Optivate
sp. zn. sukls155076/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Optivate
250 IU, 500 IU, 1000 IU
Prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Přípravek Optivate je dodáván jako prášek a rozpouštědlo pro injekční roztok.
Jedna injekční lahvička obsahuje nominálně factor VIII coagulationis humanus 250 mezinárodních jednotek (International Unit, IU), 500 IU nebo 1000 IU.
Po rekonstituci s 2,5 ml (250 IU), 5 ml (500 IU) nebo 10 ml (1000 IU) sterilizované vody na injekci přípravek Optivate obsahuje přibližně 100 IU/ml lidského koagulačního faktoru VIII.
Síla ( IU) se určuje použitím chromogenního testu podle Evropského lékopisu. Specifická aktivita přípravku Optivate odpovídá přibližně 43 IU/mg proteinu.
Přípravek Optivate také obsahuje lidský von Willebrandův faktor (vWF měřený podle aktivity ristocetinového kofaktoru) v koncentraci přibližně 430 IU, 860 IU nebo 1720 IU na jednu injekční lahvičku v balení po 250 IU, 500 IU a 1000 IU.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Injekční lahvička přípravku obsahuje bílý nebo světle žlutý prášek.
Lahvička s rozpouštědlem obsahuje čirou bezbarvou tekutinu.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII).
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Dávkování
Dávkování a doba trvání substituční léčby závisejí na závažnosti deficitu faktoru VIII, na místě a intenzitě krvácení a na klinickém stavu pacienta.
Počet podaných jednotek faktoru VIII je vyjádřen v mezinárodních jednotkách (IU), které jsou vztaženy k současnému standardu WHO pro přípravky s faktorem VIII. Aktivita faktoru VIII v plazmě je vyjádřená buďto procentuálně (s ohledem na jeho obsah v normální lidské plazmě) nebo v mezinárodních jednotkách (vztažených k mezinárodnímu standardu pro faktor VIII v krevní plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy. Výpočet potřebné dávky faktoru VIII je založen na zkušenosti, že podání 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvýší plazmatickou aktivitu faktoru VIII o 2,2 až 2,7 % normální aktivity (2,2-2,7 IU/dl). Potřebnou dávku lze určit pomocí následujícího vzorce:
potřebný počet tělesná x požadované zvýšení x 0,4
jednotek = hmotnost (kg) faktoru VIII (%) (IU/dl)
Podávané množství a frekvence podávání přípravku se vždy musí řídit klinickým účinkem v individuálním případě.
Při následujících krvácivých příhodách nesmí v příslušném období aktivita faktoru VIII v plazmě poklesnout pod uvedenou hladinu (udávanou v % normální hodnoty nebo v IU/dl). Následující tabulku lze použít jako vodítko pro dávkování přípravku při krvácivých příhodách a chirurgickém zákroku:
|
Stupeň krvácení/ Typ chirurgického zákroku |
Potřebná hladina faktoru VIII (%) (IU/dl) |
F rekvence dávkování (hodiny)/ Doba trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo dutiny ústní |
20-40 |
Opakujte každých 12 až 24 hodin. Podávejte minimálně po dobu 1 dne, až do zástavy krvácení, o které svědčí ústup bolesti, nebo do úpravy stavu. |
|
Rozsáhlejší krvácení do kloubů, svalů nebo tvorba hematomů. |
30-60 |
Opakujte infuzi každých 12 až 24 hodin po dobu 3 až 4 dní, případně déle, dokud bolest a akutního postižení nepřejdou. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infuzi každých 8 až 24 hodin až do ústupu ohrožení. |
|
Chirurgické zákroky | ||
|
Menší zákroky Včetně extrakce zubů |
30-60 |
Každých 24 hodin, nejméně 1 den, až do úpravy stavu. |
|
Velké operace |
80-100 (před a po operaci) |
Opakujte infuzi každých 8 až 24 hodin až do uspokojivého zhojení rány, poté pokračujte v léčbě po dobu nejméně dalších 7 dní takovým způsobem, aby se aktivita faktoru VIII udržovala |
v rozsahu 30 % až 60 % (IU/dl).
V průběhu léčby se doporučuje vhodným způsobem sledovat hladinu faktoru VIII a na základě takto zjištěných hodnot určovat velikost dávek a frekvenci opakování infuzí. Zejména při rozsáhlých chirurgických zákrocích je nezbytné přesné monitorování průběhu substituční terapie pomocí koagulačních testů (sledování aktivity faktoru VIII v plazmě). Odpovědi jednotlivých pacientů na podání faktoru VIII se mohou lišit dosahováním různých hodnot uzdravení (recovery in vivo) a vykazováním různého poločasu.
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na jeden kg tělesné hmotnosti v intervalech 2 až 3 dnů. V některých případech, zejména u mladších pacientů, může být nezbytné podávat přípravek častěji nebo dávky zvýšit.
Pediatrická populace
Děti do 6 let
Doporučená dávka je 17 až 30 IU/kg. Jako prevenci krvácení lze tuto dávku podávat až 3krát týdně. V klinických zkouškách byl medián dávek u dětí ve věku <6 let 24,7 IU/kg pro běžnou profylaxi a 27,6 IU/kg k léčbě krvácení.
Děti ve věku nad 6 let
Údaje o použití přípravku Optivate u dětí ve věku od 6 do 12 let jsou velmi omezené.
U pacientů je nutno pravidelně sledovat, zda nedochází k tvorbě inhibitorů faktoru VIII.
Pokud se nepodaří dosáhnout očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud se nedaří podáváním příslušné dávky zvládnout krvácení, je nutné vyšetřit, zda není přítomen inhibitor faktoru VIII. U pacientů s vysokou hladinou inhibitoru může být léčba faktorem VIII neúčinná a je nutno zvážit jiné možnosti léčby. Léčbu těchto pacientů mají řídit lékaři, kteří mají zkušenosti s péčí o pacienty s hemofilií.
Viz také bod 4.4.
Způsob podání
Rozpusťte přípravek podle pokynů uvedených v bodě 6.6. Přípravek se podává intravenózně; rychlost podávání nemá překročit 3 ml za minutu (vyšší rychlost podávání může vést k vedlejším účinkům).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku(y) nebo na kteroukoli pomocnou látku.
4.4 Zvláštní upozornění a opatření pro použití
Stejně jako všechny ostatní intravenózně podávané proteinové přípravky, může i tento přípravek vyvolávat hypersenzitivní reakce alergického typu. Přípravek obsahuje i stopová
množství jiných lidských proteinů než je faktor VIII. Pacienti musí být informováni o časných známkách hypersenzitivních reakcí, jako je vyrážka, generalizovaná kopřivka, tíseň na hrudníku, sípot, hypotenze a anafylaxe. Pokud se tyto příznaky objeví, doporučuje se pacientům, aby okamžitě přestali přípravek užívat a kontaktovali svého lékaře.
V případě šoku je nutno zahájit protišokovou léčbu podle aktuálních lékařských standardů.
Standardní opatření pro prevenci infekcí způsobených používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují výběr dárců, screeningové testování jednotlivých odběrů a směsných vzorků plazmy na specifické ukazatele infekce a zavedení účinných výrobních postupů pro inaktivaci/odstranění virů. Přesto při podávání léčivých přípravků vyrobených z lidské krve nebo plazmy nelze možnost přenosu původců infekcí zcela vyloučit. To platí i pro neznámé nebo nově objevené viry a jiné patogeny.
Přijatá opatření se pokládají za účinná u obalených virů, jako je virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV), a u neobaleného viru hepatitidy A. Přijatá opatření mohou mít omezenou účinnost u neobalených virů, jako je parvovirus B19. Infekce parvovirem B19 může být závažná u těhotných žen (infekce plodu) a u jedinců s imunodeficiencí nebo zvýšenou erytropoézou (např. při hemolytické anemii).
U pacientů, kteří dostávají pravidelně/opakovaně přípravky s faktorem VIII vyráběné z lidské plazmy, má být zváženo vhodné očkování (proti hepatitidě A a B).
Vytváření neutralizačních protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG, které působí proti prokoagulační aktivitě faktoru VIII a kvantitativně se stanovují v jednotkách BU (Bethesda Units) na 1 ml plazmy a zjišťované v modifikovaném testu. Riziko tvorby inhibitorů má vztah k expozici organizmu antihemofilickému faktoru VIII, přičemž toto riziko je nejvyšší v prvních 20 dnech expozice. Vzácně může docházet k tomu, že se inhibitory vytvoří teprve po prvních 100 dnech expozice.
Po přechodu z jednoho přípravku obsahujícího faktor VIII na jiný byly u pacientů, kteří měli v anamnéze vytváření inhibitoru, pozorovány případy recidivy tvorby inhibitoru (s nízkým titrem) po více než 100 dnech expozice. Proto se při jakékoli změně podávaného přípravku doporučuje pečlivě sledovat pacienty z důvodu možného vytváření inhibitorů.
Pacienty, kteří jsou léčeni lidským koagulačním faktorem VIII, je proto nutno pečlivě sledovat klinicky i prostřednictvím laboratorních testů z důvodu možného vytváření inhibitorů. Viz také bod 4.8 Nežádoucí účinky.
Důrazně se doporučuje, aby byl při každém podání přípravku Optivate pacientovi zaznamenán název a číslo šarže přípravku, aby byla jednoznačně zajištěna identifikace pacienta s číslem šarže přípravku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou známy žádné interakce přípravků obsahujících lidský faktor VIII s jinými léčivými přípravky.
S faktorem VIII nebyly provedeny žádné reprodukční studie na zvířatech. Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII v těhotenství a při kojení k dispozici. Proto se má v těhotenství a při kojení faktor VIII používat pouze tehdy, je-li jasně indikován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly pozorovány žádné účinky na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Občas byly při podávání přípravku pozorovány hypersenzitivní neboli alergické reakce (mimo jiné angioedém, pálení a píchání v místě aplikace, zimnice, zčervenání, generalizovaná kopřivka, bolest hlavy, vyrážka, hypotenze, letargie, nauzea, neklid, tachykardie tíseň na hrudníku, mravenčení, zvracení, sípot), které mohou v některých případech progredovat do závažné anafylaxe (včetně šoku).
Ve vzácných případech byla pozorována horečka.
U 96 pacientů v klinických studiích byly pozorovány následující nežádoucí účinky. Přibližně u 10 % pacientů lze očekávat vznik nežádoucích účinků při dlouhodobé léčbě. Jejich frekvence jsou definovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky |
Frekvence |
|
Poruchy nervového systému |
Časté | |
|
Somnolence |
Časté | |
|
Poruchy ucha a labyrintu |
Vertigo (závrať) |
Časté |
|
Poruchy kůže a podkožní tkáně |
Časté | |
|
Časté | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Ztuhlost svalů a kloubů |
Časté |
|
Celkové poruchy a reakce v místě aplikace |
Zarudnutí, vyrážka nebo bolest v místě vpichu |
Časté |
|
Periferní edém |
Časté | |
|
Třesavka (zimnice) |
Časté | |
|
Horečka (pyrexie) |
Časté |
Po uvedení na trh byly nahlášeny následující další nežádoucí účinky: kýchání, kašel, podráždění hrdla, bolest břicha a malátnost.
U pacientů s hemofilií A se mohou vytvořit neutralizující protilátky (inhibitory) proti faktoru VIII. Dojde-li k vytvoření těchto inhibitorů, projeví se to jako nedostatečná klinická odpověď. V takovém případě se doporučuje kontaktovat specializované centrum pro léčbu hemofilie. V klinickém vývojovém programu byl léčen jeden dosud neléčený pacient (previously untreated patient, PUP). Ani tento pacient, ani žádný z 95 pacientů, kteří již byli
léčeni (previously treated patients, PTP) si během klinických zkoušek nevytvořili inhibitory. Medián počtu dní expozice u těchto pacientů byl 97 dní (při rozsahu 2 až 408 dní).
Informace o bezpečnosti v souvislosti s přenosnými původci infekcí viz bod 4.4.
4.9 Předávkování
Nebyly hlášeny žádné případy předávkování lidským koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Koagulační faktory, koagulační faktor VIII,
ATC kód: B02BD02.
Komplex faktoru VIII a von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru), které mající odlišné fyziologické funkce. Po infuzi podané hemofilickému pacientovi se faktor VIII váže na von Willebrandův faktor v krevním oběhu pacienta. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX a urychluje konverzi faktoru X na aktivovanou formu faktoru X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin pak převádí fibrinogen na fibrin a může vzniknout krevní sraženina. Hemofilie A je na pohlaví vázaná dědičná porucha krevní srážlivosti v důsledku snížené hladiny faktoru VIII:C. Vede k profúznímu krvácení do kloubů, svalů nebo vnitřních orgánů, které vzniká buď spontánně, nebo v důsledku náhodně či chirurgicky vzniklých poranění. Substituční terapií je hladina faktoru VIII v plazmě zvyšována, čímž se dosahuje dočasné korekce deficitu koagulačního faktoru a korekce sklonů ke krvácení.
Von Willebrandův faktor jednak funguje jako protein, který chrání faktor VIII, jednak zprostředkuje adhezi trombocytů k místu poranění cévy, a rovněž se podílí na agregaci trombocytů.
Podle zkušeností z klinických studií malé děti, kterým byl profylakticky podáván přípravek Optivate, krvácely méně než ty, kterým byl přípravek podáván při potřebě („on demand“). Dávkování u dětí viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti přípravku Optivate byly hodnoceny u 15 pacientů s těžkou hemofilií A po bolusových dávkách 50 IU/kg. Výsledky byly následující:
Po intravenózní injekci dojde k rychlému zvýšení aktivity faktoru VIII (FVIII:C) v plazmě, po kterém následuje rychlý pokles aktivity a následně pomalejší pokles aktivity. Studie u pacientů s hemofilií A prokázaly střední poločas alfa 2,2 hodiny a střední poločas beta 12,6 hodiny. Celkově bylo in vivo dosaženo střední inkrementální výtěžnosti (incremental recovery) dané vzestupem FVIII o 2,5 IU/dl na jednu podanou IU/kg. Byla zjištěna střední doba zdržení (mean residence time, MRT) byla 17,5 hodiny (95% interval spolehlivosti [confidence interval, CI]: 16,0 až 18,9), střední plocha pod křivkou (area under the curve, AUC 0-inf) byla 17,31 hodiny. IU/ml (95% CI: 15,0 až 19,7) a střední clearance byla 3,1 ml/kg/h (95% CI: 2,7 až 3,5).
Během klinických studií bylo provedeno 309 hodnocení inkrementální výtěžnosti, přičemž všechna vycházela ze stanovení maxima FVIII:C za první hodinu (ISTH 2001). Tato hodnocení zahrnovala 27 šarží přípravku Optivate a byla provedena u 70 dospělých pacientů s těžkou hemofilií A. Celkově byly hodnoty inkrementální výtěžnosti následující:
Střední hodnota: 2,7 IU/dl na jednu podanou IU/kg
95% CI: 2,53-2,80 IU/dl na jednu podanou IU/kg
Medián: 2,6 IU/dl na jednu podanou IU/kg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Faktor VIII a von Willebrandův faktor obsažené v přípravku Optivate jsou normální složky lidské plazmy a působí stejně jako endogenní proteiny, proto testování bezpečnosti nemá význam.
Studie akutní toxicity a toxicity po opakovaném podávání na myších však prokázala, že přípravek Optivate nebyl toxický, ani při hladinách 20krát vyšších než jsou ty, které budou pravděpodobně dosaženy u člověka. V těchto studiích byla pokusným zvířatům podávána různá množství různých složek přípravku, přičemž množství jednotlivých pomocných látek byla vyšší než ta, která jsou podávána v klinických dávkách.
Provádění studií genotoxicity nebo karcinogenicity s plazmatickým koagulačním faktorem VIII s jeho přirozeným stabilizátorem von Willebrandovým faktorem i bez něho je vědecky neodůvodněné.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný Dihydrát natrium-citrátu Dihydrát chloridu vápenatého Polysorbát 20 Dihydrát trehalosy
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Pro podání je nutno výhradně užívat dodávané injekční/infuzní soupravy, neboť jinak může být léčba neúčinná v důsledku adsorpce lidského koagulačního faktoru FVIII na vnitřní povrchy některých infuzních souprav.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
1 balení přípravku Optivate obsahuje:
1 injekční lahvičku s práškem obsahující 250 IU, 500 IU nebo 1000 IU lidského koagulačního faktoru VIII. Injekční lahvičky jsou ze skla třídy I podle Ph.Eur., se zátkou z halobutylové pryže, s uzávěrem z lakovaného hliníku a odtrhávacím polypropylenovým víčkem.
1 injekční lahvičku s rozpouštědlem (sterilizovaná voda na injekci), 2,5 ml, 5 ml nebo 10 ml. Injekční lahvičky jsou ze skla třídy I podle Ph.Eur., se zátkou z halobutylové pryže a uzávěrem.
1 přepouštěcí adaptér Mix2Vial, který umožňuje snadnou a bezpečnou rekonstituci přípravku sterilizovanou vodou na injekci bez použití jehly.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
K rekonstituci přípravku Optivate používejte výhradně sterilizovanou vodu na injekci dodanou s přípravkem. K rekonstituci 250 IU, 500 IU a 1000 IU použijte 2,5 ml, 5 ml, respektive 10 ml sterilizované vody na injekci (viz ilustrace na následující stránce).
Injekční lahvičky přípravku Optivate a sterilizované vody na injekci je třeba před odtrhnutím odtrhávacího uzávěru z injekční lahvičky zahřát na 20 °C a 30 °C.
Postup pro provedení rekonstituce je následující:
|
3' |
Krok 1 •Odstraňte víčko z injekční lahvičky s přípravkem a očistěte povrch zátky tamponem navlhčeným v alkoholu. •Proveďte tento krok také s injekční lahvičkou se sterilní vodou. •Odstraňte krycí fólii z balení přepouštěcího adaptéru, ale ponechejte prostředek v obalu. | |
|
Krok 2 •Přiložte modrý konec přepouštěcího adaptéru na injekční lahvičku s vodou a zatlačte přímo dolů, až hrot pronikne pryžovou zátkou a adaptér zaklapne na své místo. •Sejměte plastový vnější obal z přepouštěcího adaptéru a zlikvidujte jej; dávejte pozor, abyste se nedotkli obnaženého konce adaptéru. | ||
|
f tm W |
Krok 3 •Lahvičku s připojeným adaptérem otočte dnem vzhůru. •Přiložte průhledný konec přepouštěcího adaptéru na injekční lahvičku s přípravkem a zatlačte přímo dolů, až hrot pronikne pryžovou zátkou a adaptér zaklapne na své místo. | |
|
i |
Krok 4 •Sterilní voda se sama nasaje do injekční lahvičky s přípravkem, protože v injekční lahvičce je podtlak. •Opatrným kroužením injekční lahvičkou přípravek důkladně promíchejte. Lahvičkou netřepejte. •Měl by vzniknout čirý nebo jemně perleťový roztok, což obvykle trvá 2 až 2 !4 minuty (nejvýše 5 minut). | |
|
. ** |
Krok 5 •Šroubovitým pohybem proti směru hodinových ručiček oddělte prázdnou lahvičku se sterilní vodou a modrou část adaptéru od průhledné části. •Zatažením za píst injekční stříkačky natáhněte do injekční stříkačky tolik vzduchu, kolik odpovídá přidávanému objemu vody. •Připojte injekční stříkačku k průhledné části přepouštěcího adaptéru Mix2Vial. •Vytlačte vzduch z injekční stříkačky do injekční lahvičky. | |
|
Krok 6 | ||
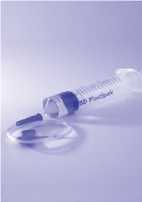
•Ihned převraťte injekční lahvičku s roztokem, který se natáhne do injekční stříkačky.
•Odpojte naplněnou injekční stříkačku od adaptéru. •Přípravek je nyní připraven k podání. Při podání používejte obvyklé bezpečné postupy. Přípravek použijte ihned po rekonstituci, nesmí být uchováván.
Poznámka: Pokud je potřeba použít více než jednu injekční lahvičku pro dosažení předepsané dávky, opakujte kroky 1 až 6; roztok z injekční lahvičky vždy natáhněte do stejné injekční stříkačky.
Přepouštěcí adaptér dodaný s přípravkem je sterilní a nelze jej použít vícekrát než jednou. Po dokončení rekonstituce jej zlikvidujte odložením do schránky na ostré předměty.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Roztok musí být čirý nebo mírně opalescentní. Nepoužívejte roztoky, které jsou zakalené nebo obsahují sediment. Rekonstituovaný přípravek před podáním prohlédněte, zda neobsahuje částice nebo není zabarvený. Přípravek podejte formou injekce co nejdříve po rekonstituci a nejpozději do jedné hodiny.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bio Products Laboratory Limited
Dagger Lane, Elstree, Hertfordshire, WD6 3BX
Velká Británie
Tel: +44 (0) 20 8957 2200
Email: info@bpl.co.uk
8. REGISTRAČNÍ ČÍSLO(-A)
75/405/15-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
26.8.2015
10. DATUM REVIZE TEXTU
26.8.2015
10