Orvatez 10 Mg/40 Mg Potahované Tablety
sp.zn.sukls238030/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Orvatez 10 mg/10 mg potahované tablety Orvatez 10 mg/20 mg potahované tablety Orvatez 10 mg/40 mg potahované tablety Orvatez 10 mg/80 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje ezetimibum 10 mg a atorvastatinum 10, 20, 40 nebo 80 mg (jako atorvastatinum calcicum trihydricum).
Pomocné látky se známým účinkem:
Jedna potahovaná tableta 10 mg/10 mg obsahuje 153 mg laktózy.
Jedna potahovaná tableta 10 mg/20 mg obsahuje 179 mg laktózy.
Jedna potahovaná tableta 10 mg/40 mg obsahuje 230 mg laktózy.
Jedna potahovaná tableta 10 mg/80 mg obsahuje 334 mg laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
Tableta 10 mg/10 mg: bikonvexní, bílá až téměř bílá, potahovaná tableta ve tvaru tobolky, velikosti 12,74 mm x 5,10 mm, na jedné straně s vyraženým „257“
Tableta 10 mg/20 mg: bikonvexní, bílá až téměř bílá, potahovaná tableta ve tvaru tobolky, velikosti 14,48 mm x 5,79 mm, na jedné straně s vyraženým „333“
Tableta 10 mg/40 mg: bikonvexní, bílá až téměř bílá, potahovaná tableta ve tvaru tobolky, velikosti 16,38 mm x 6,27 mm, na jedné straně s vyraženým „337“
Tableta 10 mg/80 mg: bikonvexní, bílá až téměř bílá, potahovaná tableta ve tvaru tobolky, velikosti 19,05 mm x 7,94 mm, na jedné straně s vyraženým „357“
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence kardiovaskulárních příhod
Přípravek Orvatez je indikován ke snížení rizika kardiovaskulárních příhod (viz bod 5.1) u pacientů s ischemickou chorobou srdeční (ICHS) a s anamnézou akutního koronárního syndromu (AKS), bez ohledu na to, zda předtím byli léčeni statinem.
Hypercholesterolemie
Přípravek Orvatez je indikován jako přídatná terapie k dietě u dospělých s primární (heterozygotní familiární a nefamiliární) hypercholesterolemií nebo smíšenou hyperlipidemií, kde je vhodné použití kombinovaného přípravku:
• u pacientů, kteří nejsou samotným statinem dostatečně kontrolováni • u pacientů, kteří se již léčí statinem a ezetimibem.
Homozygotní familiární hypercholesterolemie (HoFH)
Přípravek Orvatez je indikován jako přídatná terapie k dietě u dospělých s HoFH. Pacienti mohou dostávat i další přídatnou terapii (např. aferézu nízkodenzitního lipoproteinu [LDL]).
4.2 Dávkování a způsob podání
Dávkování
Hypercholesterolemie a/nebo ischemická choroba srdeční (s AKS v anamnéze)
Pacient musí být na odpovídající hypolipidemické dietě a v průběhu léčby přípravkem Orvatez musí v dietě pokračovat.
Dávkové rozmezí přípravku Orvatez je 10/10 mg/den až 10/80 mg/den. Obvyklá dávka je 10/10 mg jednu denně. Při zahajování léčby nebo při úpravě dávek je nutno vzít v potaz pacientovu hladinu cholesterolu v lipoproteinech s nízkou hustotou (LDL-C), rizikový status srdeční koronární choroby a odpověď na stávající cholesterol snižující léčbu.
Dávku přípravku Orvatez je nutno individuálně upravit na základě známé účinnosti různých dávkových sil přípravku Orvatez (viz bod 5.1, Tabulka 3) a odpovědi na stávající cholesterol snižující léčbu. Úpravu dávky je nutno provádět v intervalech 4 týdny nebo delších.
Homozygotní familiární hypercholesterolemie
Dávka přípravku Orvatez u pacientů s homozygotní FH je 10/10 až 10/80 mg denně. Přípravek Orvatez lze u těchto pacientů podávat jako přídatnou léčbu k jiným způsobům hypolipidemické léčby (např. LDL aferéza) nebo pokud takové způsoby léčby nejsou k dispozici.
Současné podávání se sekvestranty žlučových kyselin
Přípravek Orvatez je nutno podávat > 2 hodiny před nebo > 4 hodiny po podání sekvestrantu žlučových kyselin.
Starší osoby
U starších pacientů není nutno dávku upravovat (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Orvatez u dětí nebyla stanovena (viz bod 5.2). K dispozici nejsou žádné údaje.
Pacienti s poruchou funkce jater
Přípravek Orvatez se u pacientů s poruchou funkce jater musí podávat opatrně (viz body 4.4 a 5.2). Přípravek Orvatez je u pacientů s aktivním onemocněním jater kontraindikován (viz bod 4.3).
Pacienti s poruchou funkce ledvin
U pacientů s poruchou funkce ledvin není nutno dávku upravovat (viz bod 5.2).
Způsob podání
Přípravek Orvatez se podává perorálně. Přípravek Orvatez lze podat jako jednu dávku kdykoli během dne, s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Léčba přípravkem Orvatez je kontraindikována v těhotenství a při kojení a u žen ve fertilním věku, které nepoužívají vhodnou antikoncepci (viz bod 4.6).
Přípravek Orvatez je kontraindikován u pacientů s aktivním onemocněním jater nebo
při nevysvětlených přetrvávajících zvýšeních sérových transamináz přesahujících 3násobek horního
limitu normálu.
4.4 Zvláštní upozornění a opatření pro použití
Myopatie/rabdomyolýza
V rámci zkušeností s ezetimibem po jeho uvedení na trh byly hlášeny případy myopatie
a rabdomyolýzy. Většina pacientů, u nichž došlo k rozvoji rabdomyolýzy, užívala statin současně s ezetimibem. Rabdomyolýza byla nicméně velmi vzácně hlášena při monoterapii ezetimibem a velmi vzácně při přidání ezetimibu k jiným látkám, o nichž je známo, že jsou spojeny s vyšším rizikem rabdomyolýzy.
Přípravek Orvatez obsahuje atorvastatin. Atorvastatin, stejně jako jiné inhibitory HMG-CoA reduktázy, může ve vzácných případech mít vliv na kosterní svalstvo a způsobovat myalgii, myositidu a myopatii, která může progredovat do rabdomyolýzy, což je potenciálně život ohrožující stav vyznačující se výrazně zvýšenými hladinami kreatinkinázy (CK) (>10násobek horního limitu normálu), myoglobinemií a myoglobinurií, což může vést k selhání ledvin.
Před léčbou
U pacientů s predispozicí k rabdomyolýze je nutno předepisovat přípravek Orvatez opatrně. Hladinu CK je nutno před zahájením léčby změřit v následujících situacích:
• porucha funkce ledvin
• hypothyroidismus
• dědičné svalové poruchy v osobní nebo rodinné anamnéze
• svalová toxicita při užití statinu nebo fibrátu v anamnéze
• jaterní onemocnění a/nebo značná konzumace alkoholu v anamnéze
• starší pacient (věk >70 let), je nutno zvážit nezbytnost takového měření a to na základě přítomnosti dalších faktorů predisponujících k rabdomyolýze
• situace, kde může dojít ke zvýšení plazmatických hladin, jako jsou interakce (viz bod 4.5) a zvláštní populace, včetně genetických subpopulací (viz bod 5.2).
V takových situacích je nutno riziko uvážit ve vztahu k případnému přínosu, přičemž se doporučuje klinické sledování.
Pokud jsou při výchozím vyšetření hladiny CK významně zvýšeny (>5násobek horního limitu normálu), léčba se nemá zahajovat.
Měření kreatinkinázy
Koncentrace kreatinkinázy (CK) se nesmí měřit po namáhavém fyzickém zatížení nebo při jakékoli jiné věrohodné příčině zvýšení CK, protože se tak ztěžuje interpretace hodnot. Pokud jsou koncentrace CK významně zvýšené při výchozím vyšetření (>5násobek horního limitu normálu), je nutno k potvrzení výsledků stanovit koncentrace znovu za 5 až 7 dní.
Během léčby
• Pacienty je nutno požádat, aby neprodleně hlásili svalovou bolest, křeče nebo svalovou slabost, zvláště pokud jsou doprovázeny malátností nebo horečkou.
• Pokud se takové příznaky objeví během léčby přípravkem Orvatez, je nutno u pacienta změřit hladiny CK. Pokud se zjistí, že tyto hladiny jsou významně zvýšeny (>5násobek horního limitu normálu), musí se léčba ukončit.
• Pokud jsou svalové příznaky závažné a způsobují každodenní dyskomfort, i když jsou hladiny CK zvýšeny <5násobek horního limitu normálu, je nutno zvážit vysazení léčby.
• Pokud příznaky vymizí a hladiny CK se vrátí k normálu, lze zvážit opětovné nasazení přípravku Orvatez nebo nasazení jiného přípravku obsahujícího statin v nejnižší dávce a za pečlivého sledování.
• Přípravek Orvatez se musí vysadit, pokud dojde ke klinicky významnému zvýšení hladin CK (>10násobek horního limitu normálu) nebo pokud je diagnostikována rabdomyolýza nebo je na ni podezření.
• Velmi vzácně byl hlášen výskyt imunitně zprostředkované nekrotizující myopatie (IMNM) v průběhu nebo po ukončení léčby některými statiny. Klinicky je IMNM charakterizována perzistentní proximální svalovou slabostí a zvýšením sérové kreatinkinázy, které přetrvává navzdory přerušení léčby statiny.
Kvůli atorvastatinové složce přípravku Orvatez je riziko rabdomyolýzy zvýšeno, pokud se přípravek Orvatez podává současně s jistými léčivými přípravky, které mohou zvyšovat plazmatické koncentrace atorvastatinu, jako jsou silné inhibitory CYP3A4 nebo transportních proteinů (např. cyklosporin, telithromycin, klarithromycin, delavirdin, stiripentol, ketokonazol, vorikonazol, itrakonazol, posakonazol, a inhibitory HIV proteázy včetně ritonaviru, lopinaviru, atazanaviru, indinaviru, darunaviru atd.). Riziko myopatie může rovněž být zvýšeno při současném podávání gemfibrozilu a dalších derivátů kyseliny fibrové, bocepreviru, erythromycinu, niacinu, telapreviru nebo kombinace tipranavir/ritonavir. Pokud je to možné, je nutno místo těchto léčivých přípravků zvážit alternativní (neinteragující) terapie. (Viz bod 4.8.)
V případech, kdy je současné podávání těchto léčivých přípravků s přípravkem Orvatez nezbytné, je nutno pečlivě zvážit přínosy a rizika současného podávání. Pokud pacienti dostávají léčivé přípravky, které zvyšují plazmatické koncentrace atorvastatinu, doporučuje se nižší maximální dávka přípravku Orvatez. V případě silných inhibitorů CYP3A4 je navíc nutno zvážit nižší zahajovací dávku přípravku Orvatez a u těchto pacientů se doporučuje příslušné klinické sledování (viz bod 4.5).
Atorvastatin se nesmí podávat současně se systémovou léčbou kyselinou fusidovou nebo během 7 dnů po ukončení léčby kyselinou fusidovou. U pacientů, u kterých je systémové podání kyseliny fusidové považováno za nezbytné, se musí po dobu léčby kyselinou fusidovou přerušit léčba statinem. Byly hlášeny případy rabdomyolýzy (včetně fatálních) u pacientů užívajících současně kyselinu fusidovou a statiny (viz bod 4.5). Pacienta je třeba poučit, aby ihned vyhledal lékařskou pomoc, pokud se u něj objeví jakékoli příznaky slabosti, bolesti nebo citlivosti svalů.
Léčbu statinem je možné znovu zahájit 7 dní po poslední dávce kyseliny fusidové.
Za výjimečných okolností, kdy je potřebné dlouhodobé systémové podávání kyseliny fusidové, např. při léčbě závažných infekcí, lze v individuálních případech zvážit současné podávání přípravku Orvatez a kyseliny fusidové pod pečlivým lékařským dohledem.
Jaterní enzymy
V kontrolovaných studiích souběžného podávání u pacientů léčených ezetimibem a atorvastatinem byla pozorována po sobě jdoucí zvýšení transamináz (>3násobek horního limitu normálu) (viz bod 4.8.).
Testy jaterních funkcí je nutno provést před zahájením léčby a poté je nutno je provádět pravidelně. Testy jaterních funkcí je nutno provést u pacientů, u kterých se vyvinou jakékoli známky nebo příznaky poukazující na poškození jater. Pacienty, u kterých dojde ke zvýšení hladin transamináz, je nutno sledovat, dokud abnormalita (abnormality) nevymizí. Pokud zvýšení transamináz o více než 3násobek horní hranice normálu přetrvává, doporučuje se snížení dávky nebo vysazení přípravku Orvatez.
Přípravek Orvatez se u pacientů, kteří požívají značná množství alkoholu a/nebo mají v anamnéze onemocnění jater, musí používat opatrně.
Jaterní nedostatečnost
Kvůli neznámému účinku zvýšené expozice ezetimibu se u pacientů se středně závažnou nebo závažnou jaterní nedostatečností přípravek Orvatez nedoporučuje (viz bod 5.2).
Fibrátv
Bezpečnost a účinnost ezetimibu podávaného s fibráty nebyla stanovena, proto se současné podávání přípravku Orvatez a fibrátů nedoporučuje (viz bod 4.5).
Cyklosporin
Při zahajování léčby přípravkem Orvatez u pacientů léčených cyklosporinem je nutno postupovat opatrně. U pacientů léčených přípravkem Orvatez a cyklosporinem je nutno sledovat koncentrace cyklosporinu (viz bod 4.5).
Antikoagulancia
Pokud se přípravek Orvatez přidává k warfarinu, jinému kumarinovému antikoagulanciu nebo fluindionu, je nutno příslušným způsobem sledovat mezinárodní normalizovaný poměr (International Normalised Ratio - INR) (viz bod 4.5).
Prevence cévní mozkové příhody agresivním snížením hladin cholesterolu
V post-hoc analýze subtypů cévní mozkové příhody u pacientů bez ischemické choroby srdeční, kteří nedávno prodělali cévní mozkovou příhodu nebo tranzitorní ischemickou ataku (TIA), byla v porovnání s placebem zjištěna vyšší incidence hemoragické cévní mozkové příhody u pacientů, kteří zahájili léčbu atorvastatinem v dávce 80 mg. Zvýšené riziko bylo zejména patrné u pacientů, kteří při vstupu do studie měli předchozí hemoragickou cévní mozkovou příhodu nebo lakunární infarkt.
U pacientů s předchozí hemoragickou cévní mozkovou příhodou nebo lakunárním infarktem je poměr rizik a přínosů atorvastatinu v dávce 80 mg nejistý, přičemž potenciální riziko hemoragické cévní mozkové příhody je nutno před zahájením léčby pečlivě uvážit (viz bod 5.1).
Intersticiální plicní nemoc
U některých statinů byly hlášeny výjimečné případy intersticiální plicní nemoci, zvláště při dlouhodobé léčbě (viz bod 4.8). Příznaky mohou zahrnovat dušnost, neproduktivní kašel a celkové zhoršení zdravotního stavu (únava, úbytek tělesné hmotnosti a horečka). V případě podezření, že se u pacienta vyvinula intersticiální plicní nemoc, musí být léčba statinem vysazena.
Diabetes mellitus
Některé důkazy naznačují, že statiny zvyšují hladinu glukózy v krvi a u některých pacientů, s rizikem vzniku diabetu, mohou vyvolat hyperglykemii, která již vyžaduje diabetologickou péči. Toto riziko je však převáženo redukcí kardiovaskulárního rizika a není proto důvodem pro ukončení léčby statiny. Ohrožení pacienti (glukóza nalačno 5,6 až 6,9 mmol/l, BMI>30kg/m2, zvýšené triglyceridy v krvi, hypertenze) musí být klinicky a biochemicky monitorováni v souladu s národními doporučeními.
Pomocné látky
Přípravek Orvatez obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s nesnášenlivostí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy-galaktózy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakodynamické interakce
Atorvastatin, což je složka přípravku Orvatez, se metabolizuje prostřednictvím isoenzymu 3A4 cytochromu P450 (CYP3A4) a je substrátem transportních proteinů, např. transportéru OATP1B1 jaterní buňky. Současné podávání léčivých přípravků, které jsou inhibitory CYP3A4 nebo transportních proteinů, může vést ke zvýšeným plazmatickým koncentracím atorvastatinu a ke zvýšenému riziku myopatie. Toto riziko může být rovněž zvýšeno při současném podávání přípravku Orvatez s dalšími léčivými přípravky, které mají potenciál vyvolávat myopatii, jako jsou deriváty kyseliny fibrové a ezetimib (viz bod 4.4).
Farmakokinetické interakce
Přípravek Orvatez
Při současném podávání ezetimibu s atorvastatinem nebyly pozorovány žádné klinicky významné farmakokinetické interakce.
Účinky jiných léčivých přípravků na přípravek Orvatez Ezetimib
Antacida: Současné podávání antacid snížilo rychlost absorpce ezetimibu, nemělo však žádný vliv na biologickou dostupnost ezetimibu. Tato snížená rychlost absorpce se nepovažuje za klinicky významnou.
Cholestyramin: Současné podávání cholestyraminu zmenšilo průměrnou velikost plochy pod křivkou (area under the curve, AUC) celkového ezetimibu (ezetimib + ezetimib glukuronid) přibližně o 55 %. Postupné snížení LDL-C při přidávání přípravku Orvatez k cholestyraminu se může touto interakcí zpomalit (viz bod 4.2).
Cyklosporin: Ve studii s osmi pacienty po transplantaci ledvin s clearance kreatininu > 50 ml/min, kteří byli na stabilní dávce cyklosporinu, vedlo podávání jednotlivé 10mg dávky ezetimibu ke 3,4násobnému (rozmezí 2,3-7,9násobné) zvýšení průměrné AUC celkového ezetimibu ve srovnání se zdravou populací z kontrolní skupiny, která dostávala ezetimib samostatně, z jiné studie (n = 17).
V další studii se u pacienta po transplantaci ledviny se závažnou renální insuficiencí, který dostával cyklosporin a další mnohonásobnou terapii, projevila 12násobně vyšší expozice celkovému ezetimibu ve srovnání se souběžnými kontrolními skupinami, které dostávaly ezetimib samostatně. V crossover studii ve dvou obdobích, která se provedla se 12 zdravými jedinci, vedlo denní podávání ezetimibu v dávce 20 mg po dobu 8 dní spolu s jednorázovým podáním cyklosporinu v dávce 100 mg sedmý den k průměrnému 15% zvětšení AUC cyklosporinu (rozmezí 10% pokles až 51% zvýšení) ve srovnání s jednorázovým podáním 100mg dávky samotného cyklosporinu. Kontrolovaná studie vlivu současného podávání ezetimibu na expozici cyklosporinu u pacientů po transplantaci ledvin nebyla dosud provedena. Při nasazování přípravku Orvatez u pacientů léčených cyklosporinem je nutná opatrnost. U pacientů léčených přípravkem Orvatez a cyklosporinem je nutno sledovat koncentrace cyklosporinu (viz bod 4.4).
Fibráty: Současné podávání fenofibrátu nebo gemfibrozilu zvýšilo koncentrace celkového ezetimibu přibližně 1,5násobně, respektive 1,7násobně. I když se uvedená zvýšení nepovažují za klinicky významná, současné podávání přípravku Orvatez s fibráty se nedoporučuje (viz bod 4.4).
Atorvastatin
Inhibitory CYP3A4: Bylo prokázáno, že silné inhibitory CYP3A4 vedou k výrazně zvýšeným koncentracím atorvastatinu (viz tabulka 1 a specifické informace uvedené dále). Současnému podávání silných inhibitorů CYP3A4 (např. cyklosporinu, telithromycinu, klarithromycinu, delavirdinu, stiripentolu, ketokonazolu, vorikonazolu, itrakonazolu, posakonazolu a inhibitorů HIV proteázy, včetně ritonaviru, lopinaviru, atazanaviru, indinaviru, darunaviru atd.) je nutno se pokud možno vyhnout. V případech, kdy se současnému podávání těchto léčivých přípravků s přípravkem Orvatez vyhnout nelze, je nutno zvážit nižší zahajovací a maximální dávku přípravku Orvatez, přičemž se doporučuje příslušné klinické sledování pacienta (viz tabulka 1).
Středně silné inhibitory CYP3A4 (např. erythromycin, diltiazem, verapamil a flukonazol) mohou zvyšovat plazmatické koncentrace atorvastatinu (viz tabulka 1). Zvýšené riziko myopatie bylo pozorováno při podávání erythromycinu v kombinaci se statiny. Studie interakcí hodnotící účinky amiodaronu nebo verapamilu na atorvastatin nebyly provedeny. Je známo, že jak amiodaron, tak verapamil inhibují aktivitu CYP3A4 a současné podávání přípravku Orvatez tak může vést ke zvýšené expozici atorvastatinu. Pokud se přípravek Orvatez podává současně se středně silnými inhibitory CYP3A4, je proto nutné zvážit nižší maximální dávku přípravku Orvatez a doporučuje se příslušné klinické sledování pacienta. Příslušné klinické sledování se doporučuje po zahájení podávání inhibitoru nebo po úpravě jeho dávky.
Induktory cytochromu P450 3A4: Současné podávání atorvastatinu s induktory cytochromu P450 3A4 (např. efavirenz, rifampicin, třezalka tečkovaná) může vést k různým snížením plazmatických koncentrací atorvastatinu. V důsledku dvojího mechanismu interakce rifampicinu, (indukce
6
cytochromu P450 3A4 a inhibice transportéru OATP1B1 jatemí buňky) se současné podávání přípravku Orvatez s rifampicinem doporučuje, protože opožděné podání atorvastatinu po podání rifampicinu bylo spojeno s významným snížením plazmatických koncentrací atorvastatinu. Vliv rifampicinu na koncentrace atorvastatinu v hepatocytech však není znám a pokud se současnému podání nelze vyhnout, musí být pacienti pečlivě sledováni z hlediska účinnosti.
Inhibitory transportních proteinů: Inhibitory transportních proteinů (např. cyklosporin) mohou zvyšovat systémovou expozici atorvastatinu (viz tabulka 1). Vliv inhibice influxních transportérů jaterní buňky na koncentrace atorvastatinu v hepatocytech není znám. Pokud se současnému podávání nelze vyhnout, doporučuje se snížení dávky přípravku Orvatez a klinické sledování z hlediska účinnosti (viz tabulka 1).
Gemfibrozil/deriváty kyseliny fibrové: Podávání samotných fibrátů je příležitostně spojeno se svalovými příhodami, včetně rabdomyolýzy. Riziko těchto příhod může být současným podáváním derivátů kyseliny fibrové a atorvastatinu zvýšeno.
Ezetimib: Podávání samotného ezetimibu je spojeno se svalovými příhodami, včetně rabdomyolýzy. Riziko těchto příhod tedy může být při současném podávání ezetimibu a atorvastatinu zvýšeno. Doporučuje se vhodné klinické sledování těchto pacientů.
Kolestipol: Plazmatické koncentrace atorvastatinu a jeho aktivních metabolitů byly nižší (přibližně o 25 %), pokud se kolestipol podával současně s atorvastatinem. Nicméně účinky na lipidy byly silnější, pokud se atorvastatin a kolestipol podávaly současně, než pokud se každý z těchto léčivých přípravků podával samostatně.
Kyselina fusidová: Riziko myopatie včetně rabdomyolýzy se při současném systémovém podávání kyseliny fusidové se statiny může zvyšovat. Mechanismus této interakce (zda jde o interakci farmakodynamickou nebo farmakokinetickou nebo obě) není dosud znám. U pacientů léčených touto kombinací byla hlášena rabdomyolýza (včetně několika úmrtí).
Pokud je systémová léčba kyselinou fusidovou nezbytná, musí se po dobu léčby kyselinou fusidovou vysadit léčba atorvastatinem. Viz také bod 4.4.
Kolchicin: I když studie interakcí s atorvastatinem a kolchicinem nebyly provedeny, byly při současném podávání atorvastatinu s kolchicinem hlášeny případy myopatie, a proto při předepisování atorvastatinu s kolchicinem je nutná opatrnost.
Boceprevir: Expozice atorvastatinu byla při podávání s boceprevirem zvýšena. Pokud je potřebné současné podávání s přípravkem Orvatez, je nutno zvážit zahájení nejnižší možnou dávkou přípravku Orvatez s titrací k požadovanému klinickému účinku při sledování bezpečnosti, aniž by se přesáhla denní dávka 10/20 mg. U pacientů, kteří již přípravek Orvatez užívají, nesmí jeho dávka během současného podávání s boceprevirem přesáhnout denní dávku 10/20 mg.
Vliv přípravku Orvatez na farmakokinetiku jiných léčivých přípravků
Ezetimib
V preklinických studiích se ukázalo, že ezetimib neindukuje enzymy cytochromu P450 metabolizující léčivé látky. Nebyly pozorovány žádné klinicky významné farmakokinetické interakce mezi ezetimibem a léčivými látkami, o nichž je známo, že se metabolizují cytochromy P450 1A2, 2D6, 2C8, 2C9 a 3A4 nebo N-acetyltransferázou.
Antikoagulancia: Ve studii s 12 zdravými dospělými muži nemělo současné podávání ezetimibu (10 mg jednou denně) žádný významný vliv na biologickou dostupnost warfarinu a protrombinový čas. Po uvedení přípravku na trh se však objevily zprávy o zvýšených hodnotách International Normalised Ratio (INR) u pacientů, u nichž byl ezetimib přidán k warfarinu nebo fluindionu.
V případech, kdy se přípravek Orvatez přidá k warfarinu, jinému kumarinovému antikoagulanciu nebo fluindionu, je nutno INR řádně sledovat (viz bod 4.4).
Atorvastatin
Digoxin: Při současném podávání opakovaných dávek digoxinu a 10 mg atorvastatinu se lehce zvýšily rovnovážné koncentrace digoxinu. Pacienti užívající digoxin musí být příslušně sledováni.
Perorální kontraceptiva: Současné podávání atorvastatinu s perorálními kontraceptivy vedlo ke zvýšeným plazmatickým koncentracím norethisteronu a ethinylestradiolu.
Warfarin: V klinické studii u pacientů chronicky léčených warfarinem způsobilo během prvních 4 dnů současné podávání atorvastatinu v dávce 80 mg denně mírné prodloužení protrombinového času o asi
1,7 sekundy, který se vrátil k normálu během 15 dnů léčby atorvastatinem. I když byly hlášeny pouze velmi vzácné případy klinicky významných antikoagulačních interakcí, je nutno u pacientů léčených kumarinovými antikoagulancii protrombinový čas stanovit před zahájením léčby přípravkem Orvatez a dále dostatečně často během časné fáze léčby, aby se zajistilo, že nedojde k žádné významné změně protrombinového času. Jakmile je zdokumentován stabilní protrombinový čas, lze jej monitorovat v intervalech obvykle doporučovaných pro pacienty na kumarinových antikoagulanciích. Pokud se dávka přípravku Orvatez změní nebo se přípravek vysadí, je nutno opakovat stejný postup. Léčba atorvastatinem nebyla u pacientů neužívajících antikoagulancia spojena s krvácením ani se změnami protrombinového času.
Tabulka 1
Vliv současně podávaných léčivých přípravků na farmakokinetiku atorvastatinu
|
Současně podávaný léčivý přípravek a dávkovací režim |
Atorvastatin |
Přípravek Orvatez | |
|
Dávka (mg) |
Změna AUC& |
Klinické doporučení | |
|
Tipranavir 500 mg dvakrát denně/ ritonavir 200 mg dvakrát denně, 8 dní (14. až 21. den) |
40 mg 1. den, 10 mg 20. den |
t 9,4krát |
V případech, kdy je současné podávání s přípravkem Orvatez nezbytné, nepřekračujte dávku 10/10 mg přípravku Orvatez denně. Doporučuje se klinické sledování těchto pacientů. |
|
Cyklosporin 5,2 mg/kg/den, stabilní dávka |
10 mg jednou denně 28 dní |
t 8,7krát | |
|
Lopinavir 400 mg dvakrát denně/ ritonavir 100 mg dvakrát denně, 14 dní |
20 mg jednou denně 4 dny |
t 5,9krát |
V případech, kdy je současné podávání s přípravkem Orvatez nezbytné, doporučují se nižší udržovací dávky přípravku Orvatez. Při dávkách přípravku Orvatez přesahujících 10/20 mg se doporučuje klinické sledování těchto pacientů. |
|
Klarithromycin 500 mg dvakrát denně, 9 dní |
80 mg jednou denně 8 dní |
t 4,4krát | |
|
Současně podávaný léčivý přípravek a dávkovací režim |
Atorvastatin |
Přípravek Orvatez | |
|
Dávka (mg) |
Změna AUC& |
Klinické doporučení # | |
|
Sachinavir 400 mg dvakrát denně/ ritonavir 300 mg dvakrát denně od 5. až 7. dne, zvýšeno na 400 mg dvakrát denně 8. den, 5. až 18. den, 30 minut po podání atorvastatinu |
40 mg jednou denně 4 dny |
t 3,9krát |
V případech, kdy je současné podávání s přípravkem Orvatez nezbytné, doporučují se nižší udržovací dávky přípravku Orvatez. Při dávkách přípravku Orvatez přesahujících 10/40 mg se doporučuje klinické sledování těchto pacientů. |
|
Darunavir 300 mg dvakrát denně/ ritonavir 100 mg dvakrát denně, 9 dní |
10 mg jednou denně 4 dny |
t 3,3krát | |
|
Itrakonazol 200 mg jednou denně, 4 dny |
40 mg jedna dávka |
t 3,3krát | |
|
Fosamprenavir 700 mg dvakrát denně/ritonavir 100 mg dvakrát denně, 14 dní |
10 mg jednou denně 4 dny |
t 2,5krát | |
|
Fosamprenavir 1400 mg dvakrát denně, 14 dní |
10 mg jednou denně 4 dny |
t 2,3krát | |
|
Nelfinavir 1250 mg dvakrát denně, 14 dní |
10 mg jednou denně 28 dní |
t 1,7krátA |
Žádné specifické doporučení. |
|
Grapefruitová šťáva, 240 ml jednou denně* |
40 mg jedna dávka |
t 37 % |
Současné požívání velkých množství grapefruitové šťávy a přípravku Orvatez se nedoporučuje. |
|
Diltiazem 240 mg jednou denně, 28 dní |
40 mg jedna dávka |
t 51 % |
Po zahájení léčby nebo po úpravách dávkování diltiazemu se doporučuje příslušné sledování těchto pacientů. |
|
Erythromycin 500 mg čtyřikrát denně, 7 dní |
10 mg jedna dávka |
t 33 %A |
U těchto pacientů se doporučuje nižší maximální dávka a klinické sledování. |
|
Amlodipin 10 mg, jedna dávka |
80 mg jedna dávka |
t 18 % |
v Žádné specifické doporučení. |
|
Cimetidin 300 mg čtyřikrát denně, 2 týdny |
10 mg jednou denně 4 týdny |
i méně než 1 %a |
Žádné specifické doporučení. |
|
Antacidní suspenze hydroxidu hořečnatého a hlinitého, 30 ml čtyřikrát denně, 2 týdny |
10 mg jednou denně 4 týdny |
| 35 %a |
Žádné specifické doporučení. |
|
Efavirenz 600 mg jednou denně, 14 dní |
10 mg 3 dny |
| 41 % |
v Žádné specifické doporučení. |
|
Současně podávaný léčivý přípravek a dávkovací režim |
Atorvastatin |
Přípravek Orvatez | |
|
Dávka (mg) |
Změna AUC& |
Klinické doporučení # | |
|
Rifampicin 600 mg jednou denně, 7 dní (podávaný současně) |
40 mg jedna dávka |
t 30 % |
Pokud se současnému podávání nelze vyhnout, současné podávání přípravku Orvatez s rifampicinem se doporučuje za klinického sledování. |
|
Rifampicin 600 mg jednou denně, 5 dní (dávky odděleny) |
40 mg jedna dávka |
j 80 % | |
|
Gemfibrozil 600 mg dvakrát denně, 7 dní |
40 mg jedna dávka |
t 35 % |
Nedoporučuje se. |
|
Fenofibrát 160 mg jednou denně, 7 dní |
40 mg jedna dávka |
t 3 % |
Nedoporučuje se. |
|
Boceprevir 800 mg třikrát denně, 7 dní |
40 mg jedna dávka |
t 2,3krát |
U těchto pacientů se doporučuje nižší zahajovací dávka a klinické sledování. Dávka přípravku Orvatez nesmí během současného podávání s boceprevirem přesáhnout denní dávku 10/20 mg. |
& Údaje uvedené jako x-násobek změny představují prostý poměr mezi současným podáváním a atorvastatinem samotným (tj. 1násobek = žádná změna). Údaje uváděné jako % změny představují % rozdílu ve vztahu k atorvastatinu samotnému (tj. 0 % = žádná změna).
# Ohledně klinického významu viz body 4.4 a 4.5.
* Obsahuje jednu nebo více složek, které inhibují CYP3A4 a které mohou zvýšit plazmatické koncentrace léčivých přípravků metabolizovaných CYP3A4. Příjem jedné 240ml sklenice grapefruitové šťávy rovněž vedl ke snížení AUC aktivního orthohydroxymetabolitu o 20,4 %. Velká množství grapefruitové šťávy (více než 1,2 litru denně po 5 dní) zvýšila AUC atorvastatinu 2,5krát a AUC aktivních složek (atorvastatin a metabolity).
A Celková aktivita ekvivalentu atorvastatinu
Zvýšení je označeno jako “t”, pokles jako “j”
Tabulka 2
Vliv atorvastatinu na farmakokinetiku současně podávaných léčivých přípravků
|
Atorvastatin a dávkovací režim |
Současně podávaný léčivý přípravek |
Přípravek Orvatez | |
|
Léčivý přípravek/dávka (mg) |
Změna AUC& |
Klinické doporučení | |
|
80 mg jednou denně 10 dní |
Digoxin 0,25 mg jednou denně, 20 dní |
t 15 % |
Pacienti užívající digoxin musí být příslušně sledováni. |
|
40 mg jednou denně 22 dní |
Perorální kontraceptivum jednou denně, 2 měsíce -norethisteron 1 mg -ethinylestradiol 35 pg |
t 28 % t 19 % |
Žádné specifické doporučení. |
|
80 mg jednou denně 15 dní |
* Fenazon, 600 mg jedna dávka |
t 3 % |
Žádné specifické doporučení. |
|
10 mg jednou denně 4 dny |
Fosamprenavir 1400 mg dvakrát denně, 14 dní |
j 27 % |
Žádné specifické doporučení. |
& Údaje uváděné jako % změny představují % rozdílu ve vztahu k atorvastatinu samotnému (tj. 0 % = žádná změna).
* Současné podávání opakovaných dávek atorvastatinu a fenazonu vykázalo nevelký nebo nedetekovatelný vliv na clearance fenazonu.
Zvýšení je označeno jako “t”, pokles jako “j”
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby používat příslušná antikoncepční opatření (viz bod 4.3). Těhotenství
Ateroskleróza je chronický děj a za normálních okolností by vysazení hypolipidemických léčivých přípravků během těhotenství mělo mít minimální dopad na dlouhodobé riziko související s primární hypercholesterolemií.
Přípravek Orvatez
Během těhotenství je přípravek Orvatez kontraindikován (viz bod 4.3). O užívání přípravku Orvatez během těhotenství nejsou k dispozici žádné klinické údaje.
Současné podávání ezetimibu a atorvastatinu březím potkanům naznačilo, že ve skupině s vysokou dávkou ezetimibu/atorvastatinu došlo k na testované látce závislému zvýšení změn skeletu nazývaných “snížená osifikace sterneber”. To může souviset s pozorovaným poklesem tělesných hmotností plodů. U březích králíků byla pozorována nízká incidence deformit skeletu (spojená sternebra, spojené kaudální obratle a asymetrické změny sterneber).
Atorvastatin
Bezpečnost u těhotných žen nebyla stanovena. Kontrolované klinické studie atorvastatinu u těhotných žen nebyly provedeny. Existují vzácná hlášení vrozených anomálií po intrauterinní expozici inhibitorům HMG-CoA reduktázy. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Ezetimib
Ohledně užívání ezetimibu v těhotenství nejsou k dispozici žádné klinické údaje.
Kojení
Přípravek Orvatez je během kojení kontraindikován. Kvůli potenciálu vyvolat závažné nežádoucí účinky nesmějí ženy užívající přípravek Orvatez kojit. Studie na potkanech prokázaly, že se ezetimib vylučuje do mléka. U potkanů jsou plazmatické koncentrace atorvastatinu a jeho aktivních metabolitů podobné jako koncentrace v mléce. Není známo, zda se aktivní složky přípravku Orvatez vylučují do lidského mateřského mléka. (Viz bod 4.3.)
Fertilita
S přípravkem Orvatez nebyly žádné studie fertility provedeny.
Atorvastatin
Ve studiích na zvířatech neměl atorvastatin žádné účinky na samčí ani samičí fertilitu.
Ezetimib
Ezetimib neměl žádný účinek na fertilitu samců ani samic potkanů.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Orvatez má zanedbatelný vliv na schopnost řídit a obsluhovat stroje. Při řízení dopravních prostředků a obsluze strojů je však nutno vzít v potaz, že byla hlášena závrať.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Přípravek Orvatez (nebo současné podávání ezetimibu a atorvastatinu odpovídající přípravku Orvatez) bylo hodnoceno z hlediska bezpečnosti u více než 2400 pacientů v 7 klinických hodnoceních.
Tabulkový seznam nežádoucích účinků
Četnost nežádoucích účinků je definována následujícím způsobem: velmi časté (> 1/10), časté (> 1/100, < 1/10), méně časté (> 1/1 000, < 1/100), vzácné (> 1/10 000, < 1/1 000), velmi vzácné (< 1/10 000).
|
Přípravek Orvatez | ||
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
Infekce a infestace |
chřipka |
Méně časté |
|
Psychiatrické poruchy |
deprese; insomnie; poruchy spánku |
Méně časté |
|
Poruchy nervového systému |
závrať; dysgeuzie; bolest hlavy; parestezie |
Méně časté |
|
Srdeční poruchy |
sinusová bradykardie |
Méně časté |
|
Cévní poruchy |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Časté | |
|
abdominální dyskomfort; abdominální distenze; bolest břicha; bolest v dolní části břicha; bolest v horní části břicha; zácpa; dyspepsie; flatulence; zvýšená peristaltika; gastritida; nauzea; žaludeční dyskomfort |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
akné; urtikarie |
Méně časté |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
myalgie |
Časté |
|
artralgie; bolest zad; svalová únava; svalové spasmy; svalová slabost; bolest v končetinách |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
astenie; únava; malátnost; edém |
Méně časté |
|
Vyšetření |
zvýšení ALT a/nebo AST; zvýšení alkalické fosfatázy; zvýšení CK v krvi; zvýšení gama-glutamyltransferázy; zvýšení jaterních enzymů; abnormální testy jaterních funkcí; nárůst tělesné hmotnosti |
Méně časté |
Laboratorní hodnoty
V kontrolovaných klinických studiích byla u pacientů léčených přípravkem Orvatez incidence klinicky důležitých zvýšení sérových transamináz (ALT a/nebo AST >3násobek horního limitu normálu, opakovaně zjištěno v po sobě jdoucích dnech) 0,6 %. Tato zvýšení byla obecně asymptomatická, nebyla spojena s cholestázou a spontánně nebo po vysazení léčby se vrátila na výchozí hodnoty. (Viz bod 4.4.)
Zkušenosti z podávání po uvedení přípravku na trh a další zkušenosti z klinických studií Následující další nežádoucí účinky byly hlášeny po uvedení přípravku Orvatez na trh nebo v klinických studiích nebo během používání ezetimibu nebo atorvastatinu po uvedení na trh:
Infekce a infestace: nasofaryngitida
Poruchy krve a lymfatického systému: trombocytopenie
Poruchy imunitního systému: hypersenzitivita, včetně anafylaxe, angioedému, vyrážky a kopřivky Poruchy metabolismu a výživy: snížení chuti k jídlu; anorexie; hyperglykemie; hypoglykemie Psychiatrické poruchy: noční můry
Poruchy nervového systému: hypestezie; amnézie; periferní neuropatie Poruchy oka: rozmazané vidění; poruchy vidění Poruchy ucha a labyrintu: tinnitus; ztráta sluchu Cévní poruchy: hypertenze
Respirační, hrudní a mediastinálníporuchy: kašel; faryngolaryngeální bolest; epistaxe
Gastrointestinálníporuchy: pankreatitida; gastro-esofageální refluxní nemoc; říhání; zvracení; sucho v ústech
Poruchy jater a žlučových cest: hepatitida; cholelitiáza; cholecystitida; cholestáza; fatální a nefatální selhání jater
Poruchy kůže a podkožní tkáně: alopecie; kožní vyrážka; pruritus; erythema multiforme; angioneurotický edém; bulózní dermatitida včetně erythema multiforme, Stevens-Johnsonova syndromu a toxické epidermální nekrolýzy
Poruchy svalové a kosterní soustavy a pojivové tkáně: myopatie/rabdomyolýza; bolest šíje; otok kloubů; myositida; imunitně zprostředkovaná nekrotizující myopatie (četnost není známa) (viz bod 4.4)
Poruchy reprodukčního systému a prsu: gynekomastie
Celkové poruchy a reakce v místě aplikace: bolest na hrudi; bolest; periferní edém; pyrexie Vyšetření: pozitivní test na bílé krvinky v moči
Poranění, otravy a procedurální komplikace: tendinopatie, někdy komplikovaná rupturou
U některých statinů byly hlášeny následující nežádoucí příhody:
• sexuální dysfunkce
• výjimečné případy intersticiální plicní choroby, zvláště při dlouhodobé léčbě (viz bod 4.4)
• diabetes mellitus: frekvence výskytu bude záviset na přítomnosti nebo absenci rizikových faktorů (glukóza nalačno > 5,6 mmol/l, BMI > 30 kg/m2, zvýšení triglyceridů, hypertenze v anamnéze)
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www .sukl.cz/nahlasit-nezadouci-ucinek.
4.9 Předávkování
Přípravek Orvatez
Při předávkování je nutno přijmout symptomatická a podpůrná opatření. Je nutno provést jaterní testy a sledovat sérové hladiny CK.
Ezetimib
V klinických studiích bylo podávání ezetimibu v dávce 50 mg/den 15 zdravým jedincům po dobu
až 14 dní nebo v dávce 40 mg/den 18 pacientům s primární hyperlipidemií po dobu až 56 dní celkově dobře snášeno. Bylo popsáno několik případů předávkování; většina z nich nebyla spojena s nežádoucími účinky. Popsané nežádoucí účinky nebyly závažné. U zvířat nebyla pozorována toxicita po jednotlivých perorálně podaných dávkách 5 000 mg/kg ezetimibu potkanům a myším a 3 000 mg/kg ezetimibu psům.
Atorvastatin
V důsledku rozsáhlé vazby atorvastatinu na plazmatické proteiny se nepředpokládá, že by hemodialýza významně zvýšila clearance atorvastatinu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické účinky
Farmakoterapeutická skupina: inhibitory HMG-CoA reduktázy v kombinaci s jinými látkami upravující hladiny lipidů, ATC kód: C10BA05
Přípravek Orvatez (ezetimib/atorvastatin) je hypolipidemický léčivý přípravek, který selektivně inhibuje střevní absorpci cholesterolu a příbuzných rostlinných sterolů a inhibuje endogenní syntézu cholesterolu.
Mechanismus účinku
Přípravek Orvatez
Plazmatický cholesterol vzniká intestinální absorpcí a endogenní syntézou. Přípravek Orvatez obsahuje ezetimib a atorvastatin, dvě hypolipidemické látky se vzájemně se doplňujícími mechanismy účinku. Přípravek Orvatez snižuje zvýšený celkový cholesterol (total-C), LDL-C, apolipoprotein B (Apo B), triglyceridy (TG) a cholesterol nevysokodenzitních lipoproteinů (non-HDL-C) a zvyšuje cholesterol vysokodenzitních lipoproteinů (HDL-C) dvojitou inhibicí absorpce a syntézy cholesterolu.
Ezetimib
Ezetimib inhibuje intestinální absorpci cholesterolu. Ezetimib je účinný po perorálním podání a má mechanismus účinku, který se liší od jiných skupin látek snižujících cholesterol (např. statinů, sekvestrantů žlučových kyselin [pryskyřic], derivátů kyseliny fibrové a rostlinných sterolů). Molekulárním cílem ezetimibu je sterolový přenašeč Niemann-Pick C1 Like 1 (NPC1L1), který je odpovědný za intestinální příjem cholesterolu a fytosterolů.
Ezetimib je lokalizován na kartáčovém lemu tenkého střeva a blokuje absorpci cholesterolu; výsledkem je snížený odvod intestinálního cholesterolu do jater; statiny snižují syntézu cholesterolu v játrech a společně tyto rozdílné mechanismy zajišťují komplementární snižování cholesterolu.
Ve 2týdenní klinické studii s 18 pacienty s hypercholesterolemií inhiboval ezetimib intestinální absorpci cholesterolu o 54 % ve srovnání s placebem.
Byla provedena řada preklinických studií s cílem stanovit selektivitu ezetimibu při blokádě absorpce cholesterolu. Ezetimib inhiboval vstřebávání [14C]-cholesterolu bez vlivu na absorpci triglyceridů, mastných kyselin, žlučových kyselin, progesteronu, ethinylestradiolu nebo v tucích rozpustných vitamínů A a D.
Atorvastatin
Atorvastatin je selektivní, kompetitivní inhibitor HMG-CoA reduktázy, což je rychlost limitující enzym odpovědný za konverzi 3-hydroxy-3-methyl-glutaryl-koenzymu A na mevalonát, což je prekurzor sterolů, včetně cholesterolu. Triglyceridy a cholesterol jsou v játrech inkorporovány do lipoproteinů o velmi nízké hustotě (VLDL) a uvolňovány do plazmy k dodávce do periferních tkání. Lipoprotein o nízké hustotě (LDL) se vytváří z VLDL a je katabolizován hlavně přes receptor s vysokou afinitou k LDL (LDL receptor).
Atorvastatin snižuje plazmatický cholesterol a koncentrace lipoproteinů v séru inhibicí HMG-CoA reduktázy a následné biosyntézy cholesterolu v játrech a zvyšuje počty jaterních LDL receptorů na buněčném povrchu k zesílení příjmu a katabolismu LDL.
Atorvastatin snižuje tvorbu a počty částic LDL. Atorvastatin navozuje silný a setrvalý vzestup aktivity LDL receptorů spojený s příznivou změnou v kvalitě cirkulujících LDL částic. Atorvastatin je účinný při snižování LDL-C u pacientů s homozygotní familiární hypercholesterolemií, což je populace, která obvykle nereagovala na hypolipidemické léčivé přípravky.
Ve studii odpovědi na dávku bylo prokázáno, že atorvastatin snižuje koncentrace celkového cholesterolu (30 až 46 %), LDL-C (41 až 61 %), apolipoproteinu B (34 až 50 %) a triglyceridů (14 až 33 %), přičemž navozuje různá zvýšení HDL-C a apolipoproteinu A1. Tyto výsledky jsou konzistentní u pacientů s heterozygotní familiární hypercholesterolemií, nefamiliárními formami hypercholesterolemie a smíšenou hyperlipidemií, včetně pacientů s diabetes mellitus typu 2.
Klinická účinnost a bezpečnost
V kontrolovaných klinických studiích u pacientů s hypercholesterolemií snižoval přípravek Orvatez významně celkový cholesterol, LDL-C, Apo B a triglyceridy a zvyšoval HDL-C.
Primární hypercholesterolemie
V placebem kontrolované studii bylo 628 pacientů s hyperlipidemií randomizováno do skupin léčených placebem, ezetimibem (10 mg), atorvastatinem (10 mg, 20 mg, 40 mg nebo 80 mg) nebo do skupiny, jíž se současně podával ezetimib a atorvastatin v dávkách odpovídajících přípravku Orvatez (10/10, 10/20, 10/40 a 10/80) po dobu až 12 týdnů.
Pacienti, kteří dostávali všechny dávky přípravku Orvatez, byli srovnáváni s pacienty, kteří dostávali všechny dávky atorvastatinu. Přípravek Orvatez snižoval celkový cholesterol, LDL-C, Apo B, triglyceridy a non-HDL-C, a zvyšoval HDL-C významně více, než atorvastatin samotný. (Viz tabulka 3.)
Tabulka 3
Odpověď na přípravek Orvatez u pacientů s primární hyperlipidemií (střední hodnotaa % změny z neléčených výchozích hodnotb po 12 týdnech)
|
Léčba (denní dávka) |
N |
Celkový C |
LDL-C |
Apo B |
TGa |
HDL-C |
Non-HDL-C |
|
Souhrnná data (všechny dávky přípravku Orvatez)c |
255 |
-41 |
-56 |
-45 |
-33 |
+7 |
-52 |
|
Souhrnná data (všechny dávky atorvastatinu)c |
248 |
-32 |
-44 |
-36 |
-24 |
+4 |
-41 |
|
Ezetimib 10 mg |
65 |
-14 |
-20 |
-15 |
-5 |
+4 |
-18 |
|
Placebo |
60 |
+4 |
+4 |
+3 |
-6 |
+4 |
+4 |
|
Přípravek Orvatez podle dávky 10/10 |
65 |
-38 |
-53 |
-43 |
-31 |
+9 |
-49 |
|
10/20 |
62 |
-39 |
-54 |
-44 |
-30 |
+9 |
-50 |
|
10/40 |
65 |
-42 |
-56 |
-45 |
-34 |
+5 |
-52 |
|
10/80 |
63 |
-46 |
-61 |
-50 |
-40 |
+7 |
-58 |
|
Atorvastatin podle dávky | |||||||
|
10 mg |
60 |
-26 |
-37 |
-28 |
-21 |
+6 |
-34 |
|
20 mg |
60 |
-30 |
-42 |
-34 |
-23 |
+4 |
-39 |
|
40 mg |
66 |
-32 |
-45 |
-37 |
-24 |
+4 |
-41 |
|
80 mg |
62 |
-40 |
-54 |
-46 |
-31 |
+3 |
-51 |
a Pro triglyceridy, medián % změny výchozích hodnot b Výchozí hodnoty - bez léčby hypolipidemickým léčivým přípravkem c Přípravek Orvatez souhrnně (10/10 až 10/80 mg) významně snižoval celkový C,
LDL-C, Apo B, TG, non-HDL-C, a významně zvyšoval HDL-C v porovnání se všemi dávkami atorvastatinu souhrnně (10 až 80 mg).
V kontrolované studii, s názvem Titration of Atorvastatin Versus Ezetimibe Add-On to Atorvastatin in Patients with Hypercholesterolaemia (TEMPO), dostávalo 184 pacientů s hladinou LDL-C >2,6 mmol/l a <4,1 mmol/l a se středně vysokým rizikem ischemické choroby srdeční atorvastatin v dávce 20 mg po nejméně 4 týdny před randomizací. Pacienti, kteří neměli hladinu LDL-C <2,6 mmol/l, byli randomizováni buď do skupiny, které se podával současně ezetimib a atorvastatin (odpovídající přípravku Orvatez 10/20) nebo atorvastatin v dávce 40 mg po dobu 6 týdnů.
Přípravek Orvatez 10/20 byl při dalším snižování celkového cholesterolu významně účinnější, než zdvojení dávky atorvastatinu na 40 mg (-20 % vs. -7 %), LDL-C (-31 % vs. -11 %), Apo B (-21 % vs. -8 %), a non-HDL-C (-27 % vs. -10 %). Výsledky HDL-C a triglyceridů nebyly mezi těmito dvěma skupinami významně odlišné. Rovněž významně více pacientů léčených přípravkem Orvatez 10/20 dosáhlo LDL-C <2,6 mmol/l v porovnání s pacienty léčenými atorvastatinem v dávce 40 mg, 84 % vs. 49 %.
V kontrolované studii, s názvem The Ezetimibe Plus Atorvastatin Versus Atorvastatin Titration in Achieving Lower LDL-C Targets in Hypercholesterolaemic Pacients (EZ-PATH), dostávalo 556 pacientů s vysokým kardiovaskulárním rizikem s hladinou LDL-C >1,8 mmol/l a <4,1 mmol/l atorvastatin v dávce 40 mg po dobu nejméně 4 týdnů před randomizací. Pacienti, kteří neměli hladinu LDL-C <1,8 mmol/l, byli randomizováni buď do skupiny, které se podával současně ezetimib a atorvastatin (odpovídající přípravku Orvatez 10/40), nebo atorvastatin v dávce 80 mg po dobu 6 týdnů.
Přípravek Orvatez 10/40 byl při dalším snižování celkového cholesterolu významně účinnější, než zdvojení dávky atorvastatinu na 80 mg (-17 % vs. -7 %), LDL-C (-27 % vs. -11 %), Apo B (-18 % vs. -8 %), triglyceridů (-12 % vs. -6 %) a non-HDL-C (-23 % vs. -9 %). Výsledky HDL-C nebyly mezi těmito dvěma skupinami významně odlišné. Rovněž významně více pacientů léčených přípravkem
Orvatez 10/40 dosáhlo LDL-C <1,8 mmol/l v porovnání s pacienty léčenými atorvastatinem v dávce 80 mg, 74 % vs. 32 %.
V placebem kontrolované, 8týdenní studii bylo randomizováno 308 pacientů s hypercholesterolemií léčených atorvastatinem a kteří nedosahovali cíle ohledně LDL-C v rámci programu National Cholesterol Education Program (NCEP) (cíl ohledně LDL-C založen na výchozích hodnotách LDL-C a rizikovém statutu ischemické choroby srdeční) do skupin léčených buď ezetimibem v dávce 10 mg nebo placebem vedle již probíhající léčby atorvastatinem.
Mezi pacienty, kteří na začátku nedosahovali cíle ohledně LDL-C (přibližně 83 %), významně více pacientů léčených ezetimibem spolu s atorvastatinem dosáhlo cíle ohledně LDL-C v porovnání s pacienty léčenými placebem spolu s atorvastatinem, 67 % vs. 19 %. Ezetimib přidaný k léčbě atorvastatinem snižoval LDL-C významně více než placebo přidané k léčbě atorvastatinem, 25 % vs.
4 %. Ezetimib přidaný k léčbě atorvastatinem rovněž významně snižoval celkový cholesterol, Apo B a triglyceridy v porovnání s placebem přidaným k léčbě atorvastatinem.
V kontrolované, 12týdenní, 2fázové studii bylo randomizováno 1539 pacientů s vysokým kardiovaskulárním rizikem s hladinou LDL-C mezi 2,6 a 4,1 mmol/l , léčených atorvastatinem v dávce 10 mg denně do skupin, které dostávali: atorvastatin 20 mg, rosuvastatin 10 mg nebo přípravek Orvatez 10/10. Po 6 týdnech léčby (fáze I) byli pacienti užívající atorvastatin v dávce 20 mg, kteří nedosáhli hladiny LDL-C <2,6 mmol/l, převedeni buď na atorvastatin v dávce 40 mg nebo přípravek Orvatez 10/20 na dobu 6 týdnů (fáze II), a podobně pacienti užívající rosuvastatin v dávce 10 mg během fáze I byli převedeni buď na rosuvastatin v dávce 20 mg, nebo přípravek Orvatez 10/20.
Snížení LDL-C a srovnání mezi skupinou léčenou přípravkem Orvatez a skupinami s jinou léčbou jsou uvedeny v tabulce 4.
Tabulka 4
Odpověď na přípravek Orvatez1 2 3 u pacientů s vysokým rizikem a s hladinou LDL-C mezi 2,6 a 4,1 mmol/l léčených na začátku atorvastatinem v dávce 10 mg denně
|
Léčba |
N |
Procento změn |
y výchozích hodnot1. | ||||
|
Celkový C |
LDL-C |
Apo B |
TG1 |
HDL-C |
Non-HDL-C | ||
|
Fáze I Přechod z atorvastatinu 10 mg Orvatez 10/10 |
120 |
-13,5 |
-22,2 |
-11,3 |
-6,0 |
+0,6 |
-18,3 |
|
Atorvastatin 20 mg |
480 |
-6,4§ |
-9,54 |
-6,01 |
-3,9 |
-1,1 |
-8,1§ |
|
Rosuvastatin 10 mg |
939 |
-7,7§ |
-13,0§ |
-6,9# |
-1,1 |
+1,1 |
-10,6§ |
|
Fáze II Přechod z atorvastatinu 20 mg Orvatez 10/20 |
124 |
-10,7 |
-17,4 |
-9,8 |
-5,9 |
+0,7 |
-15,1 |
|
Atorvastatin 40 mg |
124 |
-3,8 Ě |
-6,9 Ě |
-5,4 |
-3,1 |
+1,7 |
-5,8 Ě |
|
Přechod z rosuvastatinu 10 mg Orvatez 10/20 |
231 |
-11,8 |
-17,1 |
-11,9 |
-10,2 |
+0,1 |
-16,2 |
|
Rosuvastatin 20 mg |
205 |
-4,5Ě |
-7,5 B |
-4,1Ě |
-3,2b |
+0,8 |
-6,4 B |
^ p<0,01 versus přípravek Orvatez 10/10 # p<0,05 versus přípravek Orvatez 10/10 Ě p<0,001 versus přípravek Orvatez 10/20 B p<0,05 versus přípravek Orvatez 10/20
Tabulka 4 nezahrnuje údaje srovnávající účinky přípravku Orvatez 10/10 nebo 10/20 s dávkami vyššími než atorvastatin v dávce 40 mg nebo rosuvastatin v dávce 20 mg.
V placebem kontrolované studii, s názvem Myocardial Ischaemia Reduction with Aggressive Cholesterol Lowering (MIRACL), byli pacienti s akutním koronárním syndromem (non-Q infarkt myokardu nebo nestabilní angina pectoris) randomizováni do skupiny léčené buď atorvastatinem
v dávce 80 mg/den (n = 1538) nebo placebem (n = 1548). Léčba byla zahájena během akutní fáze po přijetí do nemocnice a trvala 16 týdnů. Atorvastatin v dávce 80 mg/den poskytl 16% (p=0,048) snížení rizika kombinovaného primárního kritéria hodnocení: úmrtí z jakýchkoli příčin, nefatální infarkt myokardu, resuscitovaná srdeční zástava nebo angina pectoris s prokázanou srdeční ischemií vyžadující hospitalizaci. K tomu došlo zejména díky 26% snížení opakovaných hospitalizací kvůli angině pectoris s prokázanou srdeční ischemií (p = 0,018).
Přípravek Orvatez obsahuje atorvastatin. V placebem kontrolované studii, nazvané Anglo-Scandinavian Cardiac Outcomes Trial Lipid Lowering Arm (ASCOT-LLA), byly účinky atorvastatinu v dávce 10 mg na fatální a nefatální ischemickou chorobu srdeční hodnoceny u 10 305 hypertenzních pacientů ve věku 40 až 80 let s hladinami celkového cholestrolu <6,5 mmol/l a nejméně třemi kardiovaskulárními rizikovými faktory. Pacienti byli sledováni po medián doby 3,3 roku. Atorvastatin v dávce 10 mg významně (p<0,001) snižoval relativní riziko: fatální ischemické choroby srdeční plus nefatálního infarktu myokardu o 36 % (absolutní snížení rizika = 1,1 %); celkových kardiovaskulárních příhod a revaskularizačních procedur o 20 % (absolutní snížení rizika = 1,9 %) a celkových koronárních příhod o 29 % (absolutní snížení rizika = 1,4 %).
V placebem kontrolované studii, nazvané Collaborative Atorvastatin Diabetes Study (CARDS), byly účinky atorvastatinu v dávce 10 mg na cílové parametry kardiovaskulární choroby hodnoceny
u 2838 pacientů ve věku 40 až 75 let s diabetem typu 2, jedním nebo více kardiovaskulárními rizikovými faktory, LDL <4,1 mmol/l a triglyceridy <6,8 mmol/l. Pacienti byli sledováni po medián doby 3,9 roku. Atorvastatin v dávce 10 mg významně (p<0,05) snižoval: výskyt velkých kardiovaskulárních příhod o 37 % (absolutní snížení rizika = 3,2 %); riziko cévní mozkové příhody o 48 % (absolutní snížení rizika = 1,3 %) a riziko srdeční ischemie o 42 % (absolutní snížení rizika =
1,9 %).
Prevence kardiovaskulárních příhod
Do multicentrické randomizované dvojitě zaslepené a aktivním komparátorem kontrolované studie léčby kombinací ezetimib/simvastatin bylo zařazeno 18 144 pacientů během 10 dnů po hospitalizaci kvůli akutnímu koronárnímu syndromu (AKS; buď infarktu myokardu, nebo nestabilní angině pectoris). Všichni pacienti byli v poměru 1:1 randomizováni do skupiny léčené ezetimibem/simvastatinem 10/40 mg (n = 9 067) nebo simvastatinem 40 mg (n = 9 077) a sledováni po medián doby sledování 6,0 roku.
Průměrný věk pacientů byl 63,6 roku, 76 % tvořili muži, 84 % byli běloši a 27 % pacientů mělo diabetes mellitus. Průměrná hodnota LDL-C při příhodě, která byla kvalifikující pro zařazení do studie, byla 2,1 mmol/l (80 mg/dl) u pacientů, kteří dostávali hypolipidemickou léčbu (n = 6 390), a 2,6 mmol/l (101 mg/dl) u těch, kteří předtím nedostávali hypolipidemickou léčbu (n = 11 594). Před hospitalizací kvůli příhodě AKS kvalifikující ke vstupu do studie užívalo 34 % pacientů statin. Po jednom roce byla průměrná hodnota LDL-C u pacientů, kteří pokračovali v léčbě, 1,4 mmol/l (53,2 mg/dl) ve skupině s ezetimibem/simvastatinem a 1,8 mmol/l (69,9 mg/dl) ve skupině se samotným simvastatinem.
Primárním cílovým ukazatelem byl složený ukazatel zahrnující úmrtí z kardiovaskulárních příčin, velké koronární příhody (definované jako nefatální infarkt myokardu, popsaná nestabilní angina pectoris vyžadující hospitalizaci, nebo jakákoli koronární revaskularizace prováděná nejméně 30 dní po randomizaci) a nefatální cévní mozkovou příhodu. Studie prokázala, že léčba
18
ezetimibem/simvastatinem ve srovnání se samotným simvastatinem poskytuje rostoucí přínos ve formě snížení výskytu primárního cílového ukazatele složeného z úmrtí z kardiovaskulárních příčin, velké koronární příhody a nefatální cévní mozkové příhody (relativní snížení rizika je 6,4 %, p = 0,016). Primární cílový ukazatel se vyskytl u 2 572 z 9 067 pacientů ze skupiny s ezetimibem/simvastatinem (pravděpodobnost výskytu za 7 let byla dle Kaplan-Meierovy (KM) metody 32,72 %) a u 2 742 z 9 077 pacientů ze skupiny se samotným simvastatinem (pravděpodobnost výskytu za 7 let byla dle KM metody 34,67 %). (Viz Graf 1 a Tabulka 5.) Předpokládá se, že podobný rostoucí přínos poskytuje také kombinace ezetimibu a atorvastatinu. Celková úmrtnost se v této vysoce rizikové skupině nezměnila.
Ve studii byl pozorován celkový přínos pro všechny typy cévní mozkové příhody, nicméně bylo zaznamenáno malé nesignifikantní zvýšení rizika u hemoragické cévní mozkové příhody ve skupině s ezetimibem a simvastatinem ve srovnání se skupinou se samotným simvastatinem. Riziko hemoragické cévní mozkové příhody pro ezetimib současně podávaný se statiny s vyšší účinností nebylo hodnoceno v dlouhodobých studiích.
Léčebný účinek kombinace ezetimib/simvastatin byl obecně konzistentní napříč celkovými výsledky mnoha podskupin, dělených podle pohlaví, věku, rasy, diabetu mellitus v anamnéze, počáteční hladiny lipidů, předchozí léčby statiny, cévní mozkové příhody v anamnéze a hypertenze.
Graf 1: Účinek ezetimibu/simvastatinu na primární cílový ukazatel složený z úmrtí z kardiovaskulárních příčin, velké koronární příhody nebo nefatální cévní mozkové příhody
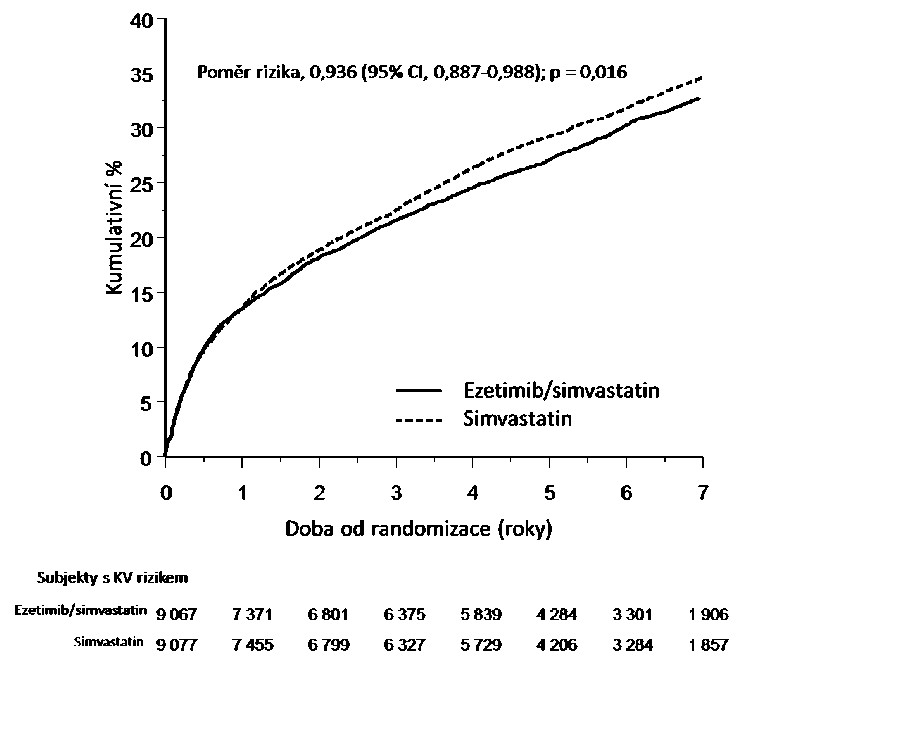
Tabulka 5: Velké kardiovaskulární příhody podle skupin u všech pacientů randomizovaných
v IMPROVE-IT
|
Výsledek |
Ezetimib/simvastatin 10/40 mga (n = 9 067) |
Simvastatin 40 mgb (n = 9 077) |
Poměr rizika (95% CI) |
p- hodnota | |
|
n K-M %c |
n |
K-M % c | |||
|
Primární složený cílový ukazatel účinnosti (Úmrtí z kardiovaskulárních příčin, velké koronární příhody a nefatální cévní mozková příhoda) |
2 572 32,72 % |
2 742 |
34,67 % |
0,936 (0,887; 0,988) |
0,016 |
|
Složky primárního složeného cílového ukazatele a vybrané cílové ukazatele účinnosti (první výskyt dané příhody |
v jakékoli | ||||
|
chvíli) | |||||
|
Úmrtí z kardiovaskulárních příčin |
537 6,89 % |
538 |
6,84 % |
1,000 (0,887; 1,127) |
0,997 |
|
Velká koronární příhoda: | |||||
|
nefatální infarkt myokardu |
945 12,77 % |
1 083 |
14,41 % |
0,871 (0,798; 0,950) |
0,002 |
|
nestabilní angina pectoris vyžadující hospitalizaci |
156 2,06 % |
148 |
1,92 % |
1,059 (0,846; 1,326) |
0,618 |
|
koronární revaskularizace po 30 dnech |
1 690 21,84 % |
1 793 |
23,36 % |
0,947 (0,886; 1,012) |
0,107 |
|
Nefatální cévní mozková příhoda |
245 3,49 % |
305 |
4,24 % |
0,802 (0,678; 0,949) |
0,010 |
a 6 % pacientů bylo titrováno na ezetimib/simvastatin 10/80 mg. b 27 % pacientů bylo titrováno na simvastatin 80 mg. c Kaplan-Meierův odhad pro 7 let.
Homozygotní familiární hypercholesterolemie (HoFH)
U pacientů s klinickou a/nebo genotypovou diagnózou HoFH byla provedena dvojitě zaslepená, randomizovaná 12týdenní studie. Údaje byly analyzovány z podskupiny pacientů (n = 36) léčených při zařazení do studie atorvastatinem v dávce 40 mg. Zvýšení dávky atorvastatinu ze 40 na 80 mg (n = 12) vedlo ke snížení LDL-C o 2 % z výchozích hodnot při podávání atorvastatinu v dávce 40 mg. Současně podávaný ezetimib a atorvastatin ekvivalentní přípravku Orvatez (10/40 a 10/80 souhrnně, n = 24) navodil snížení LDL-C o 19 % z výchozích hodnot při podávání atorvastatinu v dávce 40 mg. U těch pacientů, kteří dostávali současně ezetimib a atorvastatin odpovídající přípravku Orvatez (10/80, n = 12) bylo navozeno snížení LDL-C o 25 % z výchozích hodnot při podávání atorvastatinu v dávce 40 mg.
Po dokončení této 12týdenní studie byli vhodní pacienti (n = 35), kteří dostávali při zařazení atorvastatin v dávce 40 mg, zařazeni do skupiny, které se podával ezetimib a atorvastatin ekvivalentní přípravku Orvatez 10/40 po dobu až dalších 24 měsíců. Po nejméně 4 týdnech léčby mohla být dávka atorvastatinu zdvojena na maximální dávku 80 mg. Po uplynutí 24 měsíců přípravek Orvatez (10/40 a 10/80 souhrnně) navodil snížení LDL-C, které bylo konzistentní se snížením pozorovaným ve 12týdenní studii.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Orvatez u všech podskupin pediatrické populace při léčbě hypercholesterolemie a smíšené hyperlipidemie (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Přípravek Orvatez
Bylo prokázáno, že přípravek Orvatez je bioekvivalentní souběžnému podávání odpovídajících dávek ezetimibu a atorvastatinu v tabletách.
Absorpce
Přípravek Orvatez
Účinky vysoce tučné stravy na farmakokinetiku ezetimibu a atorvastatinu, pokud se podávají jako tablety přípravku Orvatez, jsou srovnatelné s účinky hlášenými pro jednotlivé tablety.
Ezetimib
Po perorálním podání se ezetimib rychle vstřebává a dochází k rozsáhlé konjugaci na farmakologicky aktivní fenolový glukuronid (ezetimib-glukuronid). Průměrných maximálních plazmatických koncentrací (Cmax) je dosaženo během 1 až 2 hodin u ezetimib-glukuronidu a 4 až 12 hodin u ezetimibu. Absolutní biologickou dostupnost ezetimibu nelze stanovit, protože uvedená látka je ve vodných médiích vhodných pro injekční podání prakticky nerozpustná.
Při perorálním ezetimibu ve formě 10mg tablet neměla současná konzumace jídla (s vysokým obsahem tuku a bez obsahu tuku) na biologickou dostupnost ezetimibu žádný vliv.
Atorvastatin
Atorvastatin se po perorálním podání rychle absorbuje; maximální plazmatické koncentrace (Cmax) je dosaženo za 1 až 2 hodiny. Rozsah absorpce se zvyšuje v poměru k dávce atorvastatinu. Po perorálním podání je atorvastatin v potahovaných tabletách dostupný z 95 až 99 % v porovnání s perorálním roztokem. Absolutní biologická dostupnost atorvastatinu je přibližně 12 % a systémová dostupnost inhibiční aktivity na HMG-CoA reduktázu je přibližně 30 %. Nízká systémová dostupnost se přisuzuje presystémové clearance ve sliznici gastrointestinálního traktu a/nebo metabolismu při prvním průchodu játry.
Distribuce
Ezetimib
Ezetimib a ezetimib-glukuronid jsou z 99,7 %, respektive z 88 až 92 % vázány na plazmatické bílkoviny.
Atorvastatin
Střední hodnota distribučního objemu atorvastatinu je přibližně 381 litrů. Atorvastatin je z >98 % vázán na plazmatické proteiny.
Biotransformace
Ezetimib
Ezetimib je metabolizován převážně v tenkém střevě a v játrech cestou glukuronidace (reakce II. fáze) s následným vyloučením žlučí. Minimální oxidační metabolismus (reakce I. fáze) byl pozorován u všech hodnocených živočišných druhů. Hlavními látkami vznikajícími z léčivé látky a přítomnými v plazmě jsou ezetimib a ezetimib-glukuronid, představující přibližně 10 až 20 %, respektive 80 až 90 % z celkového obsahu léčivé látky v plazmě. Ezetimib i ezetimib-glukuronid se pozvolna vylučují z plazmy s prokazatelně významným enterohepatálním oběhem. Poločas ezetimibu i ezetimib-glukuronidu je přibližně 22 hodin.
Atorvastatin
Atorvastatin se metabolizuje cytochromem P450 3A4 na ortho- a parahydroxylované deriváty a různé betaoxidační produkty. Kromě jiných cest jsou tyto produkty dále metabolizovány glukuronidací. In vitro je inhibice HMG-CoA reduktázy ortho- a parahydroxylovanými metabolity ekvivalentní inhibici navozené atorvastatinem. Přibližně 70 % obíhající inhibiční aktivity vůči HMG-CoA reduktáze se přisuzuje aktivním metabolitům.
Eliminace
Ezetimib
Po perorálním podání 14C-ezetimibu (20 mg) lidem představoval celkový ezetimib přibližně 93 % celkové radioaktivity v plazmě. Přibližně 78 % aplikované radioaktivity bylo v průběhu 10denního sběrného období zjištěno ve stolici a 11 % v moči. Po 48 hodinách nebyly v plazmě přítomny zjistitelné koncentrace radioaktivity.
Atorvastatin
Atorvastatin se eliminuje hlavně žlučí po jatemí a/nebo extrahepatální metabolizaci. Zdá se však, že léčivý přípravek nepodstupuje významný enterohepatální oběh. Střední hodnota plazmatického eliminačního poločasu u lidí je přibližně 14 hodin. Poločas inhibiční aktivity vůči HMG-CoA reduktáze je přibližně 20 až 30 hodin v důsledku příspěvku aktivních metabolitů.
Pediatrická populace
Ezetimib
Vstřebávání a metabolismus ezetimibu u dětí a dospívajících (10 až 18 let) a dospělých jsou si podobné. Na základě celkového ezetimibu neexistují mezi dospívajícími a dospělými rozdíly ve farmakokinetice. Farmakokinetické údaje pro dětskou populaci ve věku < 10 let nejsou k dispozici. Zkušenosti z klinické praxe s dětmi a dospívajícími (9 až 17 let věku) jsou omezeny na pacienty s HoFH nebo sitosterolemií.
Atorvastatin
V otevřené 8týdenní studii byli pediatričtí pacienti Tannerova stadia 1 (n = 15) a Tannerova stadia 2 (n = 24) (ve věku 6 až 17 let) s heterozygotní familiární hypercholesterolemií a výchozími hodnotami LDL-C >4 mmol/l léčeni 5mg nebo 10mg žvýkacími respektive 10mg nebo 20mg potahovanými tabletami atorvastatinu jednou denně. Ve farmakokinetickém modelu u populace s atorvastatinem byla jedinou významnou proměnnou tělesná hmotnost. Zdánlivá perorální clearance atorvastatinu u pediatrických subjektů se jevila podobná jako u dospělých, pokud byla stupňována alometricky podle tělesné hmotnosti. V celém rozmezí expozic atorvastatinu a o-hydroxyatorvastatinu byla pozorována konzistentní snížení LDL-C a celkového cholesterolu.
Starší osoby
Ezetimib
Plazmatická koncentrace celkového ezetimibu je u starších jedinců (> 65 let) přibližně dvakrát vyšší než u mladších jedinců (18 až 45 let). Snížení koncentrací LDL-C a profil bezpečnosti u starších a mladších jedinců léčených ezetimibem jsou srovnatelné.
Atorvastatin
Plazmatické koncentrace atorvastatinu a jeho aktivních metabolitů jsou u zdravých starších subjektů vyšší, než u mladých dospělých, přičemž účinky na lipidy byly srovnatelné s účinky pozorovanými u mladších populací pacientů.
Porucha funkce jater
Ezetimib
Po jednorázové 10mg dávce ezetimibu se střední hodnota AUC celkového ezetimibu u pacientů s mírnou poruchou funkce jater (Child-Pughovo skóre 5 nebo 6) ve srovnání se zdravými jedinci zvětšila přibližně 1,7násobně. Ve 14denní studii s opakovanými dávkami (10 mg denně) podávanými pacientům se středně těžkou poruchou funkce jater (Child-Pughovo skóre 7 až 9) se střední hodnota AUC celkového ezetimibu zvýšila ve srovnání se zdravými jedinci 1. den a 14. den přibližně 4násobně. U pacientů s mírnou poruchou funkce jater není nutno dávku nijak upravovat. Vzhledem k tomu, že účinky zvýšené expozice ezetimibu u pacientů se středně těžkou nebo těžkou poruchou funkce jater (Child-Pughovo skóre > 9) nejsou známy, nedoporučuje se ezetimib u uvedených skupin pacientů používat (viz body 4.2 a 4.4).
Atorvastatin
Plazmatické koncentrace atorvastatinu a jeho aktivních metabolitů jsou u pacientů s chronickým alkoholickým jaterním onemocněním (Child-Pughovo skóre B) výrazně zvýšeny (přibližně 16násobně, pokud jde o Cmax a přibližně 11násobně, pokud jde o AUC).
Porucha funkce ledvin
Ezetimib
Po jednorázové 10mg dávce ezetimibu pacientům s těžkým onemocněním ledvin (n = 8; střední hodnota CrCl < 30 ml/min/1,73 m2) se ve srovnání se zdravými jedinci (n = 9) střední hodnota AUC celkového ezetimibu zvětšila přibližně 1,5násobně.
U dalšího pacienta v této studii (po transplantaci ledviny a užívající několik léčiv včetně cyklosporinu) byla zjištěna 12násobně zvýšená expozice celkovému ezetimibu.
Atorvastatin
Onemocnění ledvin nemá žádný vliv na plazmatické koncentrace atorvastatinu a jeho aktivních metabolitů nebo jejich účinky na lipidy.
Pohlaví
Ezetimib
Plazmatické koncentrace celkového ezetimibu jsou u žen mírně vyšší (přibližně o 20 %) než u mužů.
U mužů i u žen léčených ezetimibem jsou snížení koncentrace LDL-C a profil bezpečnosti srovnatelné.
Atorvastatin
Koncentrace atorvastatinu a jeho aktivních metabolitů se u žen liší od koncentrací u mužů (ženy: přibližně o 20 % vyšší, pokud jde o Cmax a přibližně o 10 % nižší, pokud jde o AUC). Tyto rozdíly neměly žádný klinický význam, což vede k tomu, že mezi ženami a muži nejsou ohledně účinků na lipidy žádné klinicky významné rozdíly.
Polymorfismus SLCO1B1
Atorvastatin
Jaterní příjem všech inhibitorů HMG-CoA reduktázy, včetně atorvastatinu, zahrnuje transportér OATP1B1. U pacientů s polymorfismem SLCO1B1 existuje riziko zvýšené expozice atorvastatinu, což může vést ke zvýšenému riziku rabdomyolýzy (viz bod 4.4). Polymorfismus v genu kódujícím OATP1B1 (SLCO1B1 c.521CC) je spojován s 2,4krát vyšší expozicí atorvastatinu (AUC), než u jedinců bez této varianty genotypu (c.521TT). U těchto pacientů je také možný geneticky narušený jaterní příjem atorvastatinu. Možné důsledky pro účinnost nejsou známy.
5.3 Předklinické údaje vztahující se k bezpečnosti
Přípravek Orvatez
Ve tříměsíčních studiích souběžného podávání ezetimibu a atorvastatinu potkanům a psům byly pozorované toxické účinky takové, jaké se obvykle spojují se statiny. Statinové histopatologické nálezy byly omezeny na játra. Některé z toxických účinků byly výraznější než toxické účinky pozorované při podávání samotných statinů. To se přisuzuje farmakokinetickým a/nebo farmakodynamickým interakcím při současném podávání.
Současné podávání ezetimibu a atorvastatinu březím potkanům naznačilo, že ve skupině s vysokou dávkou ezetimibu/atorvastatinu (1000/108,6 mg/kg) došlo k na testované látce závislému zvýšení změn skeletu nazývaných „snížená osifikace sterneber“. To může souviset s pozorovaným poklesem tělesných hmotností plodů. U březích králíků byla pozorována nízká incidence deformit skeletu (spojená sternebra, spojené kaudální obratle a asymetrické změny sterneber).
V řadě in vivo a in vitro stanovení ezetimib podávaný samostatně nebo s atorvastatinem nevykazoval žádný genotoxický potenciál.
Ezetimib
Studie chronické toxicity ezetimibu u zvířat neprokázaly žádný cílový orgán pro toxické účinky.
U psů, jimž byl po dobu čtyř týdnů podáván ezetimib (> 0,03 mg/kg/den), se koncentrace cholesterolu ve žlučníkové žluči zvýšily 2,5- až 3,5násobně. V jednoleté studii u psů, jimž byly podávány dávky až 300 mg/kg/den, však nebyla pozorována zvýšená incidence cholelitiázy ani jiné hepatobiliární účinky. Význam těchto zjištění pro člověka není znám. Litogenní riziko při léčebném používání ezetimibu nelze vyloučit.
Dlouhodobé zkoušky kancerogenity ezetimibu byly negativní.
Ezetimib nemá žádný účinek na fertilitu samců ani samic potkanů, ani nebyla prokázána jeho teratogenita u potkanů a králíků, neměl vliv ani na prenatální a postnatální vývoj. U březích samic potkanů a králíků, jimž byly opakovaně podávány dávky 1 000 mg/kg/den, prostupoval ezetimib placentární bariérou.
Atorvastatin
Atorvastatin byl v sérii 4 in vitro testů a 1 in vivo stanovení negativní na mutagenní a klastogenní potenciál. Nebylo zjištěno, že by atorvastatin byl karcinogenní u potkanů, nicméně vysoké dávky u myší (vedoucí 6- až 11násobku AUC0-24h dosahovaných u lidí při nejvyšší doporučované dávce) vykázaly hepatocelulární adenomy u samců a hepatocelulární karcinomy u samic. Z experimentálních studií na zvířatech existují důkazy, že inhibitory HMG-CoA reduktázy mohou ovlivnit vývoj embryí a plodů. U potkanů, králíků a psů neměl atorvastatin žádné účinky na fertilitu a nebyl teratogenní, nicméně při dávkách toxických pro matku byla u potkanů a králíků pozorována fetální toxicita. Vývoj potkaních potomků byl při expozici březích samic vysokým dávkám atorvastatinu zpomalen a postnatální přežití sníženo. U potkanů existují důkazy placentárního přenosu. U potkanů jsou plazmatické koncentrace atorvastatinu podobné koncentracím v mléce. Zda se atorvastatin nebo jeho metabolity vylučují do lidského mléka, není známo.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety Vrstva ezetimibu
Sodná sůl kroskarmelózy Monohydrát laktózy Magnesium-stearát Mikrokrystalická celulóza Povidon
Natrium-lauryl-sulfát
Vrstva atorvastatinu
Mikrokrystalická celulóza Monohydrát laktózy Hyprolóza
Sodná sůl kroskarmelózy
Polysorbát 80
Uhličitan vápenatý
Magnesium-stearát
Koloidní bezvodý oxid křemičitý
Potah tablety
Hypromelóza Makrogol 8000 Oxid titaničitý (E171)
Mastek
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před kyslíkem.
6.5 Druh obalu a obsah balení
Orvatez 10 mg/10 mg, 10 mg/20 mg, 10 mg/40 mg a 10 mg/80 mg
Balení po 10, 30, 90 a 100 potahovaných tabletách v dusíkem profouknutých Al/Al blistrech (dutina z oPA-Al-PVC s víčkem z Al).
Balení po 30 x 1 a 45 x 1 potahovaných tabletách v jednodávkových dusíkem profouknutých Al/Al blistrech (dutina z oPA-Al-PVC s víčkem z Al).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
Orvatez 10 mg/10 mg potahované tablety: 31/407/14-C Orvatez 10 mg/20 mg potahované tablety: 31/408/14-C Orvatez 10 mg/40 mg potahované tablety: 31/409/14-C Orvatez 10 mg/80 mg potahované tablety: 31/410/14-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 29.10.2014
10. DATUM REVIZE TEXTU
12.4.2016
25
Současné podávání ezetimibu a atorvastatinu ekvivalentní přípravku Orvatez 10/10 nebo Orvatez 10/20
1 M-odhad (založeno na Huberově metodě; 95% interval spolehlivosti a hodnota p byly získány z přiřazení robustního regresního modelu k výchozím podmínkám léčby)
* Geometrická střední hodnota procenta změn výchozích hodnot triglyceridů byla vypočtena na základě zpětného převodu exponenciací na modelu založených středních hodnot získaných metodou nejmenších čtverců (LS) a vyjádřených jako (geometrická střední hodnota - 1) násobeno 100
p<0,001 versus přípravek Orvatez 10/10