Mononine 500 Iu
sp. zn. sukls187314/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Mononine 500 IU, prášek a rozpouštědlo pro injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně:
500 IU lidského koagulačního faktoru IX (FIX).
Mononine obsahuje přibližně 100 IU/ml faktoru IX po rekonstituci s 5 ml vody na injekci.
Účinnost (IU) je určována pomocí jednorázového testu srážlivosti podle Evropského Lékopisu. Střední specifická aktivita přípravku Mononine není menší než 190 IU/mg bílkoviny.
Vyrobeno z lidské plazmy dárců krve.
Pomocná látka se známým účinkem:
Sodík přibližně 66 mmol/l (1,5 mg/ml).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok.
Bílý prášek a čiré, bezbarvé rozpouštědlo pro injekční/infuzní roztok.
4. KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií B (kongenitální deficit faktoru IX).
4.2. Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře zkušeného s léčbou hemofilie.
Dávkování
Dávkování a délka trvání substituční léčby závisí na závažnosti deficitu faktoru IX, na místě a rozsahu krvácení a na klinickém stavu pacienta. Počet podaných jednotek faktoru IX je vyjádřen v mezinárodních jednotkách (IU), které se vztahují k současnému standardu WHO pro přípravky obsahující faktor IX. Plazmatická aktivita faktoru IX je vyjádřena buď v procentech (vzhledem k normální lidské plazmě) nebo v mezinárodních jednotkách (vztaženo na mezinárodní standard pro faktor IX v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru IX je ekvivalentní množství faktoru IX v 1 ml normální lidské plazmy.
Požadovaná léčba
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že 1 IU faktoru IX na kg tělesné hmotnosti zvýší aktivitu plazmatického faktoru IX o 1,0 % normální aktivity. Požadovaná dávka je určena pomocí následujícího vzorce:
Potřebný počet jednotek
= tělesná hmotnost (kg) x požadovaný vzestup faktoru IX (% nebo IU/dl) x 1,0
Množství, které má být podáno, metoda a frekvence podávání by vždy měla být přizpůsobena klinické účinnosti v individuálním případě.
V případě následujících hemoragických příhod by aktivita faktoru IX neměla klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu anebo IU/dl) v odpovídajícím období. Následující tabulky mohou být použity jako vodítko pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1: jednorázová intravenózní injekce | ||
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru IX v plazmě (% nebo IU/dl) |
Četnost dávek (v hodinách)/Trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo do dutiny ústní |
20-40 |
Opakovat každých 24 hodin. Nejméně 1 den, dokud nedoj de k zástavě krvácení indikované skončením bolestí, nebo ke zhojení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom |
30-60 |
Opakovat infuzi každých 24 hodin po 3-4 dny nebo déle, dokud nevymizí bolest a akutní postižení. |
|
Život ohrožující krvácení |
60-100 |
Opakovat infuzi každých 8 až 24 hodin, dokud nepomine ohrožení života. |
|
Chirurgický zákrok | ||
|
Menší včetně extrakce zubu |
30-60 |
Každých 24 hodin. Nejméně jeden den, až je dosaženo zhojení. |
|
Velké chirurgické výkony |
80-100 (před a po operaci) |
Opakovat infuzi každých 8-24 hodin až do adekvátního zhojení rány, pak pokračovat v léčbě přinejmenším po dalších 7 dnů, aby byla udržena aktivita faktoru IX od 30 % do 60 % (IU/dl). |
|
Tabulka 2: kontinuální infuze při chirurgických zákrocích | |
|
Hladiny faktoru IX potřebné pro hemostázu |
40-100% (nebo IU/dl) |
|
Počáteční dávka potřebná k dosažení potřebné hladiny |
Počáteční nárazová dávka 90 IU na kg (rozsah 75-100 IU) na kg tělesné hmotnosti nebo dávkování řízené podle hladiny pyruvátkinázy (pK-guided dosing). |
|
Frekvence podávání |
Kontinuální i.v. infuze v závislosti na clearance a na změřených hladinách faktoru IX. |
|
Trvání léčby |
Až 5 dnů. Podle povahy chirurgického zákroku může být potřebná i delší léčba. |
Profylaxe
Na dlouhodobou profylaxi krvácení u pacientů se závažnou hemofilií B se obvykle používají dávky 20 až 40 IU faktoru IX na kg tělesné hmotnosti v intervalech 3-4 dnů. V některých případech, zvláště u mladších pacientů, může být nutné použít kratší intervaly nebo vyšší dávky.
V průběhu léčby se doporučuje patřičné stanovování hladin faktoru IX, aby bylo možno řídit dávku, která má být podána a frekvenci opakovaných infuzí. Zvláště v případě větších chirurgických zákroků je nezbytné přesné monitorování substituční léčby pomocí koagulační analýzy (aktivity plazmatického faktoru IX). Odpověď na aplikaci faktoru IX se může u jednotlivých pacientů lišit. Může u nich docházet k různým hodnotám recovery in vivo a různé mohou být i poločasy.
U pacientů je třeba sledovat rozvoj inhibitorů faktoru IX.
Viz také bod 4.4.
Dříve neléčení pacienti
Bezpečnost a účinnost přípravku Mononine u dříve neléčených pacientů nebyla dosud stanovena.
Pediatrická populace
Dávkování u dětí je založeno na tělesné hmotnosti, a proto je obecně založeno na stejných pokynech, jako u dospělých. Četnost podávání má být vždy orientována na klinickou účinnost v konkrétním případě.
Způsob podání
Intravenózní podání.
Rekonstituujte přípravek tak, jak je to popsáno v bodě 6.6. Před podáním je třeba přípravek zahřát na pokojovou nebo tělesnou teplotu. Mononine se podává pomalu intravenózně tak, aby bylo možno sledovat u pacienta jakoukoliv bezprostřední reakci. Pokud dojde k jakékoli reakci, o které lze předpokládat, že je ve vztahu k podávání přípravku Mononine, je třeba snížit rychlost infuze nebo ji úplně zastavit podle toho, jak to vyžaduje klinický stav pacienta. (viz též bod 4.4.).
Jednotlivá intravenózní injekce
Proveďte venepunkci přiloženým venosetem. Připevněte injekční stříkačku na luer konec setu. Aplikujte pomalu intravenózně rychlostí, která je příjemná pro pacienta (max. 2 ml/min).
Kontinuální infuze
Mononine musí být rekonstituován vodou na injekci, jak je popsáno v bodě 6.6. Po rekonstituci se může Mononine podávat kontinuální infuzí nezředěný pomocí injekční pumpy.
Síla nezředěného, rekonstituovaného přípravku Mononine je přibližně 100 IU/ml.
Zředěný roztok lze připravit následovně:
• Zřeďte rekonstituovaný, zfiltrovaný roztok
převedením vhodného množství přípravku Mononine do požadovaného objemu fyziologického roztoku s použitím aseptické techniky.
• U ředění do 1:10 (koncentrace 10 IU faktoru IX/ml) zůstává aktivita faktoru IX stabilní po dobu 24 hodin.
• Redukce aktivity faktoru IX nastává při vyšších ředěních. Aktivita faktoru IX musí být monitorována k udržení požadované hladiny v krvi.
Příklad ředění 500 IU rekonstituovaného přípravku Mononine:
|
Požadovaná síla po naředění |
10 IU/ml |
20 IU/ml |
|
Objem rekonstituovaného přípravku Mononine |
5,0 ml |
5,0 ml |
|
Objem potřebného fyziologického roztoku |
45,0ml |
20,0 ml |
|
Dosažené ředění |
1:10 |
1:5 |
• Doporučuje se používat polyvinylchloridové (PVC) IV vaky a hadičky
• Důkladně promíchejte a zkontrolujte, zda vak nemá trhliny.
• Doporučuje se vyměnit vaky s čerstvě naředěným přípravkem Mononine každých 12-24 hodin.
Doporučená rychlost kontinuální infuze přípravku Mononine k udržení rovnovážného stavu hladiny faktoru IX na přibližně 80% je 4 IU/kg tělesné hmotnosti/hod., ale bude záviset na farmakokinetickém profilu pacienta a na požadované cílové hladině faktoru IX. U pacientů se známou clearance faktoru IX je možné rychlost infuze vypočítat individuálně.
Rychlost (IU/kg tělesné hmotnosti/hod) = clearance (ml/hod/kg tělesné hmotnosti) x požadovaný vzestup faktoru IX (IU/ml)
Bezpečnost a účinnost kontinuální infuze u dětí nebyly studovány (viz bod 4.4). Proto u dětí a dospívajících by se o kontinuální infuzi přípravku Mononine mělo uvažovat pouze tehdy, jestliže byly před chirurgickým výkonem získány farmakokinetické údaje (tj. přírůstek recovery a clearance) potřebné pro vypočítání dávkování a hladiny se musí perioperativně pečlivě sledovat.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na myší bílkovinu.
Vysoké riziko trombózy nebo diseminované intravaskulární koagulace (viz též bod 4.4 ).
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku Mononine jsou možné hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy myší bílkoviny (myší monoklonální protilátky jsou používány při procesu jeho purifikace). I když hladiny myší bílkoviny jsou velmi nízké (<50 ng myší bílkoviny na 100 IU), infuze takových bílkovin by mohla teoreticky indukovat hypersenzitivní odpověď.
Obj eví-li se příznaky přecitlivělosti, pacienti mají být poučeni, aby okamžitě přerušili používání tohoto léčivého přípravku a kontaktovali svého lékaře.
Pacienti by měli být informováni o časných příznacích hypersensitivních reakcí jako např. vyrážka, generalizovaná kopřivka, pocit tíhy na prsou, sípot, hypotenze a anafylaxe.
V případě vzniku šoku má být léčba vedena podle standardních lékařských postupů pro léčbu šoku.
Standardní dávka 2000 IU přípravku Mononine obsahuje až 30,36 mg sodíku. To je třeba vzít v úvahu u pacientů na kontrolované sodíkové dietě.
Inhibitory
Po opakovaném podávání léčivých přípravků obsahujících lidský koagulační faktor IX je třeba u pacienta zjišťovat, zda nedošlo ke vzniku neutralizujících protilátek (inhibitorů), které by měly být kvantifikovány v Bethesda jednotkách (BU) pomoci vhodného biologického testování.
V literatuře byly uvedeny zprávy ukazující na korelaci mezi výskytem inhibitorů faktoru IX a alergickými reakcemi. Proto by pacienti, u nichž došlo k alergické reakci, měli být vyšetřeni na přítomnost inhibitorů. Je třeba si uvědomit, že u pacientů s inhibitory faktoru IX je zvýšené nebezpečí anafylaxe při následné stimulaci faktorem IX.
Vzhledem k riziku vzniku alergických reakcí při použití koncentrátů faktoru IX by počáteční podání faktoru IX mělo podle rozhodnutí ošetřujícího lékaře být provedeno pod lékařským dohledem tak, aby mohla být poskytnuta odpovídající opatření při alergické reakci.
Tromboembolie
Vzhledem k potenciálnímu riziku vzniku trombotických komplikací je třeba zahájit klinické sledování časných známek trombotických příhod a konzumpční koagulopatie spolu s odpovídajícím biologickým testováním, pokud je tento přípravek podáván pacientům s jaterním onemocněním, pacientům v pooperačním období, novorozencům nebo pacientům s rizikem vzniku trombotických příhod nebo DIC. V každém z výše uvedených případů je nutné zvážit přínos léčby přípravkem Mononine a možné riziko těchto komplikací.
Kardiovaskulární příhody
Substituční terapie s FIX může u pacientů s existujícími kardiovaskulárními rizikovými faktory zvyšovat kardiovaskulární riziko.
Komplikace spojené s katetrizací
Pokud je požadován centrální žilní přístup (CVAD), je třeba zvážit riziko komplikací souvisejících s CVAD včetně lokálních infekcí, bakteriémie a trombózy v místě zavedení katetru.
Virová bezpečnost
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, screening jednotlivých odběrů a plazmatických poolů na specifické ukazatele infekce a zařazení efektivních výrobních postupů k inaktivaci/eliminaci virů. Přes všechna tato opatření při podávání léčivých přípravků vyráběných z lidské krve nebo plazmy, nelze zcela vyloučit možnost přenosu infekčních agens. To se týká také neznámých nebo nově vznikajících virů a jiných patogenů.
Rozsah opatření je účinný proti obaleným virům jako je virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV) a pro neobalené viry hepatitidy A (HAV) a parvoviru B19.
Doporučuje se zvážit vhodné očkování (hepatitida A a B) u těch pacientů, kteří pravidelně/opakovaně dostávají přípravky s faktorem IX vyrobené z lidské plazmy.
Důrazně se doporučuje, aby vždy při každém podání přípravku Mononine pacientovi byl zaznamenán název a číslo šarže použitého přípravku k zajištění možnosti dohledání spojení mezi pacientem a šarží přípravku.
Pediatrická populace
Uvedená upozornění a opatření se vztahují jak k dospělým, tak k dětem.
Neexistují žádné údaje o bezpečnosti a účinnosti pro použití kontinuální infuze u dětí, zejména není znám potenciál pro rozvoj inhibitorů (viz bod 4.2)
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravků lidského koagulačního faktoru IX s jinými léčivými přípravky.
Je málo dostupných údajů týkajících se použití s-aminokapronové kyseliny po počáteční infuzi přípravku Mononine k prevenci nebo léčbě krvácení v dutině ústní po úrazu nebo výkonu na zubech, jako je např. extrakce.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie na zvířatech nebyly s faktorem IX provedeny.
Těhotenství a kojení
Vzhledem k vzácnému výskytu hemofilie B u žen nejsou s použitím faktoru IX v průběhu těhotenství a kojení žádné zkušenosti.
Proto by měl být faktor IX v těhotenství a při kojení použit pouze tehdy, pokud je zcela jasně indikovaný.
Fertilita
Nejsou dostupné žádné údaje týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Mononine nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Následující nežádoucí účinky jsou založeny na postmarketingových zkušenostech a na údajích z odborné literatury.
Souhrn bezpečnostního profilu
Hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnici, zarudnutí kůže, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nevolnost, neklid, tachykardii, pocit tíhy na prsou, mravenčení, zvracení, sípot) byly pozorovány vzácně a mohou se v některých případech rozvinout do závažné anafylaxe (včetně šoku). V některých případech tyto reakce vyústily až do závažné anafylaxe a došlo v těsné časové souvislosti k rozvoji inhibitorů faktoru IX (viz také bod 4.4).
Nefrotický syndrom byl hlášen velmi vzácně po pokusu o navození imunitní tolerance u pacientů s hemofilií B s inhibitory faktoru IX a anamnézou alergických reakcí.
U pacientů s hemofilií B se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru IX. Pokud se takové inhibitory vyskytnou, tento stav se projeví jako nedostatečná klinická odpověď. V takových případech se doporučuje kontaktovat specializované centrum pro léčbu hemofilie.
Existuje potenciální riziko tromboembolických příhod po podání faktoru IX s vyšším rizikem u přípravků s nižší čistotou. Použití faktoru IX nízké čistoty bylo spojeno s případy infarktu myokardu, diseminované intravaskulární koagulace, žilní trombózy a plicní embolie.
Použití faktoru IX vysoké čistoty je vzácně spojeno s těmito vedlejšími účinky.
Tabulkový seznam nežádoucích účinků
Tabulka uvedená níže je podle tříd orgánových systémů klasifikace MedDRA.
Frekvence byly hodnoceny podle následující konvence: velmi časté (>1/10); (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
|
Třídy orgánových systémů podle databáze MedDRA |
Nežádoucí účinky |
Frekvence |
|
Poruchy ledvin a močových cest |
Nefrotický syndrom |
Velmi vzácné |
|
Cévní poruchy |
Tromboembolické příhody |
Není známo |
|
Celkové poruchy a reakce v místě aplikace |
Vzácné | |
|
Poruchy imunitního systému |
Přecitlivělost (alergická reakce) |
Vzácné |
|
Poruchy krve a lymfatického systému |
Inhibice FIX |
Velmi vzácné |
Popis vybraných nežádoucích účinků
V klinické studii u 2 z 51 (4%) dříve neléčených pacientů (PUPs) se vytvořily inhibitory a u jednoho z těchto pacientů to bylo spojeno ve dvou případech s anafylaktickou reakcí.
Informace o virové bezpečnosti viz bod 4.4.
Pediatrická populace
U dětí se očekává, že frekvence, typ a závažnost nežádoucích účinků budou stejné jako u dospělých. Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Při použití lidského koagulačního faktoru IX nebyly hlášeny žádné příznaky předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, krevní koagulační faktor IX.
ATC kód: B02B D04
Faktor IX je glykoprotein s jediným řetězcem a molekulární váhou asi 68,000 Daltonů. Je to koagulační faktor závislý na vitaminu K, který je syntetizován v játrech. Faktor IX je aktivován faktorem XIa ve vnitřním koagulačním systému, a faktorem VII/komplexem tkáňových faktorů v zevním koagulačním systému.
Aktivovaný faktor IX v kombinaci s aktivovaným faktorem VIII pak aktivuje faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin pak konvertuje fibrinogen na fibrin a vytváří se koagulum.
Hemofilie B je na pohlaví vázaná dědičná porucha srážlivosti krve způsobená sníženými hladinami faktoru IX. Za následek má profuzní krvácení do kloubů, svalů nebo vnitřních orgánů vznikající buď spontánně nebo v důsledku náhodného nebo chirurgického traumatu. Substituční léčbou se zvýší plazmatické hladiny faktoru IX. Tak dojde k dočasné korekci nedostatku tohoto faktoru a nápravě tendence ke krvácení.
Po rekonstituci provedené podle doporučení (viz bod 6.6) je výsledný roztok čirý bezbarvý a izotonický přípravek o neutrálním pH s asi 100x vyšší aktivitou faktoru IX, než jaká se nachází ve stejném objemu plazmy.
5.2 Farmakokinetické vlastnosti
Krátkodobá infuze přípravku Mononine u 38 pacientů s hemofilií B (recovery study) měla za následek střední přírůstkový nárůst 1,71 IU/dl IU/kg tělesné hmotnosti (rozmezí 0,85-4,66). Střední terminální poločas v podskupině 28 pacientů byl 14,9 hodin (rozmezí 7,2-22,7).
Farmakokinetické údaje přípravku Mononine byly také stanoveny u 12 pacientů (před chirurgickým zákrokem) a před léčbou kontinuální infuzi přípravku Mononine.
|
Parametr |
Studie recovery (n=38) Střední hodnota (rozmezí) |
Plánované chirurgické zákroky (n=12) Střední hodnota (rozmezí) |
|
Přírůstkový recovery (IU/dl na IU/kg) |
1,71 (0,85-4,66) |
1,21 (0,83-1,60) |
|
Terminální poločas (hod) |
14,9 (7,2-22,7)++ |
16,4 (8,7-36,6) |
|
Počáteční poločas +++ (hod) |
n.a. |
2,46 (0,34-6,2) |
|
Plocha pod křivkou (AUC)+ (hod x kg/ml) |
n.a. |
0,254 (0,147-0,408) |
|
Objem při rovnovážném stavu (ml/kg) |
n.a. |
111(77-146) |
|
Clearance (ml/hod/kg) |
n.a. |
4,27 (2,45-6,78) |
|
Průměrná doba sledování (hod) |
n.a. |
27,4 (17,7-42,6) |
+ Standardizované na 1 IU/kg dávky n.a.: nejsou k dispozici
+ + Založené na podskupině 28 pacientů
+ + + Údaje pouze od 4 z 18 pacientů. U zbývajících 8 pacientů byl sledován jednoduchý jednokompartmentový model. Distribuční proces přípravku Mononine 500IU tedy byl sledován pouze příležitostně.
Pediatrická populace
U pacientů mladších 12 let věku nejsou k dispozici žádné farmakokinetické údaje.
5.3. Předklinické údaje vztahující se k bezpečnosti
Lidský plazmatický koagulační faktor IX je normální složkou lidské plazmy a působí jako endogenní faktor IX. Zjišťování toxicity po jedné dávce nemá význam, poněvadž vyšší dávky mohou vést k přetížení oběhu.
Testování toxicity opakovaných dávek na zvířatech je neproveditelné, poněvadž u nich dojde k tvorbě protilátek na heterogenní (lidskou) bílkovinu.
Poněvadž klinické zkušenosti neposkytují žádný náznak nádorového nebo mutagenního účinku koagulačního faktoru IX z lidské plazmy, nemají experimentální studie, zvláště u heterologních druhů, význam.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Histidin,
Mannitol,
Chlorid sodný,
Kyselina chlorovodíková nebo hydroxid sodný (v malém množství na úpravu pH)
Dodávané rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky, rozpouštědly a ředidly, s výjimkou těch, které jsou uvedeny v bodě 6.1 a fyziologického roztoku.
6.3 Doba použitelnosti
2 roky.
Chemická a fyzikální stabilita při použití byla prokázána po dobu 24 hodin při teplotě <25 °C.
Z mikrobiologického hlediska má být rekonstituovaný přípravek použitý okamžitě.
Po naředění (až do 1:10) rekonstituovaného roztoku přípravku Mononine, byla stabilita prokázána až po dobu 24 hodin.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C-8°C). Chraňte před mrazem. Uchovávejte injekční lahvičku ve vnějším obalu, aby byl přípravek chráněn před světlem.
Během doby použitelnosti (pokud je přípravek uchováván ve vnějším obalu) může být uchováván při pokojové teplotě (do 25°C) po dobu jednoho měsíce, během této doby nesmí být umístěn v chladničce. Počátek uchovávání při pokojové teplotě je třeba zapsat na obalu. Na konci tohoto období musí být přípravek použitý nebo znehodnocen.
6.5 Druh obalu a obsah balení
Vnitřní obaly
500 IU prášku a 5 ml rozpouštědla v injekčních lahvičkách (sklo typ I) s uzávěrem (chlorbutylová pryž).
Balení
Jedno balení s 500 IU obsahuje:
1 injekční lahvička s práškem 1 injekční lahvička s 5 ml vody na injekci Aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 10 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Všeobecné pokyny:
- Rekonstituce a odebrání musí být prováděny za aseptických podmínek.
- Roztok je zpravidla čirý nebo lehce opalescentní. Rekonstituovaný přípravek je třeba před podáním zkontrolovat vizuálně, zda po filtraci/odebrání (viz níže) neobsahuje částečky a nemá změněnou barvu. Nepoužívejte roztoky, které jsou zkalené nebo obsahují rezidua (usazeniny/částečky).
Rekonstituce
Zahřejte rozpouštědlo na pokojovou teplotu. Sejměte vyklápěcí víčka z injekčních lahviček s přípravkem i s rozpouštědlem, očistěte pryžové zátky antiseptickým roztokem a nechte je oschnout, než otevřete balení Mix2Vial.
1
2
3
4
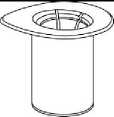
1. Otevřete balení Mix2Vial tím, že vyklopíte víčko. Nevytahujte Mix2Vial z blistru!
2. Postavte injekční lahvičku s rozpouštědlem na rovný a čistý povrch, držte ji pevně. Uchopte Mix2Vial společně s blistrem a zatlačte hrot modrého konce adaptéru rovně dolů skrz pryžovou zátku injekční lahvičky s rozpouštědlem.
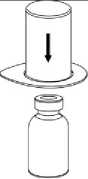
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle vzhůru. Přesvědčte se, že jste vytáhli jen blistrový obal a ne soupravu Mix2Vial.
4. Postavte injekční lahvičku s přípravkem na rovný a pevný povrch. Obraťte injekční lahvičku s rozpouštědlem spolu s nasazenou soupravou Mix2Vial a zatlačte hrot průhledného konce adaptéru přímo dolů skrz pryžovou zátku injekční lahvičky s přípravkem. Rozpouštědlo se samo automaticky nasaje do injekční lahvičky s přípravkem._
fCZJ]
5
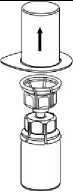
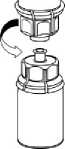
5. Uchopte jednou rukou tu část soupravy Mix2Vial, kde je injekční lahvička s přípravkem a druhou rukou tu část, kde je injekční lahvička od rozpouštědla a odšroubujte je od sebe opatrně proti směru hodinových ručiček na dvě části. Odstraňte injekční lahvičku od rozpouštědla s připojeným modrým adaptérem soupravy Mix2Vial.

6

7
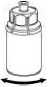
6. Jemně otáčejte injekční lahvičkou s přípravkem s připojeným průhledným adaptérem, dokud se prášek zcela nerozpustí. Netřepejte s injekční lahvičkou.
7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Zatímco je injekční lahvička s přípravkem dnem dolů, spojte injekční stříkačku pomocí koncovky Luer lock šroubováním ve směru hodinových ručiček se soupravou Mix2Vial. Vstříkněte vzduch do injekční lahvičky s přípravkem.
Natáhnutí a aplikace:

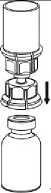
8
9
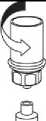
8. Zatímco držíte píst stříkačky stlačený, obraťte celý systém dnem vzhůru. Pomalým vytahováním pístu natáhněte roztok do stříkačky.
9. Po natažení roztoku do stříkačky uchopte pevně válec stříkačky (píst stále směřuje dolů) a odpojte průhledný adaptér soupravy Mix2Vial od stříkačky odšroubováním proti směru hodinových ručiček.
Aplikujte okamžitě pomalu jako intravenózní injekci nebo infuzi (viz bod 4.2).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH, 35041 Marburg, Německo
8. REGISTRAČNÍ ČÍSLO(A)
75/024/06-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25.01.2006
Datum posledního prodloužení registrace: 04.06.2008
10. DATUM REVIZE TEXTU
25.11.2015
12/12