Soolantra 10Mg/G Krém
Sp.zn. sukls180680/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Soolantra 10 mg/g krém
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden gram krému obsahuje ivermectinum 10 mg.
Pomocné látky se známým účinkem:
Jeden gram krému obsahuje 35 mg cetylalkoholu, 25 mg stearylalkoholu, 2 mg methylparabenu(E218), 1 mg propylparabenu (E216) a 20 mg propylenglykolu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Krém.
Bílý až světle žlutý hydrofilní krém.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Soolantra je indikována na topickou léčbu zánětlivých (papulopustulózních) lézí rosacey u dospělých pacientů.
4.2 Dávkování a způsob podání
Dávkování
Jedna aplikace denně, obvykle se podává v délce do 4 měsíců. Soolantru je nutné během léčebné kúry aplikovat denně. Léčebnou kúru je možné opakovat.
V případě, že nedojde ke zlepšení po 3 měsících, je třeba léčbu přerušit.
Zvláštní skupiny pacientů
Pacienti s poruchou funkce ledvin Není nutná úprava dávkování.
Pacienti s poruchou funkce jater
U pacientů s těžkou poruchou funkce jater je nutné postupovat s opatrností.
Starší pacienti
U starších pacientů nejsou doporučovány žádné specifické úpravy dávkování. (viz také bod 4.8).
Děti a dospívající
Bezpečnost a účinnost Soolantry u dětí a dospívajících mladších 18 let nebyla stanovena. Příslušné údaje nejsou dostupné.
Způsob podání Jen pro kožní podání.
Přípravek se aplikuje na kůži v množství ve velikosti hrášku na každou z pěti částí obličeje: čelo, brada, nos a obě tváře. Přípravek je třeba rozetřít v tenké vrstvě po celé kůži obličeje, přičemž je třeba vynechat oči, rty a sliznice.
Soolantru lze aplikovat pouze na obličej.
Po aplikaci přípravku je třeba umýt si ruce.
Kosmetické přípravky se na místa aplikace mohou nanést po zaschnutí přípravku.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek Soolantra nebyl hodnocen u pacientů s poruchou funkce ledvin nebo jater.
Léčivý přípravek obsahuje:
- cetylalkohol a stearylalkohol, které mohou vyvolávat lokální kožní reakce (např. kontaktní dermatitidu),
- methylparaben (E218) a propylparaben (E216), které mohou způsobovat alergické reakce (potenciálně i opožděné),
- a propylenglykol, který může způsobit podráždění kůže.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí (viz bod 5.2 Biotransformace).
Současné užívaní Soolantry s jinými topickými nebo systémově podávanými léky určenými k léčbě rosacey nebylo studováno.
V in vitro studiích bylo prokázáno, že metabolismus ivermektinu probíhá primárně prostřednictvím CYP3A4. V důsledku toho se doporučuje opatrnost při současném podávání silných inhibitorů CYP3A4, neboť se tím může významně zvýšit přítomnost léčiva v plazmě.
4.6. Fertilita, těhotenství a kojení
Údaje o podávání ivermektinu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie perorální reprodukční toxicity prokázaly, že ivermektin je u potkanů a králíků teratogenní (viz bod 5.3), avšak kvůli nízké systémové expozici po lokální aplikaci přípravku v doporučeném dávkování je riziko z hlediska bezpečnosti pro lidský plod nízké. Soolantru se nedoporučuje podávat během těhotenství.
Kojení
Ivermektin se po perorálním podání vylučuje v nízkých koncentracích do mateřského mléka. Vylučování do mateřského mléka po topické aplikaci nebylo hodnoceno. Dostupné farmakokinetické resp. toxikologické data ze studií na zvířatech také dokázaly vylučování ivermektinu do mléka. Riziko pro kojené dítě nelze vyloučit. Musí se přijmout rozhodnutí o potřebě přerušit kojení nebo přerušit, resp. vysadit terapii Soolantrou, přičemž je třeba vzít v úvahu přínos kojení pro dítě a přínos terapie pro matku.
Fertilita
Nejsou k dispozici žádné údaje o vlivu ivermektinu na fertilitu u lidí. U potkanů nebyl při podávání ivermektinu prokázán vliv na páření nebo fertilitu.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Soolantra nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8. Nežádoucí účinky
Souhrn profilu bezpečnosti
Nejčastěji uváděnými nežádoucími účinky jsou pocit pálení kůže, podráždění kůže, svědění a projevy suché kůže, které se spolu vyskytovaly u < 1 % pacientů, kterým byl tento lék aplikován v rámci klinických studií. Nežádoucí účinky bývají většinou mírné až středně těžké, přičemž po přerušení léčby se obvykle zmírní. Mezi pacienty ve věku 18-65 let a pacienty ve věku > 65 nebyly zaznamenány významné rozdíly v profilu bezpečnosti.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky jsou seřazeny podle třídy orgánových systémů a frekvence výskytu podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až<1/10), méně časté (> 1 / 1 000 až <1/100), vzácné (> 1 / 10 000 až <1 / 1,000), velmi vzácné (<1 / 10 000), není známo (z dostupných údajů nelze určit) a uváděly se v rámci klinických studií Soolantry (viz Tabulka 1).
Tabulka 1 - Nežádoucí účinky
|
Třída orsánovvch systémů |
Frekvence výskytu |
Nežádoucí účinky |
|
Nemoci kůže a podkožní tkáně |
Časté |
Pocit pálení kůže |
|
Méně časté |
Podráždění kůže, svědění projevy suché kůže |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
4.9. Předávkování
Nejsou hlášeny případy předávkování Soolantrou.
Při náhodném nebo výrazném vystavení se neznámým množstvím veterinárních lékových forem ivermektinu u lidí, ať už perorálním užitím, inhalací, injekcí nebo expozicí na povrchu těla nejčastěji byly uváděny následující nežádoucí účinky: vyrážka, otok, bolest hlavy, závratě, astenie, nauzea, zvracení a průjem.
Ostatní hlášené nežádoucí účinky zahrnují: epileptický záchvat, ataxii, dušnost, bolest břicha, parestezii, urtikarii a kontaktní dermatitidu.
V případě náhodného požití má podpůrná terapie, pokud je indikována, zahrnovat parenterální podávání tekutin a elektrolytů, podporu dýchání (v případě nutnosti kyslík a mechanická ventilace) a přípravky na kontrolu krevního tlaku v případě klinicky významné hypotenze. Pokud je to nutné, může být indukováno zvracení a/nebo laváž žaludku, a to co nejdříve, s následným podáním projímadel a jinými běžnými antidoty za účelem prevence absorpce užité látky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1. Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná dermatologika
ATC kód: D11AX22
Mechanismus účinku
Ivermektin patří do skupiny avermektinu. Protizánětlivé účinky avermektinu jsou výsledkem inhibice tvorby zánětlivých cytokinů vyvolané lipopolysacharidy. Protizánětlivé vlastnosti ivermektinu topicky aplikovaného na kůži byly pozorovány na zvířecích modelech zánětu kůže. Ivermektin také způsobuje úhyn parazitů, který nastává primárně v důsledku selektivní a vysoko afinitní vazby na glutamátem řízené chloridové kanály v nervových a svalových buňkách bezobratlých. Mechanismus účinku Soolantry v léčbě zánětlivých lézí u rosacey je neznámý, ale může souviset s protizánětlivými účinky ivermektinu jakož i s usmrcováním roztočů rodu Demodex, které se dávají do spojitosti se záněty kůže.
Klinická účinnost a bezpečnost
Soolantra aplikována jednou denně před spaním byla hodnocena v léčbě zánětlivých lézí u rosacey ve dvou randomizovaných placebem(vehikulem) kontrolovaných dvojitě zaslepených klinických studiích, které měly identický design. Tyto studie byly provedeny v souboru 1 371 pacientů ve věku 18 a více let, kteří se léčili jednou denně po dobu 12 týdnů buď Soolantrou, nebo vehikulem.
Celkem bylo 96 % pacientů kavkazské rasy a 67 % tvořily ženy. Při použití pětistupňové stupnice celkového hodnocení zkoušejícími (IGA) se stav 79 % pacientů ve výchozím stavu hodnotil jako středně těžký (IGA = 3) a u 21 % pacientů se hodnotil jako těžký (IGA = 4).
Koprimární parametry účinnosti v obou klinických studiích představovala míra úspěšnosti na základě výsledku IGA (procento pacientů „čistých“ a „téměř čistých“ po 12 týdnech studie) a absolutní změny oproti výchozímu stavu z hlediska počtu zánětlivých lézí. Stupnice IGA je založena na následujících definicích:
Tabulka 2: Stupnice celkového hodnocení zkoušejícími (IGA)
|
Stupeň |
Skóre |
Klinický popis |
|
Čistý |
0 |
Bez přítomnosti zánětlivých lézí, bez erytému |
|
Téměř čistý |
1 |
Velmi malý počet papul/pustul, přítomen velmi mírný erytém |
|
Mírný |
2 |
Několik malých papul/pustul, mírný erytém |
|
Středně těžký |
3 |
Několik malých nebo velkých papul/pustul, středně těžký erytém |
|
Těžký |
4 |
Četné malé a/nebo velké papul/pustul, těžký erytém |
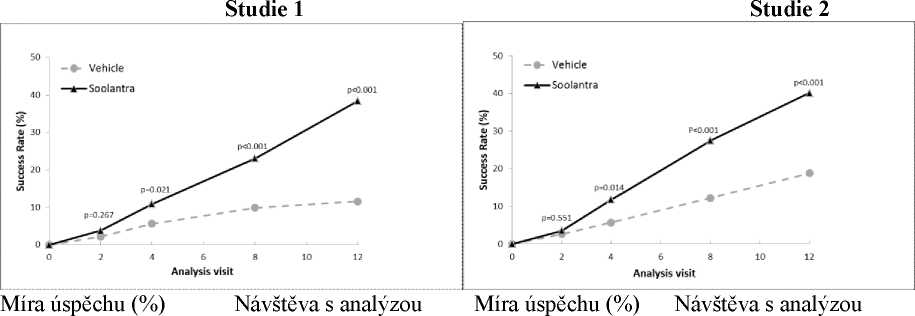
Výsledky obou klinických studií prokázaly, že Soolantra aplikována jednou denně po dobu 12 týdnů vykazovala statistickou superioritu vůči placebu (vehikulu) krému z hlediska míry úspěšnosti IGA a absolutní změny počtu zánětlivých lézí (p < 0,001, viz Tabulka 3 a Obrázky 1 - 4).
Následující tabulka a obrázky uvádějí závěry o účinnosti z obou studií.
Tabulka 3: Výsledky účinnosti
|
Studie 1 |
Studie 2 | |||
|
Soolantra (N = 451) |
Vehikulum (N = 232) |
Soolantra Vehikulum (N = 459) (N = 229) | ||
|
Celkové hodnocení zkoušejícími | ||||
|
Počet (%) pacientů čistých nebo téměř čistých podle IGA po 12 týdnech |
173 (38,4) |
27 (11,6) |
184 (40,1) |
43 (18,8) |
|
Zánětlivé léze | ||||
|
Průměrný počet zánětíivých lézí ve |
31,0 |
30,5 |
33,3 |
32,2 |
|
výchozím stavu | ||||
|
Průměrný počet zánětlivých lézí po 12 týdnech |
10,6 |
18,5 |
11,0 |
18,8 |
|
Průměrná absolutní změna (% změny) v počtu zánětlivých lézí z výchozího stavu do 12 týdnů léčby |
-20,5 (-64,9) |
-12,0 (-41,6) |
-22,2 (-65,7) |
-13,4 (-43,4) |
Obrázek 1 a 2: Časový průběh míry úspěchu IGA v týdnech
Obrázek 3 a 4: Časový průběh průměrné absolutní změny v počtu zánětlivých lézí proti výchozímu stavu
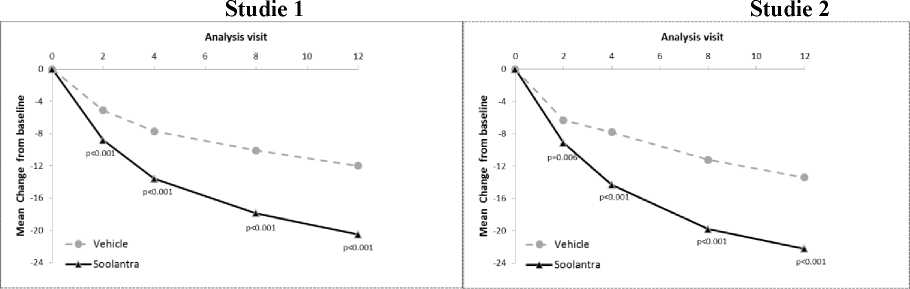
Průměrná změna proti výchozímu stavu/ Průměrná změna proti výchozímu stavu/ Návštěva s analýzou Návštěva s analýzou
Soolantra prokázala statistickou superioritu proti vehikulu krému v obou koprimárních parametrech účinnosti s časem do nástupu účinku po 4 týdnech léčby (p < 0,05).
IGA skóre se hodnotilo během 40týdenního prodloužení těchto dvou klinických studií a procenta pacientů léčených Soolantrou a dosahujících IGA skóre 0 nebo 1 pokračovaly v nárůstu až do 52 týdnů od začátku léčby. Míra úspěšnosti (IGA = 0 nebo 1) po 52 týdnech byla 71 % ve studii 1 a 76 % ve studii 2.
Účinnost a bezpečnost tohoto přípravku v terapii zánětlivých lézí u rosacey byly také hodnoceny v randomizované a aktivně kontrolované studii zaslepené z hlediska zkoušejícího. Studie byla provedena v souboru 962 pacientů ve věku 18 a více let, kteří se léčili po dobu 16 týdnů buď Soolantrou, podávanou jednou denně, nebo Metronidazolem 7,5 mg/g krému, který byl aplikován dvakrát denně. V této studii bylo 99,7 % pacientů kavkazské rasy a 65,2 % bylo žen; dle stupnice IGA byl ve výchozím stavu hodnocen stav 83,3 % pacientů jako středně těžký (IGA = 3) a stav 16,7 % pacientů jako těžký (IGA = 4) (viz obrázek 5).
Výsledky této studie prokázaly statistickou superioritu Soolantry vůči Metronidazolu 7,5 mg/g krému na primární parametr účinnosti (průměrná změna v procentu počtů zánětlivých lézí) se snížením 83,0 % pro ivermectin a 73,7 % pro metronizadol po 16 týdnech léčby oproti výchozím hodnotám (p < 0,001). Superiorita Soolantry po 16 týdnech byla potvrzena na míře úspěšnosti založené na IGA a skóre absolutní změny v počtu zánětlivých lézí (sekundární parametry (p < 0,001).
Obrázek 5
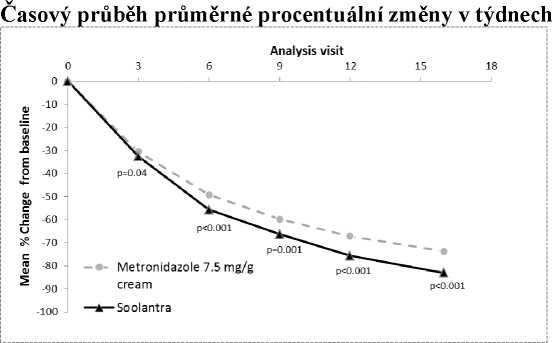
Průměrné % změny proti výchozímu stavu/Návštěva s analýzou
Přibližně 300 pacientů ve věku 65 a více let bylo léčeno v rámci všech klinických studií tohoto léku. V profilech bezpečnosti a účinnosti nebyly pozorovány žádné významné rozdíly mezi staršími pacienty a pacienty ve věku od 18 do 65 let věku.
Profil bezpečnosti, který je popsán v části 4.8, zůstal stabilní za podmínek dlouhodobé aplikace, tak jak byl pozorován během dlouhodobých terapií trvajících až jeden rok.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií Soolantry u všech podskupin pediatrické populace v léčbě papulopustulózní rosacey (informace o použití u dětí viz bod 4.2).
5.2. Farmakokinetické vlastnosti
Absorpce
Absorpce ivermektinu z přípravku Soolantra byla hodnocena v klinické studii u dospělých pacientů s těžkou papulopustulózní rosaceou v podmínkách maximální aplikace. V rovnovážném stavu (po 2 týdnech od zahájení léčby) byly nejvyšší průměrné koncentrace ivermektinu v plazmě (± standardní odchylka) dosaženy 10 ± 8 hodin po aplikaci (Cmax: 2,1 ± 1,0 ng/ml, rozsah: 0,7- 4,0 ng/ml) a nejvyšší průměrná hodnota AUC0-24h (± standardní odchylka) byla 36 ± 16 ng.h/ml (rozsah: 14-75 ng.h/ml). Hladiny systémové expozice ivermektinu dosáhly plató po dvou týdnech léčby (podmínky rovnovážného stravu). Při delším trvání léčby v rámci klinických studií fáze III byly hladiny systémové expozice ivermektinu podobné těm, které byly pozorovány po dvou týdnech léčby. V podmínkách rovnovážného stavu byly hladiny systémové expozice ivermektinu (AUC0-24h: 36 ± 16 ng.h/ml) nižší než ty, které se dosáhly po jednorázové perorální dávce 6 mg podané zdravým dobrovolníkům (AUC0-24h: 134 ± 66 ng.h/ml).
Distribuce
V in vitro studii bylo prokázáno, že ivermektin je z více než 99 % vázán na proteiny plazmy, přičemž se primárně váže na lidský sérový albumin. Nebyla pozorována žádná významná vazba ivermektinu na erytrocyty.
In vitro studie, v nichž byly použity lidské jatemí mikrozómy a rekombinantní enzymy CYP450, dokázaly, že ivermektin je primárně metabolizován CYP3A4.
In vitro studie ukazují, že ivermektin neinhibuje CYP3A4 izoenzymy 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4, 4A11 nebo 2E1. Ivermektin neindukuje expresi enzymů CYP450 (1A2, 2B6, 2C9 nebo 3A4) v kultuře lidských hepatocytů.
V rámci studie klinické farmakokinetiky s maximálním použitím přípravku byly identifikovány dva hlavní metabolity ivermektinu, které byly hodnoceny v průběhu klinických studií fáze II (3 '' - O-demethylivermektin a 4a-hydroxyivermectin). Podobně jako v případě mateřské látky, metabolity dosáhly ustáleného stavu do 2 týdnů od zahájení léčby, přičemž do 12 týdnů nebyl zaznamenán důkaz akumulace. Navíc systémová expozice metabolitem (hodnocená parametry Cmax a AUC) dosažená v rovnovážném stavu byla mnohem nižší než ta, která byla pozorována po perorálním podání ivermectinu.
Vylučování
Průměrný terminální poločas byl 6 dní (průměr: 145 hodin, rozsah 92-238 hodin), vztahoval se na pacienty, kterým byl lék aplikován jednou denně na kůži v trvání 28 dnů v rámci klinické farmakokinetické studie s maximálním použitím přípravku. Po topické aplikaci Soolantry je vylučování léčiva závislé na absorpci. Farmakokinetika ivermektinu nebyla studována u pacientů s poruchou funkce ledvin a jater.
5.3. Předklinické údaje vztahující se k bezpečnosti
Studie opakovaného podávání v délce 9 měsíců cestou dermální aplikace ivermektinu ve formě krému 10 mg/g u miniprasat neukázaly přítomnost toxických účinků nebo lokální toxicitu v rámci hladiny systémové expozice srovnatelné s klinickou expozicí.
Ivermektin neprokázal genotoxicitu v souboru in vitro a in vivo testů. Dvouletá studie karcinogenity cestou dermální aplikace krému 10 mg/g u myší neukázala vyšší výskyt nádorů.
Studie reprodukční toxicity po perorálním podání ivermektinu ukázaly teratogenní účinky u potkanů (rozštěp patra) a králíků (karpální flexury) po aplikaci vysokých dávek (hranice expozice NOAEL [úroveň bez pozorovaných nežádoucích účinků] byla minimálně 70násobná proti klinické expozici).
Neonatální toxicita v perorálních studiích u potkanů nesouvisela s expozicí in utero, ale s postnatální expozicí prostřednictvím mateřského mléka, jejímž výsledkem byly vysoké koncentrace ivermectinu v mozku a plazmě potomků. Ivermektin ve formě 10 mg/g krému prokazuje kožní dráždivost, senzibilizační a fotosensibilizační vlastnosti u morčat, není však fototoxický.
Hodnocení rizik pro životní prostředí
Ivermektin je pro bezobratlé velmi toxický, přičemž riziko bylo identifikováno pro vodní prostředí, sediment a také pro suchozemské prostředí. Pozornost je třeba věnovat prevenci kontaminace životního prostředí, zejména v případě vodního prostředí.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
Glycerol 85%
Isopropyl-palmitát
Karbomer
Dimetikon 20
Dihydrát dinatrium-edetátu
Monohydrát kyseliny citronové
Cetylalkohol
Stearylalkohol
Cetostearomakrogol
Sorbitan-stearát
Methylparaben (E218)
Propylparaben (E216)
Fenoxyethanol Propylenglykol Oleylalkohol Hydroxid sodný Čištěná voda
6.2. Inkompatibility
Neaplikovatelné.
6.3. Doba použitelnosti
2 roky.
Doba použitelnosti po prvním otevření: používejte nejdéle 6 měsíců.
6.4. Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5. Druh obalu a velikost balení
Laminované plastové bílé tuby z materiálu polyethylen (PE)/aluminium (Al)/polyethylen (PE) uzavřené:
- bílým uzávěrem z polyethylenu (HDPE) opatřeným polypropylenovým (PP) dětským bezpečnostním uzávěrem pro 15g, 30g, 45g nebo 60g tuby
- polypropylenovým (PP) bílým uzávěrem pro 2g tuby (bez dětského bezpečnostního uzávěru )
Velikosti balení: 1 tuba s obsahem 2 g, 15 g, 30 g, 45 g nebo 60 g.
Na trhu nemusí být všechny velikosti balení.
6.6. Zvláštní opatření pro likvidaci přípravku
Je třeba přijmout opatření k prevenci nebo snížení kontaminace, zejména v případě vodních zdrojů . Nepoužitý přípravek nebo odpad z něho vzniklý má být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Galderma International
Tour Europlaza, 20 avenue André Prothin - La Défense 4
La Défense Cedex 92927
Francie
8. REGISTRAČNÍ ČÍSLO
46/241/15-C
9. DATUM PRVNÍ REGISTRACE Datum první registrace: 13.5.2015
10. PRODLOUŽENÍ REGISTRACE 18.11.2015
9