Prothromplex Total Nf
sp.zn. sukls131915/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
PROTHROMPLEX TOTAL NF
Prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Léčivá látka: Prothrombinum multiplex humanum
PROTHROMPLEX TOTAL NF je prášek pro injekční roztok k intravenózní aplikaci. Jedna injekční lahvička obsahuje následující množství mezinárodních jednotek (IU) lidských koagulačních faktorů:
|
IU v lahvičce |
Po rekonstituci ve 20 ml vody na injekci IU/ml | |
|
Factor II coagulationis humanus |
480-900 |
24-25 |
|
Factor VII coagulationis humanus |
500 |
25 |
|
Factor IX coagulationis humanus |
600 |
30 |
|
Factor X coagulationis humanus |
600 |
30 |
Celkový obsah proteinu v lahvičce je 300 - 750 mg. Specifická aktivita přípravku je nejméně 0,6 IU/mg, vyjádřeno aktivitou faktoru IX.
Jedna injekční lahvička obsahuje minimálně 400 IU Proteinu C ko-purifikovaného s faktory krevního srážení.
Aktivita (IU) faktoru IX se určuje jednostupňovým koagulačním testem dle Evropského lékopisu a měří se vzhledem k mezinárodnímu standardu Světové zdravotnické organizace (WHO) pro koncentrát faktoru IX.
Aktivita (IU) faktoru II, faktoru VII a faktoru X se určuje chromogenním testem dle Evropského lékopisu a měří se vzhledem k mezinárodním standardům WHO pro koncentráty faktoru II, faktoru VII a faktoru X.
Aktivita (IU) proteinu C se určuje chromogenním testem dle Evropského lékopisu a měří se vzhledem k mezinárodnímu standardu WHO pro koncentráty proteinu C.
Pomocné látky se známým účinkem: Prothromplex Total NF obsahuje 80 mg sodíku v jedné lahvičce (stanoveno výpočtem). Dále obsahuje v jedné lahvičce sodnou sůl heparinu (max. 0,5 IU/IU faktoru IX).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok
Prášek: Bílý až nažloutlý lyofilizovaný prášek nebo kompaktní vysušená látka. Rozpouštědlo: Voda na injekci
Po rekonstituci má pH 6,5 až 7,5 a osmolalitu nejméně 240 mosmol/kg. Roztok je čirý nebo mírně opalescentní.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba krvácení a profylaxe krvácení během chirurgických výkonů u získaného deficitu koagulačních faktorů prothrombinového komplexu, jako je deficit způsobený léčbou antagonisty vitaminu K nebo v případě předávkování antagonisty vitaminu K tam, kde je zapotřebí rychlá úprava deficitu.
Léčba krvácení a profylaxe krvácení během chirurgických výkonů u vrozeného deficitu některého z koagulačních faktorů závislých na vitaminu K, pokud není k dispozici čištěný přípravek s obsahem specifického koagulačního faktoru.
Přípravek PROTHROMPLEX TOTAL NF je indikován k léčbě dospělých. Neexistují dostatečné pediatrické údaje, které doporučují podávat přípravek PROTHROMPLEX TOTAL NF dětem.
4.2 Dávkování a způsob podání Dávkování
S výjimkou léčby krvácení a perioperační profylaxe krvácení v průběhu léčby antagonisty vitamínu K je níže uvedeno pouze obecné doporučené dávkování.
Léčba by měla být zahájena pod dohledem lékaře, který má zkušenosti s léčbou poruch koagulace.
Dávkování a délka trvání substituční léčby závisí na závažnosti koagulační poruchy, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Dávkování a četnost podávání by měly být vždy stanoveny individuálně. Intervaly dávkování je nutno přizpůsobit rozdílným cirkulujícím poločasům různých koagulačních faktorů v prothrombinovém komplexu (viz bod 5.2).
Individuální požadavky na dávkování mohou být ověřeny pouze pravidelným sledováním hladin jednotlivých koagulačních faktorů v plazmě nebo pomocí testů na stanovení celkové hladiny prothrombinového komplexu (např. Quickův test, INR, prothrombinový čas) a nepřetržitým sledováním klinického stavu pacienta.
V případě velkých chirurgických výkonů je monitorování substituční terapie pomocí koagulačních testů (specifických pro jednotlivé koagulační faktory a/nebo celkových testů hladiny prothrombinového komplexu) nezbytné.
Krvácení a profylaxe krvácení během chirurgického výkonu při léčbě antagonisty vitaminu K:
Při závažném krvácení nebo před operačními výkony s vysokým rizikem krvácení je cílem dosažení normálních hodnot (hodnota Quickova testu 100%, INR 1,0).
Platí následující pravidlo: 1 IU faktoru IX/kg těl. hmotnosti zvýší hladinu Quickova testu přibližně o 1%.
Pokud podávání přípravku PROTHROMPLEX TOTAL NF vychází z měření INR - stanovení dávky závisí na INR před léčbou a cílové INR.
Dávkování uvedené níže v tabulce má být dodržováno podle doporučení uvedené v publikaci Makris et al 20011.
|
Dávkování přípravku PROTHROMPLEX TOTAL NF podle iniciálního měření INR | |
|
INR |
Dávka IU/kg (IUs vztažené k faktoru IX) |
|
2,0-3,9 |
25 |
|
4,0-6,0 |
35 |
|
>6,0 |
50 |
Korekce poruchy hemostázy, indukované antagonisty vitaminu K, přetrvává po dobu přibližně 6-8 hodin. Nicméně účinků vitamínu K, pokud je podáván současně, je obvykle dosaženo v průběhu 4-6 hodin. Tedy opakovaná léčba lidským prothrombinovým komplexem není obvykle nutná v případě, že je podán vitamín K.
Jelikož tato doporučení jsou empirická a obnova a trvání účinků se může lišit, monitorování INR v průběhu léčby je povinné.
Krvácení a profylaxe krvácení během chirurgického výkonu při vrozeném deficitu některého z koagulačních faktorů závislých na vitaminu K v případě, že není k dispozici specifický koncentrát koagulačního faktoru:
Vypočtená požadovaná léčebná dávka je založena na empirickém zjištění, že přibližně 1 IU faktoru IX na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru IX přibližně o 0,015 IU/ml; a 1 IU factoru VII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VII přibližně o 0,024 IU/ml. 1 IU faktoru II nebo X na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru II nebo X přibližně o 0,021 IU/ml.1 2
Podaná dávka specifického faktoru se vyjadřuje v mezinárodních jednotkách (IU), které jsou vztaženy k platnému standardu WHO pro jednotlivé faktory. Aktivita specifického koagulačního faktoru v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro specifický koncentrát koagulačního faktoru). Jedna mezinárodní jednotka (IU) aktivity koagulačního faktoru odpovídá množství v 1 mililitru normální lidské plazmy.
Například výpočet požadovaní dávky faktoru X je založen na empirické zkušenosti, že 1 mezinárodní jednotka (IU) faktoru X na 1 kg tělesné hmotnosti zvýší aktivitu plazmatického faktoru X přibližně o 0,017 IU/ml. Potřebná dávka se určuje podle následujícího vzorce:
Potřebné jednotky = tělesná hmotnost (kg) x požadovaný nárůst faktoru X (IU/ml) x 60
kde 60 (ml/kg) je převrácená hodnota odhadovaného zotavení.
Pokud je známo individuální zotavení, tak by měla být jeho hodnota k výpočtu použita.
Maximálníjednotlivá dávka:
Za účelem opravy INR není nutno překročit dávku 50 IU/kg. V případě, že závažnost krvácení vyžaduje vyšší dávku, musí ošetřující lékař vyhodnotit poměr riziko/přínos.
Pediatrická populace
Bezpečnost a účinnost podávání přípravku PROTHROMPLEX TOTAL NF nebyla u dětských pacientů v klinických studiích společnosti Baxter ověřena.
Způsob podání
Intravenózní podání
Přípravek PROTHROMPLEX TOTAL NF má být podáván pomalu intravenózně. Maximální rychlost podání by neměla překročit 2 ml za minutu (60 IU/min).
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Známá alergie na heparin nebo heparinem-indukovaná trombocytopenie v anamnéze.
4.4 Zvláštní upozornění a opatření pro použití
Léčba by měla být vedena pod dohledem lékaře, který má zkušenosti s léčbou poruch koagulace.
U pacientů se získaným deficitem koagulačních faktorů závislých na vitaminu K (např. indukovaném léčbou antagonisty vitaminu K) má být PROTHROMPLEX TOTAL NF používán pouze tam, kde je zapotřebí rychlá úprava hladin prothrombinového komplexu, tj. při těžkém krvácení nebo při neodkladných chirurgických výkonech. V jiných případech je zpravidla dostačující snížení dávky antagonistů vitaminu K a/nebo podání vitaminu K.
U pacientů, kteří jsou léčeni antagonisty vitaminu K pro hyperkoagulační stav, může dojít po podání infuze prothrombinového komplexu k jeho exacerbaci.
Při vrozeném deficitu kteréhokoli z faktorů závislých na vitaminu K má být použit přípravek s obsahem specifického faktoru, je-li k dispozici.
V souvislosti s přípravkem PROTHROMPLEX TOTAL NF byly hlášeny hypersenzitivní reakce včetně anafylaktických reakcí a anafylaktického šoku.
V případě alergické reakce nebo reakce anafylaktického typu musí být injekce/infuze ihned zastavena.
V případě šoku je nutno dodržovat aktuální standardní lékařské postupy pro léčbu šoku.
Tromboembólie, DIC, _fibrinolvza
Při léčbě pacientů s vrozenými nebo získanými poruchami koagulace koncentráty lidského prothrombinového komplexu včetně přípravku PROTHROMPLEX TOTAL NF, a to zejména při opakovaném dávkování, existuje riziko vzniku trombózy a diseminované intravaskulární koagulace (DIC).
V souvislosti s podáním přípravku PROTHROMPLEX TOTAL NF byly hlášeny arteriální a venózní tromboembolické příhody včetně infarktu myokardu, cévní mozkové příhody (např. mrtvice), plicní embolie a DIC.
Toto riziko může být vyšší při léčbě izolovaného deficitu F VII, protože ostatní koagulační faktory závislé na vitaminu K s delšími poločasy se mohou akumulovat ve výrazně vyšším množství než je normální.
Pacienti léčení koncentráty lidského prothrombinového komplexu je třeba pečlivě sledovat s ohledem na známky a příznaky intravaskulární koagulace nebo trombózy. Vzhledem k riziku tromboembolických komplikací je třeba během podávání koncentrátů lidského prothrombinového komplexu zvláště pečlivě sledovat
• pacienty s anamnézou ischemické choroby srdeční
• pacienty s jaterním onemocněním
• pacienty před a po operaci
• novorozence
• nebo jiné pacienty s rizikem tromboembolických příhod či diseminované intravaskulámí koagulace.
U všech těchto stavů je nutno zvážit potenciální přínos léčby vzhledem k riziku těchto komplikací.
Virová bezpečnost
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a určité výrobní kroky odstraňující nebo deaktivující viry. Přesto nemůže být při podávání léčiv vyráběných z lidské krve nebo plazmy zcela vyloučena možnost přenosu infekce. To se vztahuje též na jakékoli neznámé nebo vznikající viry či jiné patogeny.
Přijatá opatření jsou považována za účinná u obalených virů jako HIV, HBV a HCV, a u neobaleného viru HAV.
Přijatá opatření mohou mít omezenou účinnost na neobalené viry jako je parvovirus B19. Infekce parvovirem B19 může být nebezpečná pro těhotné ženy (fetální infekce) a pro jedince, kteří mají sníženou funkci imunitního systému nebo zvýšenou erytropoézu (např. hemolytická anémie).
Pokud jsou přípravky připravené z lidské krve nebo plazmy podávány pravidelně/opakovaně, musí být uvažováno o vhodné vakcinaci (hepatitida A a B).
V zájmu pacienta se při každé aplikaci přípravku Prothromplex Total NF důrazně doporučuje zaznamenat název a číslo šarže přípravku na přiložený samolepicí štítek, aby bylo možné dohledat u pacienta použitou šarži přípravku.
Sodík
Přípravek PROTHROMPLEX TOTAL NF obsahuje 80 mg sodíku v jedné lahvičce nebo 0,13 mg sodíku v mezinárodní jednotce přípravku PROTHROMPLEX TOTAL NF (tj. dávka 50 IU/kg tělesné hmotnosti obsahuje 6,5 mg sodíku/kg tělesné hmotnosti; dávka 35 IU/kg tělesné hmotnosti obsahuje 4,6 mg sodíku/kg tělesné hmotnosti a dávka 25 IU/kg tělesné hmotnosti obsahuje 3,3 mg sodíku / kg tělesné hmotnosti (stanoveno výpočtem)). To je třeba vzít v úvahu u pacientů na kontrolované dietě s nízkým obsahem sodíku.
Heparin
Heparin může vyvolat alergické reakce a pokles počtu krevních buněk, což může ovlivnit koagulační systém. Pacienti s heparinem indukovanými alergickými reakcemi v anamnéze by neměli užívat léčiva s obsahem heparinu.
Pediatrická populace:
Neexistují dostatečné údaje, které doporučují podávat přípravek PROTHROMPLEX TOTAL NF dětem.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Přípravky lidského prothrombinového komplexu působí proti účinkům léčby antagonisty vitaminu K. Nebyly provedeny žádné studie interakcí.
Interference s biologickými testy:
Při provádění koagulačních testů citlivých na heparin u pacientů léčených vysokými dávkami lidského prothrombinového komplexu je nutno vzít v úvahu obsah heparinu jako složky přípravku.
4.6 Fertilita, těhotenství a kojení
Vliv přípravku PROTHROMPLEX TOTAL NF na fertilitu nebyl v kontrolovaných klinických studíí stanoven.
Bezpečnost použití koncentrátů lidského prothrombinového komplexu u těhotných nebo kojících žen nebyla stanovena.
Adekvátní údaje o podávání přípravku PROTHROMPLEX TOTAL NF těhotným a kojícím ženám nejsou k dispozici.
Studie na zvířatech nejsou vhodné k posouzení bezpečnosti během těhotenství, vývoje embrya a plodu, porodu a postnatálního vývoje. Proto má být PROTHROMPLEX TOTAL NF během těhotenství a kojení používán pouze tehdy, je-li to jednoznačně indikováno.
Informace týkající se rizika infekce parvovirem B19 u těhotných žen viz bod 4.4.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nejsou k dispozici údaje o účincích na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu Poruchy imunitního systému:
Během léčby koncentráty lidského prothrombinového komplexu, včetně léčby přípravkem PROTHROMPLEX TOTAL NF, se mohou vyvinout cirkulující inhibitory, které následně inhibují jeden či více faktorů lidského prothrombinového komplexu. Pokud se tyto inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď na léčbu.
Cévní _ poruchy
Po podání lidského prothrombinového komplexu existuje riziko tromboembolických příhod (viz bod 4.4).
Informace o virové bezpečnosti viz bod 4.4.
Tabulkový souhrn nežádoucích účinků
Akutní infarkt myokardu, žilní trombóza a pyrexie uvedené v tabulkovém přehledu nežádoucích účinků níže byly hlášeny v jedné klinické studii s přípravkem PROTHROMPLEX TOTAL NF u pacientů (n=61) se získaným deficitem koagulačních faktorů prothrombinového komplexu (II, VII, IX, X), u nichž dávka perorálních antikoagulancií způsobila zvrat stavu. Ostatní nežádoucí účinky zahrnuté v tabulce byly hlášeny pouze při postmarketingovém použití.
Nežádoucí účinky v níže uvedené tabulce jsou seřazeny podle tříd orgánového systému MedDRA (TOS) (verze 15.1), pak podle preferovaného termínu.
Frekvence výskytu jsou definovány jako: velmi časté (> 1/10) časté (> 1/100 až < 1/10) méně časté (> 1/1 000 až < 1/100) vzácné (> 1/10 000 až < 1/1 000) velmi vzácné (< 1/10 000) není známo (z dostupných údajů nelze určit).
V této tabulce jsou zahrnuty nežádoucí účinky z postmarketinkového použití. Kategorie frekvence byla stanovena statisticky na předpokladu, že každý nežádoucí účinek se mohl objevit v klinické studii s 61 pacienty.
|
Třída orgánového systému |
Nežádoucí účinek |
Frekvence |
|
Poruchy krve a |
Diseminovaná intravaskulární koagulace |
Časté |
|
Třída orgánového systému |
Nežádoucí účinek |
Frekvence |
|
lymfatického systému |
Inhibitory proti jednomu nebo více faktorům prothrombinového komplexu (faktory II, VII, IX, X) | |
|
Poruchy imunitního systému |
Anafylaktický šok Anafylaktická reakce Přecitlivělost |
Časté |
|
Poruchy nervového systému |
Cerebrovaskulární příhoda Bolest hlavy |
Časté |
|
Srdeční poruchy |
Srdeční selhání Akutní infarkt myokardu |
Časté |
|
Cévní poruchy |
Arteriální trombóza Žilní trombóza Hypotenze Návaly |
Časté |
|
Respirační, hrudní a mediastinální poruchy |
Sípot |
Časté |
|
Gastrointestinální poruchy |
Časté | |
|
Poruchy kůže a podkožní tkáně |
Kopřivka Eerytematózní vyrážka Svědění |
Časté |
|
Poruchy ledvin a močových cest |
Nefrotický syndrom |
Časté |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
‘rozvinutý u pacientů s vrozeným deficitem faktorů
Reakce třídy
Poruchy kůže a podkožní tkáně: angioedém, parestezie Celkové poruchy a reakce v místě aplikace: reakce v místě infuze Poruchy nervového systému: letargie Psychiatrické poruchy: neklid
Pediatrická populace
Informace o pediatrické populaci viz bod 4.2.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www .sukl .cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Použití vysokých dávek přípravků s obsahem lidského prothrombinového komplexu bylo spojováno s případy infarktu myokardu, diseminované intravaskulární koagulace, žilní trombózy a plicní
embolie. Proto je v případě předávkování zvýšené riziko rozvoje tromboembolických komplikací nebo diseminované intravaskulární koagulace.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika: koagulační faktory IX, II, VII a X v kombinaci.
ATC kód: B02BD01.
Koagulační faktory II, VII, IX a X, které se tvoří v játrech a jsou závislé na vitaminu K, se běžně označují jako prothrombinový komplex.
Faktor VII je zymogen aktivní serinové proteázy, faktoru VIIa, který iniciuje vnitřní kaskádu koagulace. Komplex tkáňového faktoru a faktoru VIIa aktivuje koagulační faktory X a IX, přičemž dochází k tvorbě faktorů IXa a Xa. Další aktivací koagulační kaskády se aktivuje prothrombin (faktor II) a přeměňuje se na thrombin. Působením thrombinu se fibrinogen konvertuje na fibrin a tím vzniká krevní sraženina. Normální tvorba thrombinu je zásadní také pro funkce destiček jako součásti primární hemostázy.
Izolovaný závažný deficit faktoru VII vede ke snížené tvorbě thrombinu a může vyvolat krvácení způsobené poruchou tvorby fibrinu a primární hemostázy. Izolovaný deficit faktoru IX je jednou z klasických hemofilií (hemofilie B). Izolované deficity faktoru II nebo faktoru X jsou velmi vzácné, ale v závažné formě vyvolávají sklon ke krvácení podobně jako u klasické hemofilie.
Získaný deficit koagulačních faktorů prothrombinového komplexu závislých na vitaminu K se objevuje během léčby antagonisty vitaminu K. Pokud je deficit závažný, projevuje se sklonem ke krvácení, charakterizovaným retroperitoneálním nebo cerebrálním krvácením spíše než krvácením do svalů a kloubů. Těžká jaterní insuficience také způsobuje významné snížení hladin prothrombinového komplexu a klinicky sklon ke krvácení, který je však často komplexní díky simultánně probíhající intravaskulární koagulaci nízkého stupně, nízkým hladinám destiček, deficitu inhibitorů koagulace a narušené fibrinolýze.
Podání koncentrátů lidského prothrombinového komplexu působí zvýšení plazmatických hladin koagulačních faktorů závislých na vitaminu K a dočasnou úpravu poruch koagulace u pacientů s deficitem jednoho nebo více těchto faktorů.
Pediatrická populace
Neexistují dostatečné údaje, které doporučují podávat přípravek PROTHROMPLEX TOTAL NF dětem.
5.2 Farmakokinetické vlastnosti
Poločas
40-60 hodin 3-5 hodin 16-30 hodin 30-60 hodin
Koagulační faktor
Faktor II Faktor VII Faktor IX Faktor X
5.3 Předklinické údaje vztahující se k bezpečnosti
Faktory lidského prothrombinového komplexu (v koncentrátu) jsou normální složkou lidské plazmy a chovají se stejně jako endogenní koagulační faktory.
Testování toxicity po podání jedné dávky nemá význam vzhledem k tomu, že vyšší dávky vedou k objemovému přetížení.
Studie toxicity po opakovaném podání nejsou u zvířat proveditelné, protože se tvoří heterologní proteiny a interferují s následnými testy.
Lidské koagulační faktory nejsou považovány za karcinogenní nebo mutagenní, proto nejsou experimentální studie, zvláště u heterologních druhů, pokládány za nutné.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
chlorid sodný
dihydrát natrium-citrátu
sodná sůl heparinu 0,2 - 0,5 IU/IU FIX
koncentrát lidského antithrombinu III 15 - 30 IU v jedné lahvičce (0,75 - 1,5 IU/ml)
Rozpouštědlo: voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6. Pro rekonstituci se má používat pouze přiložená souprava a pro injekci/infuzi pouze dodaná injekční/infuzní souprava, protože může dojít k selhání léčby v důsledku adsorpce koagulačního faktoru na vnitřní povrch některého injekčního/infuzního setu.
Stejně jako u všech přípravků obsahujících koagulační faktory může být účinnost a snášenlivost narušena mísením s jinými léčivy. Proto se doporučuje propláchnout před a po podání přípravku PROTHROMPLEX TOTAL NF žilní vstup fyziologickým roztokem.
6.3 Doba použitelnosti
3 roky
Během uvedené doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C), po jedno období trvající nejvýše 6 měsíců. Zahájení a ukončení doby uchovávání při pokojové teplotě má být vyznačeno na obalu přípravku. Po uchovávání při pokojové teplotě se přípravek PROTHROMPLEX TOTAL NF nesmí vracet do chladničky (2°C - 8°C) ale musí být během 6 měsíců použit nebo zlikvidován.
Chemická a fyzikální stabilita přípravku byla doložena po dobu 3 hodin při teplotě 20 - 25°C. Z mikrobiologického hlediska by měl být přípravek PROTHROMPLEX TOTAL NF použit ihned po rekonstituci, protože přípravek neobsahuje žádná konzervační činidla. Není-li ihned použit, jsou doba uchovávání a podmínky před použitím v odpovědnosti uživatele. Připravený přípravek k použití se nesmí vracet do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (při 2°C až 8°C). Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
Prášek je dodáván v jednorázových injekčních lahvičkách z bezbarvého skla hydrolytické třídy II. Rozpouštědlo je dodáváno v inj. lahvičkách z neutrálního bezbarvého skla hydrolytické třídy I. Obě injekční lahvičky jsou uzavřeny zátkou z butylové pryže.
Obsah balení:
1 inj. lahvička přípravku PROTHROMPLEX TOTAL NF - prášek pro injekční roztok 1 inj. lahvička vody na injekci 20 ml
1 triple set (1 zavzdušňovací jehla, 1 infuzní set (motýlek), 1 jehla k jednorázovému použití), 1 filtrační jehla, 1 převodní jehla
Velikost balení: 1 x 600 IU + 20 ml rozpouštědla
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
K rekonstituci použijte pouze přiložený set.
Přípravek PROTHROMPLEX TOTAL NF se rekonstituuje bezprostředně před podáním.
Roztok je čirý nebo mírně opalescentní. Zakalené roztoky nebo roztoky obsahující usazeniny nepoužívejte.
Rekonstituce prášku pro přípravu injekčního roztoku:
Použijte aseptický postup!
1. Zahřejte neotevřenou inj. lahvičku obsahující rozpouštědlo (vodu na injekci) na pokojovou nebo tělesnou teplotu (maximálně 37°C).
2. Odstraňte ochranná víčka z inj. lahvičky s práškem a z inj. lahvičky s rozpouštědlem (obr. A) a dezinfikujte pryžové zátky obou inj. lahviček.
3. Pootočením odstraňte ochranný kryt z j ednoho konce přiložené převodní j ehly. Odkrytou j ehlu vpíchněte přes pryžovou zátku do inj. lahvičky s rozpouštědlem (obr. B a C).
4. Odstraňte ochranný kryt z druhého konce převodní jehly; nedotýkejte se přitom odkrytého konce!
5. Převraťte inj. lahvičku s rozpouštědlem nad inj. lahvičku s práškem a propíchněte volným koncem převodní jehly pryžovou zátku inj. lahvičky s práškem (obr. D). Rozpouštědlo se pomocí vakua samo natáhne do inj. lahvičky s práškem.
6. Oddělte obě inj. lahvičky od sebe vytažením převodní jehly spolu s inj. lahvičkou rozpouštědla z inj. lahvičky s práškem (obr. E). Injekční lahvičkou s práškem jemně zatřepejte, aby se urychlilo rozpouštění.
7. Po úplném rozpuštění prášku vpíchněte přiloženou zavzdušňovací jehlu (Obr. F), čímž všechna pěna opadne. Zavzdušňovací jehlu vyjměte.
Injekce/infuze:
Použijte aseptický postup!
Rekonstituovaný přípravek má být před podáním vizuálně vždy zkontrolován s ohledem na obsah
částic či změnu zabarvení.
1. Sejměte ochranný kryt z přiložené filtrační jehly pootočením a povytažením a nasaďte jehlu na sterilní jednorázovou inj. stříkačku. Natáhněte roztok do inj. stříkačky (obr. G).
2. Odpojte filtrační jehlu od injekční stříkačky a roztok aplikujte pomalu intravenózně (maximální rychlost podávání infuze/injekce: 2 ml/min).
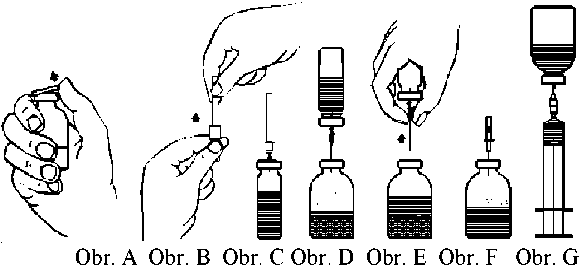
Po podání zlikvidujte všechny použité jehly společně se stříkačkou a/nebo infuzním setem do schránky na ostré předměty, aby se zabránilo riziku pro jiné osoby.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Do 30.11.2016
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
Od 1.12.2016
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
75/474/93-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16.6.1993
Datum posledního prodloužení registrace: 29.8.2015
10. DATUM REVIZE TEXTU
27.7.2016
11/11
1. Makris M, Watson HG: The Management of Coumarin-Induced Over-Anticoagulation. Br.J.Haematol. 2001;114: 271-280.
Ostermann H, Haertel S, Knaub S, Kalina U, Jung K, Pabinger I. Pharmacokinetics of Beriplex P/N prothrombin complex concentrate in healthy volunteers. Thromb Haemost. 2007;98(4):790-797.