koncentracemi může však stoupat riziko vzniku nežádoucích účinků souvisejících s infuzí.
Připravený roztok může být také použit pro perorální podání.
Léčebné indikace pro intravenózní a perorální podání jsou různé. Obě cesty podání nelze zaměnit. Informace o přípravě roztoku jsou uvedeny v bodě 6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním.
4.3 Kontraindikace
Hypersenzitivita na vankomycin nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a zvláštní opatření pro použití Upozornění:
V případě závažných akutních hypersensitivních reakcí (např. anafylaxe) musí být léčba vankomycinem okamžitě přerušena a musí být zahájena opatření obvyklá pro naléhavé případy (např. podání antihistaminik, kortikosteroidů, a pokud je to nezbytné, i umělé dýchání).
Rychlé podání ve formě bolusu (tj. po dobu několika minut) může být spojeno s těžkou hypotenzí (včetně šoku a vzácně i srdeční zástavy), s reakcí jako po histaminu, jako je makulopapulózní nebo erytematózní vyrážka (tzv. syndrom rudého muže nebo syndrom červeného krku). Vankomycin je třeba infundovat pomalu ve zředěném roztoku (2,5 až 5,0 g/l) rychlostí nejvýše 10 mg/min po dobu nejméně 60 minut, aby se zamezilo reakcím spojeným s rychlou infuzí. Po přerušení infuze tyto reakce obvykle rychle pominou.
Nefrotoxicita: vankomycin musí být u pacientů se selháním ledvin používán s opatrností, protože při dlouhodobě vysoké koncentraci v krvi je riziko rozvoje jeho toxických účinků mnohem vyšší. Při léčbě těchto pacientů a u pacientů, kteří užívají současně jiné nefrotoxické látky (např. aminoglykosidy) musí být pečlivě monitorována funkce ledvin a je nutné postupovat podle příslušných dávkovacích schémat, aby se snížilo riziko nefrotoxicity na minimum (viz bod 4.2).
Ototoxicita: ototoxicita, která může být přechodná nebo trvalá, byla hlášena u pacientů s předchozím ohluchnutím po příliš vysokých intravenózních dávkách vankomycinu nebo jako důsledek současně podávaných jiných ototoxických léčivých látek, jako jsou aminoglykosidy. Hluchotě může předcházet tinnitus. Zkušenosti s jinými antibiotiky naznačují, že hluchota může být progresivní i přes ukončení léčby. Pro snížení rizika ototoxicity se doporučuje pravidelně monitorovat hladiny vankomycinu v krvi a periodicky testovat sluchové funkce.
Klinicky významné sérové koncentrace byly hlášeny u některých pacientů léčených pro aktivní pseudomembranózní kolitidu způsobenou C. difficile po opakovaných perorálních dávkách vankomycinu. Proto může být u těchto pacientů vhodné sledovat sérové koncentrace vankomycinu.
Upozornění:
Vankomycin silně dráždí tkáně a vyvolává nekrózu v místě vpichu, jestliže je podán intramuskulárně. Bolest a tromboflebitida se může objevit u řady pacientů, kteří dostávají vankomycin, a může být případně i závažná. Četnost a závažnost tromboflebitidy je možné minimalizovat pomalou aplikací látky ve zředěném roztoku (viz bod 6.6) a pravidelným střídáním místa infuze.
Vankomycin má být používán s opatrností u pacientů s alergickou reakcí na teikoplanin, protože byly hlášeny zkřížené hypersenzitivní reakce mezi vankomycinem a teikoplaninem.
Vankomycin může prohloubit depresi myokardu vyvolanou anestetikem. V průběhu anestezie se dávky vankomycinu musí podávat dobře naředěné a pomalu, za pečlivého monitorování srdečních funkcí. Aby se umožnila adaptace na změnu polohy, je třeba změnu polohy odložit na dobu po skončení infuze.
U pacientů, kteří dostávají vankomycin, je třeba monitorovat v pravidelných intervalech hematologické hodnoty a funkci ledvin, stejně jako sluch.
S prodlouženou dobou užívání se doporučuje pravidelné sledování hladin vankomycinu v krvi v průběhu léčby, zejména u pacientů s poruchou funkce ledvin nebo sluchovým postižením, nebo pokud jsou současně podávány ototoxické nebo nefrotoxické látky, jako například aminoglykosidy. V takových případech má být funkce ledvin pravidelně sledována a dávkování se má upravit tak, aby odpovídalo
5
případnému snížení funkce ledvin.
U pacientů s poruchou sluchové funkce, u pacientů současně užívajících ototoxické léky a u pacientů s renální dysfunkcí je nutné pravidelné sledování sluchových funkcí.
Pediatrická populace:
U předčasně narozených dětí a malých dětí může být nezbytné potvrzení cílových sérových hladin vankomycinu. Současné podávání vankomycinu a anestetik u dětí bylo spojeno se vznikem erytému a zčervenáním jako po histaminu. Je-li podání vankomycinu nutné pro chirurgickou profylaxi, je vhodné aplikovat anestetika až po dokončení infuze vankomycinu.
Používání u starších osob:
Přirozený úbytek glomerulární filtrace se zvyšujícím se věkem může vést ke zvýšení sérových hladin vancomycinu. Starší lidé jsou zvláště citliví na poškození sluchu a sluchové funkce pacientů nad 60 let mají být pravidelně sledovány. Je třeba se vyvarovat souběžného nebo následného podání jiných nefrotoxických látek.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Jiné potenciálně nefrotoxické nebo ototoxické léčivé látky
Souběžné nebo následné podávání vankomycinu s jinými potenciálně neurotoxickými nebo nefrotoxickými látkami, zejména s gentamicinem, amfotericinem B, streptomycinem, neomycinem, kanamycinem, amikacinem, tobramycinem, viomycinem, bacitracinem, polymyxinem B, kolistinem a cisplatinou mohou potencovat nefrotoxicitu a/nebo ototoxicitu vankomycinu, takže je třeba pacienta pečlivě kontrolovat.
Pro synergické působení (např. s gentamicinem) je v těchto případech třeba maximální dávku vankomycinu snížit na 500 mg každých 8 hodin.
Anestetika
Současné podávání vankomycinu a anestetik bylo spojeno s erytémem, zčervenáním jako po histaminu a s anafylaktoidními reakcemi. Tyto reakce mohou být redukovány podáním vankomycinu 60 minut před indukcí anestezie.
Mvorelaxancia
Podává-li se vankomycin v průběhu chirurgického výkonu anebo bezprostředně po něm, může být účinek (nervosvalová blokáda) současně použitých myorelaxancií (např. sukcinylcholinu) zesílen nebo prodloužen.
4.6 Fertilita, těhotenství a kojení
Adekvátní bezpečnostní údaje o podávání vankomycinu ženám během těhotenství nejsou k dispozici. Studie reprodukční toxicity u zvířat nenaznačují jakékoliv účinky na embryonální/fetální vývoj nebo na průběh těhotenství (viz bod 5.3).
Vankomycin však proniká placentou a nelze vyloučit potenciální riziko ototoxického a nefrotoxického ovlivnění plodu a novorozence. Vankomycin se proto může v průběhu těhotenství podávat, pouze pokud je jednoznačně indikován a po pečlivém zvážení očekávaného prospěchu a možného rizika.
Kojení
Vankomycin se vylučuje do mateřského mléka, a proto by se v období kojení měl podávat pouze tehdy, jestliže jiná antibiotika selhala. Vankomycin je třeba podávat kojícím matkám opatrně, protože je
6
možnost výskytu nežádoucích účinků u kojenců (poruchy střevní flory s průjmy, osídlení kvasinkám podobnými organismy a možná senzibilizace).
Po zvážení důležitosti vankomycinu pro kojící matku důležitá, je třeba rozhodnout, zda se má kojení ukončit.
Fertilita
Fertilitní studie (ať už u mužů nebo žen) nebyly prováděny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vancomycin nemá žádný nebo jen zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Nežádoucí účinky, uvedené níže, jsou definovány za použití MedDRA klasifikace a dle systému orgánového třídění jako: velmi časté (> 1/10), časté (> 1/100, < 1/10), méně časté (> 1/1000, < 1/100), vzácné (>1/10 000, < 1/1000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Mezi nejčastější nežádoucí účinky patří zánět žil a pseudoalergické reakce v souvislosti s příliš rychlým intravenózním podáním vankomycinu.
|
Třída orgánových systémů |
Frekence výskytu |
|
Poruchy krve a lymfatického systému |
Vzácné - trombocytopenie - neutropenie - agranulocytóza - eosinofilie |
|
Poruchy imunitního systému |
Vzácné - anafylaktoidní reakce - hypersenzitivní reakce |
|
Poruchy ucha a labyrintu |
Méně časté - přechodná nebo trvalá ztráta sluchu Vzácné - tinnitus - závratě |
|
Srdeční poruchy |
Velmi vzácné - srdeční zástava |
|
Cévní poruchy |
Časté - pokles krevního tlaku - tromboflebitida Vzácné - vaskulitida (zahrnující leuko-cytoklastickou/ hypersenzitivní vaskulitidu) |
|
Respirační, hrudní a mediastinální |
Časté |
|
poruchy |
- dušnost, stridor |
|
Gastrointestinální poruchy |
Vzácné - nauzea Velmi vzácné - pseudomembranózní enterokolitida po intravenózním podání |
|
Poruchy kůže a podkožní tkáně |
Časté - vyrážka a zánět sliznic - svědění - kopřivka Velmi vzácné - exfoliativní dermatitida - Stevens-Johnsonův syndrom - Lyellův syndrom - bulózní dermatitida vyvolaná lineární IgA Není známo - léková reakce s eozinofilií a systémovými příznaky (DRESS) |
|
Poruchy ledvin a močových cest |
Časté - renální insuficience s primárními projevy zvýšené koncentrace kreatininu nebo močoviny v séru Vzácné - intersticiální nefritida - akutní selhání ledvin |
|
Celkové poruchy a reakce v místě |
Časté - zarudnutí horní poloviny těla a obličeje - bolest a křeče hrudních a zádových svalů Vzácné - léková horečka - třesavka |
|
aplikace |
Komentář [KM1]: Prosím naformátovat do tabulky dle common verze
Během infuze nebo krátce po rychlé infuzi může dojít k anafylaktické reakci. Projevy zahrnují hypotenzi, dušnost, urtikárii nebo pruritus. Reakce se zmírní, když je infuze zastavena, a poté obvykle zcela vymizí mezi 20 minutami až 2 hodinami po zastavení infuze.
Ototoxicita byla primárně pozorována u pacientů, kteří dostávali vysoké dávky přípravku nebo byli současně léčeni jinými ototoxickými přípravky nebo pokud měli preexistující poruchu funkce ledvin nebo sluchu.
Po perorálním podání existuje možnost absorpce vankomycinu v místě léze trávicího traktu a riziko výše zmíněných nežádoucích účinků nelze vyloučit.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Toxicita v důsledku předávkování byla hlášena. Podání dávky 500 mg i.v. dvouletému dítěti mělo za následek smrtelnou intoxikaci. Podání celkem 56 g během 10 dní dospělému pacientovi způsobilo selhání ledvin. U určitých vysoce rizikových stavů (např. v případě závažného poškození ledvin) se mohou objevit vysoké hladiny vankomycinu v séru a ototoxické/nefrotoxické účinky.
Opatření při předávkování
• Specifické antidotum není známo.
• Je nutná symptomatická terapie a udržování renálních funkcí.
Vankomycin se hemodialýzou nebo peritoneální dialýzou odstraňuje špatně. Ke snížení koncentrací vankomycinu v séru byla použita hemofiltrace nebo hemoperfuze polysulfonovými pryskyřicemi.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
ATC klasifikace:
Farmakoterapeutická skupina: jiná antibakteriální léčiva, glykopeptidová antibiotika,
ATC kód: J01XA01.
Mechanismus účinku
Vankomycin je glykopeptidové antibiotikum. Vankomycin má baktericidní účinek na proliferující bakterie prostřednictvím inhibice biosyntézy buněčné stěny tvorbou komplexů a peptidoglykanovými prekurzory. Kromě toho narušuje permeabilitu bakteriální buněčné membrány a RNA syntézu.
Mechanismus vzniku rezistence
Rezistence vůči vankomycinu může být založena na těchto mechanismech:
• Změna cílové struktury: Tato forma rezistence se objevila v posledních několika letech, zejména u druhu Enterococcus faecium. Tato změna je důsledkem nahrazení terminální D-alanin-D-alanin funkce postranního řetězce aminokyselin v prekurzoru mureinu D-Ala-D-laktátem, což vede ke značnému snížení afinity k vankomycinu.
• U stafylokoků je snížená citlivost nebo rezistence na vankomycin založená na nadprodukci prekurzorů mureinu, na které se vankomycin váže.
Neexistuje zkřížená rezistence mezi vankomycinem a dalšími druhy antibiotik, dochází ale ke zkřížené rezistenci s jinými glykopeptidovými antibiotiky, jako je teikoplanin. Sekundární vznik rezistence během léčby je vzácný.
V některých zemích jsou rostoucí případy rezistence pozorovány především u enterokoků, nárůst je obzvláště alarmující u multirezistentních kmenů Enterococcus faecium.
Synergismus
Kombinace vankomycinu s aminoglykosidovým antibiotikem synergicky působí proti četným kmenům Staphylococcus aureus, neenterokokových D-streptokoků, enterokoků a streptokoků skupiny viridans. Kombinace vankomycinu s cefalosporinem synergicky působí proti některým oxacilin-rezistentním kmenům Staphylococcus epidermidis a kombinace vankomycinu s rifampicinem působí synergicky na kmeny Staphylococcus epidermidis a má i částečně synergický efekt proti některým kmenům Staphylococcus aureus. Protože kombinace vankomycinu s cefalosporinem může mít také antagonistický efekt u některých kmenů Staphylococcus epidermidis a v kombinaci s rifampicinem u některých kmenů Staphylococcus aureus, je vhodné ověření synergismu před podáním. Je třeba získat bakteriální vzorky pro kultivaci s cílem izolace a identifikace vyvolávajících mikrobů a zjištění jejich citlivosti na vankomycin.
Hraniční hodnoty
Hraniční hodnoty minimální inhibiční koncentrace (MIC) stanovené Evropským výborem pro testování antimikrobiální citlivosti (EUCAST) jsou pro Staphylococcus spp. a Streptococcus spp. citlivé < 2 mg/l a rezistentní > 2 mg/l; pro koaguláza-negativní stafylokoky citlivé < 4 mg/L a resistentní > 4 mg/L; pro Enterococcus spp. citlivé < 4 mg/l a rezistentní > 4 mg/l; pro nepříbuzné druhy citlivé < 2 mg/l a rezistentní > 4 mg/l.
Citlivost
Prevalence získané rezistence se může lišit geograficky a v čase pro vybrané kmeny a je vhodné vzít v úvahu i místní informace o rezistenci zejména při léčbě závažných infekcí. Podle potřeby má být využito odborného poradenství, zejména jestliže je lokální výskyt rezistence takový, že prospěšnost léčiva je přinejmenším u některých typů infekce sporná.
Vankomycin má úzké spektrum účinku.
BĚŽNĚ CITLIVÉ KMENY Aerobní grampozitivní mikroorganizmy
Staphylococcus aureus Koaguláty- negativní Staphylococcus Staphylococcus spp Streptococcus pneumoniae Streptococcus spp Enterococcus spp Anaerobní mikroorganizmy Clostridium difficile
KMENY, U NICHŽ ZÍSKANÁ REZISTENCE MŮŽE BÝT PROBLÉM
Enterococcus faecium
PŘIROZENĚ REZISTENTNÍ ORGANIZMY
Gramnegativní mikroorganizmy, mykobakterie, houby
5.2 Farmakokinetické vlastnosti Distribuce
Po intravenózním podání se vankomycin distribuuje téměř do všech tkání a difunduje do pleurální, perikardiální, ascitické a synoviální tekutiny i do srdečního svalu a srdečních chlopní. Dosahuje zde srovnatelných koncentrací jako v krevní plazmě. Údaje o koncentracích v kosti (ve spongióze, v kompaktě) jsou velmi rozdílné. Zdánlivý distribuční objem za rovnovážného stavu se udává na hodnotě 0,43 (až 0,9) l/kg. Skrz nezanícené meningy prochází vankomycin hematoencefalickou bariérou jen v nepatrné míře. Vankomycin je z 30 až 55 % a někdy i více vázán na plazmatické bílkoviny.
Po perorálním opakovaném podávání vankomycinu byly pozorovány u pacientů léčených pro pseudomembranózní kolitidu způsobenou Clostridium difficile plazmatické koncentrace vankomycinu.
Vylučování
Vankomycin se metabolizuje pouze v malém rozsahu. Po parenterálním podání se vylučuje téměř kompletně jako mikrobiologicky aktivní látka (přibližně 75-90 % během 24 hodin) glomerulární filtrací ledvinami. Biliární exkrece je nevýznamná (méně než 5 % podané dávky).
U pacientů s normální funkcí ledvin je poločas v séru zhruba 4-6 (5-11) hodin, u dětí 2,2-3 hodiny. Při poruše renálních funkcí se poločas vankomycinu může značně prodloužit (až na 7,5 dne). V takových případech je vzhledem k ototoxicitě vankomycinu indikováno monitorování plazmatických koncentrací.
Průměrné plazmatické koncentrace po i.v. infuzi 1 g vankomycinu po dobu 60 minut byly kolem 63 mg/l ke konci infuze, zhruba 23 mg/l za 2 hodiny a asi 8 mg/l po 11 hodinách.
Plazmatická clearance vankomycinu úzce koreluje s glomerulární filtrací.
Celková systémová a renální clearance vankomycinu může být u starších pacientů snížena. Jak ukazují studie u anefrických pacientů, metabolická clearance se zdá být velmi malá.
U člověka dosud nebyly identifikovány žádné metabolity vankomycinu.
Podává-li se vankomycin v průběhu peritoneální dialýzy intraperitoneálně, vstoupí do systémové cirkulace v průběhu 6 hodin cca 60 % podaného množství. Po i.p. podání vankomycinu v dávce 30 mg/kg tělesné hmotnosti je dosaženo sérových hladin přibližně 10 mg/l.
Po perorálním podání se vysoce polární vankomycin prakticky neabsorbuje. Po perorální aplikaci se objeví v aktivní formě ve stolici, a představuje proto vhodné chemoterapeutikum při pseudomembranózní a stafylokokové kolitidě.
Vankomycin snadno přestupuje přes placentu a je distribuován do pupečníkové krve.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinická data neodhalila žádné zvláštní riziko pro člověka na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaných dávkách.
Omezené údaje o mutagenních účincích vykazují negativní výsledky, dlouhodobé studie u zvířat nejsou, pokud jde karcinogenní potenciál, k dispozici. Ve studiích teratogenity, kde laboratorní potkani a králíci dostávali dávky přibližně odpovídající dávce u lidí přepočtené na tělesný povrch (mg/m2), nebyly žádné přímé či nepřímé teratogenní účinky pozorovány.
Studie na zvířatech sledující použití v perinatálním/postnatální období a studie účinků na fertilitu nejsou k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Roztok kyseliny chlorovodíkové 0,5 mol/l (k úpravě pH)
6.2 Inkompatibility
Roztok vankomycinu má nízké pH. To může způsobit chemickou nebo fyzikální nestabilitu po smísení s jinými látkami. Proto by měla být u každého parenterálního roztoku před použitím vizuálně zkontrolována případná přítomnost sraženin a změna zabarvení. Mísení s alkalickými roztoky je třeba se vyhnout.
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch uvedených v bodu 6.6.
6.3 Doba použitelnosti
3 roky
Doba použitelnosti rekonstituovaného roztoku
Chemická a fyzikální stabilita přípravku po rekonstituci ve vodě na injekci byla prokázána po dobu 48 hodin při teplotě 25 °C a po dobu 96 hodin při teplotě 2-8°C.
Doba použitelnosti naředěného roztoku
Chemická a fyzikální stabilita naředěného roztoku (0,9% roztokem chloridu sodného nebo 5% roztokem glukózy) připraveného k použití byla prokázána po dobu 48 hodin při teplotě 25 °C a po dobu 96 hodin při teplotě 2-8 °C.
Z mikrobiologického hlediska má být infuzní roztok použit okamžitě. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by tato doba neměla být delší než 24 hodin při teplotě 2° až 8 °C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
Doba použitelnosti rekonstituovaného roztoku k perorálnímu podání Rekonstituovaný roztok je nutné použít ihned.
6.4 Zvláštní opatření pro uchovávání
Prášek
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Rekonstituovaný a naředěný roztok Podmínky uchovávání viz bod 6.3.
6.5 Druh obalu a velikost balení
Injekční lahvička z bezbarvého skla typu II s bromobutylovou pryžovou zátkou a hliníkovým víčkem s plastovým flip-off uzávěrem.
Velikost balení: 1, 5, 10 nebo 20 injekčních lahviček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním Příprava infuzního roztoku
Přípravek musí být rekonstituován a vzniklý koncentrát musí být před podáním dále naředěn. Vancomycin Mylan 500 mg: Obsah jedné lahvičky se rozpustí v 10 ml vody na injekci.
Vancomycin Mylan 1000 mg: Obsah jedné lahvičky se rozpustí ve 20 ml vody na injekci.
Jeden ml rekonstituovaného roztoku obsahuje 50 mg vankomycinu.
Podmínky uchovávání rekonstituovaného léčivého přípravku, viz bod 6.3.
Vhodná rozpouštědla pro další ředění vankomycinu jsou voda na injekci, 5 % roztok glukózy a 0,9 % roztok chloridu sodného.
V závislosti na způsobu podání se liší i způsob ředění.
Intermitentní infuze:
Vancomycin Mylan 500 mg: Rekonstituované roztoky, které obsahují 500 mg vankomycinu musí být dále naředěny alespoň 100 ml rozpouštědla. Požadovaná dávka se podává intravenózní infuzí rychlostí ne více než 10 mg/min po dobu alespoň 60 minut.
Vancomycin Mylan 1000 mg: Rekonstituované roztoky, které obsahují 1000 mg vankomycinu musí být dále naředěny nejméně 200 ml rozpouštědla. Požadovaná dávka se podává intravenózní infuzí rychlostí ne více než 10 mg/min po dobu alespoň 60 minut.
Kontinuální infuze:
Kontinuální infuze má být použita pouze v případě, kdy léčba intermitentní infuzi není možná. 1000 mg až 2000 mg vankomycinu (což odpovídá 2 až 4 lahvičkám rekonstituovaného roztoku) se naředí v dostatečném množství některého výše uvedených vhodných rozpouštědel tak, aby bylo možné podat požadovanou denní dávku v infuzi trvající 24 hodin.
Podmínky uchovávání ředěných roztoků viz bod 6.3.
U rekonstituovaného a ředěného roztoku musí být před použitím vizuálně zkontrolována případná přítomnost pevných částic a změna zabarvení. Lze použít pouze čirý, bezbarvý nebo světle žlutý roztok bez pevných částic.
Příprava perorálního roztoku
Rekonstituovaný roztok lze dále naředit 30 ml vody a pacient/ka roztok buď vypije, nebo mu/jí může být podán nazogastrickou sondou.
Likvidace
Lahvičky jsou určeny pouze pro jednorázové použití. Nepoužitý přípravek musí být zlikvidován. Všechen nepoužitý přípravek nebo odpadní materiál musí být zlikvidovány v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Mylan S.A.S.
117 allée des Parcs 69 800 Saint Priest Francie
8. REGISTRAČNÍ ČÍSLA
Vancomycin Mylan 500 mg: 15/354/11-C Vancomycin Mylan 1000 mg: 15/355/11-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25.5.2011
Datum posledního prodloužení registrace: 19.5.2016
10. DATUM REVIZE TEXTU
13.7.2016
13