Lutrate Depot 3,75 Mg
Sp.zn.sukls31482/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Lutrate Depot 3,75 mg
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým uvolňováním
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje leuprorelini acetas 3,75 mg (což odpovídá leuprorelinum 3,57 mg). Jeden ml připravené suspenze obsahuje leuprorelini acetas 1,875 mg.
Pomocné látky se známým účinkem:
Jedna injekční lahvička obsahuje 1,3 - 2,2 mg (< 1 mmol) sodíku (jako sodnou sůl karmelosy). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým uvolňováním. Prášek: bílý až téměř bílý prášek.
Rozpouštědlo: čirý bezbarvý roztok bez viditelných částic (pH 5,0 - 7,0).
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Lutrate Depot je určen k paliativní léčbě lokálně pokročilého nebo metastatického karcinomu prostaty.
4.2 Dávkování a způsob podání
Dávkování
Obvyklá doporučená dávka přípravku Lutrate Depot je 3,75 mg v jedné depotní injekci podávané v měsíčních intervalech formou jednorázové intramuskulární injekce.
Lutrate Depot musí být aplikován pod dohledem lékaře či kvalifikovaného zdravotnického pracovníka.
Depotní léková forma přípravku Lutrate Depot obsahuje dávku umožňující průběžné uvolňování leuprorelin-acetátu v průběhu jednoho měsíce. Lyofilizát má být rekonstituován a podán v jedné intramuskulární injekci v měsíčních intervalech. Přípravek nesmí být podán intraarteriálně či intravenózně. Mikrosférický prášek Lutrate Depot má být rekonstituován bezprostředně před aplikací intramuskulární injekce. Stejně jako u ostatních léčivých přípravků podávaných injekčně, je třeba místo vpichu pravidelně měnit.
Léčba přípravkem Lutrate Depot nemá být přerušena, projeví-li se remise či zlepšení stavu.
Odezva na léčivý přípravek Lutrate Depot má být sledována pomocí pravidelného měření hladin testosteronu a prostatického specifického antigenu (PSA) v séru. Klinické studie ukázaly, že se hladina testosteronu během prvních 4 dnů léčby zvýšila u většiny nekastrovaných pacientů. V průběhu dalších 3-4 týdnů se hladina snížila až na kastrační úroveň. Dále se kastrační úroveň (testosteron méně než 0,5 ng/ml) udržovala po celou dobu trvání terapie.
Pokud se odezva pacienta nejeví jako optimální, je vhodné ověřit, zda hladiny testosteronu v séru dosáhly kastrační úrovně či na ní setrvávají. Na počátku léčby se občas objeví přechodné zvýšení hladin kyselé fosfatázy, hodnoty se však obvykle vrátí k normálním či téměř normálním hodnotám do 4. týdne léčby.
Délka trvání léčby
Přípravek Lutrate Depot se podává jako intramuskulární injekce aplikovaná jednou měsíčně.
Zvláštní skupiny pacientů
Pediatrická populace
Bezpečnost a účinnost přípravku Lutrate Depot u pediatrických pacientů nebyla stanovena. Proto se nedoporučuje užití přípravku Lutrate Depot u dětí a dospívajících, dokud nebudou údaje o bezpečnosti a účinnosti dostupné.
Renální/hepatální nedostatečnost
Farmakokinetické vlastnosti přípravku Lutrate Depot nebyly stanoveny u pacientů s poruchou funkce jater či ledvin.
Starší pacienti
V klinických studiích provedených s přípravkem Lutrate Depot byl průměrný věk subjektů 71,6 ± 9,2 let. Údaje o přípravku tedy reflektují farmakokinetiku, účinnost a bezpečnost přípravku Lutrate Depot u této skupiny pacientů.
Způsob podání
Přípravek Lutrate Depot může být podán pouze intramuskulárně. Nepodávejte jej žádnou jinou cestou. Pokud by byl přípravek náhodně podán subkutánně, pak musí být pacient pečlivě sledován, neboť údaje o podání přípravku Lutrate Depot jinou cestou než intramuskulární nejsou k dispozici. Informace o rekonstituci léčivého přípravku před podáním jsou uvedeny v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, analoga hormonu uvolňujícího luteinizační hormon (LHRH) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. V lékařské literatuře byly popsány anafylaktické reakce na syntetický LHRH nebo agonisty LHRH.
Předchozí orchiektomie.
Přípravek Lutrate Depot nesmí být užíván v monoterapii u pacientů s nádorem prostaty a současně prokázanou kompresí míchy nebo míšními metastázami.
Přípravek Lutrate Depot je kontraindikován u žen.
Přípravek Lutrate Depot je kontraindikován u pediatrických pacientů.
4.4 Zvláštní upozornění a opatření pro použití
V počátečních fázích léčby přípravkem Lutrate Depot se může, stejně jako u jiných agonistů LHRH, objevit přechodné zvýšení hladin testosteronu. V některých případech to může být spojeno se vzplanutím nebo exacerbací růstu nádoru, což má za následek dočasné zhoršení symptomů nádoru prostaty. Tyto příznaky obvykle v průběhu léčby vymizí. Vzplanutí samotné se může v některých případech projevit systémovými nebo neurologickými příznaky (jako je bolest kostí ...). Při použití jiných agonistů LHRH byly popsány případy orchiatrofie a gynekomastie.
Léčba přípravkem má být ihned ukončena, objeví-li se u pacienta známky či symptomy naznačující anafylaxi/anafylaktickou reakci (dyspnoe, astma, rinitida, angioneurotický edém nebo otok hlasivek, hypotenze, urtikarie, vyrážka, pruritus nebo intersticiální pneumonitida). Pacienti mají být před zahájením léčby poučeni o nutnosti přerušit léčbu a kontaktovat lékaře, jakmile se u nich objeví některý z výše uvedených symptomů. Pacienti, u nichž se již projevila hypersensitivita na leuprolid, mají být pečlivě monitorování a přípravek Lutrate Depot jim nemá být znovu podán.
U pacientů léčených leuprorelin-acetátem byly pozorovány jednotlivé případy ureterální obstrukce (s nebo bez hematurie) a komprese míchy nebo metastatické vertebrální léze, které mohou přispět k paralýze s nebo bez fatálních komplikací. Léčba pacientů s rizikem ureterální obstrukce, komprese míchy či metastatických vertebrálních lézí má být důkladně zvážena a pacienti mají být v prvních týdnech léčby pečlivě monitorováni. U těchto pacientů pak má být zvážena profylaktická léčba antiandrogeny.
Pokud se objeví urologické nebo neurologické komplikace, je třeba aplikovat příslušná specifická opatření.
U pacientů léčených agonisty GnRH, jako je leuprorelin-acetát, je zvýšené riziko rozvoje deprese, která může být i závažná. Pacienti mají být s ohledem na toto riziko informováni a v případě výskytu depresivních symptomů léčeni odpovídajícím způsobem.
V lékařské literatuře bylo popsáno snížení kostní denzity u mužů, kteří podstoupili orchiektomii nebo kteří byli léčeni agonisty LHRH. Zavedení antiandrogenní terapie do léčebného režimu sice snižuje ztrátu kostní hmoty, ale zvyšuje riziko jiných nežádoucích účinků, jako jsou problémy se srážením krve či edém. Pokud jsou antiandrogeny podávány delší dobu, je třeba věnovat velkou pozornost kontraindikacím a opatřením spojeným s tímto užíváním. Léčba pacientů s rizikem osteoporózy nebo s osteoporózou v anamnéze má být důkladně zvážena a pacienti mají být pečlivě monitorováni v průběhu léčby leuprorelin-acetátem.
Po podání leuprorelin-acetátu byla hlášena hepatální dysfunkce a ikterus se zvýšenými hladinami jaterních enzymů. Proto je nutné pečlivé sledování a případná aplikace příslušných opatření.
Odezva na terapii přípravkem Lutrate Depot má být sledována pomocí klinických parametrů a pravidelným měřením hladin testosteronu a PSA v séru.
U pacientů se mohou projevit změny metabolismu (např. intolerance glukózy nebo zhoršení již existujícího diabetu), hypertenze, změny tělesné hmotnosti nebo kardiovaskulární poruchy. Jak lze očekávat u této skupiny léků, může se rozvinout nebo zhoršit již existující diabetes, a tedy může být zapotřebí častějších kontrol hladiny krevní glukózy u diabetických pacientů v průběhu léčby přípravkem Lutrate Depot. Před započetím léčby je nutno pečlivě zvážit léčbu pacientů s rizikem metabolických nebo kardiovaskulárních poruch a pak je adekvátně monitorovat v průběhu androgenní deprivační terapie. Léčba leuprorelin-acetátem má za následek potlačení hypofýzo-gonádového systému. Výsledky diagnostických testů hypofyzárních gonádotropních a gonádových funkcí provedených během a po léčbě leuprorelin-acetátem mohou být zkresleny.
U pacientů léčených leuprorelin-acetátem bylo hlášeno prodloužení protrombinového času.
Po podání leuprorelin-acetátu byly hlášeny křeče. Tyto případy se týkaly pacientů, kteří měli v anamnéze křeče, epilepsii, cerebrovaskulární poruchy, anomálie nebo tumory centrálního nervového systému a pacientů, kteří současně užívali přípravky, které mohou vyvolat křeče, jako je např. bupropion a selektivní inhibitory zpětného vychytávání serotoninu (SSRI). Křeče byly také hlášeny u pacientů bez přítomnosti výše uvedených podmínek.
Leuprorelin-acetát má být podáván s příslušnými opatřeními v případě kardiovaskulárního onemocnění (včetně kongestivního srdečního selhání), tromboembolismu, edému, deprese a hypofyzární apoplexie.
Leuprorelin-acetát má být podáván s opatrností pacientům s prokázanými poruchami krvácivosti, trombocytopenií nebo léčených antikoagulanty.
Pozor si mají dávat aktivně sportující, protože přípravek Lutrate Depot obsahuje látku, která může dávat pozitivní výsledek při dopingové kontrole.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v lahvičce. Je tedy v podstatě bez obsahu sodíku.
Androgen-deprivační léčba může prodlužovat QT interval.
Před zahájením léčby přípravkem Lutrate Depot má lékař zvážit poměr přínosů a rizik, včetně rizika Torsade de pointes, u pacientů s rizikovými faktory pro prodloužení QT intervalu v anamnéze a u pacientů souběžně užívajících léčivé přípravky, které mohou prodlužovat QT interval (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
S leuprorelin-acetátem nebyly provedeny žádné farmakokinetické studie interakcí. Vzhledem k tomu, že leuprorelin-acetát je peptid, jenž je primárně rozkládán peptidázou a nikoliv enzymy cytochromu P-450, jak je uvedeno ve specifických studiích, a také vzhledem k faktu, že léčivý přípravek se váže na plazmatické proteiny jen z přibližně 46 %, dá se předpokládat, že lékové interakce by se neměly objevovat.
Kvůli souvislosti androgen-deprivační léčby a prodloužení QT intervalu má být pečlivě zvážena souběžná léčba přípravkem Lutrate Depot s léčivými přípravky, o kterých je známo, že prodlužují QT interval, a léčba přípravky, které mohou vyvolat Torsade de pointes, jako antiarytmika třídy I A (např. chinidin, disopyramid), třídy III (např. amiodaron, sotalol, dofetilid, ibutilid), methadon, moxifloxacin, antipsychotika a další (viz bod 4.4.).
4.6 Fertilita, těhotenství a kojení
Přípravek Lutrate Depot není určen pro podávání ženám.
Injekce leuprorelin-acetátu může při podání v těhotenství způsobit poškození plodu.
Spontánní abort po aplikaci léčiva během těhotenství tedy nelze vyloučit.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Schopnost řídit a obsluhovat stroje může být ovlivněna z důvodů poruch vidění a závratí.
4.8 Nežádoucí účinky
Pokud není uvedeno jinak, je následující bezpečnostní profil přípravku Lutrate Depot založen na výsledcích klinické studie fáze III, v níž byli pacienti s nádorem prostaty léčeni 6 intramuskulárními jednoměsíčními dávkami přípravku Lutrate Depot a sledováni až po celkovou dobu 26 týdnů. Většina hlášených nežádoucích účinků byly obvyklé reakce spojené s terapií suprese testosteronu.
Nejčastěji hlášené nežádoucí účinky přípravku Lutrate Depot jsou návaly horka, bolest v místě vpichu, podráždění v místě vpichu, noční pocení a bolest hlavy.
Následující nežádoucí účinky z klinických studií byly klasifikovány dle jednotlivých tříd orgánových systémů a podle klesající incidence (velmi časté > 1/10, časté > 1/100 až < 1/10, méně časté > 1/1 000 až < 1/100, vzácné > 1/10 000 až < 1/1 000 a velmi vzácné < 1/10 000).
Tabulka 1
Frekvence výskytu nežádoucích účinků během terapie přípravkem Lutrate Depot
|
Třídy orgánových systémů |
Frekvence výskytu |
Nežádoucí účinek |
|
Poruchy metabolismu a výživy |
Časté |
Zvýšená chuť k jídlu |
|
Méně časté |
Anorexie, hypercholesterolemie, hyperlipidemie | |
|
Psychiatrické poruchy |
Méně časté |
Poruchy spánku, insomnie, snížené libido, změny nálady a deprese* |
|
Poruchy nervového systému |
Časté | |
|
Méně časté |
Somnolence | |
|
Poruchy ucha a labyrintu |
Méně časté | |
|
Cévní poruchy |
Velmi časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hyperbilirubinemie |
|
Poruchy kůže a podkožní tkáně |
Časté |
Hyperhidróza, noční pocení, studený pot |
|
Méně časté |
Periorbitální edém, urtikarie, pruritus | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté | |
|
Méně časté |
Artralgie, svalové spazmy, bolest v končetinách | |
|
Poruchy ledvin a močových cest |
Méně časté |
Retence moči, inkontinence, polakisurie |
|
Poruchy reprodukčního systému a prsu |
Časté |
Erektilní dysfunkce |
|
Méně časté |
Otok prsou, citlivost prsou, poruchy ejakulace | |
|
Srdeční a cévní poruchy |
Není známo |
Prodloužení QT intervalu (viz body 4.4 a 4.5) |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Únava, astenie, pyrexie, lokální nežádoucí účinky (viz tabulka 2) |
|
Méně časté |
Slabost, pocity horka a chladu, pocit roztřesenosti | |
|
Vyšetření |
Méně časté |
Zvýšení hladin AST, ALT, zvýšená hladina bilirubinu, zvýšená hladina gama-glutamyltransferázy |
* Během post-marketingového sledování byly změny nálady a deprese při dlouhodobém podávání klasifikovány jako časté nežádoucí účinky.
S ohledem na závažnost, 98 % všech nežádoucích účinků spojených s léčbou bylo mírného nebo středně závažného stupně. Jako mírné bylo hlášeno 89 % návalů horka a jako středně závažné 9 %. Dva případy (0,2 %) návalů horka byly hlášeny jako závažné.
Během studie bylo 29 pacienty (18,1 %) hlášeno celkem 35 lokálních nežádoucích účinků v místě aplikace.
Lokální nežádoucí účinky po podání přípravku Lutrate Depot jsou typické i pro ostatní podobné přípravky podávané formou intramuskulární injekce. Nejčastěji hlášené nežádoucí účinky byly bolest, podráždění, diskomfort, pohmožděniny a erytém v místě vpichu. Jako méně časté nežádoucí účinky byly hlášeny reakce v místě vpichu, otok, poranění a hemoragie (Tabulka 2).
Tabulka 2
Četnost výskytu lokálních nežádoucích účinků u pacientů během léčby přípravkem Lutrate Depot
%
|
v místě aplikace - frekvence | ||
|
Časté |
Bolest v místě vpichu |
8,1 |
|
Podráždění v místě vpichu |
4,4 | |
|
Diskomfort v místě vpichu |
1,9 | |
|
Erytém v místě vpichu |
1,3 | |
|
Pohmoždění v místě vpichu |
1,3 | |
|
Méně časté |
Reakce v místě vpichu |
0,6 |
|
Otok v místě vpichu |
0,6 | |
|
Poranění v místě vpichu |
0,6 | |
|
Hemoragie v místě vpichu |
0,6 |
* Subjekty mohou být zařazeny pod více než jednu kategorii.
Při opakovaném podávání přípravku Lutrate Depot byly hlášeny jako opakující se nežádoucí účinky otok (0,6 %), bolest (0,6 %), zhmoždění (0,6 %) a podráždění (0,6 %). Tyto reakce byly hlášeny jako nezávažné a mírné. Žádný pacient nepřerušil léčbu z důvodu lokálních nežádoucích účinků.
V průběhu fáze I klinické studie (CRO-02-43) se zdravými dobrovolníky a jednorázově podávaným přípravkem Lutrate Depot GP-Pharm 7,5 mg byl hlášen jeden případ zatvrdnutí v místě vpichu.
Další nežádoucí účinky hlášené v průběhu léčby leuprorelin-acetátem zahrnovaly impotenci, pokles libida (obojí v důsledku deprivace testosteronu), periferní edém, pulmonální embolismus, palpitaci, myalgii, svalovou slabost, třesavku, dyspnoi, periferní vertigo, vyrážku, amnézii, poruchy vidění a citlivost kůže. Vzácně byl po podání krátkodobě i dlouhodobě působících agonistů LHRH hlášen infarkt v místech již existujících adenomů hypofýzy. Vzácně byly hlášeny případy trombocytopenie a leukopenie. Hlášeny byly změny v toleranci glukózy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Klinická data o akutním předávkování přípravkem Lutrate Depot nebo leuprorelin-acetátem nejsou dostupná. V klinických studiích při subkutánním denním podávání leuprorelin-acetátu pacientům s nádorem prostaty nevyvolaly dávky 20 mg/den až po dobu 2 let žádné jiné nežádoucí účinky než ty, které byly pozorovány při dávce 1 mg/den.
Ve studiích na zvířatech vyvolaly dávky až 500násobně vyšší než humánní doporučená denní dávka dyspnoi, sníženou aktivitu a lokální podráždění v místě vpichu.
V případě předávkování mají být pacienti pečlivě monitorováni a léčba má být symptomatická a podpůrná.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Endokrinní terapie. Hormony a příbuzné látky. Analoga gonadotropin-releasing hormonu, ATC kód: L02AE02.
Chemický název leuprorelin-acetátu je 5-oxo-L-prolyl-L-histidyl-L-tryptofyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-L-prolyl-ethylamid.
Po perorálním podání je leuprorelin-acetát neaktivní vzhledem ke špatné membránové permeabilitě a téměř kompletní inaktivaci intestinálními proteolytickými enzymy.
Leuprorelin-acetát je silný agonista LHRH při krátkodobé nebo intermitentní léčbě, avšak při kontinuálním, nepulzatilním, podání analoga LHRH indukují inhibici gonadotropinové sekrece a supresi testikulární steroidogeneze.
Po navázání na LHRH receptory hypofýzy vyvolá leuprorelin-acetát počáteční zvýšení cirkulujících hladin luteinizačního hormonu (LH) a folikuly stimulujícího hormonu (FSH), což vede k rychlému vzestupu hodnot testosteronu a dihydrotestosteronu. Avšak po 5 až 8 dnech od podání přípravku způsobí analoga LHRH desensitizaci LHRH receptorového komplexu a/nebo potlačení funkce adenohypofýzy. Vzhledem k tomu, že se na povrchu buněk nachází méně receptorů, je buněčná stimulace snížena a syntéza a sekrece gonadotropinu je nižší. Po několika týdnech terapie agonistou LHRH je sekrece LH a FSH nakonec potlačena. V důsledku toho Leydigovy buňky ve varlatech přestávají produkovat testosteron a koncentrace testosteronu v séru v průběhu 2 až 4 týdnů po zahájení léčby klesá na kastrační úroveň (méně než 0,5 ng/ml).
Do otevřené, multicentrické klinické studie s opakovanými dávkami přípravku Lutrate Depot bylo zahrnuto 160 pacientů s nádorem prostaty bez předchozí systémové protinádorové terapie, hormonální terapie nádoru prostaty, prostatické operace nebo orchiektomie. Ve studii byla hodnocena účinnost a bezpečnost přípravku Lutrate Depot při podání pacientům s nádorem prostaty, kteří mohli mít prospěch z androgenní deprivační terapie. Přípravek Lutrate Depot byl podáván intramuskulárně v 6 dávkách jednou měsíčně.
Hladiny testosteronu byly monitorovány v různých dnech v průběhu 168 dnů. Podle očekávání, střední hladiny testosteronu po první injekci rychle vzrostly z výchozí hladiny (4,119 ± 1,341 ng/ml) a dosáhly maximálních hladin (Cmax) 6,598 ± 2,249 ng/ml třetí den. Po dosažení maxima hladiny testosteronu klesaly a do 21. dne 78,7 % hodnotitelných pacientů dosáhlo kastrační úrovně (definována jako méně než 0,5 ng/ml testosteronu). Do 28. dne dosáhlo kastrační úroveň 96,8 % pacientů a 73,1 % dosáhlo hladiny < 0,2 ng/m (Obrázek 1).
Obrázek 1
Střední (± SD) plazmatická hladina testosteronu v průběhu léčby 6 i.m. injekcemi přípravku Lutrate Depot podávanými jednou za měsíc
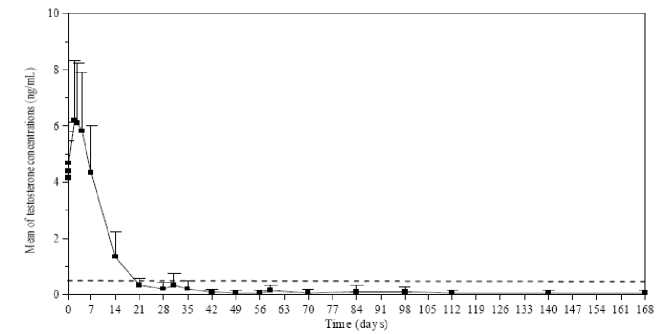
Sekundární cíl účinnosti zahrnoval měření sérových koncentrací LH, FSH a PSA. Do 14 dnů a čtvrtého dne po první injekci přípravku Lutrate Depot, klesla průměrná sérová hladina LH a FSH pod výchozí koncentrace. Od 28. dne koncentrace zůstávaly hluboko pod výchozí hodnotou koncentrace až do konce studie. Střední sérové hladiny PSA v průběhu léčby postupně klesaly (první měsíc) a dále zůstávaly pod výchozí hladinou až do konce studie. V průběhu studie však byla pozorována široká interindividuální variabilita v koncentracích PSA.
Frekvence zvýšených hladin testosteronu byla 11,8 %, z toho u tzv. „mini flare“ výkyvů hladiny byla frekvence 10,5 %. Nebyly hlášeny žádné nežádoucí účinky spojené s léčbou, které by naznačovaly klinické známky flare testosteronu (retence moči, komprese míchy nebo zhoršení bolesti kostí) u žádného pacienta se zvýšenou hladinou testosteronu.
5.2 Farmakokinetické vlastnosti
Absorpce
Po třech injekcích podávaných jednou za měsíc přípravku Lutrate Depot pacientům s nádorem prostaty (n = 12) byla maximální plazmatická koncentrace leuprorelin-acetátu v těchto 3 cyklech podobná. Po prvním podání (Den 0 - 28) byla Cmax 13 145,6 ± 3 070,6 pg/ml. Střední čas pro dosažení Cmax (Tmax) byl 0,04 dne, což odpovídá 0,96 h (rozmezí 0,96 - 4,08 h).
Distribuce
Studie distribuce nebyly s přípravkem Lutrate Depot provedeny. Avšak, u zdravých mužských dobrovolníků byl střední distribuční objem v ustáleném stavu leuprorelin-acetátu po i.v. bolusu 1,0 mg 27 l. Při hodnocení in vitro kolísala vazba na lidské plazmatické proteiny od 43 % do 49 %.
Metabolismus
Studie metabolismu nebyly s přípravkem Lutrate Depot provedeny. Avšak, podání dávky 1,0 mg leuprorelin-acetátu zdravým mužským dobrovolníkům jako i.v. bolus ukázalo, že střední systémová clearance byla 7,6 l/h s konečným eliminačním poločasem přibližně 3 hodiny - založeno na 2kompartmentovém modelu.
Předpokládá se, že leuprorelin je metabolizován na menší inaktivní peptidy, které mohou být vyloučeny nebo dále katabolizovány.
Eliminace
Studie eliminace nebyly s přípravkem Lutrate Depot provedeny. Avšak, po podání leuprorelin-acetátu 3 pacientům bylo méně než 5 % dávky vyloučeno močí v nezměněné formě a ve formě metabolitu M-I.
Zvláštní skupiny pacientů
Renální/hepatální nedostatečnost
Farmakokinetika léčivého přípravku nebyla u pacientů s poruchou funkce ledvin či jater stanovena.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podání a genotoxicity provedených s leuprorelin-acetátem neodhalily žádné zvláštní riziko pro člověka.
Dle očekávání v souladu se známými farmakologickými vlastnostmi, prokázaly neklinické studie reverzibilní účinky na reprodukční systém. Ve studiích reprodukční toxicity nebyla zjištěna žádná teratogenita leuprorelin-acetátu. U králíků však byla pozorována embryotoxicita/letalita.
Studie kancerogenity provedené na potkanech, jimž byl leuprorelin-acetát podáván subkutánně (0,6 - 4 mg/kg/den), ukázaly nárůst hypofyzárních adenomů v závislosti na dávce. Dále bylo pozorováno signifikantní zvýšení, ale nezávislé na dávce, výskytu adenomů pankreatických Langerhansových buněk u samic a testikulárních intersticiálních adenomů u samců, přičemž nejvyšší incidence byla zjištěna u skupiny, jíž byla podávána nízká dávka. Podávání leuprorelin-acetátu vedlo k inhibici růstu určitých hormon-dependentních nádorů (prostatické tumory u potkaních samců Noble a Dunning a DMBA-indukované tumory mléčných žláz u potkaních samic). Ve studiích kancerogenity na myších však tyto účinky pozorovány nebyly. Studie kancerogenity s přípravkem Lutrate Depot nebyly provedeny.
Studie in vitro a in vivo ukázaly, že leuprorelin-acetát není mutagenní. Studie mutagenity s přípravkem Lutrate Depot nebyly provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Lyofilizát (injekční lahvička):
Polysorbát 80 Mannitol (E 421)
Sodná sůl karmelosy (E 466)
Triethyl-citrát Polyglaktin (1:1)
Rozpouštědlo (předplněná injekční stříkačka):
Mannitol (E 421)
Hydroxid sodný (k úpravě pH)
Kyselina chlorovodíková 35% (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Pro rekonstituci přípravku Lutrate Depot nesmí být použito jiné rozpouštědlo, než sterilní rozpouštědlo dodávané s přípravkem.
6.3 Doba použitelnosti
3 roky (neotevřené)
Po rekonstituci rozpouštědlem použijte vzniklou suspenzi okamžitě.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
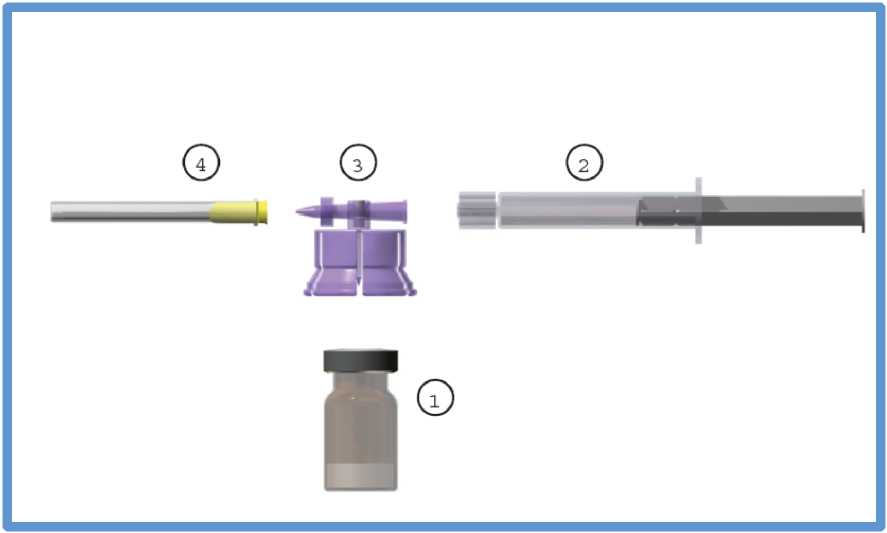
Komerční kit obsahuje:
1. Jednu (1) skleněnou injekční lahvičku (sklo hydrolytické třídy I) obsahující 3,75 mg leuprorelin-acetátu ve formě lyofilizovaného prášku, injekční lahvička je uzavřená brombutylovou zátkou a hliníkovým uzávěrem s modrým plastikovým odtrhovacím víčkem.
2. Jednu (1 ) skleněnou předplněnou injekční stříkačku (sklo hydrolytické třídy I) obsahující 2 ml rozpouštědla, uzavřenou elastomerovým uzávěrem.
3. Jeden (1) adaptační systém - polykarbonát/HDPE.
4. Jednu (1) sterilní jehlu velikosti 20G.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Způsob podání
Injekční lahvička s přípravkem Lutrate Depot ve formě mikrosférického prášku má být rekonstituována těsně před podáním intramuskulární injekce. Nutné je zajištění aseptické techniky.
Rekonstituovaný přípravek je mléčně bílá suspenze.
Pro rekonstituci přípravku Lutrate Depot nesmí být použito žádné jiné rozpouštědlo než rozpouštědlo přiložené v balení.
Před podáním musí být přípravek vytemperován na pokojovou teplotu.
Přípravek Lutrate Depot rekonstituujte dle následujících instrukcí:

2

i
Nasaďte adaptační systém (fialový) na lahvičku až do slyšitelného zacvaknutí.
3
Připojte bílou úchytku k injekční stříkačce s rozpouštědlem. Sejměte gumový vršek z injekce a připojte ji k adaptačnímu systému.
5

Držte injekční stříkačku, dobře spojenou s lahvičkou, ve vzpřímené poloze a zvolna
S lahvičkou, stále spojenou s injekční stříkačkou, jemně třepejte po dobu asi jedné minuty do získání
6

Otočte celý systém dnem vzhůru a opatrně vytahujte píst, abyste natáhli suspenzi léčiva z lahvičky
stlačujte píst injekční stříkačky, abyste přemístili všechno rozpouštědlo do lahvičky.
homogenní mléčně bílé suspenze.
do injekční stříkačky.
7

Otáčením horní části adaptačního systému proti směru hodinových ručiček odpojte injekční stříkačku a jehlu. Léčivý přípravek je připraven k aplikaci.
8

Očistěte místo vpichu tampónky vlhčenými alkoholem a nechte kůži oschnout. Aplikujte injekci suspenze intramuskulárně do vnějšího horního kvadrantu hýžďového svalu.
Na stěnách injekční lahvičky mohou ulpět zbytky přípravku, což není na závadu. Během výroby přípravku je injekční lahvička naplněna nadbytkem produktu, aby se zajistila konečná dávka 3,75 mg leuprorelin-acetátu.
Přípravek je určen pro jednorázové použití. Veškerý zbylý roztok musí být zlikvidován.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Angelini Pharma Osterreich GmbH Gewerbestrasse 18 - 20 Gewerbegebiet Klein-Engersdorf 2102 Bisamberg Rakousko
8. REGISTRAČNÍ ČÍSLO
44/616/12-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21.11.2012
Datum posledního prodloužení registrace: 9.6.2016
10. DATUM REVIZE TEXTU
3.8.2016
11