Spiriva Respimat 2,5 Mikrogramu
Sp.zn.sukls180686/2015 SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Spiriva Respimat 2,5 mikrogramu, roztok k inhalaci
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Podaná dávka je tiotropium 2,5 mikrogramu na 1 vstřik (2 vstřiky tvoří 1 léčivou dávku) a odpovídá tiotropii bromidum monohydricum 3,124 mikrogramům.
Podaná dávka je dávka, která po průchodu náustkem je pacientovi k dispozici.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Roztok k inhalaci
Čirý bezbarvý roztok k inhalaci
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
CHOPN
Tiotropium je indikováno k udržovací bronchodilatační léčbě ke zmírnění příznaků u pacientů s chronickou obstrukční plicní nemocí (CHOPN).
Přípravek Spiriva Respimat je indikován k přídatné bronchodilatační léčbě dospělých pacientů s astmatem, kteří jsou léčeni kombinací inhalačních kortikosteroidů (> 800 pg budesonidu/den nebo ekvivalentní dávky jiného inhalačního kortikosteroidu) spolu s dlouhodobě působícími beta2-agonisty, a kteří prodělali jednu nebo více těžkých exacerbací v předešlém roce.
4.2 Dávkování a způsob podání
Dávkování
Léčivý přípravek je určen pouze k inhalaci. Náplň lze vložit a použít pouze s inhalátorem Respimat (viz 4.2).
Jednu léčivou dávku tvoří dva vstřiky z inhalátoru Respimat.
Doporučená dávka přípravku pro dospělé je 5 mikrogramů tiotropia podaná dvěma vstřiky z inhalátoru Respimat jednou denně, ve stejnou denní dobu.
Doporučená dávka nesmí být překročena.
Při léčbě astmatu se plný přínos léčby projeví až po několika dávkách léčivého přípravku.
Zvláštní skupiny pacientů
Starší pacienti mohou užívat tiotropium-bromid v doporučených dávkách.
Pacienti s poruchou funkce ledvin mohou užívat tiotropium-bromid v doporučených dávkách. Dávkování u pacientů se středně těžkým až těžkým postižením renálních funkcí (clearance kreatininu < 50 ml/min) viz 4.4 a 5.2.
Pacienti s poruchou jaterních funkcí mohou užívat tiotropium-bromid v doporučených dávkách (viz 5.2).
Pediatrická populace CHOPN
Neexistuje žádné relevantní použití přípravku Spiriva Respimat u dětí a dospívajících do 18 let věku. Cystická fibróza
Účinnost a bezpečnost přípravku Spiriva Respimat nebyla stanovena (viz body 4.4 a 5.1).
Účinnost a bezpečnost přípravku Spiriva Respimat u dětí a dospívajících nebyla dosud stanovena. Způsob podání
K zajištění správného podání léčivého přípravku musí být pacientovi ukázáno, jak správně používat inhalátor, lékařem nebo jiným specialistou.
Návod k použití a zacházení
Úvod
Spiriva Respimat (tiotropii bromidum). Seznamte se s tímto návodem k použití před tím, než začnete přípravek Spiriva Respimat používat.
Inhalátor budete používat pouze JEDNOU DENNĚ. Při každém použití proveďte DVA VSTŘIKY.
Kryt
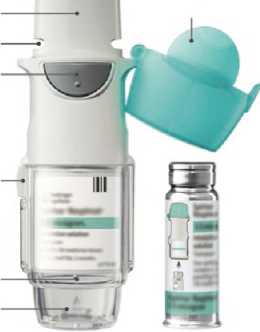
Zásobník
Ná ustek Vzduchový ventil
Tlačítko uvolňující dávku
Bezpečnostní pojistka
Průhledný vnější obal Prorážecí část
• Pokud jste přípravek Spiriva Respimat nepoužíval(a) déle než 7 dní, proveďte jeden vstřik s inhalátorem otočeným náustkem směrem k zemi.
• Pokud jste přípravek Spiriva Respimat nepoužíval(a) déle než 21 dnů, opakujte kroky 4 až 6
v bodě „Příprava inhalátoru k prvnímu použití“, dokud se neobjeví zřetelný obláček. Pak znovu třikrát zopakujte kroky 4 až 6.
• Nedotýkejte se prorážecí části uvnitř průhledného vnějšího obalu.
Jak o přípravek Spiriva Respimat pečovat
Očištění náustku včetně jeho vnitřní kovové součásti provádějte pouze vlhkou látkou nebo papírovým kapesníkem, a to nejméně jedenkrát týdně.
Funkci inhalátoru přípravku Spiriva Respimat neovlivňují žádné menší změny barvy náustku.
V případě potřeby otřete vnější část inhalátoru přípravku Spiriva Respimat vlhkým hadříkem.
Kdy si obstarat nový inhalátor Spiriva Respimat

• Inhalátor Spiriva Respimat obsahuje 60 vstřiků (30 léčivých dávek), pokud jej používáte podle pokynů (dva vstřiky jednou denně).
• Indikátor dávek přibližně ukazuje zbývající množství léku.
• Když ukazatel vstoupí do červené oblasti stupnice, znamená to, že léčba vystačí přibližně ještě na 7 dní (14 vstřiků) a je třeba si obstarat lékařský předpis na nový inhalátor.
• Jakmile indikátor počtu dávek dosáhne konce červené stupnice, inhalátor Spiriva Respimat se automaticky uzamkne - nelze již podat další dávku. V tomto okamžiku již průhledným vnějším obalem nelze otáčet. 1
Příprava inhalátoru k prvnímu použití
|
1. Sejměte průhledný vnější obal • Kryt zůstává uzavřen. • Stiskněte bezpečnostní pojistku a současně druhou rukou stáhněte průhledný vněj ší obal směrem dolů. |
^ Bezpečnostní pojistka í , Průhledný vnéjSí obal ty |
|
2. Vložte zásobník • Zasunujte zásobník úzkým koncem do inhalátoru. • Zásobník je nutno zasunovat silně tlakem proti pevnému povrchu až zacvakne na své místo. • Po vložení do inhalátoru již zásobník nevytahujte. |
wS?11' |
|
3. Vraťte průhledný vnější obal na své místo • Vraťte průhledný vnější obal na své místo, až zacvakne. • Znovu již průhledný vnější obal neodstraňujte. |
' Průhledný vnéjSí obal |
|
4. Otočte průhledný vnější obal • Kryt zůstává uzavřen. • Otáčejte průhledným vnějším obalem ve směru šipek na štítku, dokud se neozve cvaknutí (půl otáčky). |
B ' m, |
|
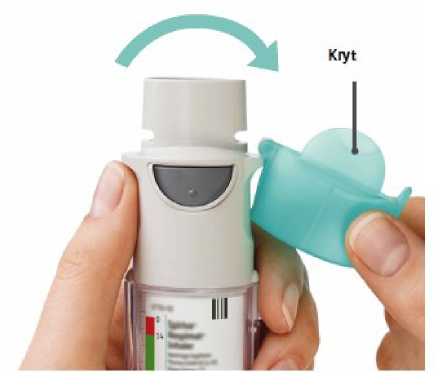
5. Otevřete kryt • Otevřete kryt tak, aby plně odskočil. |
Kryt

6. Stiskněte
• Namiřte inhalátor směrem k zemi.
• Stiskněte tlačítko uvolňující dávku.
• Uzavřete kryt.
• Opakujte kroky 4-6, dokud se neobjeví zřetelný obláček.
• Jakmile se objeví zřetelný obláček, opakujte kroky 4-6 ještě třikrát.
Váš inhalátor je nyní připraven k použití. Tyto kroky nebudou mít žádný vliv na počet dávek, které budete mít k dispozici. Po přípravě bude Váš inhalátor schopen dodat 60 vstřiků (30 léčivých dávek).

OTÁČEJTE
• Kryt zůstává uzavřen.
• OTÁČEJTE průhledným vnějším obalem ve směru šipek na štítku, dokud se neozve cvaknutí (půl otáčky).

OTEVŘETE
• OTEVŘETE kryt tak, aby plně odskočil.
STISKNETE
• Pomalu a plně vydechněte
• Sevřete rty těsně kolem konce náustku, aniž byste jimi překryl(a) vzduchový ventil. Nasměrujte inhalátor směrem k zadní vnitřní části Vašeho krku.
• Během pomalého a hlubokého nádechu ústy STISKNETE prstem tlačítko uvolňující dávku a pokračujte v pomalém nádechu tak dlouho jak je to pro Vás možné.
• Zadržte dech na 10 sekund nebo na tak dlouho, jak je to pro Vás možné.
• Opakujte kroky „Otáčejte“, „Otevřete“, „Stiskněte“ celkem pro 2 vstřiky.
• Přiklopte víčko, dokud nebudete potřebovat Váš inhalátor znovu použít.
Vzduchový
ventil
4.3 Kontraindikace
Spiriva Respimat je kontraindikován u pacientů s hypersenzitivitou na tiotropium-bromid, atropin nebo jeho deriváty, např. ipratropium nebo oxitropium, nebo na kteroukoli pomocnou látku (viz 6.1).
4.4 Zvláštní upozornění a opatření pro použití
Tiotropium-bromid, bronchodilatans podávané jednou denně k udržovací léčbě, nelze použít jako zahajovací léčbu akutních záchvatů bronchospasmu nebo k úlevě od akutních příznaků. V případě akutního záchvatu je třeba použít rychle účinkující beta2-agonisty.
Přípravek Spiriva Respimat nemá být u pacientů s astmatem používán jako monoterapie (lék první linie). Astmatičtí pacienti musí být poučeni o nutnosti pokračovat beze změny v protizánětlivé léčbě inhalačními kortikosteroidy i po zahájení léčby přípravkem Spiriva Respimat, a to i v případě, že se jejich příznaky zlepší.
Po podání tiotropium-bromidu, roztoku k inhalaci, se mohou vyskytnout časné alergické reakce.
Vzhledem k anticholinergním vlastnostem je tiotropium-bromid nutno podávat s opatrností u pacientů s glaukomem s úzkým úhlem, hyperplazií prostaty nebo obstrukcí hrdla močového měchýře.
Inhalačně podávané léky mohou vyvolat bronchospasmus způsobený inhalací.
Tiotropium je nutno podávat s opatrností pacientům s nedávno prodělaným infarktem myokardu (před méně než 6 měsíci); u jakékoliv nestabilní nebo život ohrožující srdeční arytmie nebo srdeční arytmie vyžadující intervenci nebo změnu farmakoterapie v průběhu posledního roku; při hospitalizaci pro srdeční selhání (NYHA třídy III nebo IV) během posledního roku. Tito pacienti byli vyloučeni z klinických studií a na tyto stavy může mít vliv anticholinergní mechanizmus účinku.
Vzhledem k tomu, že u pacientů se středně těžkým až těžkým postižením renálních funkcí (clearance kreatininu < 50 ml/min) dochází ke zvyšování plazmatické koncentrace, může být tiotropium-bromid těmto pacientům podáván jen v případech, kdy očekávaný přínos léčby převyšuje její potenciální rizika. S léčbou pacientů s těžkým postižením renálních funkcí nejsou dlouhodobé zkušenosti (viz 5.2).
Pacienti musí být upozorněni, aby jim sprej nevnikl při aplikaci do očí. Musí být poučeni o tom, že by mohlo dojít k vyvolání nebo ke zhoršení glaukomu s úzkým úhlem, bolestem očí nebo očním obtížím, přechodnému rozmazanému vidění, vizuálnímu haló nebo duhovému vidění spojenému se zarudnutím očí v důsledku překrvení spojivky a otoku rohovky. Pokud se některé z kombinací těchto očních příznaků objeví, musí pacienti užívání tiotropium-bromidu ukončit a ihned vyhledat pomoc lékaře.
Sucho v ústech, které se vyskytuje při léčbě anticholinergiky, může být při dlouhodobé expozici spojeno se zubním kazem.
Tiotropium-bromid nesmí být užíván častěji než jednou denně (viz 4.9).
Přípravek Spiriva Respimat se nedoporučuje u pacientů s cystickou fibrózou (CF). Pokud je přípravek Spiriva Respimat používán pacienty s cystickou fibrózou, může dojít ke zhoršení projevů a symptomů cystické fibrózy (například závažné nežádoucí účinky, plicní exacerbace, infekce dýchacího traktu).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ačkoliv nebyly prováděny oficiální studie zabývající se lékovými interakcemi, byl tiotropium-bromid podáván současně s jinými léky obvykle užívanými při léčbě CHOPN a astmatu, včetně beta2-agonistů, methylxantinů, perorálních a inhalačních kortikosteroidů, antihistaminik, mukolytik, modifikátorů leukotrienů, kromonů a léčby pomocí anti-IgE a to bez vzniku klinických projevů lékových interakcí.
Nebylo zjištěno, že podávání dlouhodobě působících beta-agonistů nebo inhalačních kortikosteroidů mění míru expozice tiotropia.
Současné podávání tiotropium-bromidu s jinými léky obsahujícími anticholinergika nebylo zkoumáno, a proto se nedoporučuje.
4.6 Fertilita, těhotenství a kojení
Existuje velmi omezené množství údajů o použití tiotropia u těhotných žen. Studie provedené u zvířat nenaznačují přímé nebo nepřímé zdraví škodlivé účinky s ohledem na reprodukční toxicitu při klinicky významných dávkách (viz bod 5.3). Jako preventivní opatření se upřednostňuje nepodávat přípravek Spiriva Respimat během těhotenství.
Kojení
Není známo, zda je tiotropium-bromid vylučován do lidského mateřského mléka. Navzdory studiím prováděným na hlodavcích, které prokázaly, že k vylučování tiotropium-bromidu do mateřského mléka dochází jen ve velmi malém množství, není používání přípravku Spiriva Respimat v průběhu kojení doporučeno. Tiotropium-bromid je dlouhodobě působící látka. Při rozhodování, zda pokračovat/ukončit kojení nebo pokračovat/ukončit léčbu přípravkem Spiriva Respimat, je třeba brát v úvahu prospěch kojení pro dítě vzhledem k prospěchu léčby přípravkem Spiriva Respimat pro matku.
Fertilita
Klinické údaje týkající se plodnosti nejsou pro tiotropium k dispozici. Neklinické studie s tiotropiem neprokázaly žádné známky nežádoucích účinků na plodnost (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit nebo obsluhovat stroje nebyly provedeny. Výskyt závratí nebo rozmazaného vidění může ovlivnit schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Mnoho z uvedených nežádoucích účinků může být připsáno anticholinergním účinkům tiotropium-bromidu.
Tabulkový souhrn nežádoucích účinků
Frekvence přiřazené níže uvedeným nežádoucím účinkům vycházejí z hrubé incidence nežádoucích účinků (tj. příhody přisouzené účinkům tiotropia) ve skupině léčené tiotropiem ze 7 placebem kontrolovaných klinických studií u pacientů s CHOPN (3282 pacientů) a 6 placebem kontrolovaných klinických studií u pacientů s astmatem (1256 pacientů). Doba léčby se pohybovala od 4 týdnů do 1 roku.
Četnost výskytu nežádoucích účinků je definována následujícím způsobem:
Velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Třídy orgánových systémů podle databáze MedDRA |
Frekvence CHOPN |
Frekvence |
|
Poruchy metabolismu a výživy | ||
|
Dehydratace |
Není známo |
Není známo |
|
Poruchy nervového systému | ||
|
Závratě |
Méně časté |
Méně časté |
|
Bolesti hlavy |
Méně časté |
Méně časté |
|
Vzácné |
Méně časté | |
|
Poruchy oka | ||
|
Glaukom |
Vzácné |
Není známo |
|
Třídy orgánových systémů podle databáze MedDRA |
Frekvence CHOPN |
Frekvence |
|
Zvýšený nitrooční tlak |
Vzácné |
Není známo |
|
Rozmazané vidění |
Vzácné |
Není známo |
|
Srdeční poruchy | ||
|
Fibrilace síní |
Vzácné |
Není známo |
|
Palpitace |
Vzácné |
Méně časté |
|
Supraventrikulární tachykardie |
Vzácné |
Není známo |
|
Vzácné |
Není známo | |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Méně časté |
Méně časté | |
|
Faryngitida |
Méně časté |
Méně časté |
|
Dysfonie |
Méně časté |
Méně časté |
|
Epistaxe |
Vzácné |
Není známo |
|
Bronchospasmus |
Vzácné |
Méně časté |
|
Vzácné |
Není známo | |
|
Zánět dutin |
Není známo |
Není známo |
|
Gastrointestinální poruchy | ||
|
Sucho v ústech |
Časté |
Časté |
|
Zácpa |
Méně časté |
Vzácné |
|
Kandidóza úst a hltanu |
Méně časté |
Méně časté |
|
Dysfagie |
Vzácné |
Není známo |
|
Refluxní choroba jícnu |
Vzácné |
Není známo |
|
Zubní kazy |
Vzácné |
Není známo |
|
Gingivitida |
Vzácné |
Vzácné |
|
Glossitida |
Vzácné |
Není známo |
|
Stomatitida |
Není známo |
Vzácné |
|
Střevní obstrukce, včetně paralytického ileu |
Není známo |
Není známo |
|
Není známo |
Není známo | |
|
Poruchy kůže a podkožní tkáně, poruchy imunitního | ||
|
systému | ||
|
Kožní vyrážka |
Méně časté |
Vzácné |
|
Třídy orgánových systémů podle databáze MedDRA |
Frekvence CHOPN |
Frekvence |
|
Pruritus |
Méně časté |
Vzácné |
|
Angioneurotický edém |
Vzácné |
Vzácné |
|
Kopřivka |
Vzácné |
Vzácné |
|
Kožní infekce, vředy na kůži |
Vzácné |
Není známo |
|
Suchá kůže |
Vzácné |
Není známo |
|
Přecitlivělost (včetně náhlých reakcí) |
Není známo |
Vzácné |
|
Anafylaktická reakce |
Není známo |
Není známo |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Otoky kloubů |
Není známo |
Není známo |
|
Poruchy ledvin a močových cest | ||
|
Retence moči |
Méně časté |
Není známo |
|
Méně časté |
Není známo | |
|
Infekce močových cest |
Vzácné |
Není známo |
Popis vybraných nežádoucích účinků
Často zaznamenanými nežádoucími účinky v kontrolovaných klinických studiích u pacientů s CHOPN byly anticholinergní nežádoucí účinky, jako je sucho v ústech, které se vyskytlo u přibližně 2,9 % pacientů. Suchost v ústech byla zaznamenána u 1,2% pacientů s astmatem.
V 7 klinických studiích s CHOPN bylo sucho v ústech důvodem k přerušení léčby u 3 z 3282 pacientů (0,1%) léčených tiotropiem. V 6 klinických studiích s astmatem (u 1256 pacientů) nebyla hlášena žádná přerušení léčby z důvodu sucha v ústech.
Závažné nežádoucí účinky související s anticholinergním působením zahrnují glaukom, zácpu, střevní obstrukci včetně paralytického ileu a retenci moči.
Jiné zvláštní skupiny pacientů
Výskyt anticholinergních nežádoucích účinků se může zvyšovat s rostoucím věkem pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: http://www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Vysoké dávky tiotropium-bromidu mohou vyvolat anticholinergní příznaky a projevy.
Nicméně po inhalaci jednotlivé dávky až 340 mikrogramů tiotropium-bromid nebyly u zdravých dobrovolníků zaznamenány žádné systémové anticholinergní nežádoucí účinky. Mimo sucha v ústech/hrdle a sucha nosní sliznice nebyly zaznamenány žádné významné nežádoucí účinky při podávání dávky až 40 pg tiotropia v roztoku k inhalaci po dobu 14 dní u zdravých jedinců s výjimkou zřetelného snížení sekrece slin od 7. dne.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná inhalační léčiva k terapii onemocnění spojených s obstrukcí dýchacích cest, anticholinergika ATC kód: R03B B04
Mechanismus účinku
Tiotropium-bromid je dlouhodobě působící specifický antagonista muskarinových receptorů. Má podobnou afinitu k jednotlivým subtypům muskarinových receptorů M1 až M5. V dýchacích cestách se tiotropium-bromid kompetitivně a reverzibilně váže na M3 receptory hladké svaloviny průdušek, antagonizuje cholinergní (bronchokonstrikční) účinky acetylcholinu, což vede uvolnění hladkého svalstva průdušek. Účinek byl závislý na dávce a trval déle než 24 hodin. Tiotropium-bromid je jako dusíkaté kvartérní anticholinergikum při inhalačním podání topicky (broncho-) selektivní a vykazuje přijatelné terapeutické rozmezí předtím, než dojde k výskytu systémových anticholinergních účinků.
Farmakodynamické účinky
Disociace tiotropia zvláště z M3 receptorů je velmi pomalá, vykazuje významně delší poločas disociace než ipratropium. Disociace z receptorů M2 je rychlejší než z receptorů M3, z čehož byla ve funkčních in vitro studiích vyvozena (za kinetické kontroly) receptorová selektivita k subtypu M3 oproti subtypu M2. Klinickým korelátem vysoké účinnosti, velmi pomalé receptorové disociace a topické inhalační selektivity je významná a dlouhodobá bronchodilatace u pacientů s CHOPN a astmatem.
Klinická účinnost a bezpečnost u CHOPN
Klinický vývojový program Fáze III zahrnoval dvě roční, dvě 12týdenní a dvě 4týdenní randomizované dvojitě slepé studie, prováděné u 2901 pacientů s CHOPN (1038 z nich užívalo tiotropium v dávce 5 pg). Roční program se skládal ze dvou studií kontrolovaných placebem. Dvě 12týdenní studie byly kontrolovány jak aktivně (ipratropiem), tak placebem. Ve všech těchto šesti studiích byly hodnoceny plicní funkce. Vedle toho obě roční studie zahrnovaly hodnocení ukazatelů zdravotního stavu, a to dyspnoe, kvality života v souvislosti se zdravotním stavem a vliv na exacerbace.
Studie kontrolované placebem
Plicní _ funkce
Podání tiotropia v roztoku k inhalaci zajistilo v jedné denní dávce významné zlepšení plicních funkcí (usilovně vydechnutý objem za 1 sekundu a usilovná vitální kapacita) během 30 minut po aplikaci první dávky ve srovnání s placebem (Průměrné zlepšení FEV1 ve 30. minutě 0,113 litrů, 95% interval spolehlivosti (CI):0,102-0,125 litrů,p<0,0001).
Zlepšení plicních funkcí bylo zachováno po dobu 24 hodin v rovnovážném stavu ve srovnání s placebem (průměrné zlepšení FEV1: 0,122 litrů, 95% interval spolehlivosti (CI):0,106-0,138 litrů,
p<0,0001).
Farmakodynamického rovnovážného stavu bylo dosaženo během jednoho týdne.
Spiriva Respimat významně zlepšil ranní a večerní hodnoty PEFR (vrcholová výdechová rychlost) hodnocené v denních záznamech pacientů v porovnání s placebem (průměrné zlepšení PEFR:průměrné zlepšení ráno 22 l/min, 95%CI: 18-55 l/min, p<0,0001; večer 26 l/min, 95%CI: 23-30 l/min, p<0,0001). Podávání přípravku Spiriva Respimat vedlo ve srovnání s placebem ke sníženému užívání úlevové bronchodilatační terapie (průměrné snížení užití úlevové medikace bylo 0,66 použití během dne, 95%CI: 0,51-0,81 použití během dne, p<0,0001).
Bronchodilatační účinek přípravku Spiriva Respimat přetrval po dobu 1 roku podávání bez známek vzniku tolerance.
Dyspnoe, kvalita života ve vztahu ke zdraví, exacerbace CHOPN v dlouhodobých lročních studiích Dyspnoe
Přípravek Spiriva Respimat významně zlepšil dušnost (hodnoceno pomocí indexu TDI - Transition Dyspnea Index) v porovnání s placebem (průměrné zlepšení 1,05 jednotek; 95%CI: 0,73-1,38 jednotek, p<0,0001). Toto zlepšení přetrvávalo po celou dobu léčby.
Kvalita života ve vztahu ke zdraví
Zlepšení celkového průměrného skóre hodnocení kvality života pacientů (za použití Respiračního dotazníku nemocnice Sv. Jiří - St. George's Respiratory Questionnaire) mezi přípravkem Spiriva Respimat a placebem bylo na konci dvou jednoročních studií 3,5 jednotek (95% CI: 2,1-4,9, p<0,0001). Pokles o 4 jednotky je v současnosti považován za klinicky významný.
Exacerbace CHOPN
Ve třech randomizovaných dvojitě zaslepených a placebem kontrolovaných klinických studiích s trváním v délce jednoho roku vedla léčba přípravkem Spiriva Respimat k významnému snížení rizika exacerbací CHOPN ve srovnání s placebem. Exacerbace CHOPN byla definována jako „komplex nejméně dvou respiračních příhod/příznaků trvající po dobu tří dnů nebo delší, které si vyžádaly změnu v léčbě (předepsání antibiotik a/nebo systémových kortikoidů a/nebo významnou změnu předepsané respirační medikace)”.
Léčba přípravkem Spiriva Respimat vedla ke snížení rizika hospitalizace z důvodu exacerbace CHOPN (významnému v přiměřeně mocné rozsáhlé studii exacerbací).
Složená analýza dvou studií fáze III a oddělená analýza dodatečné studie exacerbací je uvedena v tabulce 1. Jako souběžná léčba byla povolena veškerá respirační medikace s výjimkou anticholinergik a beta-sympatomimetik s dlouhodobým účinkem, tj. rychle působící beta-sympatomimetika, inhalační kortikoidy a xanthiny. Ve studii exacerbací byla navíc povolena beta-sympatomimetika s dlouhodobým účinkem.
Tabulka 1: Statistická analýza exacerbací CHOPN a exacerbací CHOPN s hospitalizací u pacientů se středně těžkou až velmi těžkou CHOPN
|
Studie (NSpiriva, Nplacebo) |
Cíl studie |
Spiriva Respimat |
Placebo |
Redukce rizika v % (95% CI)a |
p |
|
Jednoletá studie fáze III, složená analýzad (670, 653) |
Dny do první exacerbace CHOPN |
160a |
86a |
29 (16 až 40)b |
<0,0001b |
|
Průměrná frekvence incidence exacerbací na pacient-rok |
0,78c |
1,00c |
22 (8 až 33)c |
0,002c | |
|
Doba do první exacerbace CHOPN s hospitalizací |
25 (-16 až 51)b |
0,20b | |||
|
Průměrná frekvence incidence exacerbací s hospitalizací na pacient-rok |
0,09c |
0,11c |
20 (-4 až 38) c |
0,096c |
|
Studie (NSpiriva, Nplacebo) |
Cíl studie |
Spiriva Respimat |
Placebo |
Redukce rizika v % (95% CI)a |
p |
|
Jednoletá studie exacerbací fáze IIIb (1939, 1953) |
Dny do první exacerbace CHOPN |
169a |
119a |
31 (23 až 37)b |
<0,0001b |
|
Průměrná frekvence incidence exacerbací na pacient-rok |
0,69c |
0,87c |
21 (13 až 28)c |
<0,0001c | |
|
Doba do první exacerbace CHOPN s hospitalizací |
27 (10 až 41)b |
0,003b | |||
|
Průměrná frekvence incidence exacerbací s hospitalizací na pacient-rok |
0,12c |
0,15c |
19 (7 až 30)c |
0,004c |
a Doba do první příhody: dny s léčbou, při které 25% pacientů mělo nejméně jednu exacerbaci CHOPN/exacerbaci CHOPN s hospitalizací. Ve studii A 25% pacientů užívajících placebo mělo exacerbaci do 112. dne, zatímco u přípravku Spiriva Respimat mělo 25% pacientů exacerbaci do 173. dne (p=0,09); ve studii B 25% pacientů užívajících placebo mělo exacerbaci do 74. dne, zatímco u přípravku Spiriva Respimat mělo25% pacientů exacerbaci do 149. dne (p<0,0001).
b Poměry rizika byly stanoveny pomocí Coxova proporčního modelu rizika. Redukce rizika v procentech je 100 (1 - poměr rizika).
c Poissonovská regrese. Redukce rizika je 100 (1 - poměr výskytu).
d Složení bylo stanoveno při návrhu designu studií. Cíle studie exacerbací byly významně zlepšeny v individuálních analýzách dvou jednoletých studií.
Dlouhotrvající studie kontrolovaná aktivní látkou
Za účelem srovnání účinnosti a bezpečnosti přípravku Spiriva Respimat a Spiriva HandiHaler (5,711 pacientů používalo přípravek Spiriva Respimat; 5,694 pacientů používalo přípravek Spiriva HandiHaler) byla provedena dlouhodobá rozsáhlá randomizovaná dvojitě zaslepená aktivní látkou kontrolovaná studie s délkou trvání až 3 roky. Primárními cíli byly čas do první exacerbace CHOPN, čas do úmrtí ze všech příčin, a v podstudii (u 906 pacientů) byl hodnocen parametr through FEVi (hodnota před podáním dávky).
Čas do první exacerbace CHOPN byl v průběhu studie u přípravku Spiriva Respimat a Spiriva HandiHaler numericky srovnatelný (poměr rizik (Spiriva Respimat/Spiriva HandiHaler) 0,98 s 95% intervalem spolehlivosti 0,93 až 1,03). Medián počtu dní do první exacerbace CHOPN byl 756 dní u přípravku Spiriva Respimat a 719 dní pro Spiriva HandiHaler.
Bronchodilatační účinek přípravku Spiriva Respimat se udržel po dobu 120 týdnů a byl srovnatelný s účinkem pozorovaným u přípravku Spiriva HandiHaler. Průměrný rozdíl v parametru through FEVi mezi přípravkem Spiriva Respimat a přípravkem Spiriva HandiHaler byl -0,010 litru (95% interval spolehlivosti -0,038 až 0,018 litru).
V postmarketingové studii TIOSPIR srovnávající přípravky Spiriva Respimat a Spiriva HandiHaler byla mortalita ze všech příčin včetně následného sledování vitálního stavu u přípravku Spiriva Respimat a Spiriva HandiHaler srovnatelná (poměr rizik (Spiriva Respimat/Spiriva HandiHaler) 0,96, 95% interval spolehlivosti 0,84 - 1,09). Expozice léčivé látce odpovídala 13,135 a 13,050 paciento-rokům.
V placebem kontrolovaných studiích se sledováním vitálního stavu do konce zamýšlené léčebné periody vykázal přípravek Spiriva Respimat numerický nárůst mortality ze všech příčin ve srovnání
s placebem (poměr frekvence (95% interval spolehlivosti) 1,33 (0,93, 1,92), s léčebnou expozicí vůči přípravku Spiriva Respimat 2574 paciento-roků; rozdíl v mortalitě byl pozorován u pacientů se známými poruchami srdečního rytmu. Přípravek Spiriva HandiHaler vykázal 13% snížení rizika úmrtí ((poměr rizik zahrnující následné sledování vitálního stavu (tiotropium/placebo) = 0,87; 95% interval spolehlivosti 0,76 až 0,99)). Léčebná expozice vůči přípravku Spiriva HandiHaler byla 10927 paciento-roků. Ani u podskupiny pacientů se známými poruchami srdečního rytmu ve studii s přípravkem Spiriva HandiHaler kontrolované placebem ani ve studii TIOSPIR srovnávající přípravky Spiriva HandiHaler a Spiriva Respimat nebyl zaznamenán rozdíl v riziku mortality.
Klinická účinnost a bezpečnost u astmatu
Klinický program fáze III u perzistujícího astmatu zahrnoval dvě jednoleté randomizované, dvojitě zaslepené, placebem kontrolované studie, které zahrnovaly celkem 907 pacientů s astmatem (z nich 453 používalo přípravek Spiriva Respimat) na kombinaci IKS (> 800 pg budesonidu/den nebo ekvivalent) a beta2-agonisty s dlouhodobým účinkem (LABA). Primárním sledovaným cílem ve studiích byly hodnoty funkce plic a těžké exacerbace.
Studie astmatu PrimoTinA
Ve dvou jednoročních studiích u pacientů s částečnou nebo nedostatečnou kontrolou astmatu i při udržovací léčbě přinejmenším IKS (> 800 pg budesonidu/den nebo ekvivalent) plus LABA, vykázal přípravek Spiriva Respimat klinicky významné zlepšení funkce plic oproti placebu, když byl podáván jako přídatná léčba k léčbě IKS/LABA.
Ve 24. týdnu bylo průměrné zlepšení hodnoty vrcholového FEV1 0,110 litru (95% CI: 0,063 až 0,158 litru, p<0,0001), minimální hodnota FEV1 se v průměru zlepšila o 0,093 litru (95% CI: 0,050 až 0,137 litru, p<0,0001). Zlepšení plicních funkcí ve srovnání s placebem se udrželo po dobu 24 hodin.
Ve studiích PrimoTinA u pacientů s částečnou nebo nedostatečnou kontrolou astmatu (n = 453) léčených kombinací IKS plus LABA snížila přídatná léčba tiotropiem riziko těžkých exacerbací astmatu o 21% v porovnání se skupinou pacientů léčených IKS plus LABA plus placebo (n = 454). Snížení rizika průměrného počtu těžkých exacerbací astmatu na pacienta a rok bylo 20%.
To bylo podpořeno 31% snížením rizika zhoršení astmatu a 24% snížením rizika průměrného počtu příhod zhoršení astmatu na pacienta a rok (viz tabulka 2).
Tabulka 2: Exacerbace u pacientů s částečnou nebo nedostatečnou kontrolou astmatu léčených IKS (> 800 pg budesonidu/den nebo ekvivalent) plus LABA (Studie PrimoTinA)
|
Studie |
Cíl |
Spiriva Respimat, přídatná léčba u pacientů léčených alespoň IKSa/LABA (n = 453) |
Placebo, přídatná léčba u pacientů léčených alespoň IKSa/LABA (n = 454) |
% snížení rizika (95% CI)a |
Hodnota p |
|
Dvě 1-roční studie fáze III, souhrnná analýza |
Počet dní do 1. těžké exacerbace astmatu |
282c |
226c |
H (0, 38) |
0,0343 |
|
Průměrný počet těžkých exacerbací astmatu/pacient-rok |
0,530 |
0,663 |
(0, 36) |
0,0458 | |
|
Počet dní do 1. zhoršení astmatu |
315c |
181c |
H (18, 42) |
<0,0001 | |
|
Průměrný počet příhod zhoršení astmatu/pacient-rok |
2,145 |
2,835 |
(9, 37) |
0,0031 |
a > 800 pg budesonidu/den nebo ekvivalent
b Poměr rizik, interval spolehlivosti a hodnoty p byly získány pomocí Coxova modelu proporcionálních rizik s léčbou coby parametrem účinnosti. Procentuální snížení rizika je 100 (1 - poměr rizik). c Čas do první příhody: počet dní léčby, kdy 25%/50% pacientů prodělalo nejméně jednu těžkou exacerbaci/zhoršení astmatu Poměr četností byl stanoven pomocí Poissonovy regrese s logaritmem expozice (v letech) coby vyvažujícím parametrem. Procentuální snížení rizika je 100 (1-poměr četnosti).
Pediatrická populace
CHOPN
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Spiriva Respimat u všech podskupin pediatrické populace v rámci diagnózy CHOPN (viz bod 4.2 pro informace o pediatrickém použití).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Spiriva Respimat v léčbě astmatu u jedné nebo více podskupin pediatrické populace (viz bod 4.2 pro informace o pediatrickém použití).
Klinická účinnost a bezpečnost u cystické fibrózy (CF):
Klinický vývojový program u CF zahrnoval 3 multicentrické studie s 959 pacienty ve věku 5 měsíců a vyšším. Pacienti do 5 let věku používali nástavec (AeroChamber Plus®) s obličejovou maskou a byli zahrnuti pouze do hodnocení bezpečnosti. Dvě pivotní studie (studie fáze II pro stanovení dávky a konfirmační studie fáze III) srovnávaly vliv na plicní funkce (procento predikované hodnoty FEV1 AUC0-4h a FEV1 hodnoty) přípravku Spiriva Respimat (tiotropium 5 pg: 469 pacientů) oproti placebu (315 pacientů) ve 12 týdnů trvajících randomizovaných dvojitě zaslepených periodách. Studie fáze III zahrnovala také dlouhodobou otevřenou rozšířenou studii s trváním až 12 měsíců. V těchto studiích byly jako souběžná léčba povoleny veškeré respirační medikace kromě anticholinergik, tj. beta-agonisté s dlouhodobým účinkem, mukolytika a antibiotika.
Účinky na plicní funkce jsou uvedeny v tabulce 3. Nebylo pozorováno žádné významné zlepšení příznaků a zdravotního stavu (exacerbace hodnoceny podle Dotazníku na respirační a systémové příznaky (Respiratory and Systemic Symptoms Questionnaire) a Dotazníku na kvalitu života při cystické fibróze (Cystic Fibrosis Questionnaire)).
Tabulka 3: Upravený průměrný rozdíl od placeba pro absolutní změny od výchozího stavu po 12 týdnech_
|
Fáze II |
Fáze III | |||||
|
Všichni pacienti (nSpiriva = l76, nplacebo = 168) |
Všichni pacienti (nSpiriva = 293, nplacebo = 147) |
< 11 let věku (nSpiriva = 95, nplacebo = 47) |
> 12 let věku (nSpiriva = 198, nplacebo = 100) | |||
|
průměr (95% CI) |
hodnota p |
průměr (95% CI) |
hodnota p |
průměr (95% CI) |
průměr (95% CI) | |
|
FEV1AUC0-4h (% predikované hodnoty) a absolutní změny |
3,39 (1,67; 5,12) |
< 0,001 |
1,64 (-0,27; 3,55) |
0,092 |
-0,63 (-4,58; 3,32) |
2,58 (0,50; 4,65) |
|
FEV1AUC0-4h (litry) absolutní změny |
0,09 (0,05; 0,14) |
< 0,001 |
0,07 (0,02; 0,12) |
0,010 |
0,01 (-0,07; 0,08) |
0,10 (0,03; 0,17) |
|
Trough FEV1 (% predikované hodnoty) a absolutní změny |
2,22 (0,38; 4,06) |
0,018 |
1,40 -0,50; 3,30 |
0,150 |
-1,24 (-5,20; - 271) |
2,56 (0,49; 4,62) |
|
Trough FEV1 |
0,06 |
0,028 |
0,07 |
0,012 |
-0,01 |
0,10 |
|
Fáze II |
Fáze III | |||||
|
Všichni pacienti (nSpiriva = 176, nplacebo = 168) |
Všichni pacienti (nSpiriva = 293, nplacebo = 147) |
< 11 let věku (nSpiriva = 95, nplacebo = 47) |
> 12 let věku (nSpu™ = 198, nplacebo = 100) | |||
|
průměr (95% CI) |
hodnota p |
průměr (95% CI) |
hodnota p |
průměr (95% CI) |
průměr (95% CI) | |
|
(litry) absolutní změny |
(0,01; 0,11) |
(0,02; 0,12) |
(-0,08; 0,06) |
(0,03; 0,17) | ||
Co-primámí sledovaní cíloví ukazatelé
Všechny nežádoucí účinky pozorované ve studiích u pacientů s CF jsou známé nežádoucí účinky tiotropia (viz bod 4.8). Nejčastěji pozorovanými nežádoucími účinky, které byly považovány za související během 12 týdnů trvající dvojitě zaslepené periody, byly kašel (4,1%) a sucho v ústech (2,8%).
Počet a procento pacientů, kteří hlásili nežádoucí účinky vyžadující zvláštní pozornost u onemocnění cystickou fibrózou bez ohledu na souvislost s tiotropiem, jsou uvedeny v tabulce 4. Počet projevů a symptomů, které se manifestují u cystické fibrózy, během terapie tiotropiem statisticky nevýznamně vzrostl, zejména u pacientů < 11 let věku.
Tabulka 4: Procento pacientů s nežádoucími účinky vyžadující zvláštní pozornost u onemocnění cystickou fibrózou podle věkových skupin během 12 týdnů léčby bez ohledu na souvislost s terapií tiotropiem (souhrnná data ze studie fáze II a fáze III)
|
< 11 let věku |
> 12 let věku | |||
|
nplacebo 96 |
nSpriva 158 |
nplacebo 215 |
nSpriva 307 | |
|
7,3 |
7,0 |
5,1 |
6,2 | |
|
Zácpa |
1,0 |
1,9 |
2,3 |
2,6 |
|
Distální intestinální obstrukční syndrom |
0,0 |
0,0 |
1,4 |
1,3 |
|
Infekce respiračního traktu |
34,4 |
36,7 |
28,4 |
28,3 |
|
Zvýšené množství sputa |
1,0 |
5,1 |
5,6 |
6,2 |
|
Exacerbace |
10,4 |
14,6 |
18,6 |
17,9 |
„Syndrom distální intestinální obstrukce“ a „zvýšené množství sputa“ jsou preferovanými termíny MedDRA. „Infekce respiračního traktu“ představují termín MedDRA vyšší úrovně pro označení skupiny. „Bolest břicha“, „zácpa“ a „exacerbace“ jsou soubory preferovaných termínů MedDRA.
U 34 pacientů (10,9 %) randomizovaných do skupiny užívající placebo a 56 pacientů (12 %) randomizovaných do skupiny používající přípravek Spiriva Respimat se vyskytla závažná nežádoucí příhoda.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Spiriva Respimat v podskupině pediatrických pacientů mladších než 1 rok.
5.2 Farmakokinetické vlastnosti
a) Všeobecný úvod
Tiotropium-bromid je achirální kvartérní amoniová sloučenina špatně rozpustná ve vodě. Podává se ve formě roztoku k inhalaci prostřednictvím inhalátoru Respimat. Přibližně 40% inhalované dávky se ukládá v cílovém orgánu, tj. v plicích, zbývající množství v gastrointestinálním traktu. Některé z níže uvedených farmakokinetických údajů byly zjištěny při podávání dávek vyšších, než jsou doporučené dávky léčebné.
b) Všeobecné vlastnosti léčivé látky po podání léčivého přípravku
Absorpce: Údaje o vylučování močí naznačují, že po inhalaci mladými zdravými dobrovolníky dosáhne systémové cirkulace přibližně 33% inhalované dávky. Perorální roztoky tiotropium-bromidu mají absolutní biologickou dostupnost 2-3%. Neočekává se ovlivnění vstřebávání této kvartérní amoniové sloučeniny potravou.
Maximální plazmatické koncentrace tiotropia byly zaznamenány 5-7 minut po inhalaci.
Za rovnovážného stavu byly dosaženy vrcholové plazmatické hladiny tiotropia u pacientů s CHOPN 10,5 pg/ml a rychle klesaly multikompartmentovým způsobem. Minimální plazmatické koncentrace v rovnovážném stavu činily 1,60 pg/ml.
Vrcholové plazmatické koncentrace tiotropia v rovnovážném stavu 5,15 pg/ml bylo dosaženo 5 minut po podání stejné dávky pacientům s astmatem.
Systémová expozice tiotropia po inhalaci inhalátorem Respimat byla podobná expozici tiotropia inhalováného pomocí inhalátoru HandiHaler.
Distribuce: Léčivo je ze 72% vázáno na plazmatické proteiny a jeho distribuční objem je 32 l/kg. Lokální koncentrace v plicích nejsou známy, ale způsob podávání vede k podstatně vyšším koncentracím v plicích. Studie u potkanů ukázaly, že tiotropium neproniká ve významném množství přes hematoencefalickou bariéru.
Biotransformace: Rozsah biotransformace je nízký. To je zřejmé z vylučování močí ze 74% nezměněné sloučeniny po intravenózním podání mladým zdravým dobrovolníkům. Ester tiotropium-bromid je neenzymaticky štěpen na alkohol (N-methylskopin) a na kyselinu (dithienylglykolová kyselina), které na muskarinové receptory nepůsobí.
Studie in vitro s jaterními mikrozomy a lidskými hepatocyty ukazují, že jistá část léku (<20% dávky po intravenózním podání) je metabolizována oxidací závislou na cytochromu P 450 (CYP) a následnou konjugací s glutathionem na řadu metabolitů II. řádu.
In vitro studie s jaterními mikrozomy ukazují, že tuto enzymatickou cestu lze inhibovat inhibitory CYP 2D6 (a 3A4), chinidinem, ketokonazolem a gestodenem. CYP 2D6 a 3A4 jsou tedy zapojeny do metabolické cesty, která odpovídá za vylučování menšího podílu dávky. Tiotropium-bromid dokonce i v koncentracích překračujících terapeutické koncentrace neinhibuje CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 a 3A jaterních mikrozomů lidí.
Eliminace: Efektivní poločas tiotropia nastává u zdravých dobrovolníků a pacientů s CHOPN mezi 27-45 hodinami po inhalaci. Efektivní poločas byl u pacientů s astmatem 34 hodin. Celková clearance po intravenózním podání u mladých zdravých dobrovolníků byla 880 ml/min. Intravenózně podané tiotropium je vylučováno hlavně močí v nezměněné formě (74%). Po inhalačním podání roztoku pacientům s CHOPN činí vylučování močí v rovnovážném stavu 18,6 % (0,93 pg) dávky, zbytek představuje zejména nevstřebaný lék ze střeva, vylučovaný stolicí. Po inhalačním podání roztoku zdravým dobrovolníkům činí vylučování močí 20,1 - 29,4 % dávky, zbytek představuje zejména nevstřebaný lék ze střeva vylučovaný stolicí. U pacientů s astmatem se 11,9% (0,595 pg) dávky vylučuje v nezměněné formě močí po dobu 24 hodin po podání dávky za rovnovážného stavu. Renální clearance tiotropia překračuje clearanci kreatininu, což svědčí o jeho sekreci do moči.
Při dlouhodobém inhalačním podávání jednou denně u pacientů s CHOPN bylo dosaženo farmakokinetického rovnovážného stavu do 7. dne bez jeho další kumulace.
Linearita/nelinearita: Tiotropium vykazuje v terapeutickém rozmezí lineární farmakokinetiku nezávislou na lékové formě.
c) Vlastnosti léku při podávání zvláštním skupinám pacientů
Starší pacienti: Jak lze očekávat u všech přednostně renálně vylučovaných léků, je pokročilý věk spojený s poklesem renální clearance tiotropia (z 347 ml/min u pacientů s CHOPN ve věku < 65 let na 275 ml/min u pacientů s CHOPN ve věku > 65 let). Pokročilý věk nevedl k odpovídajícímu zvýšení
AUC0-6,ss nebo hodnot Cmaxss. Nebylo zjištěno, že by se expozice vůči tiotropiu měnila s věkem astmatických pacientů.
Pacienti s poruchou funkce ledvin: Při inhalačním podávání tiotropia jednou denně u pacientů s CHOPN v rovnovážném stavu měla mírná porucha funkce ledvin (clearance kreatininu CLCr 50-80 ml/min) za následek lehké zvýšení hodnoty AUC0-6ss (o 1,8 - 30 % vyšší) a podobně byla ovlivněna i hodnota Cmaxss ve srovnání s pacienty s normální funkcí ledvin (CLCr > 80 ml/min).
U pacientů s CHOPN se středně těžkou až těžkou poruchou fUnkce ledvin (clearance kreatininu < 50 ml/min) vedlo intravenózní podání jedné dávky tiotropia ke zdvojnásobení jeho celkové expozice (82% nárůst AUC0-4h a 52% nárůst Cmax) ve srovnání s pacienty s normální funkcí ledvin, což bylo potvrzeno plazmatickými koncentracemi po inhalaci suchého prášku.
U astmatických pacientů s mírnou poruchou funkce ledvin (CLCr 50-80 ml/min) nevedla inhalace tiotropia k významnému zvýšení expozice ve srovnání s pacienty s normální funkcí ledvin.
Pacienti s poruchou funkce jater: Nepředpokládá se, že by jaterní insuficience měla nějaký významný vliv na farmakokinetiku tiotropia. Tiotropium je přednostně eliminováno renálně (74% u mladých zdravých dobrovolníků) a prostým neenzymatickým esterickým štěpením na farmakologicky neaktivní metabolity.
Japonští pacienti s CHOPN: Ve zkřížené srovnávací studii byla průměrná vrcholová plazmatická koncentrace tiotropia v rovnovážném stavu 10 minut po podání dávky inhalačně o 20 % až 70 % vyšší u japonských pacientů s CHOPN, v porovnání s bělošskou populací. Nebyl však zaznamenán žádný signál vyšší mortality nebo kardiálního rizika u japonských pacientů ve srovnání s bělošskou populací. Pro další etnika nebo rasy jsou k dispozici jen nedostačující farmakokinetické údaje.
Pediatrická populace :
Do programu klinických studií s pacienty s CHOPN nebyli zařazeni žádní pediatričtí pacienti (viz bod 4.2). Pediatričtí pacienti byli hodnoceni jako součást klinického programu s pacienty s CF, který zahrnoval také dospělé pacienty.
Po inhalaci 5 pg tiotropia byla plazmatická hladina tiotropia u pacientů s CF od 5 let věku 10,1 pg/ml 5 minut po podání dávky v rovnovážném stavu, a poté rychle klesala. Podíl dávky dostupný u pacientů s CF do 5 let věku, kteří používali nástavec a masku, byl přibližně 3-4x nižší, než podíl dávky pozorovaný u pacientů s CF od 5 let věku. Expozice tiotropia byla u pacientů s CF do 5 let věku závislá na tělesné hmotnosti.
d) Farmakokinetický(é)/farmakodynamický(é) vztah(y)
Neexistuje žádná přímá souvislost mezi farmakokinetickými a farmakodynamickými vlastnostmi.
5.3 Předklinické údaje vztahující se k bezpečnosti
Mnoho účinků pozorovaných v průběhu konvenčních studií zabývajících se farmakologickou bezpečností, toxicitou při opakovaném podávání a reprodukční toxicitou může být vysvětleno anticholinergními vlastnostmi tiotropium-bromidu. Typickými účinky pozorovanými u zvířat byla snížená konzumace potravy, snížení přírůstku tělesné hmotnosti, sucho v ústech a nosu, snížená tvorba slz a slin, mydriáza a zrychlení srdeční frekvence.
Dalšími významnými změnami zaznamenanými v průběhu studií toxicity při opakovaném podávání bylo mírné podráždění dýchacích cest u potkanů a myší projevující se rinitidou a změnami epitelu nosní dutiny a hrtanu, prostatitida spolu s proteinovými depozity a litiáza močového měchýře potkanů.
U mladých potkanů při expozici od 7. postnatálního dne do dosažení pohlavní zralosti byly pozorovány stejné přímé a nepřímé farmakologické změny jako ve studiích toxicity při opakovaném
podávání, podobně jako rinitida. Nebyla zaznamenána žádná systémová toxicita ani žádné toxikologicky významné účinky na klíčové parametry vývoje, vývoj průdušnice nebo klíčových orgánů.
Škodlivé účinky ve vztahu k březosti, vývoji embrya/plodu, porodu nebo postnatálnímu vývoji bylo možno zaznamenat pouze při dávkách toxických pro matky. Tiotropium-bromid nebyl teratogenní pro potkany ani pro králíky. Ve všeobecných reprodukčních studiích a studiích fertility u potkanů nebyl zjištěn při jakémkoliv dávkování žádný náznak nežádoucího účinku na plodnost nebo schopnost rozmnožování, jak u léčených rodičů, tak i u jejich potomků.
Respirační (podráždění) a urogenitální (prostatitida) změny a reprodukční toxicita byly pozorovány při lokální i systémové expozici více než pětkrát vyšší než při expozici terapeutické. Studie zabývající se genotoxicitou a kancerogenním potenciálem neodhalily pro člověka žádné zvláštní riziko.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Benzalkonium-chlorid Dinatrium-edetát Čištěná voda
Kyselina chlorovodíková 3,6% (k úpravě pH)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
Od prvního použití: 3 měsíce
6.4 Zvláštní opatření pro uchovávání Chraňte před mrazem.
6.5 Druh obalu a obsah balení
Druh a materiál nádobky, která je v kontaktu s přípravkem:
Roztok je naplněn do polyethylen/polypropylenového zásobníku s polypropylenovým víčkem se vsazeným silikonovým těsnicím kroužkem. Zásobník je vložen do hliníkové nádobky.
Velikosti balení a dodávané prostředky:
Jednoduché balení: 1 inhalátor Respimat a 1 zásobník, poskytující 60 vstřiků (30 léčivých dávek)
Dvojité balení: 2 jednoduchá balení, každé obsahující 1 inhalátor Respimat a 1 zásobník, poskytující 60 vstřiků (30 léčivých dávek)
Trojité balení: 3 jednoduchá balení, každé obsahující 1 inhalátor Respimat a 1 zásobník, poskytující 60 vstřiků (30 léčivých dávek)
Osminásobné balení: 8 jednoduchých balení, každé obsahující 1 inhalátor Respimat a 1 zásobník, poskytující 60 vstřiků (30 léčivých dávek)
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Německo
8. REGISTRAČNÍ ČÍSLO(A)
14/666/07-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
24.10.2007 / 6.8.2012
10. DATUM REVIZE TEXTU
11.3.2016
20/20
Přípravek Spiriva Respimat je nutno zlikvidovat nejpozději po třech měsících používání od přípravy pro první použití, a to i v případě, že lék nebyl zcela spotřebován.