Sporanox
sp. zn sukls267071/2012
sp. zn. sukls115130/2014 a sukls205661/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
SPORANOX 10 mg/ml
perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml perorálního roztoku obsahuje itraconazolum 10 mg Pomocné látky se známým účinkem: sorbitol.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok.
Žlutý až světle jantarový čirý roztok s charakteristickou vůní po třešních.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
SPORANOX perorální roztok je určen:
• K léčbě orální a/nebo ezofageální kandidózy HIV pozitivních nebo jinak imunodeficitních pacientů.
• K profylaxi hlubokých mykotických infekcí u pacientů s hematologickými malignitami nebo pacientů podstupujících transplantaci kostní dřeně a u pacientů, u nichž lze očekávat neutropenii (tj. < 500 buněk/pl).
4.2 Dávkování a způsob podání
Pro optimální vstřebávání itrakonazolu je nezbytné užívat perorální roztok SPORANOX nalačno (pacientovi je doporučeno nejíst a nepít ještě nejméně jednu hodinu po užití roztoku).
Při léčbě orální a/nebo ezofageální kandidózy je zapotřebí roztok „poválet“ v ústech přibližně 20 sekund před polknutím, po polknutí ústa nevyplachovat.
Léčba orální a/nebo ezofageální kandidózy
200 mg (2 odměrky) denně rozděleně ve dvou dávkách nebo v jedné dávce po dobu 1 týdne. Pokud se nedostaví po jednom týdnu terapeutická odpověď, má léčba pokračovat ještě další týden.
Léčba orální a/nebo ezofageální kandidózy rezistentní na flukonazol
100 - 200 mg (1 - 2 odměrky) dvakrát denně po 2 týdny. Pokud se nedostaví po dvou týdnech terapeutická odpověď, má léčba pokračovat další 2 týdny. Jestliže se nedostaví známky zlepšení, nemá být denní dávka 400 mg podávána déle než 14 dní.
Profylaxe mykotických infekcí
5 mg/kg denně (0,5 ml/kg denně) podáváno rozděleně ve dvou dávkách. V klinických studiích bylo zahájeno profylaktické podávání bezprostředně před cytostatickou léčbou a obvykle jeden týden před transplantací. Léčba pokračovala do obnovy neutrofilů (tj. > 1 000 buněk/^l).
Empirická léčba febrilních neutropenických pacientů se suspektní systémovou mykózou Léčba perorálním roztokem SPORANOX 200 mg (20 ml) dvakrát denně až do vymizení klinicky významných příznaků neutropenie. Bezpečnost a účinnost přípravku SPORANOX u empirické léčby febrilních pacientů u suspektních systémových mykóz při užívání převyšujícím 28 dní nejsou známy.
Pediatrická populace
Klinická data týkající se léčby pediatrických pacientů perorálním roztokem SPORANOX jsou omezená. Roztok SPORANOX není doporučen u pediatrických pacientů pouze v případě, pokud potenciální přínos převýší možná rizika. Viz bod 4.4 Zvláštní upozornění a opatření pro použití.
Profylaxe mykotických infekcí: Údaje o účinnosti u neutropenických dětí nejsou dostupné. Omezená zkušenost bezpečného podání existuje pro dávku 5 mg/kg denně (0,5 ml/kg denně) rozděleně ve dvou dávkách. Incidence nežádoucích účinků jako jsou např. průjem, bolesti břicha, zvracení, horečka, vyrážka a mukozitida byla vyšší než u dospělých. Je však obtížné rozlišit, zda lze nežádoucí účinky přičítat perorálnímu roztoku SPORANOX nebo důsledku chemoterapie.
Starší osoby
Klinická data týkající se léčby perorálním roztokem SPORANOX u starších osob jsou omezená. Perorální roztok SPORANOX je doporučen u starších osob pouze v případě, pokud potenciální přínos léčby převýší možná rizika. Obecně je doporučeno, aby při stanovení dávkování u starších osob byla vzata v úvahu vyšší frekvence snížených jaterních, ledvinových nebo srdečních funkcí a souběžných onemocnění nebo terapií jinými přípravky (viz bod 4.4).
Porucha funkce jater
O perorálním užívání itrakonazolu pacienty s poruchou funkce jater jsou k dispozici omezené údaje. Užívání tohoto léku touto skupinou pacientů má být věnována zvýšená pozornost. (Viz bod 5.2 Farmakokinetické vlastnosti, Zvláštní skupiny pacientů, Porucha funkce jater).
Porucha funkce ledvin
O perorálním užívání itrakonazolu pacienty s poruchou funkce ledvin jsou k dispozici omezené údaje. U některých pacientů s poruchou funkce ledvin může být expozice itrakonazolu nižší. Užívání tohoto léku touto skupinou pacientů má být věnována zvýšená pozornost a má být zvážena úprava dávkování.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Současné užívání následujících léčiv je s perorálním roztokem SPORANOX kontraindikováno (viz rovněž bod 4.5).
• Substráty metabolizované CYP3A4, které mohou prodloužit QT interval, např. astemizol, bepridil, cisaprid, dofetilid, levacetylmethadol (levomethadyl), mizolastin, pimozid, chinidin, sertindol a terfenadin jsou s perorálním roztokem SPORANOX kontraindikovány. Současné užívání může způsobit zvýšení plazmatických koncentrací těchto substrátů, což může vést ke QT prolongaci a vzácnému výskytu torsade de pointes.
• Inhibitory HMG-CoA reduktázy metabolizované CYP3A4 jako atorvastatin, simvastatin a lovastatin.
• Triazolam a perorální midazolam.
• Námelové alkaloidy, jako dihydroergotamin, ergometrin (ergonovin), ergotamin a methylergometrin (methylergonovin).
Eletriptan.
• Nisoldipin.
• SPORANOX perorální roztok nemá být užíván pacienty s prokázanou ventrikulámí dysfunkcí, jako je např. městnavé srdeční selhání (CHF) nebo CHF v anamnéze kromě život ohrožujících nebo jiných závažných infekcí (viz bod 4.4).
SPORANOX perorální roztok nesmí být užíván v těhotenství kromě život ohrožujících situací (viz bod 4.6).
Pokud užívají perorální roztok SPORANOX ženy ve fertilním věku, mají používat antikoncepční opatření. V účinných antikoncepčních opatřeních je nezbytné pokračovat až do příští menstruace po ukončení léčby perorálním roztokem SPORANOX.
4.4 Zvláštní upozornění a opatření pro použití
Zkřížená hypersenzitivita
Informace o zkřížené hypersenzitivitě mezi itrakonazolem a ostatními azolovými antimykotiky nejsou k dispozici. U pacientů s hypersenzitivitou k ostatním azolům je při předepisování perorálního roztoku SPORANOX zapotřebí zvýšené opatrnosti.
Účinky na srdce
Ve studii s přípravkem SPORANOX I.V. u zdravých dobrovolníků bylo pozorováno přechodné asymptomatické snížení ejekční frakce levé srdeční komory. Ejekční frakce se do aplikace další infuze normalizovala. Klinický význam tohoto nálezu pro perorální lékové formy není znám.
U itrakonazolu byl prokázán negativní inotropní účinek a v souvislosti s podáním přípravku SPORANOX byly hlášeny případy městnavého srdečního selhání. Srdeční selhání bylo častěji hlášeno při spontánních hlášeních u celkové denní dávky 400 mg než u nižších celkových denních dávek, což naznačuje, že riziko srdeční slabosti se může s celkovou denní dávkou itrakonazolu zvyšovat.
SPORANOX má být užíván u pacientů s městnavým srdečním selháním nebo městnavým srdečním selháním v anamnéze pouze, pokud prospěch léčby zřetelně převýší riziko. Při tomto individuálním posouzení prospěchu a rizika mají být zohledněny faktory jako závažnost indikace, dávkovací režim (např. celková denní dávka) a individuální rizikové faktory svědčící pro rozvoj městnavého srdečního selhání. K těmto rizikovým faktorům patří srdeční onemocnění, jako např. ischemická choroba srdeční a chlopenní vady; závažné plicní onemocnění jako např. chronická obstrukční choroba bronchopulmonální; renální selhání a další stavy provázené otoky. Tito pacienti mají být informováni o známkách a příznacích městnavého srdečního selhání, mají být léčeni s opatrností, v průběhu léčby mají být monitorovány známky a příznaky městnavého srdečního selhání a pokud se tyto známky nebo příznaky během léčby objeví, SPORANOX má být vysazen.
Blokátory kalciového kanálu mohou mít negativní inotropní účinky, které mohou být aditivní k účinkům itrakonazolu. Itrakonazol může dále inhibovat metabolismus blokátorů kalciového kanálu.
Z tohoto důvodu je zapotřebí opatrnosti při současném užívání itrakonazolu a blokátorů kalciového kanálu z důvodu zvýšeného rizika CHF.
Účinky na činnost jater
V souvislosti s užíváním přípravku SPORANOX se velmi vzácně vyskytly případy závažné hepatotoxicity, včetně fatálního akutního jaterního selhání. Většinu z těchto případů tvoří pacienti s předchozím jaterním onemocněním, kteří byli léčeni pro systémové indikace, vykazovali další závažná onemocnění a/nebo užívali jiné hepatotoxické léky. U některých pacientů nebyly patrné zřejmé rizikové faktory jaterního onemocnění. Některé z těchto případů byly pozorovány během prvního měsíce léčby, z toho některé v prvním týdnu. U pacientů léčených přípravkem SPORANOX má být zváženo monitorování jatemích funkcí. Pacienti mají být poučeni o tom, že mají bezodkladně hlásit svému lékaři známky a příznaky svědčící pro hepatitidu, k nimž patří anorexie, nauzea, zvracení, únava, bolest v břiše nebo tmavá moč. U těchto pacientů má být neprodleně ukončena léčba přípravkem SPORANOX a má být provedeno vyšetření jaterních testů.
Jsou k dispozici omezené údaje u perorální formy itrakonazolu u pacientů s poruchou funkce jater. Opatrnost je nutná, pokud je přípravek užíván těmito pacienty. Je doporučeno, aby pacienti s poruchou funkce jater byli pečlivě monitorováni, pokud užívají itrakonazol. Při zahájení léčby u pacientů s cirhózou jater s jinými přípravky metabolizovanými CYP3A4, je třeba vzít v úvahu prodloužený eliminační poločas itrakonazolu pozorovaný po jedné perorální dávce v klinickém hodnocení s tobolkami itrakonazolu.
U pacientů se zvýšenými nebo abnormálními hodnotami jaterních enzymů nebo s aktivním jaterním onemocněním nebo po předchozí zkušenosti s hepatotoxicitou jiných léků léčba perorálním roztokem SPORANOX není vůbec doporučena, jedině v případě, že se jedná o možnost závažného ohrožení života a zahájena pouze, pokud očekávaný přínos převýší riziko jaterního poškození. V těchto případech je nutné monitorování jaterních funkcí u pacientů s existujícími abnormalitami jaterních funkcí, nebo po předchozí zkušenosti jaterní toxicity s jinými přípravky (viz bod 5.2 Farmakokinetické vlastnosti, Zvláštní skupiny pacientů, Porucha funkce jater).
Cystická fibróza
U pacientů s cystickou fibrózou bylo pozorováno kolísání terapeutických hladin v rovnovážném stavu při podávání perorálního roztoku v dávce 2,5 mg/kg 2x denně. Koncentrace v rovnovážném stavu > 250 ng/ml byly dosaženy přibližně u 50 % subjektů starších 16 let, avšak nikoli u pacientů mladších 16 let. Pokud se při léčbě perorálním roztokem SPORANOX nedostaví léčebná odpověď, je zapotřebí pacienta převést na alternativní léčbu.
Pediatrická populace
Vzhledem k omezeným klinickým údajům o použití perorálního roztoku SPORANOX u pediatrických pacientů lze přípravek podávat dětem pouze, pokud potenciální přínos léčby převýší možná rizika.
Starší osoby
Vzhledem k omezeným klinickým údajům o použití perorálního roztoku SPORANOX u starších pacientů lze přípravek podávat těmto pacientům pouze, pokud potenciální přínos léčby převýší možná rizika. Obecně je doporučeno, aby při stanovení dávkování u starších osob byla vzata v úvahu vyšší frekvence snížených jaterních, ledvinových nebo srdečních funkcí a souběžných onemocnění nebo terapií jinými přípravky (viz bod 4.4).
Porucha funkce ledvin
O perorálním užívání itrakonazolu pacienty s poruchou funkce ledvin jsou k dispozici omezené údaje. Užívání přípravku touto skupinou pacientů má být věnována zvýšená pozornost.
Léčba pacientů se závažnou neutropenií
Perorální roztok SPORANOX nebyl sledován při léčbě orální a/nebo ezofageální kandidózy u pacientů se závažnou neutropenií. Vzhledem k farmakokinetickým vlastnostem (viz bod 5.2) se nedoporučuje používat perorální roztok SPORANOX k zahájení léčby pacientů při bezprostředním riziku systémové kandidózy.
Ztráta sluchu
U pacientů léčených itrakonazolem byla hlášeno přechodná nebo trvalá ztráta sluchu. Mnoho těchto hlášení zahrnovalo současné podávání chinidinu, které je kontraindikováno (viz body 4.3 a 4.5). Ztráta sluchu se většinou vrátí k normálu po ukončení léčby, ale u některých pacientů může přetrvávat.
Neuropatie
V případě neuropatie, která může souviset s perorálním roztokem SPORANOX, má být léčba vysazena.
Zkřížená rezistence
Kmeny Candida spp. rezistentní na flukonazol nemohou být považovány za citlivé na itrakonazol. U systémové kandidózy má být před zahájením léčby itrakonazolem proveden test citlivosti.
Zaměnitelnost
Nedoporučuje se, aby SPORANOX perorální roztok byl používán zaměnitelně s jinými lékovými formami itrakonazolu.Je to z důvodu lékové expozice, která je při užití stejné dávky přípravku vyšší u perorálního roztoku než u tobolek, při užití stejné dávky přípravku.
Potenciál k interakcím
Souběžné užívání určitých přípravků s itrakonazolem může mít za následek změnu v účinnosti itrakonazolu a/nebo souběžně užívaného přípravku, život ohrožující situaci a/nebo náhlé úmrtí Přípravky, které jsou kontraindikovány, nejsou doporučeny anebo mají být užívány s opatrností spolu s itrakonazolem, jsou uvedeny v bodě 4.5.
Perorální roztok SPORANOX obsahuje sorbitol. Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Itrakonazol je metabolizován převážně cytochromem CYP3A4. Ostatní substance, které se podílí na této metabolické dráze, nebo mění aktivitu CYP3A4 mohou ovlivnit farmakokinetiku itrakonazolu. Podobně i itrakonazol může změnit farmakokinetiku dalších látek, které sdílejí tuto metabolickou cestu. Itrakonazol je silný inhibitor CYP3A4 a inhibitor P-glykoproteinu. Při současném užívání těchto přípravků mají být konzultovány příslušné informace o metabolické cestě a možné přizpůsobení dávkování.
Přípravky, které mohou snižovat plazmatické koncentrace itrakonazolu
Současné podávání itrakonazolu spolu s enzymovými induktory CYP3A4 může snížit biologickou dostupnost itrakonazolu, což může vést k tomu, že účinnost může být do značné míry snížena. Příkladem jsou:
• Antibakteriální látky: isoniazid, rifabutin, (viz také bod Přípravky, jejichž plazmatické koncentrace může itrakonazol snižovat), rifampicin.
• Antikonvulziva: karbamazepin (viz také bod Přípravky, které mohou snižovat plazmatické koncentrace itrakonazolu), fenobarbital, fenytoin.
• Antivirotika: efavirenz, nevirapin.
Z tohoto důvodu není doporučena kombinace itrakonazolu se silnými enzymovými induktory. Souběžnému užívání těchto přípravků se má vyvarovat 2 týdny před léčbou a během léčby itrakonazolem, pokud benefit převáží rizika potenciálně snížené účinnosti itrakonazolu. Během současného užívání má být sledována antifungální aktivita, a pokud je to nutné, je třeba zvýšit dávku itrakonazolu.
Přípravky, které mohou zvyšovat plazmatické koncentrace itrakonazolu
Silné inhibitory CYP3A4 mohou zvyšovat biologickou dostupnost itrakonazolu. Příkladem jsou:
• Antibakteriální látky: ciprofloxacin, klarithromycin, erythromycin.
• Antivirotika: ritonavirem potencovaný darunavir, ritonavirem potencovaný fosamprenavir, indinavir (viz také bod Přípravky, které mohou snížit plazmatické koncentrace itrakonazolu), ritonavir (viz také bod Přípravky, které mohou snížit plazmatické koncentrace intrakonazolu) a telaprevir.
Tyto přípravky mají být užívány s opatrností, pokud jsou společně používány s perorálním roztokem itrakonazolu. Pacienti, kteří musí užívat itrakonazol souběžně se silnými inhibitory CYP3A4 mají být pečlivě monitorováni pro příznaky zvýšeného nebo prodlouženého účinku farmakologických efektů itrakonazolu a v případě potřeby je nutné snížit dávku itrakonazolu. Je vhodné stanovit plazmatické koncentrace itrakonazolu.
Přípravky, jejichž plazmatické koncentrace může itrakonazol zvyšovat
Itrakonazol a jeho hlavní metabolit hydroxy - itrakonazol mohou inhibovat metabolismus léků metabolizovaných CYP3A4 a mohou inhibovat P-glykoprotein, což může vést ke zvýšené plazmatické koncentraci těchto přípravků nebo jejich aktivních metabolitů, pokud jsou užívány spolu s itrakonazolem. Tyto zvýšené plazmatické koncentrace mohou zvýšit nebo prodloužit jak léčebný účinek, tak i nežádoucí účinky těchto přípravků. CYP3A4 metabolizuje přípravky, které prodlužují QT interval a mohou být kontraindikovány s itrakonazolem, protože jejich kombinace může vést k ventrikulární tachyarytmii včetně výskytu torsade de pointes a potenciálně fatální arytmie. Jakmile je léčba ukončena, plazmatické koncentrace itrakonazolu se během 7 až 14 dní snižují v závislosti na dávce a délce léčby na téměř nezjistitelnou koncentraci. U pacientů s cirhózou nebo u pacientů užívajících inhibitory CYP3A4 může plazmatická koncentrace klesat pozvolna. To je především důležité na počátku léčby s přípravky, u kterých je metabolismus ovlivněn itrakonazolem.
Vzájemně působící přípravky jsou rozděleny do kategorií takto:.
- Kontraindikované: V žádném případě nesmějí být užívány spolu s itrakonazolem a také ještě dva týdny po ukončení léčby itrakonazolem.
- Nejsou doporučené: Užívání těchto přípravků se má vyvarovat během léčby a až dva týdny po ukončení léčby itrakonazolem, pokud potenciální přínos převýší možná rizika nežádoucích účinků. Jestliže není možné se vyhnout souběžnému užívání, je doporučeno provádět monitorování klinických příznaků a symptomů zvýšených nebo prodloužených účinků nebo nežádoucích účinků vzájemně se ovlivňujících přípravků, a pokud je to nutné je třeba snížit dávkování nebo přerušit užívání. Je vhodné stanovit plazmatické koncentrace itrakonazolu.
- Užívání s opatrností: Je doporučeno pečlivé monitorování, pokud je přípravek souběžně užívaný s itrakonazolem. Během souběžného užívání se doporučuje provádět u pacientů monitorování klinických příznaků a symptomů souvisejících se zvýšením nebo prodloužením účinku nebo nežádoucích účinků vzájemně se ovlivňujících přípravků, a pokud je to nutné je třeba snížit dávkování nebo přerušit užívání. Pokud je to vhodné, doporučuje se stanovit plazmatické koncentrace.
Příklady přípravků, u kterých mohou být zvýšené plazmatické hodnoty při užívání itrakonazolu, uvedené dle třídy přípravku s doporučením týkající se souběžného podávání s itrakonazolem.
|
Třída přípravku |
Kontraindikované |
Nedoporučené |
Užívané s opatrností |
|
Alfa blokátory |
tamsulosin | ||
|
Analgetika |
levacetylmethadol (levomethadyl) methadon |
fentanyl |
alfentanil, buprenorfin IV a sublinguální oxykodon sufentanil |
|
Antiarytmika |
disopyramid dofetilid dronedaron chinidin |
digoxin | |
|
Antibakteriální látky |
telithromycin - u pacientů s vážnou poruchou funkcí jater nebo ledvin |
rifabutina |
telithromycin |
|
Antikoagulancia a antiagregancia |
tikagrelor |
apixaban rivaroxaban |
kumariny cilostazol dabigatran |
|
Antikonvulziva |
karbamazepina | ||
|
Antidiabetika |
repaglinid, saxagliptin | ||
|
Antiparazitika a Antiprotozoa |
halofantrin |
prazikvantel | |
|
Antihistaminika |
astemizol mizolastin terfenadin |
bilastin ebastin | |
|
Antimigrenika |
námelové alkaloidy, jako je dihydroergotamin ergometrin (ergonovin) ergotamin methylergometrin (methylergonovin) |
eletriptan | |
|
Antineoplastika |
irinotekan |
axitinib dabrafenib dasatinib ibrutinib nilotinib sunitinib trabektedin |
bortezomib busulfan docetaxel erlotinib gefitinib imatinib ixabepilon lapatinib ponatinib trimetrexát vinka alkaloidy |
|
Antipsychotika Anxiolytika a Hypnotika |
lurasidon perorální midazolam pimozid sertindol triazolam |
alprazolam aripiprazol brotizolam buspiron haloperidol midazolam IV perospiron kvetiapin ramelteon risperidon | |
|
Antivirotika |
simeprevir |
maravirok indinavirb ritonavirb |
|
Třída přípravku |
Kontraindikované |
Nedoporučené |
Užívané s opatrností |
|
sachinavir | |||
|
Betablokátory |
nadolol | ||
|
Blokátory kalciového kanálu |
bepridil felodipin lerkanidipin nisoldipin |
další dihydropyridiny, verapamil | |
|
Kardiovaskulární látky Ostatní látky |
ivabradin ranolazin |
aliskiren sildenafil užívaný k léčbě plicní hypertenze |
bosentan riocigvát |
|
Diuretika |
eplerenon | ||
|
Gastrointestinální přípravky |
cisaprid domperidon |
aprepitant | |
|
Immunosupresiva |
everolimus |
budesonid ciklesonid cyklosporin dexamethason flutikason methylprednisolon rapamycin (také známý jako sirolimus) takrolimus temsirolimus | |
|
Přípravky regulující lipidy |
lovastatin simvastatin |
atorvastatin | |
|
Přípravky k léčbě onemocnění respiračního traktu |
salmeterol | ||
|
SSRI Tricyklická a příbuzná antidepresiva |
reboxetin | ||
|
Urologické přípravky |
fesoterodin - u pacientů mírnou až vážnou poruchou poruchou jater solifenacin - u pacientů s vážnou poruchou funkce ledvin, nebo mírnou až vážnou poruchou funkce jater |
darifenacin vardenafil |
fesoterodin imidafenacin oxybutynin sildenafil k léčbě erektilní dysfunkce solifenacin tadalafil tolterodin |
|
Ostatní |
kolchicin, u pacientů s renálním nebo jaterním poškozením |
kolchicin konivaptan tolvaptan |
alitretinoin (perorální formulace) cinakalcet mozavaptan |
a Viz také bod Přípravky, které mohou snižovat plazmatické koncentrace itrakonazolu a Viz také bod Přípravky, které mohou zvyšovat plazmatické koncentrace itrakonazolu
Přípravky, jejichž plazmatické koncentrace může itrakonazol snižovat
Užívání itrakonazolu společně s nesteroidním antirevmatikem meloxikamem může snížit plazmatickou koncentraci meloxikamu. Užívání meloxikamu je doporučeni s opatrností, pokud je užíván společně s itrakonazolem, jejich účinky nebo nežádoucí účinky se mají monitorovat. Pokud je meloxikam užíván společně s itrakonazolem, je doporučena, pokud je to nezbytné, úprava dávkování meloxikamu.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
SPORANOX nesmí být užíván v těhotenství kromě život ohrožujících situací, kdy potenciální přínos pro matku převýší možné riziko poškození plodu (viz bod 4.3).
Ve studiích se zvířaty prokázal itrakonazol reprodukční toxicitu (viz bod 5.3).
Informace o užívání přípravku SPORANOX během těhotenství jsou omezené. V rámci postmarketingového sledování byly hlášeny případy kongenitálních abnormalit zahrnujících skeletální, urogenitální, kardiovaskulární a oftalmologické malformace včetně chromozomálních a mnohočetných malformací. Kauzální vztah s přípravkem SPORANOX nebyl prokázán.
Epidemiologické údaje o použití přípravku SPORANOX během prvního trimestru gravidity - většinou u krátkodobě léčené vulvovaginální kandidózy - nesvědčí o zvýšeném riziku malformací ve srovnání s kontrolními subjekty, které nebyly podrobeny žádné známé teratogenní látce. U modelu u potkanů bylo prokázáno, že itrakonazol prochází placentou.
Ženy ve fertilním věku
Ženy ve fertilním věku, které užívají perorální roztok SPORANOX mají používat antikoncepční opatření. V účinných antikoncepčních opatřeních je nezbytné pokračovat až do příští menstruace po ukončení léčby přípravkem SPORANOX. U těchto žen mají být pravidelně monitorovány jatemí funkce až do ukončení léčby.
Kojení
Velmi malé množství itrakonazolu je vylučováno do mateřského mléka. Očekávaný prospěch léčby perorálním roztokem SPORANOX má být proto posouzen oproti možnému riziku kojení. V případě pochybností nemá pacientka kojit.
Fertilita
Viz Neklinické informace týkajících se informací o fertilitě zvířat relevantní k itrakonazolu a hydroxypropyl-P-cyklodextrinu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
S přípravkem nebyly provedeny studie účinku na řízení a obsluhování strojů. V některých případech se mohou objevit nežádoucí účinky jako závratě, poruchy zraku a ztráta sluchu (viz bod 4.8 Nežádoucí účinky). Toto je třeba vzít v úvahu při řízení a obsluhování strojů.
4.8 Nežádoucí účinky
Klinické studie
Bezpečnost perorálního roztoku SPORANOX byla hodnocena u 889 pacientů, v šesti dvojitě zaslepených a čtyřech otevřených klinických studiích. 624 pacientů z 889 léčených perorálním roztokem SPORANOX ve dvojitě zaslepených klinických studiích. Všech 889 pacientů užívalo minimálně jednu dávku perorálního roztoku SPORANOX k léčbě orofaryngeálních a ezofageálních kandidóz a poskytlo bezpečnostní údaje. Nežádoucí účinky (ADR) hlášené u >1% pacientů užívajících perorální roztok SPORANOX v těchto klinických studiích jsou uvedeny v Tabulce 1.
|
Tabulka 1: Nežádoucí účinky hlášené u pacientů léčených perorálním roztokem SPORANOX v 10 klinických studiích s incidencí > 1 % | |
|
Třídy orgánových systémů Nežádoucí účinek |
SPORANOX perorální roztok % (N=889) |
|
Poruchy nervového systému | |
|
3,6 | |
|
Dysgeuzie |
1,5 |
|
Závrať |
1,1 |
|
Respirační, hrudní a mediastinální poruchy | |
|
1,8 | |
|
Gastrointestinální poruchy | |
|
9,1 | |
|
8,2 | |
|
5,2 | |
|
Abdominální bolest |
4,5 |
|
1,0 | |
|
Poruchy kůže a podkožní tkáně | |
|
2,5 | |
|
Celkové poruchy a reakce v místě aplikace | |
|
Pyrexie |
5,2 |
Nežádoucí účinky hlášené u <1% pacientů léčených perorálním roztokem SPORANOX v těchto klinických studiích jsou uvedeny v Tabulce 2.
Tabulka 2: Nežádoucí účinky hlášené u <1% pacientů
léčených perorálním roztokem SPORANOX v 10 klinických
studiích_
Třídy orgánových systémů
Nežádoucí účinek_
Poruchy krve a lymfatického systému_
Leukopenie_
T rombocytopenie_
Poruchy imunitního systému_
Hypersenzitivita_
Poruchy metabolismu a výživy_
Hypokalemie_
Poruchy nervového systému_
Hypestézie_
Periferní neuropatie_
Parestézie_
Poruchy ucha a labyrintu_
Tinitus_
Srdeční potíže_
Srdeční selhání_
Gastrointestinální potíže_
Zácpa_
Poruchy jater a žlučových cest_
Tabulka 2: Nežádoucí účinky hlášené u <1% pacientů
léčených perorálním roztokem SPORANOX v 10 klinických studiích_
Jatemí selhání_
Hyperbilirubinemie_
Poruchy kůže a podkožní tkáně_
Pruritus_
Kopřivka_
Poruchy svalové a kosterní soustavy a pojivové tkáně_
Artralgie_
Myalgie_
Poruchy reprodukčního systému a prsu_
Menstruační potíže_
Celkové poruchy a reakce v místě aplikace_
Edém
Pediatrická populace
Bezpečnost perorálního roztoku SPORANOX byla hodnocena u 250 pacientů ve věku od 6 měsíců do 14 let, kteří se účastnili pěti otevřených klinických studií. Tito pacienti užívali minimálně jednu dávku perorálního roztoku SPORANOX pro léčbu plísňových infekcí a poté poskytli údaje.
Na základě sběru údajů z těchto klinických studií byly často hlášené nežádoucí účinky (ADR) u pediatrických pacientů: zvracení (36,0 %), pyrexie (30,8 %), průjem (28,4 %), záněty sliznice (23,2 %), vyrážka (22,8 %), abdominální bolest (17,2 %), nauzea (15,6 %), hypertenze (14,0 %) a kašel (11,2 %). Všeobecně byly nežádoucí účinky u pediatrické populace podobné těm, jaké byly pozorované u dospělých jedinců, ale incidence je u pediatrické populace vyšší.
Postmarketingové sledování
Nežádoucí účinky identifikované během postmarketingového sledování s přípravkem SPORANOX
(všechny lékové formy) jsou uvedeny v Tabulce 3, frekvence jsou stanoveny podle následující
konvence:
velmi časté > 1/10;
časté > 1/100 a < 1/10;
méně časté > 1/1000 a < 1/100;
vzácné > 1/10 000 a < 1/1000;
velmi vzácné < 1/10 000, není známo (z dostupných údajů nelze určit) .
(Hlášení četnosti výskytu nežádoucích účinků je založeno na spontánních hlášeních a nepředstavuje přesnější odhady incidence, které mohou být získány z klinických a epidemiologických studií.)
|
Tabulka 2: Postmarketingové hlášení nežádoucích účinků | |
|
Poruchy krve a lymfatického systému | |
|
Méně časté |
Leukopenie, neutropenie, trombocytopenie |
|
Poruchy imunitního systému | |
|
Není známo |
Sérová choroba, angioneurotický edém, anafylaktické reakce, anafylaktoidní reakce hypersenzitivita* |
|
Poruchy metabolismu a výživy | |
|
Méně časté |
Hypokalemie |
|
Není známo |
Hypertriacylglycerolemie |
|
Poruchy nervového systému | |
|
Časté | |
|
Méně časté |
Periferní neuropatie* závratě |
|
Není známo |
Parestézie, hypestézie, třes |
|
Poruchy oka | |
|
Méně časté |
Poruchy vidění včetně rozmazaného vidění a diplopie |
|
Poruchy ucha |
a labyrintu |
|
Není známo |
Tinitus, přechodná nebo trvalá ztráta sluchu* |
|
Srdeční poruchy | |
|
Není známo |
Městnavé srdeční selhání* |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté | |
|
Není známo |
Plicní edém |
|
Gastrointestinální poruchy | |
|
Časté | |
|
Méně časté |
Dyspepsie, zácpa |
|
Není známo |
Pankreatitida |
|
Poruchy jater a žlučových cest | |
|
Časté |
Zvýšení hodnot jaterních enzymů |
|
Méně časté |
Hepatitida, hyperbilirubinemie |
|
Není známo |
Závažná hepatotoxicita (včetně některých fatálních případů akutního selhání jater)* |
|
Poruchy kůže |
a podkožní tkáně |
|
Časté | |
|
Méně časté | |
|
Není známo |
Toxická epidermální nekrolýza, Stevens-Johnsonův syndrom, akutní generalizovaná ekzematózní pustulóza, erythema multiforme, exfoliativní dermatitida, leukocytoklastická vaskulitida, kopřivka, alopecie, fotosenzitivita |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Není známo |
Myalgie, artralgie |
|
Poruchy ledvin a močových cest | |
|
Není známo |
Polakisurie, inkontinence moči |
|
Poruchy reprodukčního systému a prsu | |
|
Není známo |
Menstruační poruchy, erektilní dysfunkce |
|
Celkové poruchy a reakce v místě aplikace | |
|
Časté |
Pyrexie |
|
Méně časté |
Otok |
viz bod 4.4 Zvláštní upozornění a opatření pro použití
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování Příznaky a známky
Všeobecně nežádoucí účinky hlášené s předávkováním byly shodné s těmi, které byly hlášeny při užívání itrakonazolu (viz bod 4.8).
Léčba
V případě předávkování je nutné přistoupit k podpůrným opatřením. Itrakonazol nelze odstranit hemodialýzou. Specifické antidotum není k dispozici.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antimykotika pro systémové použití, triazolové deriváty ATC kód: J02AC02
Mechanismus účinku
Studie in vitro prokázaly, že itrakonazol narušuje v buňkách mikromycet syntézu ergosterolu. Ergosterol představuje životně důležitou membránovou komponentu mykotických buněk. Narušení jeho syntézy vede v konečném důsledku k antimykotickému účinku.
Vztah farmakokinetiky a farmakodynamiky
Vztah farmakokinetiky a farmakokinetiky itrakonazolu a triazolů je všeobecně špatně pochopený. Farmakodynamické účinky
Mikrobiologie
Itrakonazol, triazolový derivát, vykazuje široké spektrum účinnosti.
Hraniční hodnoty itrakonazolu byly stanoveny pouze CLSI pro Candida spp. z povrchových mykotických infekcí (CLSI M27-A2), hraniční hodnoty pro metodiku EUCAST nebyly stanoveny). Metodou CLSI byly stanoveny tyto hraniční hodnoty: citlivé < 0,125 pg/ml, citlivé, závislé na dávce 0,25 - 0,5 pg/ml a rezistentní > 1 pg/ml. Interpretační hraniční hodnoty nebyly stanoveny CLSI pro vláknité mikromycety.
Hraniční hodnoty EUCAST pro itrakonazol byly stanoveny pro Aspergillus flavus, A. fumigatus, A. nidulans a A. terreus, a jsou následující: citlivé < 1 mg/l , rezistentní > 2 mg/l. Metodou EUCAST musí být ještě stanoveny hodnoty pro itrakonazol a Candida spp .
Studie in vitro prokazují, že itrakonazol inhibuje růst širokého spektra plísní patogenních pro člověka obvykle v koncentracích < 1 pg/ml. K nim patří:
Candida spp. (včetně Candida albicans, Candida tropicalis, Candida parapsilosis, a Candida dubliniensis), Aspergillus spp., Blastomyces dermatitidis, Cladosporium spp., Coccidioides immitis, Cryptococcus neoformans, Geotrichum spp., Histoplasma spp., včetně H. capsulatum, Paracoccidioides brasiliensis, Penicillium marneffei, Sporothrix schenckii a Trichosporon spp. Itraconazol také prokazují in vitro účinek protit Epidermophyton floccosum, Fonsecaea spp., Malassezia spp., Microsporum spp., Pseudallescheria boydii, Trichophyton spp. a různé další kvasinky a plísně.
Candida Krusei, Candida glabrata a Candida guillermondii jsou všeobecně nejméně susceptibilní druhy Candida, včetně některých izolátů, které in vitro vykazují průkaznou rezistenci k itrakonazolu.
Hlavní druhy plísní, které nejsou inhibovány itrakonazolem, jsou Zygomycetes (např. Rhizopus spp., Rhizomucor spp., Mucor spp. a Absidia spp ), Fusarium spp., Scedosporium spp. a Scopulariopsis spp.
Ukazuje se, že rezistence k azolům se vyvíjí pomalu a je často výsledkem několika genetických mutací. Popsaný mechanismus zahrnuje nadměrnou expresi ERG11, který kóduje cílový enzym 14a-demethylázu, bodové mutace v ERG11 vedoucí ke snížené cílové afinitě a/nebo nadměrnou expresi přenašeče vedoucí ke zvýšenému efluxu. Zkřížená rezistence mezi jednotlivými azoly byla pozorována v rámci Candida spp., přestože rezistence k jednomu azolu neznamená nutně rezistenci k ostatním azolům. Byly hlášeny kmeny Aspergillus fumigatus rezistentní k itrakonazolu.
5.2 Farmakokinetické vlastnosti
Obecné farmakokinetické údaje
Vrcholových plazmatických koncentrací itrakonazolu je dosaženo za 2,5 hod po podání perorálního roztoku. Itrakonazol je podroben rozsáhlé jaterní metabolizaci na četné metabolity. Farmakokinetika itrakonazolu je nelineární a v důsledku toho vykazuje po opakovaném podání kumulaci v plazmě. Rovnovážného stavu je obecně dosaženo přibližně za 15 dní s hodnotami cmax a AUC 4krát až 7krát vyššími, než ty co byly pozorované po jednorázovém podání. Rovnovážného stavu s hodnotami cmax
2,0 pg/ml je dosaženo po perorálním podání 200 mg 1x denně. Výsledný biologický poločas itrakonazolu je obecně po jednorázovém podání v rozmezí 16 až 28 hodin a zvyšuje se po opakovaném podání na 34 až 42 hodin. V průběhu 7 až 14 dní po ukončení léčby klesají plazmatické koncentrace v závislosti na dávce a délce léčby, až na nedetekovatelné koncentrace. Celková průměrná plazmatická clearance následně po intravenózním podání je 278 ml/min. Celková průměrná plazmatická clearance itrakonazolu klesá ve vysokých dávkách díky saturačnímu mechanismu jaterního metabolismu.
Absorpce
Po podání perorálního roztoku je itrakonazol rychle absorbován. Vrcholových plazmatických koncentrací itrakonazolu je dosaženo za 2,5 hodiny po podání perorálního roztoku na lačno. Pozorovaná absolutní biologická dostupnost itrakonazolu je v nasyceném stavu přibližně 55 % a zvyšuje se o 30 % při užití nalačno.
Expozice itrakonazolu je vyšší u perorálního roztoku než u tobolek, jestliže jsou užívány stejné dávky (viz bod 4.4).
Distribuce
Většina itrakonazolu přítomného v plazmě je vázána na plazmatické proteiny (99,8 %), hlavní vazebnou složkou je albumin (99,6 % pro hydroxylované metabolity). Itrakonazol vykazuje rovněž značnou afinitu k lipidům. Pouze 0,2 % itrakonazolu přítomného v plazmě se vyskytuje ve volné formě. Itrakonazol je distribuován do rozsáhlého kompartmentu (> 700 l), což svědčí o extenzivní distribuci do tkání: koncentrace v plicích, ledvinách, játrech, kostech, žaludku, slezině a ve svalech byly nalezeny 2 - 3x vyšší než odpovídající plazmatické koncentrace, vychytávání v keratinových tkáních, zvláště v kůži, je až 4x vyšší než v plazmě. Koncentrace v mozkomíšní tekutině jsou daleko nižší než v plazmě, ale byla dokázána účinnost proti infekcím přítomným v mozkomíšní tekutině
Metabolismus
Itrakonazol je extenzivně metabolizován játry na velký počet metabolitů. Ze studií in vitro vyplývá, že hlavním enzymem zodpovědným za metabolismus itrakonazolu je CYP3A4.
Hlavním metabolitem je hydroxy-itrakonazol, který má in vitro antimykotický účinek srovnatelný s itrakonazolem; plazmatické koncentrace těchto metabolitů jsou přibližně dvojnásobné než u itrakonazolu.
Vylučování
Itrakonazol je vylučován především ve formě neúčinných metabolitů močí (35 %) a stolicí (54 %). Renální vylučování itrakonazolu a jeho aktivního metabolitu hydroxy - itrakonazolu představuje méně než 1 % intravenózní dávky. Na základě perorální radioaktivně značené dávky, se pohybuje exkrece nezměněného léčiva stolicí v rozmezí 3 % až 18 % dávky.
Redistribuce itrakonazolu z keratinových tkání se zdá být zanedbatelnou, eliminace z těchto tkání je závislá na epidermální regeneraci. Na rozdíl od plazmy přetrvávají koncentrace v kůži 2 - 4 týdny po ukončení 4týdenní léčby a v nehtovém keratinu - kde může být itrakonazol prokázán již 1 týden po zahájení léčby - přetrvávají nejméně 6 měsíců po ukončení tříměsíční léčby.
Zvláštní skupiny pacientů
Porucha funkce jater
Itrakonazol je metabolizován převážně v játrech. Studie farmakokinetiky byly provedeny u 6 zdravých jedinců a u 12 pacientů s cirhózou, kterým bylo podáno jednorázově 100 mg itrakonazlou ve formě tobolek. Statisticky významné snížení cmax (o 47 %) a dvojnásobné zvýšení eliminačního poločasu itrakonazolu (37 versus 16 hodin) bylo zaznamenáno u pacientů s cirhózou ve srovnání se zdravými jedinci. Nicméně celková expozice itrakonazolu založená na AUC byla stejná u pacientů s cirhózou a zdravých subjektů. U pacientů s cirhózou nejsou k dispozici údaje o dlouhodobém užívání itrakonazolu (viz body 4.2 a 4.4).
Porucha funkce ledvin
O perorálním užívání itrakonazolu pacienty s poruchou funkce ledvin jsou k dispozici omezené údaje. Studie farmakokinetiky byly provedeny s jednou 200 mg dávkou intrakonazolu (čtyři 50mg tobolky), která byla podávána třem skupinám pacientů s poruchou funkce ledvin (uremie: n=7; hemodialýza: n= 07; kontinuální ambulantní peritoneální dialýza: n=5). U pacientů s uremií s průměrnou clearancí kreatininu 13 m/min x 1,73 m2, byla mírně snížena expozice založená na AUC v porovnání s parametry u normální populace. Tato studie neprokázala významný vliv hemodialýzy nebo kontinuální peritoneální dialýzy na farmakokinetiku itrakonazolu (Tmax, cmax, a AUC0-8h). Časové profily plazmatických koncentrací prokázaly širokou interindividuální variabilitu u všech těchto tří skupin.
Po jednorázovém intravenózním podání byl průměrný poločas itrakonazolu u pacientů se střední (definováno v této studii jako CrCl 50-79 ml/min), mírnou (definováno v této studii jako CrCl 20-49 ml/min), a vážnnou poruchou funkce ledvin (definováno v této studii jako CrCl <20 ml/min), podobný jako u zdravých jedinců (rozsah hodnot 42 -49 hodin versus 48 hodin u pacientů s poruchou funkce ledvin a zdravých jedinců vzájemně). Celková expozice itrakonazolu založená na AUC byla snížená u pacientů s mírným a vážným poškozením funkce ledvin přibližně o 30 % a 40 %v porovnání s jedinci s normální funkcí ledvin.
U pacientů s poruchou funkce ledvin nejsou dostupné údaje při dlouhodobém užívání itrakonazolu. Dialýza neměla efekt na poločas nebo clearanci itrakonazolu nebo hydroxy-itrakonazolu (viz také body 4.2 a 4.4).
Pediatrická populace
U pediatrické populace jsou dostupné omezené údaje o farmakokinetice itrakonazolu. Studie farmakokinetiky u dětí a dospívajících nebo intravenózním roztokem. Individuální dávky kapslí a perorálního roztoku v rozmezí 1,5 až 12,5 mg/kg/den byly podávány jednou nebo dvakrát denně. Intravenózně bylo podáváno buď 2,5 mg/kg v jedné dávce nebo 2,5 mg/kg v jedné dávce. U stejné denní dávky přineslo podání 2x denně v porovnání s jedinou denní dávkou vrcholové a spodní koncentrace srovnatelné s dospělými při jedné denní dávce. U AUC itrakonazolu nebyl pozorován význam v závislosti na věku a celkové tělesné kondici, zatímco bylo zaznamenáno nevýrazné spojení mezi věkem a distribučním objemem cmax a konečnou eliminací itrakonazolu.
Hydroxypropyl-beta-cyklodextrin
Perorální biologická dostupnost hydroxypropyl-beta-cyklodextrinu je dána rozpustností itrakonazolu v perorálním roztoku je průměrně nižší než 0,5 % a je podobná jako u samostatného hydroxypropyl-beta-cyklodextrinu. Tato nízká biologická dostupnost hydroxypropyl-beta-cyklodextrinu není změněna přítomností potravy a je podobná po jednorázovém a opakovaném podávání.
5.3 Předklinické údaje vztahující se k bezpečnosti
Itrakonazol
Itrakonazol byl testován standardním souborem preklinických studií.
Studie akutní toxicity itrakonazolu u myší, laboratorních potkanů, morčat a psů prokázaly široké bezpečnostní rozmezí. Studie (sub)chronické toxicity po perorálním podání u laboratorních potkanů a psů byly zaměřeny na některé cílové orgány nebo tkáně: kůru nadledvinek, játra a mononukleární fagocytární systém a rovněž na poruchy lipidového metabolismu projevující se přítomností xantomových buněk v různých orgánech.
Histologická vyšetření kůry nadledvinek prokázala u vysokých dávek reverzibilní otok s buněčnou hypertrofií retikulární a fascikulární zóny, někdy doprovázené ztenčením glomerulární zóny. Při vysokých dávkách byly nalezeny reverzibilní jaterní změny. Mírné změny byly pozorovány v sinusoidních buňkách a byla pozorována vakuolizace hepatocytů, která svědčí o buněčné dysfunkci, avšak bez zjevné hepatitidy nebo hepatické nekrózy. Histologické změny mononukleárního fagocytárního systému jsou charakterizovány zejména makrofágy se zvýšeným bílkovinným materiálem v různých parenchymatózních tkáních.
Po dlouhodobém podávání itrakonazolu juvenilním psům byla pozorována všeobecně nižší kostní minerální denzita.
Ve třech toxikologických studiích s laboratorními potkany vyvolal itrakonazol kostní defekty. Vyvolané defekty zahrnují sníženou aktivitu kostních plotének, ztenčení zona compactata dlouhých kostí a zvýšení kostní fragility.
Karcinogenita a mutagenita
Itrakonazol není primární karcinogen u laboratorních potkanů nebo myší. U samců laboratorních potkanů se však vyskytla vyšší incidence sarkomu měkkých tkání, která je přičítána zvýšení nenádorových chronických zánětlivých reakcí pojivové tkáně jako důsledku zvýšených hladin cholesterolu a cholesterolóze pojivové tkáně. Nebyly zjištěny známky mutagenního potenciálu itrakonazolu.
Reprodukční toxicita
Na dávce závislé zvýšení maternální toxicity, embryotoxicity a teratogenity u laboratorních potkanů a myší bylo nalezeno při vysokých dávkách itrakonazolu. U laboratorních potkanů spočívá teratogenita v rozsáhlých skeletálních defektech; u myší spočívá ve výhřezu mozku a makroglosii.
Fertilita
V průběhu léčby itrakonazolem nebylo zjištěno primární ovlivnění fertility.
Hydroxypropyl-beta-cyclodextrin (HP-P-CD) - hydroxypropylbetadex
Studie toxicity po jednorázovém a opakovaném podání u myší, laboratorních potkanů a psů prokázaly široké rozmezí bezpečnosti po perorálním i intravenózním podání HP-P-CD. Většina účinků byla ve skutečnosti adaptivní (histologické změny v urinárním traktu, změkčení stolice vzhledem k osmotické retenci vody v tlustém střevě, aktivace mononukleárního fagocytárního systému) a vykázala dobrou reverzibilitu. Nepatrné jaterní změny byly prokázány při 30x vyšších dávkách HP-P-CD než navrhovaných pro člověka.
Perorální podávání juvenilním bíglům při dávce HP-P-CD na 1200 mg/kg po dobu až 13 týdnů se 4týdenní rekonvalescencí bylo klinicky dobře tolerováno bez žádných účinků zaznamenaných ve srovnání s kontrolními zvířaty v laboratoři nebo s histopatologickým vyšetřením.
Karcinogenita a mutagenita
Ve studii u myší nebyla prokázána žádná primární karcinogenita.
Při studiu karcinogenity u laboratorních potkanů byla prokázána zvýšená incidence neoplazmatického bujení v tlustém střevě (5 000 mg/kg/den) a v exokrinních buňkách pankreatu (od 500 mg/kg/den).
Ve srovnání k tělesnému povrchu odpovídá expozice HP-P-CD člověku, při doporučeném klinickém dávkování perorálního roztoku SPORANOX, přibližně 1,7násobku expozice nejnižší dávky ve studii karcinogenity u laboratorních potkanů při dávce 500 mg/kg/den.
Mírně zvýšený výskyt adenokracinomu tlustého střeva spojený s hypertrofickými /hyperplastickými zánětlivými změnami střevní sliznice způsobené zvýšenou osmolalitou HP-P-CD, je považován za méně klinicky relevantní.
Vznik pankreatických tumorů je podmíněn mitogenní aktivitou cholecystokininu u laboratorních potkanů. To nebylo prokázáno při studiu karcinogenity u myší ani ve 12měsíční studii u psů ani ve dvouleté studii u samic opic makaků obecných. Mitogenní aktivita cholecystokininu nebyla prokázána u člověka. Nicméně klinická relevance těchto zjištění není použitelná.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Hydroxypropylbetadex, sorbitol 70% nekrystalizující, propylenglykol, kyselina chlorovodíková 35%, třešňové aroma, karamel, dihydrát sodné soli sacharinu, roztok hydroxidu sodného 1mol/l a čištěná voda.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky v originálním balení,
1 měsíc po prvním otevření.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Hnědá skleněná lahvička o obsahu 150 ml s pojistným šroubovacím uzávěrem (polypropylen/LDPE), plastovým krytem s odměrkou (2,5 ml, 5 ml, 10 ml), krabička.
Velikost balení: 150 ml
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
SPORANOX perorální roztok je opatřen bezpečnostním uzávěrem proti otevření lahvičky dětmi. Lahvičku lze otevřít takto: zatlačit plastikový šroubovací klobouček směrem dolů a současně jím otáčet proti směru hodinových ručiček.
Spolu s perorálním roztokem přípravku SPORANOX je dodávána dávkovací odměrka.
Použijte odměrku tak, jak je umístěna na lahvičce. Ujistěte se, že část odměrky s vyznačenou stupnicí (ta, která přiléhá nejméně k lahvičce) je směrem vzhůru; to je část odměrky, kterou máte naplnit. Pokud šipka po straně směřuje vzhůru, správná strana je nahoře.
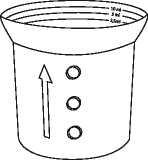
Výdej přípravku vázán na lékařský předpis.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag s.r.o., Karla Engliše 3201/6, 150 00 Praha 5, Česká republika
8. REGISTRAČNÍ ČÍSLO
26/1097/97-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19.11. 1997
Datum posledního prodloužení registrace: 28.1.2015
10. DATUM REVIZE TEXTU
22.4.2015
19/19