Vancomycin Actavis 500 Mg
Sp. zn. sukls162350/2015 SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Vancomycin Actavis 500 mg
prášek pro koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje vancomycinum 500 mg (jako vancomycini hydrochloridum), což odpovídá 500 000 IU
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok Bílý až krémový porézní koláč.
Přibližná hodnota pH roztoku po rekonstituci je 3.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Vankomycin je indikován dospělým, novorozencům, dětem od 1 měsíce do 12 let a dospívajícím starším 12 let.
Intravenózní podání roztoku vankomycinu je indikováno k léčbě níže uvedených těžkých infekcí, které jsou vyvolány citlivými grampozitivními bakteriemi a které nelze léčit jinými antibiotiky, nebo jsou rezistentní na jiná antibiotika, např. peniciliny a cefalosporiny (viz bod 5.1):
- endokarditida
- infekce kostí (osteomyelitida)
- pneumonie
- infekce měkkých tkání.
Jestliže je to vhodné, vankomycin se má podávat s jinými antibakteriálními látkami. Toto zejména platí při léčbě endokarditidy.
Vankomycin lze použít při peroperační profylaxi bakteriální endokarditidy, u pacientů s vysokým rizikem rozvoje bakteriální endokarditidy, kteří se podrobují velkým chirurgickým výkonům (např. srdeční a cévní výkony apod.) a nemohou dostat vhodná beta-laktamová antibiotika.
Pozornost je třeba věnovat oficiálním doporučením o správném použití antibakteriálních látek.
4.2 Dávkování a způsob podání
Dávkování
Pacienti vyžadující omezený příjem tekutin mohou dostávat roztok v koncentraci 500mg/50 ml nebo 1000 mg/100
ml. U těchto vyšších koncentrací se může zvýšit riziko nežádoucích účinků vyvolaných infuzí. Infuzí vyvolané nežádoucí účinky se mohou objevit při jakékoliv rychlosti nebo koncentraci.
Intravenózní podání (infuze) u pacientů s normální funkcí ledvin Dospělí a dospívající starší 12 let
Doporučená denní intravenózní dávka je 2000 mg; rozdělená do 500 mg dávek každých 6 hodin nebo 1000 mg každých 12 hodin.
Při bakteriální endokarditidě je obecně přijatý režim 1000 mg (1 g) vankomycinu intravenózně každých 12 hodin po dobu 4 týdnů buď samotného, nebo v kombinaci s jinými antibiotiky. Delší léčba může být vyžadována v závislosti na rozvoji patogenu. Je třeba vzít v úvahu národní doporučení.
Peroperační profylaxe bakteriální endokarditidy
běžně doporučená dávka pro dospělé 1 g (1000 mg) vankomycinu intravenózně před chirurgickým výkonem (při úvodu do anestezie) a v závislosti na trvání a typu výkonu se dávka 1 g vankomycinu i.v. může podat za 12 hodin po chirurgickém výkonu. Je třeba vzít v úvahu národní doporučení.
Pediatrická populace Děti od 1 měsíce do 12 let
Obvyklé intravenózní dávkování je 10 mg/kg tělesné hmotnosti v jedné dávce podávaných každých 6 hodin (celkové denní dávkování 40 mg/kg tělesné hmotnosti). Jednotlivé dávky mají být podávány nejméně po dobu 60 minut.
Novorozenci (donošení)
Ve věku 0-7 dní: Počáteční dávka 15 mg/kg tělesné hmotnosti, následně 10 mg/kg tělesné hmotnosti každých 12 hodin.
Ve věku 7-30 dní: Počáteční dávka 15 mg/kg tělesné hmotnosti, následně 10 mg/kg tělesné hmotnosti každých 8 hodin.
Jednotlivé dávky mají být podávány nejméně 60 minut. U těchto pacientů je vhodné pečlivě sledovat koncentraci vankomycinu v séru.
Bylo hlášeno, že k dosažení terapeutických koncentrací v séru u těhotných pacientek mohou být nutné významně vyšší dávky ( viz bod 4.6).
Starší pacienti
Vzhledem k poklesu funkce ledvin může být nutné snížení dávky (viz níže).
Obézní pacienti
Může být potřeba upravit obvyklé denní dávky.
Pacienti s poruchou funkce jater
Nejsou žádné údaje o snížení dávky u pacientů s jaterní insuficiencí.
Pacienti s poruchou ledvin
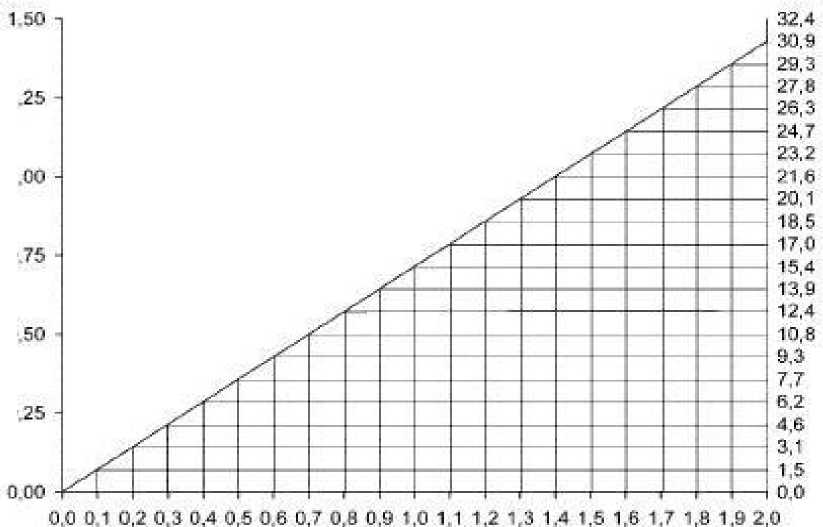
U pacientů s poruchou funkce ledvin se dávkování vankomycinu musí upravit, aby nedošlo k toxickým hladinám v séru. Hladiny vankomycinu v séru mají být pravidelně sledovány.
U většiny pacientů s poruchou funkce ledvin lze ke stanovení potřebné dávky použít následující monogram založený na clearance kreatininu:
|
%
’o
u
bQ
>
"O
O
-a
<N
t>0
S,
s
s
'3
>
-c3
Q
Clearance kreatininu (ml/min/kg)
Dávkovači nomogram pro vankomycin u pacientů s poruchou funkce ledvin
Nomogram neplatí pro funkčně anefrické pacienty na dialýze. Pro tyto pacienty má být podána zahajovací dávka 15 mg /kg tělesné hmotnosti k dosažení okamžitých terapeutických hladin v séru. Požadovaná udržovací dávka pro udržení stabilních hladin je 1,9 mg /kg tělesné hmotnosti za 24 hodin. Poté jsou vhodné individuální udržovací dávky od 250 mg do 1000 mg u pacientů s výraznou poruchou funkce ledvin a dávka může být podávána raději jedenkrát za několik dní než denně. Při anurii se doporučuje dávka 1 g každý sedmý až desátý den.
Při použití polysulfonátových membrán pro hemodialýzu („vysoký tok dialýzy“), poločas rozpadu vankomycinu se snižuje. U pacientů s pravidelnou hemodialýzou mohou být nezbytné udržovací dávky.
Pokud je k dispozici pouze hladina kreatininu v séru, lze clearance kreatininu vypočítat podle následujícího vzorce:
Muži: Tělesná hmotnost (kg) x (140 - věk (roky))
72 x kreatinin v séru (mg/100 ml)
Ženy: 0,85 x hodnota vypočítaná podle výše uvedeného vzorce.
Monitorování koncentrace vankomycinu v séru
Koncentrace vankomycinu v séru se má sledovat od druhého dne léčby bezprostředně před podáním příští dávky a jednu hodinu po infuzi. Terapeutické hladiny vankomycinu v krvi mají být jednu hodinu po ukončení infuze mezi 30 a 40 mg/l (maximálně 50 mg/l), minimální hladina (krátce před dalším podáním) v rozmezí 5 -10 mg/l.
Normálně mají být koncentrace sledovány dvakrát nebo třikrát týdně.
Délka léčby
Délka trvání léčby závisí na závažnosti infekce stejně jako na klinickém a bakteriologickém vývoji. Způsob podání
Parenterálně se vankomycin podává pouze pomalou intravenózní infuzí (ne více než 10 mg/min - po dobu nejméně 60 min), která je dostatečně zředěná (nejméně 100 ml na 500 mg nebo nejméně 200 ml na 1000 mg).
Pacientům s omezeným příjmem tekutin se podává roztok v koncentraci 500 mg/50 ml nebo 1000mg/100 ml.
Použití vyšších koncentrací může zvýšit riziko vzniku nežádoucích účinků souvisejících s infuzí. Příhody v souvislosti s infuzí se však mohou vyskytnout při jakékoli rychlosti nebo koncentraci.
Podmínky pro rekonstituci a naředění léčivého přípravku před podáním viz bod 6.6.
4.3 Kontraindikace Hypersenzitivita na léčivou látku.
4.4 Zvláštní upozornění a opatření pro použití
Rychlé podání ve formě bolusu (tj. po dobu několika minut) může být spojeno s těžkou hypotenzí, včetně šoku a vzácně i srdeční zástavou, příznaky podobnými účinku histaminu, a makulopapulární nebo erytematózní vyrážkou („syndrom rudého muže“ nebo „syndrom červeného krku“). Vankomycin je třeba podávat infuzí ve zředěném roztoku po dobu nejméně 60 minut, aby se zamezilo reakcím spojeným s rychlou infuzí. Po přerušení infuze obvykle tyto reakce rychle pominou (viz body 4.2 a 4.8).
V případě těžkých akutních hypersenzitivních reakcí (např. anafylaxe), musí být léčba vankomycinem ihned přerušena a zahájena obvyklá neodkladná opatření.
Vankomycin se má používat s opatrností u pacientů s alergickou reakcí na teikoplanin, protože byly popsány reakce zkřížené hypersenzitivity mezi vankomycinem a teikoplaninem.
Vzhledem k jeho ototoxickým a nefrotoxickým účinkům se má vankomycin podávat s opatrností u pacientů s poruchou funkce ledvin a dávka se má snížit podle stupně renálního poškození. Riziko toxicity se zvyšuje vysokou koncentraci v krvi nebo prodlouženou terapií. Pravidelně mají být monitorovány hladiny vankomycinu v krvi a prováděny testy na renální funkce.
Ototoxicita, která může být přechodná či trvalá (viz bod 4.8), byla hlášena u pacientů s již přítomnou hluchotou, kteří dostali nadměrně vysoké intravenózní dávky nebo byli současně léčeni jinou ototoxickou léčivou látkou, jako je aminoglykosid. Hluchotě může předcházet tinitus. Zkušenosti s jinými antibiotiky ukazují, že hluchota může progredovat přes vysazení léčby. Ke snížení rizika ototoxicity se mají pravidelně stanovovat hladiny vankomycinu v krvi a doporučuje se provádět pravidelné kontroly funkce sluchu.
Vankomycin se nemá podávat pacientům se ztrátou sluchu v minulosti. Pokud se léčivý přípravek u těchto pacientů použije, dávku je třeba upravovat pravidelným stanovením hladiny léku v krvi.
Pediatrická populace
U nedonošených dětí a kojenců je vhodné ověřit požadované sérové koncentrace vankomycinu. Současné podávání vankomycinu a anestetik bývá u dětí spojeno s erytémem a návaly podobnými reakcím na histamin..
Starší pacienti
Přirozený pokles glomerulární filtrace s rostoucím věkem, pokud se neupraví dávka, může vést ke zvýšení sérových koncentrací vankomycinu (viz bod 4.2).
Bezpečnostní opatření
Pravidelné sledování hladin vankomycinu v krvi je indikováno při dlouhodobém podávání zvláště u pacientů s poruchou funkce ledvin nebo poruchou sluchu a také při souběžném podávání neurotoxických nebo ototoxických látek.
Dávky se mají titrovat podle sérových hladin. Je třeba kontrolovat hladiny vankomycinu v krvi a pravidelně vyšetřovat funkci ledvin. Obecně se doporučuje monitorovat koncentrace 2-3x týdně.
Pacientům s hraniční funkcí ledvin a osobám starším 60 let je třeba provádět opakovaně vyšetření sluchu a hladin vankomycinu v krvi. Všem pacientům léčených tímto lékem se mají pravidelně provádět hematologická vyšetření, vyšetření moče a testy renálních funkcí.
Vankomycin výrazně dráždí tkáně a v místě vpichu způsobuje nekrózy po intramuskulárním podání; proto musí být podáván intravenózně. U mnohých pacientů, kteří podávají vankomycin, se vyskytuje bolest v místě vpichu a tromboflebitida, někdy v závažné podobě.
Četnost a závažnost tromboflebitidy lze minimalizovat pomalým podáváním a zředěním roztoku (2,5 až
5,0 g/l) a střídáním místa infuze.
Dlouhodobé používání vankomycinu může vést k přerůstání a necitlivých organismů. Pacienta je třeba pečlivě sledovat.. Dojde-li během léčby k superinfekci, mají být přijata příslušná opatření. Ve vzácných případech byla hlášena pseudomembranózní kolitida způsobené C. difficile, která postihuje pacienty léčené intravenózním vankomycinem.
Byly hlášeny případy zkřížené hypersenzitivity. Proto musí být vankomycin podáván velmi obezřetně u pacientů se známou hypersenzitivitou na teikoplanin
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Anestetika
Současné podávání vankomycinu a anestetik bylo spojováno s erytémem, návaly podobnými reakcím na histamin a s anafylaktoidními reakcemi.
Bylo hlášeno, že četnost reakcí souvisejících s infuzí se zvyšuje při současném podávání anestetik. Účinky související s infuzí lze snížit podáním vankomycinu v podobě infuze trvající déle než 60 minut před úvodem do anestezie.
Jiná potenciálně nefrotoxická nebo ototoxická léčiva
Při současném nebo následném systémovém nebo lokálním podávání vankomycinu s jinými potenciálně ototoxickými, neurotoxickými nebo nefrotoxickými látkami, jako jsou amfotericin B, aminoglykosidy, bacitracin, polymyxin B, kolistin, viomycin nebo cisplatin, sledovat, jestli se nezvyšuje riziko nefrotoxicity nebo ototoxicity.
Myorelaxancia
Při současném podání vankomycinu a neuromuskulárních blokátorů se zvyšuje riziko neuromuskulární blokády.
4.6 Fertilita, těhotenství a kojení
Dosud nejsou k dispozici odpovídající bezpečnostní údaje o podávání vankomycinu těhotným ženám. Studie reprodukční toxicity na zvířatech neprokázaly účinky na vývoj embrya, plodu nebo na období březosti (viz bod 5.3).
Vankomycin však proniká placentou a nelze vyloučit potenciální riziko ototoxického a nefrotoxického ovlivnění embrya a novorozence. Vankomycin se proto může v průběhu těhotenství podávat, pouze pokud je to nezbytně nutné a po pečlivém zvážení očekávaného prospěchu a možného rizika.
Kojení
Vankomycin se vylučuje do mateřského mléka. Vankomycin je třeba podávat kojícím matkám opatrně, protože je možnost výskytu nežádoucích účinků u novorozenců (poruchy střevní flory a průjmy, přerůstání kvasinek a možná senzibilizace).
Po zvážení významu tohoto léčivého přípravku pro kojící matku, je třeba rozhodnout, zda se má kojení ukončit.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vankomycin nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
V každé skupině jsou nežádoucí účinky uváděny podle jejich klesající závažnosti. Níže uvedené nežádoucí účinky jsou definovány s použitím frekvence podle MedDRA a třídy orgánových systémů podle databáze MedDRA:
velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (>1/10 000 až < 1/1000); velmi vzácné (< 1/10000); není známo (z dostupných údajů nelze určit).
Intravenózní infuze
Nejčastější nežádoucí účinky jsou flebitida a pseudoalergické reakce při příliš rychlé intravenózní infuzi vankomycinu. (viz bod 4.4).
Poruchy krve a lymfatického systému
Vzácné (>1/10 000 až < 1/1000): trombocytopenie, neutropenie, agranulocytóza, eosinofilie.
Poruchy imunitního systému
Vzácné (>1/10 000 až < 1/1000): anafylaktické reakce, hypersenzitivní reakce.
Poruchy ucha a labyrintu
Méně časté (> 1/1000 až < 1/100): přechodná anebo trvalá ztráta sluchu.
Vzácné ( >1/10 000 až < 1/1000): tinitus, závratě.
Srdeční poruchy
Velmi vzácné (< 1/10000): srdeční zástava.
Cévní poruchy
Časté (> 1/100 až < 1/10): pokles krevního tlaku, tromboflebitida.
Vzácné ( >1/10 000 až < 1/1 000) : vaskulitida.
Respirační, hrudní a mediastinální poruchy Časté (> 1/100 až < 1/10): dyspnoe, stridor.
Gastrointestinální poruchy
Vzácné (>1/10 000 až < 1/1000): nauzea, průjem.
Velmi vzácné (< 1/10000): pseudomembranózní enterokolitida.
Poruchy kůže a podkožní tkáně
Časté (> 1/100 až < 1/10): exantém a zánět sliznic, pruritus, kopřivka.
Velmi vzácné (< 1/10000): exfoliativní dermatitida, Stevens-Johnsonův syndrom, lineární IgA bulózní dermatitida, Lyellův syndrom.
Poruchy ledvin a močových cest
Časté (> 1/100 až < 1/10): renální insuficience projevující se primárně zvýšenou hladinou kreatininu v séru
Vzácné (>1/10 000 až < 1/1000): intersticiální nefritida, akutní selhání ledvin
Celkové poruchy a reakce v místě aplikace
Časté (> 1/100 až < 1/10): flebitida, zarudnutí horní poloviny těla a obličeje, bolesti a spazmy hrudních a zádových svalů
Vzácné (>1/10 000 až < 1/1000): léková horečka, třesavka.
Poruchy imunitního systému
Není známo: Během postmarketingového sledování byla hlášena léková reakce s eosinofilií a systémovými příznaky (DRESS).
Příhody související s infuzí
Během rychlé infuze vankomycinu nebo krátce po jejím ukončení se mohou objevit anafylaktoidní reakce včetně poklesu krevního tlaku, dušnosti, kopřivky nebo svědění. Také se může objevit zčervenání horní poloviny těla (syndrom rudého muže), bolest a křeče hrudních a zádových svalů. Tyto reakce obvykle vymizí po ukončení podávání infuze, obvykle do 20 minut až 2 hodin. Vankomycin se má podávat pomalou infuzí (déle než 60 minut - viz bod 4.4). Ototoxicita může být reverzibilní nebo trvalá a byla hlášena hlavně u pacientů s vysokými dávkami vankomycinu a s poruchou sluchu v anamnéze a při současné léčbě jinými ototoxicky působícími léky, jako např. aminoglykosidy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Doporučuje se podpůrná péče s udržováním glomerulární filtrace. Vankomycin se z krve špatně odstraňuje pomocí hemodialýzy nebo peritoneální dialýzy. Hemoperfuze s použitím pryskyřice Amberlit XAD-4 má podle hlášení limitovaný přínos.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Glykopeptidová antibiotika,
ATC kód: J01XA01.
Mechanizmus účinku
Vankomycin je tricyklické glykopeptidové antibiotikum, které inhibuje syntézu buněčné stěny u citlivých bakterií vazbou o vysoké afinitě na D-alanyl-D-alaninový konec prekurzovoých jednotek buněčné stěny. Lék je pro dělící se mikroorganizmy baktericidní.
Vztah mezi farmakokinetikou a farmakodynamikou
Aktivita vankomycinu se považuje za závislou na čase s plochou pod křivkou (AUC), tzn., že antimikrobiální aktivita závisí na délce doby, kdy hladina léku převýší minimální inhibiční koncentraci (MIC) cílového mikroorganizmu.
Mechanizmus rezistence
Získaná rezistence na glykopeptidy je založená na získání různých van komplexů genů a změně D-alanyl-D-alaninového cíle na D-alanyl-D-laktát nebo D-alanyl-D-serin, které se na vankomycin vážou slabě, protože chybí kritické místo pro vazbu vodíku. Byla hlášena zkřížená rezistence s teikoplaninem u některých van genů. Van geny se vzácně nachází u Staphylococcus aureus, kde jsou změny struktury buněčné stěny středně citlivé, což je obvykle heterogenní.
Citlivost
Vankomycin je účinný proti grampozitivním bakteriím. Gramnegativní mikroorganizmy jsou rezistentní.
Prevalence získané rezistence se může lišit geograficky a s časem pro určité kmeny a je třeba sledovat lokální informace o rezistenci, zejména při léčbě závažných infekcí. Jestliže je to nutné, je třeba si vyžádat radu odborníka, v případě, že lokální prevalence rezistence je taková, že využití látky v několika posledních případech infekce je diskutabilní.
Hraniční hodnoty
Doporučení EUCAST (Evropský výbor pro testování antimikrobiální citlivosti)
|
Citlivý |
Rezistentní | |
|
Staphylococcus spp. |
<2 mg/l |
>2 mg/ l |
|
Enterococcus spp. |
<4 mg/ l |
> 4 mg/ l |
|
Streptococcus spp |
< 2 mg/ l |
> 2 mg/ l |
|
Streptococcus pneumoniae |
< 2 mg/ l |
> 2 mg/ l |
|
Grampozitivní anaeroby |
< 2 mg/ l |
> 2 mg/ l |
|
Bez vztahu ke kmeni * |
< 2 mg/ l |
> 4 mg/ l |
* Hraniční hodnoty bez vztahu k bakteriálnímu kmeni byly stanoveny zejména na základě farmakokinetických a farmakodynamických dat a jsou nezávislé na distribuci MIC u konkrétních kmenů. Lze je použít pouze u kmenů, u kterých nebyly stanoveny hraniční hodnoty specifické pro kmen a pro kmeny, kde se nedoporučuje provádět testování citlivosti.
Třídy_
Běžně citlivé kmeny Grampozitivní
Enterococcus faecalis.
Staphylococcus aureus Staphylococcus coagulase negative
Streptococcus spp.
Streptococcus pneumoniae
Clostridium spp._
Kmeny, u nichž může být získaná rezistence problém
Enterococcus ^ faecium_
Přirozeně rezistentní
Gramnegativní mikroorganizmy
Chlamydia spp.
Mycobacteria Mycoplasma spp.
Rickettsia spp._
5.2 Farmakokinetické vlastnosti
Absorpce
Vankomycin se podává intravenózně k léčbě systémových infekcí. U pacientů s normální funkcí ledvin intravenózní infuze vícenásobných dávek 1000 mg vankomycinu (15 mg/kg) po dobu 60 minut vytvoří
přibližné průměrné plazmatické hladiny 50-60 pg/ml, 20-25 pg/ml a 5-10 pg/ml okamžitě po podání, 2 hodiny a 11 hodin po dokončení infuze. Intravenózní infuze vícenásobných dávek obsahující 500 mg po 30 minutách vytvoří průměrné plazmatické koncentrace kolem 40-50 mg/l, 19-20 mg/l a 10-11 mg/l okamžitě po podání, za 2 hodiny a 6 hodin po dokončení infuze. Plazmatické hladiny po podání vícenásobných dávek jsou podobné koncentracím po jednotlivé dávce.
V případě perorálního podání vysoce polární vankomycin není prakticky absorbován. Po perorálním podání se objeví v aktivní formě ve stolici, a proto je vhodný pro chemoterapii pseudomembranózní kolitidy a stafylokokové kolitidy.
Distribuce
U sérových koncentrací vankomycinu 10 mg/l až 100 mg/l je přibližně 30-55% léku vázáno na plazmatické bílkoviny, měřeno ultrafiltrací.
Po intravenózním podání vankomycin-hydrochloridu byly nalezeny inhibiční koncentrace v pleurálních, perikardiálních, ascitických a synoviálních tekutinách, v moči a v tekutinách z peritoneální dialýzy a ve tkáních síňového ouška.
U nezanícených meningů vankomycin prochází hematoencefalickou bariéru pouze v malém rozsahu. Eliminace
Eliminační poločas vankomycinu u pacientů s normální funkcí ledvin je 4 až 6 hodin. V prvních 24 hodinách se přibližně 80% podané dávky vyloučí glomerulární filtrací močí. Porucha funkce ledvin zpožďuje vylučování vankomycinu. U anefrických pacientů je průměrný poločas 7,5 dnů.
Metabolizmus léčivého přípravku je velmi malý. Přibližně 35-65% intraperitoneální dávky podaného vankomycinu během peritoneální dialýzy je systematicky absorbováno během 6 hodin. Sérových koncentrací přibližně 8 mg/l je dosaženo po intraperitoneální injekci 30 mg/kg vankomycinu. I když vankomycin při hemodialýze nebo peritoneální dialýze není vylučován efektivně, byla hlášena zvýšená clearence vankomycinu u hemoperfuze a hemofiltrace. Celková systémová a renální clearence vankomycinu může být snížena v závislosti na věku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po opakovaném podávání neodhalily žádné zvláštní riziko pro člověka.
Omezené údaje o mutagenních účincích ukazují negativní výsledky. Dlouhodobé studie na zvířatech o kancerogenním potenciálu nejsou k dispozici.
Ve studiích teratogenity, ve kterých byly potkanům a králíkům podávány dávky zhruba odpovídající dávkám u lidí na základě tělesného povrchu (mg/m2), nebyly pozorovány přímé ani nepřímé teratogenní účinky.
Studie na zvířatech v perinatálním a postnatálním období a studie z hlediska účinků na fertilitu nejsou k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Žádné
6.2 Inkompatibility
Roztok vankomycinu má nízké pH. To může způsobit chemickou nebo fyzikální nestabilitu po mísení s jinými látkami. Je třeba se vyvarovat mísení s alkalickými roztoky. Každý roztok k parenterálnímu užití je nutné před užitím vizuálně zkontrolovat na přítomnost sraženin a na změnu zbarvení.
Tento léčivý přípravek se nesmí mísit s jinými roztoky , s výjimkou těch, které jsou uvedené v bodě 6.6.
6.3 Doba použitelnosti
Doba použitelnosti originálního balení 2 roky.
Doba použitelnosti rekonstituovaného koncentrátu Rekonstituovaný koncentrát se má naředit bezprostředně po přípravě.
Doba použitelnosti naředěného roztoku
Z mikrobiologického hlediska má být přípravek použit okamžitě.
6.4 Zvláštní opatření pro uchovávání Prodejní balení
Uchovávejte při teplotě do 25 °C.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Rekonstituovaný koncentrát a naředěný roztok
Podmínky uchovávání rekonstituovaného a naředěného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
10ml injekční lahvička z bezbarvého skla třídy I, s chlorobutylovou zátkou třídy I potaženou silikonem a šedým hliníkovým/polypropylenovým odtrhovacím uzávěrem.
Velikost balení: 1 injekční lahvička
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Prášek se musí rekonstituovat a výsledný koncentrát před použitím dále naředit.
Příprava rekonstituovaného roztoku
Obsah jedné injekční lahvičky s 500 mg se naředí v 10 ml sterilní vody na injekci.
Vzhled rekonstituovaného roztoku
Čirý, bezbarvý až světle žlutý roztok, prakticky prostý vláken a viditelných částic.
1 ml rekonstituovaného roztoku obsahuje vancomycinum 50 mg.
Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
Příprava konečného naředěného infuzního roztoku
Rekonstituované roztoky obsahující vankomycin v koncentraci50 mg/ml se dále naředí podle způsobu podání.
Vhodná rozpouštědla jsou:
chlorid sodný 9 mg/ml (0,9 %), glukóza 50 mg/ml (5 %), 0,9% roztok chloridu sodného (9 mg/ml) a 5% roztok glukózy (50 mg/ml) nebo Ringer acetát.
Před podáním musí být rekonstituovaný a naředěný roztok vizuálně zkontrolován na přítomnost částic a na změnu zbarvení. Použit může být pouze čirý a bezbarvý až světle žlutý roztok bez vláken a viditelných částic.
Intermitentní infuze
Rekonstituovaný roztok obsahující 500 mg vankomycinu (50 mg/ml) se musí dál naředit s minimálně 100 ml ředidla okamžitě po rekonstituci.
Koncentrace vankomycinu v infuzním roztoku nesmí přesahovat 5 mg/ml.
Potřebná dávka se podává pomalu intravenózní infuzí rychlostí nejvýše 10 mg/min, po dobu minimálně 60 minut nebo i déle.
Podmínky uchovávání naředěného léčivého přípravku viz bod 6.3.
Likvidace
Injekční lahvičky jsou určené k jednorázovému podání. Nepoužitý přípravek se musí zničit.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Actavis Group PTC ehf.
Reykjavikurvegur 76-78 220 Hafnarfjordur Island
8. REGISTRAČNÍ ČÍSLO(A)
15/115/15-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25.2.2015
10. DATUM REVIZE TEXTU
26.5.2016
11