Tachyben I.v. 25 Mg Injekční Roztok
sp. zn. sukls55384/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. Název přípravku
Tachyben i.v. 25 mg injekční roztok Tachyben i.v. 50 mg injekční roztok
Tachyben i.v. 100 mg koncentrát pro přípravu infuzního roztoku
2. Kvalitativní a kvantitativní složení Tachyben i.v. 25 mg injekční roztok
5 ml injekčního roztoku obsahuje 25 mg urapidilum Tachyben i.v. 50 mg injekční roztok 10 ml injekčního roztoku obsahuje 50 mg urapidilum Tachyben i.v. 100 mg koncentrát pro přípravu infuzního roztoku 20 ml injekčního roztoku obsahuje 100 mg urapidilum Úplný seznam pomocných látek viz bod 6.1
3. Léková forma
Tachyben i.v. 25 mg injekční roztok
Injekční roztok. Po naředění může být použit jako infuzní roztok.
Tachyben i.v. 50 mg injekční roztok
Injekční roztok. Po naředění může být použit jako infuzní roztok.
Tachyben i.v. 100 mg koncentrát pro přípravu infuzního roztoku Koncentrát pro přípravu infuzního roztoku
Popis přípravku: čirý, bezbarvý roztok bez viditelných částic. pH 5,6 - 6,6
4. Klinické údaje
4.1 Terapeutické indikace
Hypertenzní krize (tj. kritický vzestup krevního tlaku), těžké a velmi těžké formy hypertenze, hypertenze rezistentní na běžnou terapii.
Kontrolované snižování krevního tlaku u hypertenzních pacientů při operaci nebo v pooperačním údobí.
4.2 Dávkování a způsob podání
Dávkování: -Hypertenzní krize, těžké, respektive velmi těžké formy hypertenze a hypertenze rezistentní na běžnou terapii:
1. intravenózní injekce
Injekčně se podává zvolna 10-50 mg urapidilu i.v. za stálé kontroly krevního tlaku. Antihypertenzní účinek je možno očekávat během 5 minut po podání. Podání přípravku urapidil i.v. 25 nebo urapidil i.v. 50 lze podle výše krevního tlaku zopakovat.
2. Pomalá intravenózní infúze nebo kontinuální infúze pomocí perfúzoru.
Infúze se používá k udržení poklesu krevního tlaku dosaženého podáním injekce urapidilu, Pokyny k ředění přípravku před použitím viz 6.6.
Nejvyšší kompatibilní množství jsou 4 mg urapidilu na 1 ml infúzního roztoku.
Rychlost podávání
Rychlost podávání volíme podle individuální reakce krevního tlaku. Počáteční doporučená rychlost je 2 mg/min.
Udržovací dávka
Průměrně 9 mg/hod, vztaženo na 250 mg urapidilu v 500 ml infúzního roztoku odpovídá 1 mg = 44 kapek = 2,2 ml infúzního roztoku.
- Kontrolované snižování krevního tlaku u hypertenzních pacientů při operaci nebo po ní Intravenozní infúze nebo perfúzor se používá k udržování krevního tlaku dosaženého injekcí Dávkovací schéma:
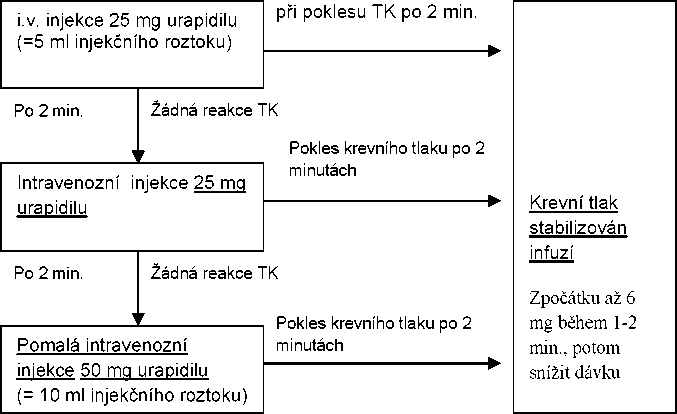
Poznámka
Tachyben i.v. se podávají ležícímu pacientovi intravenózně form ou injekce nebo infúze. Přípravek se aplikuje formou jednorázové nebo opakova né! injekce nebo pomalé intravenózní infúze. Injekce lze kombinovat s následnou infúzí.
Starší pacienti
U starších pacientů je třeba přistupovat k podávání antihypertenziv se zvýšenou opatrností a na počátku léčby je podávat v nižších dávkách, protože citlivost vůči těmto přípravkům bývá v těchto případech často změněná.
Pacienti s ledvinnými nebo jaterními funkčními poruchami
U pacientů s renálními a/nebo jaterními poruchami může být potřebné snížení dávky urapidilu.
Pediatrická populace
Bezpečnost a účinnost intravenózního urapidilu u dětí ve věku mezi 0 a 18 lety nebyla stanovena. Proto nemůže být doporučeno podávání ani příslušné dávkování.
Délka léčby
Z toxikologického hlediska je délka léčby 7 dní považována za bezpečnou; obecně tato doba nemá být pro parenterální léčbu intravenózními antihypertenzivy překročena. Parenterální léčbu je při opětovném vzestupu tlaku možné zopakovat. Z parenterální akutní terapie je možné přejít na jiné perorálně podávané antihypertenzivum.
4.3 Kontraindikace
Tachyben i.v. se nesmí podávat při přecitlivělosti na urapidil nebo pomocné látky obsažené v přípravku, Tachyben i.v. se nesmí podávat u stenózy aortálního istmu a AV zkratu (vyjma dialyzačního zkratu, který je hemodynamicky neúčinný).
4.4 Zvláštní upozornění a opatření pro použití Zvláště pečlivý dohled je nutný u pacientů:
- se srdečním selháním způsobeným funkčním poškozením mechanického původu, např. stenózou aortální či mitrální chlopně, pulmonální embolií, nebo poruchou srdeční akce z důvodu onemocnění perikardu
- u pacientů s poruchami jaterních funkcí
- u pacientů se středně těžkou až těžkou poruchou funkce ledvin
- u starších pacientů
- u pacientů, kteří jsou současně léčeni cimetidinem (viz bod 4.5).
Jestliže před přípravky Tachyben i.v. bylo podáváno jiné antihypertenzivum, je
třeba vyčkat dostatečně dlouho na nástup jeho účinku. Dávkování přípravku Tachyben i.v. je
třeba odpovídajícím způsobem zredukovat.
Prudký pokles krevního tlaku by mohl vyvolat bradykardii nebo zástavu srdce.
Protože v přípravku je obsažen propylen glykol, mohou být po podání přípravku Tachyben i.v. pozorovány příznaky podobné opilosti.
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) na jednu dávku, je tedy v podstatě bez obsahu sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Antihypertenzní účinek Tachyben i.v. může být zesílen současným užíváním blokátorů alfa-adrenergních receptorů včetně těch, které se podávají v urologických indikacích, vasodilatancií a jiných antihypertenziv, a v případech způsobených hypovolemií (např. při průjmu, zvracení) a alkoholem.
Při současném podávání baclofenu je třeba opatrnosti, protože baclofen může zvýšit antihypertenzní účinky.
Cimetidin podávaný současně inhibuje metabolismus urapidilu. Při současném podávání cimetidinu je možné zvýšení sérového maxima urapidilu o 15%, a proto je nutno zvažovat snížení dávky urapidilu.
Pozornost je třeba věnovat současnému podávání:
- imipraminu (má antihypertenzivní účinek a je riziko ortostatické hypotenze)
- neuroleptik (mají antihypertenzivní účinek a je riziko ortostatické hypotenze)
- kortikoidů (mohou způsobit snížení antihypertenzivní účinnosti retencí sodíku)
Jelikož dosud neexistují dostatečné údaje o kombinované léčbě s ACE inhibitory, tato léčba se v současné době nedoporučuje.
4.6 Fertilita, těhotenství a kojení
Urapidil i.v. se nedoporučuje, protože doposud nejsou k dispozici zkušenosti týkající se podávání u těhotných žen. Studie na zvířecích modelech prokázaly reprodukční toxicitu bez teratogenního potenciálu (viz bod 5.3). Možné riziko u lidské populace není známo, protože přenos výsledků studií na lidskou populaci je limitován,
Kojení
Údaje o vylučování urapidilu do mléka nejsou známy a proto při léčbě urapidilem nelze kojení doporučit.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Tento přípravek má malý vliv na schopnost řídit a obsluhovat stroje. Odpověď na léčbu je individuální. Týká se to hlavně období začátku léčby, změny dávkování, a v kombinaci s alkoholem.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Většina nežádoucích účinků je způsobena náhlým poklesem krevního tlaku, ale dle zkušeností vymizí během několika minut i během dlouhodobé infúze; ukončení léčby musí být uváženo v závislosti na závažnosti nežádoucích účinků.
Tabulkový souhrn nežádoucích účinků
Níže uvedená tabulka používá MedDRA třídy orgánových systémů.
Četnost výskytu je vyjádřena podle konvence MedDRA:
Velmi časté: (>1/10)
Časté: (>1/100 až <1/10)
Méně časté (>1/1000 až <1/100)
Vzácné (>1/10000 až < 1/1000), včetně jednotlivých hlášení Není známo (z dostupných údajů nelze určit)
|
Frekvence Třídy Orgánových systémů |
velmi časté (>1/10) |
časté (>1/100 , <1/10) |
méně časté (> 1/1 000 , < 1/100) |
vzácné (> 1/10 000 , < 1/1 000) |
Velmi vzácné (<1/10,000) |
neznámé (z dostupných dat nelze určit) |
|
Poruchy krve a lymfatického systému |
trombocytopenie | |||||
|
Srdeční poruchy |
palpitace, tachykardie, bradykardie, pocit tlaku na hrudi, respirační tíseň, srdeční dysrytmie | |||||
|
Gastrointestinální poruchy |
nausea | |||||
|
Celkové a jinde nezařazené poruchy a lokální |
astenie |
|
reakce po podání | ||||||
|
Poruchy nervového systému |
závratě, bolesti hlavy. | |||||
|
Psychiatrické poruchy |
neklid | |||||
|
Poruchy reprodukčního systému a choroby prsů |
priapismus | |||||
|
Respirační, hrudní a mediastinální poruchy |
nasální kongesce | |||||
|
Poruchy kůže a podkoží |
profuzní pocení |
alergické reakce jako svědění, vyrážky, exanthém |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www. sukl. cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Symptomy předávkování
Příznaky předávkování jsou závratě, posturální snížení krevního tlaku a kolaps únava a snížení reakční rychlosti.
Léčba v případě předávkování
Nadměrný pokles krevního tlaku může být zmírněn zvednutím nohou a doplňováním objemu. Pokud jsou tyto prostředky nedostatečné, mohou být podány pomalou i.v. injekcí vazokonstrikční látky při monitorování krevního tlaku. Ve velmi vzácných případech je nutná intravenózní injekce katecholaminů (např. adrenalin, 0.5-1.0 mg naředěný 10 ml fyziologického roztoku).
5. Farmakologické vlastnosti
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: ANTIADRENERGNÍ LÁTKY S PERIFERNÍM ÚČINKEM, ALFA-ADRENERGNÍ receptoroví antagonisté.
ATC kód: C02C A06
Urapidil vyvolává vyvážený pokles systolického a diastolického krevního tlaku na základě snížení periferního odporu.
Srdeční frekvence zůstává konstantní.
Srdeční objem se nemění. Následkem vyššího afterload se může snížený srdeční objem zvýšit.
Mechanizmus účinku
Urapidil má centrální i periferní účinek.
- Na periferii: Urapidil blokuje převážně postsynaptické alfa-receptory a inhibuje tak vazokonstrikční působení katecholaminů.
- Centrálně: Urapidil má také centrální účinky. Moduluje aktivitu oběhových regulačních center; na základě toho je blokován reflexní vzestup tonu sympatiku nebo dochází k jeho celkovému snižování.
5.2 Farmakokinetické vlastnosti
Po intravenózním podání 25 mg urapidilu je pozorován dvojfázový průběh (iniciální distribuční fáze, terminální eliminační fáze) koncentrace v séru. Distribuční fáze má poločas asi 35 minut. Distribuční objem je 0,8 l/kg (0,6-1,2 l/kg).
Urapidil je metabolizován především v játrech. Hlavním metabolitem je urapidil hydroxylovaný na 4. pozici fenylového jádra, který nemá významnou antihypertenzní účinnost. O-demethylovaný metabolit urapidilu má stejnou biologickou aktivitu jako urapidil, ale vyskytuje se jen ve velmi malých množstvích.
Eliminace urapidilu a jeho metabolitů u lidí je až z 50-70% renální cestou, z čehož asi 15% podané dávky činí farmakologicky aktivní urapidil, zbytek se vylučuje stolicí převážně jako para-hydroxylovaný metabolit urapidilu, který nemá antihypertenzní účinky. Sérový poločas po intravenózní bolusové injekci je 2,7 h (1,8-3,9 hod). Vazba urapidilu (v lidském séru) na proteiny krevní plazmy je asi 80%. Tato relativně nízká vazba na proteiny by mohla objasňovat, proč dosud nebyly zaznamenány interakce urapidilu s léky, které mají vysokou vazbu na proteiny krevní plazmy.
U starších pacientů a pacientů s pokročilým selháním jater a/nebo ledvin se snižuje distribuční objem a clearance a plazmatický poločas je delší.
Látka prostupuje hemato-encefalickou bariérou a prochází placentou.
5.3 Předklinické údaje vztahující se k bezpečnosti
Akutní toxicita
Studie akutní toxicity urapidil hydrochloridu byly provedeny na myších a potkanech. Hodnoty LD50 (vztažené k urapidil bázi) byly mezi 508 a 750 mg/kg tělesné hmotnosti po perorálním podání a mezi 140 a 260 mg/kg tělesné hmotnosti po intravenózním podání. Hlavními toxickými symptomy byly: sedace, ptóza, snížení motility, ztráta obranných reflexů, hypothermie, dyspnoe, cyanóza, tremor a křeče vedoucí ke smrti.
Chronická toxicita/Subchronická toxicita
Chronická toxicita byla studována na potkanech po perorálním podání s krmivem po dobu 6 a 12 měsíců s dávkami až 250 mg/kg tělesné hmotnosti/den. Byly pozorovány následující účinky: sedace, ptóza, snížení váhového přírůstku, prodloužení cyklů mezi říjemi a snížení hmotnosti dělohy.
Chronická toxicita byla studována u psů po dobu 6 a 12 měsíců s dávkami až 64 mg/kg tělesné hmotnosti. Dávky 30 mg/kg tělesné hmotnosti/den a vyšší způsobily sedaci, nadměrné slinění a tremor. U psů nebyly pozorovány žádné klinické či histopatologické změny.
Mutagenní a tumorigenní potenciál
Urapidil neprokázal žádné mutagenní vlastnosti ve studiích na bakteriích (Amesův test, host-mediated assay), s lidskými lymfocyty a v kostní dřeni při metafázovém testu na myších. Test na opravy DNA na potkaních hepatocytech byl negativní.
Studie karcinogenity u myší a potkanů trvající 18 a 24 měsíců nepřinesly žádné informace o tumorigenním potenciálů s významem pro člověka. Speciální studie na myších a potkanech ukázaly, že urapidil zvyšuje hladinu prolaktinu a u hlodavců zvýšený prolaktin stimuluje růst prsní tkáně. Na základě dostupných informací se výskyt tohoto účinku u člověka po podání terapeutických dávek neočekává a v klinických studiích nebyl pozorován.
Reprodukční toxicita
Studie reprodukční toxicity na potkanech, myších a králících nepřinesly žádné důkazy o teratogenním účinku.
Studie na laboratorních potkanech a králících prokázaly reprodukční toxicitu urapidilu. Nežádoucí účinky spočívaly ve snížené frekvenci zabřeznutí u potkanů, sníženém přírůstku tělesné hmotnosti a příjmu potravy a tekutin u březích ramlic, sníženém přežívání potkaních plodů a sníženém přežívání novorozených laboratorních potkanů a jejich sníženém váhovém přírůstku.
Prodloužení cyklu říje u samic potkanů bylo pozorováno v reprodukčních studiích i ve studiích chronické toxicity. Tento účinek, spolu se snížením hmotnosti dělohy pozorovaným v chronických studiích, je považován za důsledek zvýšení prolaktinu, ke kterému dochází u hlodavců po léčbě urapidilem. Plodnost samic potkanů nebyla poškozena.
Vzhledem k podstatným rozdílům mezi zvířecími druhy však neexistují důkazy, že by tato zjištění měla význam pro člověka. V dlouhodobých klinických studiích nebyl zaznamenán žádný vliv na ženský hypofyzárně-gonádový systém.
6. Farmaceutické údaje
6.1 Seznam pomocných látek
Propylenglykol
Dihydrát dihydrogenfosforečnanu sodného Dihydrát hydrogenfosforečnanu sodného Kyselina chlorovodíková 35%
Hydroxid sodný 4%, Kyselina chlorovodíková 3,7%
Voda na injekci
6.2 Inkompatibility
T ento přípravek se nesmí mísit s jinými přípravky s výjimkou těch, které jsou zmíněny v 6.6.
S tímto přípravkem nesmí být současně podávány následující substance (nebo roztoky pro přípravu infuze):
alkalické injekční a infúzní roztoky- vzhledem ke kyselé povaze injekčního roztoku by mohlo dojít ke vzniku zákalu nebo k vyvločkování
6.3 Doba použitelnosti
3 roky
Po prvním otevření:
Chemická a fyzikální stabilita byla prokázána po 50 hodinách při 15-25°C.
Z mikrobiologického hlediska by měl být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti
uživatele a normálně by doba neměla být delší než 24 hodin při 2 až 8 oC, pokud rekonstituce/ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30°C.
Podmínky uchovávání naředěného přípravku viz 6.3.
6.5 Druh obalu a obsah balení
Ampule z bezbarvého skla (typ I Ph. Eur.).
Velikost balení: 5 amp.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Ampule 100 mg mohou být použity pouze pro stabilizaci krevního tlaku infuzí.
Pro zahajovací léčbu jsou k dispozici ampule o obsahu 25 mg a 50 mg urapidilu. Toto dávkování může být také použito po naředění pro intravenozní infuzi.
Ředění musí být provedeno za aseptických podmínek.
Před aplikací musí být zkontrolována vizuálně barva roztoku a nepřítomnost částic. Smí být použit pouze čirý a bezbarvý roztok.
Příprava naředěného roztoku Intravenozní infuze
K 500 ml jednoho z kompatibilních roztoků přidejte 250 mg urapidilu (2 ampule 100 mg urapidilu a 1 ampuli 50 mg urapidilu).
Perfúzor
100 mg urapidilu je nataženo do perfúzoru a rozpuštěno v 50 ml jednoho z kompatibilních roztoků.
Kompatibilní roztoky pro ředění:
- chlorid sodný 9 mg/ml (0.9%) infuzní roztok
- Glukóza 50 mg/ml (5%) infuzní roztok
- Glukóza 100 mg/ml (10%) infuzní roztok
Pro jednorázové použití.
Nepoužitý roztok a infuzní sety musí být likvidovány v souladu s místními požadavky.
7. Držitel rozhodnutí o registraci
EVER Neuro Pharma GmbH Oberburgau 3, A-4866 Unterach Rakousko
Telefon: +43 7665 2055 0 Telefax: +43 7665 2055 910
8. Registrační číslo(a)
Tachyben i. v. 25 mg injekční roztok: 58/242/12-C Tachyben i.v. 50 mg injekční roztok: 58/243/12-C
Tachyben i.v. 100 mg koncentrát pro přípravu infuzního roztoku: 58/244/12-C
9. Datum první registrace/Prodloužení registrace Datum první registrace: 4.4.2012
10. Datum revize textu 16.8.2015
9