Panadol Pro Děti 24 Mg/Ml Jahoda
sp. zn. sukls78048/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Panadol pro děti 24 mg/ml jahoda perorální suspenze
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml suspenze obsahuje paracetamolum 24 mg, což odpovídá paracetamolum 120 mg v 5 ml suspenze.
Pomocné látky:
směs sodných solí parabenů (E 215, E 217, E 219), nekrystalizující sorbitol 70 %, roztok maltitolu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální suspenze.
Bezbarvá až bílá nebo nahnědlá průsvitná suspense s jahodovým aroma.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Panadol pro děti 24 mg/ml jahoda je určený k:
A. tlumení mírné až středně silné bolesti včetně:
■ bolesti při prořezávání zubů
■ bolesti zubů
■ bolesti hlavy
■ bolesti v krku provázející záněty horních cest dýchacích
B. snížení horečky provázející chřipku, akutní záněty horních cest dýchacích a infekční choroby dětského věku, jako jsou např. spalničky, zarděnky, plané neštovice, spála a příušnice, a ke snížení zvýšené teploty po očkování.
4.2 Dávkování a způsob podání
Tento léčivý přípravek je určen pro perorální podání.
Pacient má užívat nejnižší účinnou dávku po nejkratší dobu nutnou ke zlepšení příznaků.
Děti ve věku 2 - 3 měsíce:
Dětem ve věku 2 - 3 měsíce se Panadol pro děti 24 mg/ml jahoda podává pouze po očkování k symptomatické úlevě od horečky.
Jednotlivá dávka je 2,5 ml (120 mg/5 ml). Odstup mezi dávkami je 6 hodin. Pokud horečka přetrvává i po druhé dávce, je potřeba vyhledat lékaře.
Děti ve věku 3 měsíců - 12 let:
Jednotlivá dávka pro děti od 3 měsíců do 12 let je 10 - 15 mg/kg.
|
Tělesná hmotnost |
Věk |
Dávka | |
|
5 - 6 kg |
3 - 6 měsíců |
3 ml |
(72 mg paracetamolu) |
|
7 - 8 kg |
4 ml |
(96 mg paracetamolu) | |
|
9 - 10 kg |
6 - 12 měsíců |
5 ml |
(120 mg paracetamolu) |
|
11 - 13 kg |
1 - 2 roky |
6 ml |
(144 mg paracetamolu) |
|
14 - 16 kg |
2 - 3 roky |
8 ml |
(192 mg paracetamolu) |
|
17 - 20 kg |
3 - 6 let |
10 ml |
(240 mg paracetamolu) |
|
21 - 25 kg |
13 ml |
(312 mg paracetamolu) | |
|
26 - 33 kg |
6 - 12 let |
16 ml |
(384 mg paracetamolu) |
|
34 - 40 kg |
20 ml |
(480 mg paracetamolu) | |
Správná dávka se určí pomocí uvedené tabulky podle tělesné hmotnosti dítěte. Pokud není jistota o tělesné hmotnosti dítěte, použije se k určení dávky přípravku věk dítěte.
Celková denní dávka nesmí přesáhnout 60 mg/kg tělesné hmotnosti.
Přípravek může být podáván opakovaně podle potřeby s odstupem jednotlivých dávek nejlépe 6 hodin. Minimální interval mezi dvěma dávkami jsou 4 hodiny. Nepodávají se více než 4 dávky během 24 hodin.
Přípravek se dítěti nemá podávat déle než 3 dny bez porady s lékařem.
Pacienti s poruchou funkce ledvin:
Při renální insuficienci je nutné upravit dávkování:
■ při glomerulární filtraci 50 - 10 ml/min se podávají jednotlivé dávky v intervalu nejméně 6 hodin;
■ při glomerulární filtraci pod 10 ml/min se podávají v intervalu 8 hodin.
Pacienti s poruchou funkce jater:
U pacientů se sníženou funkcí jater nemají být podávány maximální dávky a interval mezi jednotlivými dávkami by měl být nejméně 6 hodin. Podávání přípravku dětem se závažnou poruchou funkce jater je kontraindikováno (viz bod 4.3).
Způsob podávání:
Přípravek je dodáván v balení s dávkovacím aplikátorem.
Před použitím je nutno obsah lahvičky protřepat a odšroubovat uzávěr. Dávkovací aplikátor se vloží do vložky v hrdle lahvičky, lahvička se otočí dnem vzhůru a vytažením pístu se aplikátor naplní požadovaným množstvím suspenze.
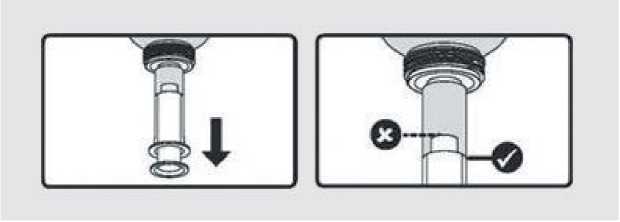
Pro správně odměřenou dávku musí být nejširší část pístu v jedné linii se správnou kalibrační značkou (v ml) na válci dávkovacího aplikátoru (tj. kde končí hladina suspenze: viz značka „V" na obrázku).
Je-li stanovená dávka větší než 10 ml, odměření se opakuje podle potřeby. Aplikátor se po použití propláchne teplou vodou a nechá vyschnout.
4.3 Kontraindikace
■
■
■
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Závažná hepatocelulární insuficience.
Akutní hepatitida.
4.4 Zvláštní upozornění a zvláštní opatření pro použití
Obsahuje paracetamol. Rodiče je třeba upozornit, aby dětem nebyly podávány současně jiné přípravky obsahující paracetamol. Souběžné podávání by mohlo vést k předávkování.
Předávkování paracetamolem může způsobit selhání jater vedoucí až k potřebě transplantace jater nebo smrti.
Pacienti s diagnostikovanou poruchou funkce jater nebo ledvin se musí před zahájením užívání tohoto léčivého přípravku poradit s lékařem.
Při podávání pacientům s poruchou funkce jater a u pacientů dlouhodobě (nad 10 dní) užívajících vyšší dávky paracetamolu se doporučuje pravidelná kontrola jaterních testů. Nebezpečí předávkování a poškození jater v souvislosti s paracetamolem je vyšší u pacientů se základním jaterním onemocněním.
Paracetamol by měl být užíván se zvýšenou opatrností při deficitu enzymu glukóza-6-fosfátdehydrogenázy, hemolytické anemii a u dětí s poruchou funkce ledvin (viz bod 4.2). Při dlouhodobé léčbě nelze vyloučit možnost poškození ledvin.
U pacientů s deplecí glutathionu, jako jsou významně podvyživení či anorektičtí pacienti, při velmi nízkém BMI nebo chroničtí těžcí alkoholici, byly hlášeny případy poruchy funkce až selhání jater. U stavů s deplecí glutathionu, jako je např. sepse, může použití paracetamolu zvyšovat riziko metabolické acidózy.
Přípravek je určen k léčbě dětí, pokud by však byl výjimečně užíván dospělým, je třeba upozornit, že po dobu léčby se nesmějí pít alkoholické nápoje.
Paracetamol může být již v dávkách nad 6 - 8 g denně hepatotoxický. Jaterní poškození se však může vyvinout i při nižších dávkách, pokud spolupůsobí induktory jaterních enzymů nebo jiné hepatotoxické léky.
Přípravek obsahuje směs sodných solí parabenů, které mohou způsobit alergické reakce (pravděpodobně zpožděné).
Přípravek obsahuje roztok maltitolu a nekrystalizující sorbitol 70 % (666,5 mg v 5 ml suspenze). Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Antikoagulační efekt warfarinu nebo jiných kumarinových derivátů může být současným dlouhodobým a pravidelným podáváním paracetamolu zesílený spolu se zvýšeným rizikem krvácení. Občasné užívání nemá signifikantní efekt.
Rychlost absorpce paracetamolu může být zvýšena metoklopramidem nebo domperidonem. Cholestyramin naopak absorpci paracetamolu může snižovat.
Současné dlouhodobé užívání paracetamolu a kyseliny acetylsalicylové nebo dalších nesteroidních protizánětlivých léčivých přípravků může vést k poškození ledvin.
Hepatotoxické látky mohou zvýšit možnost kumulace a předávkování paracetamolem.
Paracetamol zvyšuje plazmatickou hladinu kyseliny acetylsalicylové a chloramfenikolu.
Probenecid ovlivňuje vylučování a koncentraci paracetamolu v plazmě.
Induktory mikrosomálních enzymů (rifampicin či fenobarbital) mohou zvýšit toxicitu paracetamolu v důsledku vzniku vyššího podílu toxického epoxidu při jeho biotransformaci.
Paracetamol může snížit biologickou dostupnost lamotriginu s možným snížením jeho účinku, a to z důvodu možné indukce jeho metabolismu v játrech.
Současné podávání paracetamolu a zidovudinu zvyšuje riziko neutropenie.
Současné podávání paracetamolu a isoniazidu zvyšuje riziko hepatotoxicity.
Žádné interakce klinického významu při občasném použití u dětí však nebyly u tohoto přípravku dosud pozorovány.
4.6 Fertilita, těhotenství a kojení
Přípravek je určen pro použití v pediatrii k léčbě dětí do 12 let, pokud by však byl užíván dospělou ženou, platí následující informace:
Paracetamol prochází placentární bariérou. Studie u zvířat ani u člověka nenaznamenaly žádné riziko paracetamolu s ohledem na těhotenství nebo embryo-fetální vývoj.
Paracetamol je vylučován do mateřského mléka. Studie u člověka nezaznamenaly u paracetamolu žádné riziko s ohledem na kojení nebo pro kojené dítě.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Neuplatňuje se, přípravek je určen k podávání dětem. Paracetamol neovlivňuje pozornost a schopnost soustředění.
4.8 Nežádoucí účinky
Nežádoucí účinky paracetamolu jsou při dodržování terapeutických dávek vzácné.
Nežádoucí účinky z historických dat z klinických studií vycházejí z malých expozicí pacientů a nejsou časté. V souladu s těmito zjištěními, nežádoucí účinky hlášené po uvedení přípravku na trh při použití terapeutických dávek jsou uvedeny v tabulce níže podle orgánových systémů a frekvence jejich výskytu. Frekvence byla odhadnuta ze spontánních hlášení po uvedení na trh.
|
Vyšetření |
vzácné (> 1/10 000 až < 1/1 000) |
zvýšení hladiny kreatininu v séru |
|
Poruchy krve a lymfatického systému |
velmi vzácné (< 1/10 000) |
trombocytopenie, agranulocytóza, leukopenie hemolytická anemie |
|
Respirační, hrudní a mediastinální poruchy |
velmi vzácné (< 1/10 000) |
bronchospasmus (analgetické astma) u predisponovaných pacientů (se senzitivitou i kyselinu acetylsalicylovou nebo jiná NSAID) |
|
Poruchy ledvin a močových c< |
velmi vzácné (< 1/10 000) |
při dlouhodobé léčbě nelze vyloučit možnost poškození ledvin (viz bod 4.4) |
|
Poruchy kůže a podkožní tkár |
vzácné ( > 1/10 000 až < 1/1 000) |
kopřivka |
|
Poruchy imunitního systému |
velmi vzácné (< 1/10 000) |
anafylaxe kožní hypersenzitivní reakce včetně kožní vyrážky, angioedému a závažných kožních reak (SJS, TEN a AGEP) |
|
Poruchy jater a žlučových ces |
vzácné (> 1/10 000 až < 1/1 000) |
zvýšená hladina jaterních aminotransferáz |
|
velmi vzácné (< 1/10 000) |
jaterní dysfunkce |
Hlášení podezření na nežádoucí účinky:
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Předávkování již relativně nízkými dávkami paracetamolu (8 - 15 g v závislosti na tělesné hmotnosti pacienta) může mít za následek závažné poškození jater a někdy akutní renální tubulární nekrózu a selhání jater s potřebou jejich transplantace jater nebo může vést ke smrti.
Do 24 hodin se může objevit nauzea, zvracení, letargie a pocení. Bolest v břiše může být prvním příznakem jaterního poškození a vzniká za 1 - 2 dny. Může vzniknout jaterní selhání, encefalopatie, kóma až smrt. Komplikace selhání jater představuje acidóza, edém mozku, krvácivé projevy, hypoglykémie, hypotenze, infekce a renální selhání. Prodloužení protrombinového času je indikátorem zhoršení funkce jater a proto se doporučuje jeho monitorování. Pacienti, kteří užívají induktory enzymů (karbamazepin, fenytoin, barbituráty, rifampicin) nebo mají abúzus alkoholu v anamnéze, jsou více náchylní k poškození jater. K akutnímu renálnímu selhání může dojít i bez přítomnosti závažného poškození jater. Jinými projevy intoxikace jsou poškození myokardu a pankreatitida.
Léčba předávkování:
V případě předávkování paracetamolem je nezbytná okamžitá lékařská pomoc, i když nejsou přítomny žádné příznaky předávkování.
Je nutná hospitalizace. Vyvolání zvracení, výplach žaludku, zvláště byl-li paracetamol požit před méně než 4 hodinami. Poté je nutné podat methionin (2,5 g p.o.), dále jsou vhodná podpůrná opatření. Podání aktivního uhlí z důvodů snížení gastrointestinální absorpce je sporné. Doporučuje se monitorování plazmatické koncentrace paracetamolu. Specifické antidotum acetylcystein je nutno podat do 8 -15 hodin po otravě, příznivé účinky však byly pozorovány i při pozdějším podání. Acetylcystein se většinou podává dospělým, dospívajícím a dětem i.v. v 5% glukóze v počáteční dávce 150 mg/kg tělesné hmotnosti během 15 minut. Potom 50 mg/kg v infúzi 5 % glukózy po dobu 4 hodin a dále 100 mg/kg do 16. resp. 20. hodiny od zahájení terapie. Acetylcystein lze podat i p.o. do 10 hodin od požití toxické dávky paracetamolu v dávce 70 - 140 mg/kg 3 krát denně. U velmi těžkých otrav je možná hemodialýza či hemoperfúze.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná analgetika a antipyretika, anilidy.
ATC skupina: N02BE01
Paracetamol je analgetikum - antipyretikum bez protizánětlivého účinku a s dobrou gastrointestinální snášenlivostí (nedráždí žaludek). Vhodné u dospělých pacientů i v pediatrii. Mechanismus účinku je pravděpodobně podobný účinku působení kyseliny acetylsalicylové a je závislý na inhibici prostaglandinů primárně v centrálním nervovém systému. Tato inhibice je však selektivní.
Absence periferní inhibice prostaglandinů dává paracetamolu důležité farmakologické vlastnosti, jako je např. zachování protektivních prostaglandinů v gastrointestinálním traktu. Paracetamol je proto vhodný zejména pro pacienty s anamnézou onemocnění nebo užívající současnou medikaci, kde je periferní inhibice prostaglandinů nežádoucí (jako jsou např. pacienti s anamnézou gastrointestinálního krvácení nebo starší pacienti).
Paracetamol neovlivňuje glykémii a je vhodný i u diabetiků. Nemá vliv na hladinu kyseliny močové a její vylučování do moči. Paracetamol lze podat ve všech případech, kde jsou kontraindikovány salicyláty.
5.2 Farmakokinetické vlastnosti Absorpce:
Paracetamol je rychle a téměř úplně vstřebáván z gastrointestinálního traktu. Koncentrace v plasmě dosahuje vrcholu za 15 - 60 minut po podání per os. Biologický poločas v plasmě je 1 - 4 hodiny po terapeutických dávkách. Při závažné jaterní insuficienci dochází k jeho prodloužení až na 5 hodin. Při insuficienci ledvin se poločas neprodlužuje, ale protože vázne vylučování ledvinami je třeba dávky paracetamolu redukovat.
Distribuce:
Paracetamol je relativně rovnoměrně distribuován do většiny tělesných tekutin.
Vazba na plasmatické bílkoviny je při terapeutických koncentracích minimální. Při koncentracích zaznamenaných při akutní intoxikaci však může být vázáno až 20 - 30 %.
Biotransformace:
Paracetamol je metabolizován hlavně v játrech. Dvě hlavní metabolické cesty jsou glukurokonjugace a sulfokonjugace. Poslední cesta je rychle saturována při dávkách vyšších, než jsou terapeutické dávky. Méně významná cesta, katalyzovaná cytochromem P 450, vede k tvorbě intermediárního produktu, který je za normálních podmínek užívání rychle detoxikován redukovaným glutathionem a eliminován močí po konjugaci s cysteinem nebo kyselinou merkapturovou. Během masivního předávkování je množství tohoto toxického metabolitu vždy zvýšeno.
Eliminace:
Exkrece je prakticky výlučně renální ve formě konjugovaných metabolitů (převážně ve formě glukuronidů a suflátů). Po podání terapeutických dávek se během 24 hodin v moči objeví 90 - 100 % podané látky. Ve formě nezměněného paracetamolu se vylučuje méně než 5 % podané dávky.
5.3 Předklinické údaje vztahující se k bezpečnosti přípravku
LD 50 u myši: p.o. 338 mg/kg, i.p. 500 mg/kg.
Předklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity, hodnocení kancerogenního potenciálu a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
karbomer 974 P, xanthanová klovatina, jahodové aroma (obsahuje přírodní a připravované aromatické látky, aromatické látky totožné s přírodními, tokoferol-alfa (E 307), nekrystalizující sorbitol 70 %, roztok maltitolu, sukralosa, draselná sůl acesulfamu, směs sodných solí parabenů (E 215, E 217, E 219), dihydrát dinatrium-edetátu, hydroxid sodný (k úpravě pH), kyselina jablečná (k úpravě pH), čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti 2 roky
Po prvním otevření: 1 rok.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu při teplotě do 30 °C. Chraňte před mrazem.
6.5 Druh obalu a obsah balení
Hnědá PET lahvička s bílým PP dětským bezpečnostním a pojistným uzávěrem s HDPE vložkou a PE vložkou v hrdle lahvičky pro použití dávkovacího aplikátoru, 10 ml pístový dávkovací aplikátor (bílý PE píst a bezbarvá průhledná PP pipeta dělená modře po 0,5 ml), krabička.
Velikost balení: 60 ml, 100 ml, 200 ml.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
GlaxoSmithKline Consumer Healthcare Czech Republic s.r.o.
Hvězdova 1734/2c, 140 00 Praha 4 - Nusle, Česká republika
8. REGISTRAČNÍ ČÍSLO
07/261/92-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18. 3. 1992
Datum posledního prodloužení registrace: 16.3.2016
10. DATUM REVIZE TEXTU
23.3.2016
8/8