Kyprolis
Tento dokument je příbalovou informací k léčivému přípravku, který je předmětem specifického léčebného programu. Příbalová informace byla předložena žadatelem o specifický léčebný program. Nejedná se o dokument schválený Státním ústavem pro kontrolu léčiv.
ÚPLNÁ INFORMACE PRO PRESKRIPCI: OBSAH1
1. INDIKACE A POUŽITÍ
2. DÁVKOVÁNÍ A ZPŮSOB PODÁNÍ
2.1 Pokyny pro dávkování
2.2 Sledování hydratace a tekutin
2.3 Premedikace dexamethasonem
2.4 Úprava dávkování na základě toxicity
2.5 Zvláštní upozornění pro podávání
2.6 Rekonstituce a příprava pro intravenózní podání
3. DÁVKOVAČI' FORMY A SÍLY
4. KONTRAINDIKACE
5. ZVLÁŠTNÍ UPOZORNĚNÍ A OPATŘENÍ PRO POUŽITÍ
5.1 Srdeční zástava, městnavé srdeční selhání, ischemie myokardu
5.2 Plicní hypertenze
5.3 Plicní komplikace
5.4 Infuzní reakce
5.5 Syndrom nádorového rozpadu
5.6 Trombocytopenie
5.7 Jaterní toxicita a jaterní selhání
5.8 Embryofetální toxicita
6. NEŽÁDOUCÍ ÚČINKY
6.1 Bezpečnostní zkušenosti z klinických studií
7. LÉKOVÉ INTERAKCE
8. POUŽITÍ U SPECIÁLNÍCH POPULACÍ PACIENTŮ
8.1.Těhotenství
8.3 Kojící ženy
8.4 Použití u pediatrické populace
8.5 Použití u starších lidí
8.6 Porucha funkce ledvin
8.7 Porucha funkce jater
8.8 Poškození srdce
10. PŘEDÁVKOVÁNÍ
11. POPIS
12. KLINICKÁ FARMAKOLOGIE
12.1 Mechanismus účinku
12.2 Farmakodynamika
12.3 Farmakokinetika
13. NEKLINICKÁ TOXIKOLOGIE
13.1 Karcinogeneze, mutageneze a postižení fertility
13.2 Toxikologie a/anebo farmakologie u zvířat
14. KLINICKÉ STUDIE
14.1 Relaps mnohočetného myelomu
16. JAK SE LÉK DODÁVÁ, UCHOVÁVÁNÍ LÉKU A ZACHÁZENÍ S NÍM
16.1 Jak se lék dodává
16.2 Uchovávání léku a zacházení s ním
17. INFORMACE O PORADENSKÉ SLUŽBĚ PRO PACIENTY
Kapitoly nebo podkapitoly vynechané v úplné informaci pro preskripci nejsou v tomto seznamu uvedeny.
1. INDIKACE A POUŽITÍ
KYPROLIS je indikován k léčbě pacientů s mnohočetným myelomem, kteří již prodělali nejméně dvě léčby včetně léčby bortezomibem a imunomodulační látkou a byla u nich prokázána progrese onemocnění 60. den nebo během 60 dní po dokončení poslední léčby. Lék je schválen na základě míry odpovědi na léčbu [viz Klinické studie (14.1)]. Klinický přínos, jako je zlepšení přežití nebo zlepšení symptomů, nebyl ověřen.
2. DÁVKOVÁNÍ A ZPŮSOB PODÁVÁNÍ
2.1 Pokyny pro dávkování
KYPROLIS se podává intravenózně během 2 až 10 minut dva po sobě jdoucí dny každý týden po dobu tří týdnů (1., 2., 8., 9., 15. a 16. den), po nichž následuje přestávka trvající 12 dní (17. až 28. den). Každé 28denní období se považuje za jeden léčebný cyklus (tabulka č. 1)
Doporučená dávka přípravku KYPROLIS v 1. cyklu je 20 mg/m2/den. v případě snášenlivosti léku v 1. cyklu se v 2. cyklu dávka zvýší na 27 mg/m2/den a pokračuje se v ní i v dalších cyklech. V léčbě se může pokračovat až do progrese choroby nebo do vzniku nepřijatelné toxicity [viz Dávkování a způsob podání (2.4)].
Dávka se vypočítá s použitím skutečné plochy povrchu těla pacienta na začátku léčby. Pacienti s plochou povrchu těla nad 2,2 m2 by měli dostat dávku náležící pro plochu povrchu těla 2,2 m2. Úpravy dávky nejsou nutné při změnách tělesné váhy menších nebo rovných 20 %.
Tab. 1 Dávkovači režim přípravku KYPROLIS u pacientů s mnohočetným myelomem
|
KYPROLIS (20 mg/m2): |
1. cyklus | |||||||||
|
1. týden |
2. týden |
3. týden |
4. týden | |||||||
|
1. den |
2. den |
3.-7. den |
8. den |
9. den |
10.-14. den |
15. den |
16. den |
17. -21. den |
22.-28. den | |
|
20 |
20 |
Nepodá se |
20 |
20 |
Nepodá se |
20 |
20 |
Nepodá se |
Nepodá se | |
|
KYPROLIS (27 mg/m2): |
2. cyklus a další cykly3 | |||||||||
|
1. týden |
2. týden |
3. týden |
4. týden | |||||||
|
1. den |
2. den |
3.-7. den |
8. den |
9. den |
10.-14. den |
15. den |
16. den |
17.- 21. den |
22.-28. den | |
|
27 |
27 |
Nepodá se |
27 |
27 |
Nepodá se |
27 |
27 |
Nepodá se |
Nepodá se | |
a V případě snášenlivosti v předchozím cyklu
2.2 Sledování hydratace a tekutin
Při léčbě přípravkem KYPROLIS je nutné pacienty hydratovat, aby se snížilo riziko renální toxicity a syndromu rozpadu nádoru (TLS) [viz Zvláštní upozornění a opatření pro použití (5.5)]. Během léčby je třeba udržovat přiměřený stav objemu tekutin a pečlivě monitorovat biochemické parametry. Před každou dávkou v 1. cyklu se intravenózně podá 250 - 500 ml fyziologického roztoku nebo jiné vhodné
tekutiny. Po aplikaci přípravku KYPROLIS se podle potřeby podá intravenózně dalších 250 - 500 ml vhodné tekutiny. V dalších cyklech se podle potřeby pokračuje v intravenózní hydrataci. V tomto období je rovněž třeba monitorovat pacienty z hlediska nadměrného podání tekutin [viz Zvláštní upozornění a opatření pro použití (5.1)].
2.3 Premedikace dexamethasonem
Před všemi dávkami přípravku KYPROLIS v 1. cyklu a před všemi dávkami přípravku KYPROLIS v prvním cyklu, ve kterém se zvýší dávka na 27 mg/m2 se pacienti premedikují dexamethasonem 4 mg perorálně nebo intravenózně ke snížení výskytu a závažnosti infuzních reakcí [viz Zvláštní upozornění a opatření pro použití (5.4)]. Premedikace dexamethasonem (4 mg perorálně nebo intravenózně) se obnoví v případě rozvoje nebo opětovného výskytu těchto symptomů v následujících cyklech.
2.4 Úprava dávkování na základě toxicity
Doporučená opatření a úpravy dávek jsou uvedeny v tabulce č. 2.
Tab. 2 Úpravy dávek z důvodu toxicity při léčbě přípravkem KYPROLIS
|
Hematologická toxicita |
Doporučené opatření |
|
• Neutropenie 3.a nebo 4. stupně • Trombocytopenie 4. stupně [viz Zvláštní upozornění a opatření pro použití (5.6)] |
• Vysaďte dávku léku. • Pokud se stav zcela upraví před další plánovanou dávkou, pokračujte na stejné úrovni dávky. • Pokud se stav upraví na neutropenii 2. stupně nebo trombocytopenii 3. stupně, snižte dávku o jednu úroveň (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2). • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
|
Nehematologická toxicita |
Doporučené opatření |
|
Srdeční toxicita 3.nebo 4. stupeň, začátek nebo zhoršení: • městnavého srdečního selhání; • snížené funkce levé komory; • nebo ischemie myokardu [viz Zvláštní upozornění a opatření pro použití (5.1)] |
• Vysaďte lék do úpravy stavu nebo návratu na původní stav. • Po úpravě zvažte, zda je vhodné zahájení léčby přípravkem KYPROLIS sníženou dávkou (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2). • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
|
Plicní hypertenze [viz Zvláštní upozornění a opatření pro použití (5.2)] |
• Vysaďte lék do úpravy stavu nebo návratu na původní stav. • Léčbu znovu zahajte dávkou podávanou před příhodou nebo sníženou dávkou (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2) podle zvážení lékaře. • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
|
Plicní komplikace • 3. nebo 4. stupeň [viz Zvláštní upozornění a opatření pro použití (5.3)] |
• Vysaďte lék do úpravy stavu nebo návratu na původní stav. • Zvažte opětovné zahájení při další plánované léčbě dávkou sníženou o jednu úroveň (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2). |
|
• V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. | |
|
Jaterní toxicita • Zvýšení transamináz, bilirubinu 3. nebo 4. stupně nebo další jaterní abnormality [viz Zvláštní upozornění a opatření pro použití (5.7)] |
• Vysaďte lék do úpravy stavu nebo návratu na původní stav. • Po úpravě zvažte, zda je vhodné zahájení léčby přípravkem KYPROLIS. Léčba může být znovu zahájena sníženou dávkou (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2} za častého monitorování jaterních funkcí. • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
|
Renální toxicita • Kreatinin v séru rovný nebo vyšší než dvojnásobek vstupní hodnoty [viz Nežádoucí účinky (6.1)] |
• Vysaďte lék do doby úpravy funkce ledvin na 1. stupeň nebo na výchozí hodnotu a sledujte funkce ledvin. • Jestliže lze poruchu přisoudit přípravku KYPROLIS, znovu zahajte nejbližší plánovanou léčbu sníženou dávkou (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2) podle zvážení lékaře. • Jestliže nelze poruchu přisoudit přípravku KYPROLIS, znovu zahajte léčbu dávkou použitou před příhodou. • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
|
Periferní neuropatie • 3. nebo 4. stupeň [viz Nežádoucí účinky (6.1)] |
• Vysaďte lék do úpravy stavu nebo návratu na původní stav. • Léčbu znovu zahajte dávkou podávanou před příhodou nebo sníženou dávkou (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2) podle zvážení lékaře. • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
|
Jiné • Nehematologická toxicita 3. nebo 4. stupně |
• Vysaďte lék do úpravy stavu nebo návratu na původní stav. • Zvažte opětovné zahájení při další plánované léčbě dávkou sníženou o jednu úroveň (z 27 mg/m2 na 20 mg/m2 NEBO z 20 mg/m2 na 15 mg/m2). • V případě snášenlivosti se snížená dávka může podle zvážení lékaře zvýšit na předchozí dávku. |
3 Kritéria pro nežádoucí účinky podle National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) verze 3.0
2.5 Zvláštní upozornění pro podávání
Množství přípravku KYPROLIS, který je obsažen v jedné lahvičce na jedno použití (60 mg carfilzomibu) může být vyšší než je potřebná dávka. Při výpočtu potřebného množství je nutná opatrnost, aby se zabránilo předávkování.
KYPROLIS se nesmí míchat a ani podávat v infuzi s jinými léčivými přípravky.
Linka pro intravenózní podání se má propláchnout fyziologickým roztokem nebo 5% glukózou bezprostředně před a po podání přípravku KYPROLIS. KYPROLIS se nemá podat jako bolus. KYPROLIS se má podat během 2 až 10 minut.
2.6 Rekonstituce a příprava pro intravenózní podání
Lahvičky s přípravkem KYPROLIS neobsahují antimikrobiální konzervační látky a jsou určeny pouze pro jednorázové použití. Neotevřené lahvičky s přípravkem KYPROLIS jsou stabilní až do data uvedeném na obale, pokud se uchovávají pří teplotě 2°C - 8°C (36°F až 46°F). Rekonstituovaný roztok obsahuje carfilzomib v koncentraci 2 mg/ml. Před rekonstitucí je nutné si přečíst úplné pokyny pro přípravu.
Postup při rekonstituci/přípravě
1. Těsně před použitím vyjměte lahvičku z lednice.
2. Asepticky rekonstituujte každou lahvičku pomalým injikováním 29 ml sterilní vody pro injekce, USP, roztok směřujte na VNITŘNÍ STĚNU LAHVIČKY, aby se minimalizovala tvorba pěny.
3. Lahvičkou jemně točte a/anebo jí obracejte asi 1 minutu nebo do úplného rozpuštění koláče nebo prášku. Lahvičkou NETŘEPEJTE, aby se netvořila pěna. Pokud se vytvoří pěna, nechte roztok v lahvičce v klidu po dobu 2 až 5 minut, dokud pěna nezmizí.
4. Po rekonstituci je KYPROLIS připravený k intravenóznímu podání. Rekonstituovaný přípravek by měl být čirý a bezbarvý roztok. Jestliže je přítomna jakákoliv změna barvy nebo částice, rekonstituovaný přípravek nepoužívejte.
5. Při podání v intravenózním vaku odeberte z lahvičky vypočtenou dávku [viz Dávkování a způsob podání (2.1)] a rozpusťte ji v 50 ml 5% glukózy injekční roztok, USP pro intravenózní vaky.
6. Lahvičku obsahující nepoužitou část přípravku ihned vyhoďte.
V tabulce č. 3 uvádíme stabilitu rekonstituovaného přípravku KYPROLIS při různých teplotách
a v různých obalech.
Tab. 3: Stabilita rekonstituovaného přípravku KYPROLIS
|
Podmínky uchovávání rekonstituovaného Přípravku KYPROLIS |
Stabilita2 podle druhu obalu | ||
|
Lahvička |
Stříkačka |
l.v. vak (D5Wb) | |
|
V lednici (2°C - 8°C; 36°F - 46°F) |
24 hodin |
24 hodin |
24 hodin |
|
Při pokojové teplotě (15°C - 30°C; 59°F - 86°F) |
4 hodiny |
4 hodiny |
4 hodiny |
3 Celková doba od rekonstituce do podání nemá být delší než 24 hodin b5% roztok glukózy pro injekce, USP
4. KONTRAINDIKACE
Žádné
5. ZVLÁŠTNÍ UPOZORNĚNÍ A OPATŘENÍ PRO POUŽITÍ
5.1 Srdeční zástava, městnavé srdeční selhání, ischemie myokardu
Byl popsán případ úmrtí, který se vyskytl za 1 den po podání přípravku KYPROLIS. Po podání přípravku KYPROLIS se vyskytly případy vzniku nebo zhoršení již přítomného městnavého srdečního selhání se sníženou funkcí levé komory nebo ischemií myokardu. U 7 % pacientů byly hlášené příhody srdečního selhání (např. městnavé srdeční selhání, plicní edém, pokles ejekční frakce). Je nutné sledování pacientů z hlediska srdečních komplikací a jejich rychlá léčba. U pacientů se srdečními příhodami 3. nebo 4. stupně se má KYPROLIS vysadit až do jejich úpravy a zvážit, zda znovu zahájit léčbu přípravkem KYPROLIS na základě vyhodnocení poměru přínosů a rizik [viz Dávkování a způsob podání (2.4)]. Do klinických studií nebyli zařazeni pacienti se srdečním selháním třídy III a IV podle klasifikace New York Heart Association, s infarktem myokardu v minulých 6 měsících a poruchami srdečního vedení, které není kontrolováno farmakologickou léčbou. Tito pacienti mohou mít zvýšené riziko srdečních komplikací.
5.2 Plicní hypertenze
Plicní hypertenze byla hlášena u 2 % pacientů léčených přípravkem KYPROLIS a u méně než 1 % pacientů byla 3. nebo vyššího stupně. Je nutné vyšetření srdce zobrazovacími metodami, případně dalšími testy. V případě výskytu plicní hypertense se má KYPROLIS vysadit do jejího odeznění nebo návratu k výchozímu stavu a na základě vyhodnocení poměru přínosů a rizik zvážit, zda znovu zahájit léčbu přípravkem KYPROLIS [viz Dávkování a způsob podání (2.4)].
5.3 Plicní komplikace
Dušnost byla hlášena u 35 % pacientů zařazených do klinických studií. Dušnost 3. stupně se vyskytla v 5 %. Nebyl hlášen žádný případ dušnosti 4. stupně a vyskytl se 1 případ úmrtí (5. stupeň). Pacienty je třeba sledovat a dušnost neprodleně léčit; léčba přípravkem KYPROLIS se má vysadit do odeznění symptomů nebo návratu k výchozímu stavu [viz Dávkování a způsob podání (2.4) a Nežádoucí účinky 6.1)1
5.4 Infuzní reakce
Infuzní reakce byly charakterizovány celým spektrem systémových projevů, jako je horečka, třesavka, artralgie, myalgie, zčervenání v obličeji, otok obličeje, zvracení, slabost, dušnost, hypotenze, synkopa, pocit tíhy na hrudi nebo angína pectoris. Tyto reakce se mohou vyskytnout bezprostředně nebo až do 24 hodin po podání přípravku KYPROLIS. Před podáním přípravku KYPROLIS se má podat dexamethason, aby se snížil výskyt a závažnost reakcí [viz Dávkování a způsob podání (2.3)]. Pacienty je třeba informovat o rizicích a symptomech a o nutnosti obrátit se na lékaře v případě výskytu projevů infuzní reakce [viz Informace o poradenské službě pro pacienty (17)].
5.5 Syndrom rozpadu nádoru
Syndrom rozpadu nádoru se po podání přípravku KYPROLIS rozvinul u méně než 1 % pacientů. Pacienti s mnohočetným myelomem a velkou zátěží nádorem se mají považovat za rizikovější pro syndrom rozpadu nádoru. Před podáním přípravku KYPROLIS je třeba zajistit, aby byli pacienti dobře hydratováni [viz Dávkování a způsob podání (2.2)]. Během léčby je třeba pacienty sledovat na výskyt syndromu rozpadu nádoru a případně ihned léčit. Léčba přípravkem KYPROLIS se má přerušit do odeznění syndromu rozpadu nádoru [viz Dávkování a způsob podání (2.4)].
5.6 Trombocytopenie
KYPROLIS způsobuje trombocytopenii s nejnižším počtem trombocytů kolem 8. dne každého 28denního cyklu a úpravou na výchozí hodnotu do začátku příštího 28denního cyklu. U pacientů s mnohočetným myelomem se trombocytopenie vyskytla v 36 % a 4. stupeň trombocytopenie byl přítomný u 10 % nemocných. Trombocytopenie po podání přípravku KYPROLIS vedla ke snížení dávky léku u 1 % pacientů a k přerušení léčby přípravkem KYPROLIS došlo u méně než 1 % pacientů. Při léčbě přípravkem KYPROLIS je nutné často sledovat počet trombocytů. V případě klinické indikace se dávka sníží, případně se přeruší léčba [viz Dávkování a způsob podání (2.4)].
5.7 Jaterní toxicita a jatemí selhání
Byly hlášeny (< 1 %) případy jaterního selhání včetně smrtelných případů. KYPROLIS může způsobit zvýšení hladin sérových transamináz a bilirubinu. U pacientů se zvýšením transamináz a bilirubinu 3. stupně nebo vyšším, případně s jinými jaterními abnormalitami se má KYPROLIS vysadit až do jejich úpravy nebo návratu na výchozí hodnoty. Po úpravě se zváží, zda je vhodné opětovné zahájení léčby přípravkem KYPROLIS. Jaterní enzymy je nutné často monitorovat [viz Dávkování a způsob podání (2.4) a Nežádoucí účinky (6.1)].
5.8 Embryofetální toxicita
Pokud se KYPROLIS podá těhotné ženě, může poškodit plod na základě svého mechanismu účinku a nálezů na zvířatech. Neexistují dostačující a řádně kontrolované studie u těhotných žen užívajících KYPROLIS. Carfilzomib způsobil embryofetální toxicitu u březích samic králíků v dávkách, které byly nižší než u pacientů užívajících doporučenou dávku.
Ženám v reprodukčním věku se má doporučit, aby po dobu léčby přípravkem KYPROLIS neotěhotněly. Pokud se tento lék používá v těhotenství nebo pokud pacientka během léčby tímto lékem otěhotní, je nutné pacientku informovat o potenciálním riziku na plod [viz Použití u speciálních populací pacientů (8.1)1
6. NEŽÁDOUCÍ ÚČINKY
V dalších oddílech informace pro preskripci budou podrobněji probrány tyto nežádoucí účinky:
• Srdeční zástava, městnavé srdeční selhání, ischemie myokardu [viz Zvláštní upozornění a opatření pro použití (5.1)]
• Plicní hypertenze [viz Zvláštní upozornění a opatření pro použití (5.2)]
• Plicní komplikace [viz Zvláštní upozornění a opatření pro použití (5.3)]
• Infuzní reakce [viz Zvláštní upozornění a opatření pro použití (5.4)]
• Syndrom rozpadu nádoru [viz Zvláštní upozornění a opatření pro použití (5.5)]
• Trombocytopenie [viz Zvláštní upozornění a opatření pro použití (5.6)]
• Jaterní toxicita a jaterní selhání [viz Zvláštní upozornění a opatření pro použití (5.7)]
Nejčastěji hlášené nežádoucí účinky přípravku KYPROLIS (výskyt 30 % nebo vyšší) pozorované v klinických studiích u pacientů s mnohočetným myelomem byly únava, anemie, nauzea, trombocytopenie, dušnost, průjem a pyrexie.
6.1 Bezpečnostní zkušenosti z klinických studií
Jelikož klinické studie se provádějí za velmi různých podmínek, míru nežádoucích účinků pozorovaných v klinických studiích s lékem nelze přímo porovnat s mírou v klinických studiích s jiným lékem a nemusí ani odrážet míry pozorované v lékařské praxi.
Celkem 526 pacientů s relapsem mnohočetného myelomu, případně s refrakterním mnohočetným myelomem dostávalo KYPROLIS vmonoterapii nebo s dexamethasonem podaným před dávkou přípravku KYPROLIS. Medián léčby činil 4 cykly a medián kumulativní dávky přípravku KYPROLIS byl
993,4 mg.
Úmrtí ze všech příčin do 30 dnů od poslední dávky přípravku KYPROLIS bylo zaznamenáno u 37 z 526 (7 %) pacientů. Úmrtí, která nebylo možné přičíst progresi onemocnění, byly kardiální u 5 pacientů (akutní koronární syndrom, srdeční zástava, srdeční porucha), orgánové selhání u 4 pacientů (multiorgánové selhání, jaterní selhání, selhání ledvin), infekce u 4 nemocných (sepse, pneumonie, bakteriální infekce dýchacích cest), dusnost a nitrolební krvácení vždy u 1 pacienta a 1 pacient byl nalezen mrtvý z neznámé příčiny.
Závažné nežádoucí účinky byly hlášené u 45 % pacientů. Nejčastější závažné nežádoucí účinky byly pneumonie (10 %), akutní selhání ledvin (4 %), pyrexie (3 %) a městnavé srdeční selhání (3 %). Nežádoucí účinky, které vedly k vysazení přípravku KYPROLIS, se vyskytly u 15 % pacientů a jednalo se o městnavé srdeční selhání (2 %), srdeční zástavu, dušnost, zvýšenou hladinu kreatininu v krvi a akutní selhání ledvin (vždy u 1 %).
Nežádoucí účinky, které se vyskytly v 10 a více procentech, jsou uvedeny v tabulce č. 4.
Tab. 4 Výskyt nežádoucích účinků vyskytujících se u > 10 % pacientů s mnohočetným myelomem
léčených přípravkem KYPROLIS
|
Příhoda |
Pacienti {N = 526), [n (%)] | ||
|
Všechny stupně3 |
Příhody 3. stupně |
Příhody 4. stupně | |
|
Únava |
292 (55,5) |
38 (7,2) |
2 (0,4) |
|
Anemie |
246 (46,8) |
111 (21,1) |
7(1,3) |
|
236 (44,9) |
7 (1,3) |
0 | |
|
Trombocytopenie |
191 (36,3) |
69 (13,1) |
54 (10,3) |
|
Dusnost |
182 (34,6) |
25 (4,8) |
1 (0,2)b |
|
172 (32,7) |
4 (0,8) |
1 (0,2) | |
|
Pyrexie |
160 (30,4) |
7 (1,3) |
2 (0,4) |
|
Infekce horních cest dýchacích |
149 (28,3) |
17 (3,2) |
0 |
|
145 (27,6) |
7 (1,3) |
0 | |
|
137 (26,0) |
1 (0,2) |
0 | |
|
Zvýšení hladiny kreatininu v krvi |
127 (24,1) |
13 (2,5) |
1 (0,2) |
|
Lymfopenie |
126 (24,0) |
84 (16,0) |
11(2,1) |
|
Periferní otoky |
126 (24,0) |
3 (0,6) |
0 |
|
117 (22,2) |
5 (1,0) |
0 | |
|
Zácpa |
110 (20,9) |
1 (0,2) |
0 |
|
Neutropenie |
109 (20,7) |
50 (9,5) |
4 (0,8) |
|
106 (20,2) |
15 (2,9) |
0 | |
|
94 (17,9) |
0 |
0 | |
|
84 (16,0) |
1 (0,2) |
0 | |
|
Artralgie |
83 (15,8) |
7 (1,3) |
0 |
|
Svalové spasmy |
76 (14,4) |
2 (0,4) |
0 |
|
Hypertenze |
75 (14,3) |
15 (2,9) |
2 (0,4) |
|
Astenie |
73 (13,9) |
12 (2,3) |
1 (0,2) |
|
Hypokalemie |
72 (13,7) |
14 (2,7) |
3 (0,6) |
|
Hypomagnesemie |
71 (13,5) |
2 (0,4) |
0 |
|
Leukopenie |
71 (13,5) |
27 (5,1) |
1 (0,2) |
|
Bolest končetin |
70 (13,3) |
7 (1,3) |
0 |
|
67 (12,7) |
52 (9,9) |
3 (0,6)b | |
|
Zvýšení hladiny aspartát aminotransferázy |
66(12,5) |
15 (2,9) |
1 (0,2) |
|
Závratě |
66(12,5) |
5 (1,0) |
1 (0,2) |
|
Hypestesie |
64 (12,2) |
3 (0,6) |
0 |
|
63 (12,0) |
1 (0,2) |
0 | |
|
Bolest |
63 (12,0) |
12 (2,3) |
1 (0,2) |
|
Hyperglykemie |
62 (11,8) |
16 (3,0) |
3 (0,6) |
|
Bolest hrudní stěny |
60 (11,4) |
3 (0,6) |
0 |
|
Hyperkalcemie |
58(11,0) |
13 (2,5) |
8 (1,5) |
|
Hypofosfatemie |
55 (10,5) |
24 (4,6) |
3 (0,6) |
|
Hyponatremie |
54 (10,3) |
31 (5,9) |
3 (0,6) |
a Kritéria pro nežádoucí účinky podle National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) verze 3.0
b U jedné příhody se jednalo o závažnost 5. stupně
Popis vybraných lékových nežádoucích účinků
Renální příhody
Nejčastějšími renálními nežádoucími účinky byly vzestup hladiny kreatininu v krvi (24 %) a selhání ledvin (9 %), které byly většinou 1. nebo 2. stupně závažnosti. Renální nežádoucí účinky 3. stupně se vyskytly u 6 % a 4. stupně u 1 % nemocných. Přerušení léčby pro zvýšenou hladinu kreatininu v krvi a pro akutní selhání ledvin bylo zaznamenáno vždy po 1 %. U 1 pacienta došlo k úmrtí pro souběžnou sepsi a zhoršení funkce ledvin [viz Dávkování a způsob podání (2.4)].
Periferní neuropatie
Periferní neuropatie (včetně všech příhod periferní senzorické neuropatie a periferní motorické neuropatie) se vyskytla u 14 % pacientů zařazených do klinických studií. Periferní neuropatie 3. stupně se vyskytla u 1 % pacientů. Závažné příhody periferní neuropatie se vyskytly u méně než 1 % pacientů a vedly ke snížení dávky léku u méně než 1 % pacientů a vysazení léčby rovněž u méně než 1 % nemocných. Léčba se má vysadit nebo přerušit podle výše uvedených doporučení [viz Dávkování a způsob podání (2.4)].
Infekce virem herpes zoster
Reaktivace infekce virem herpes zoster byla hlášena u 2 % pacientů. U pacientů s infekcí virem herpes zoster v anamnéze se má zvážit antivirová profylaxe.
7. LÉKOVÉ INTERAKCE
Carfilzomib se primárně metabolizuje aktivací peptidázy a epoxidové hydrolázy. Díky tomu je farmakokinetický profil carfilzomibu jen s malou pravděpodobností ovlivněn souběžným podáváním inhibitorů a induktorů cytochromu P450. Nepředpokládá se, že by carfilzomib ovlivňoval expozici dalších léků [viz Klinická farmakologie (12.3)].
8. POUŽITÍ U SPECIÁLNÍCH POPULACÍ PACIENTŮ 8.1. Těhotenství
Těhotenství kategorie D [viz Zvláštní upozornění a opatření pro použití (5.8)]
Ženám v reprodukčním věku se má doporučit, aby po dobu léčby přípravkem KYPROLIS neotěhotněly. Pokud se KYPROLIS podá těhotné ženě, může poškodit plod na základě svého mechanismu účinku a nálezů na zvířatech. Carfilzomib způsobil embryofetální toxicitu u březích samic králíků v dávkách, které byly nižší než u pacientů užívajících doporučenou dávku. Pokud se tento lék používá v těhotenství nebo pokud pacientka během léčby tímto lékem otěhotní, je nutné pacientku informovat o potenciálním riziku na plod.
Carfilzomib byl podáván intravenózně březím samicím potkanů a králíků v období organogeneze v dávkách 0,5, 1 a 2 mg/kg/den potkanům a 0,2, 0,4 a 0,8 mg/kg/den králíkům. Carfilzomib nebyl u žádné testované dávky teratogenní. U králíků byl zjištěn vzestup preimplantační ztráty při dávce > 0,4 mg/kg/den a vzestup u časných resorpcí a poimplantační ztráty a pokles váhy plodů u dávek 0,8 mg/kg/den, které jsou toxické pro matky. Dávky testované na králících v hodnotách 0,4 mg/kg/den je zhruba o 20 % vyšší a dávka 0,8 mg/kg/den o 40 % vyšší než je doporučená dávka 27 mg/m2 u člověka na základě plochy tělesného povrchu.
8.3 Kojící ženy
Není známo, zda se KYPROLIS vylučuje do lidského mateřského mléka. Jelikož se do lidského mateřského mléka vylučují mnohé léky a vzhledem k potenciálu závažných nežádoucích účinků přípravku KYPROLIS u kojených dětí je nutné se rozhodnout, zda přestat s kojením nebo vysadit lék, přičemž je nutné vzít v úvahu důležitost léku pro matku.
8.4 Použití u pediatrické populace
Bezpečnost a účinnost přípravku KYPROLIS u pediatrických pacientů nebyla stanovena.
8.5 Použití u starších lidí
Ve studiích s přípravkem KYPROLIS nebyly pozorovány významné rozdíly mezi pacienty mladšími 65 let a pacienty ve věku 65 let a staršími z hlediska bezpečnosti a účinnosti.
8.6 Porucha funkce ledvin
Farmakokinetika a bezpečnost přípravku KYPROLIS byly hodnoceny ve studii fáze 2 u pacientů s normální funkcí ledvin a u pacientů s mírnou, středně těžkou a těžkou poruchou funkce ledvin a dále u pacientů podstupujících chronickou dialyzační léčbu. V průměru byli pacienti léčeni po dobu 5,5 cyklů a dostávali KYPROLIS v dávce 15 mg/m2 v 1. cyklu, 20 mg/m2 v 2. cyklu a 27 mg/m2 v 3. cyklu a dalších cyklech. Farmakokinetika a bezpečnost přípravku KYPROLIS nebyly ovlivněny stupněm výchozí poruchy funkce ledvin, a to včetně pacientů v chronickém dialyzačním programu. Jelikož nebyly studovány koncentrace přípravku KYPROLIS při dialýze, lék by se měl podávat až po dialýze [viz Klinická farmakologie (12.3)].
8.7 Porucha funkce jater
Bezpečnost, účinnost a farmakokinetika přípravku KYPROLIS nebyly hodnoceny u pacientů se vstupní poruchou funkce jater. Z klinických studií s přípravkem KYPROLIS byli vyřazeni pacienti s těmito laboratorními hodnotami: ALT a AST > trojnásobek horní hranice normálních hodnot a bilirubin > dvojnásobek horní hranice normálních hodnot [viz Klinická farmakologie (12.3)].
8.8 Poškození srdce
Pacienti se srdečním selháním třídy III a IV podle klasifikace New York Heart Association nebyli do klinických studií zařazeni. Bezpečnost u této skupiny pacientů nebyla hodnocena.
10. PŘEDÁVKOVÁNÍ
Není známo specifické antidotum pro léčbu předávkování přípravkem KYPROLIS. V případě předávkování pacienta sledujte a zabezpečte příslušnou podpůrnou péči.
KYPROLIS (carfilzomib) pro injekce je protinádorová látka dostupná pouze pro intravenózní použití. KYPROLIS je sterilní, bílý až bělavý lyofilizovaný prášek a je k dispozici v lahvičkách na jedno použití. Každá lahvička přípravku KYPROLIS obsahuje 60 mg carfilzomibu, 3000 mg sulfobutyleter beta-cyklodextrinu, 57,7 mg kyseliny citrónové a hydroxid sodný na úpravu pH (cílové pH 3,5).
Carfilzomib je modifikovaný tetrapeptidyl epoxid izolovaný jako krystalická volná báze. Chemický název carfilzomibu je (2S)-N-((S)-l-((S)-4-metyl-l-((R)-2-metyloxiran-2-yl)-l-oxopentan-2-ylcarbamoyl)-2-fenyletyl)-2-((S)-2-(2-morfolinoacetamido)-4-fenylbutanamido)-4-metylpentanamid. Strukturní vzorec carfilzomibu je následující:
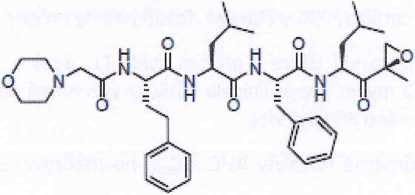
Carfilzomib je krystalická látka o molekulární hmotnosti 719,9. Jeho molekulární vzorec je C40H57N5O7. Carfilzomib je prakticky nerozpustný ve vodě a velmi slabě rozpustný za kyselých podmínek.
12. KLINICKÁ FARMAKOLOGIE
12.1 Mechanismus účinku
Carfilzomib je tetrapeptidický epoxyketonový inhibitor proteazomu, který se ireverzibilně váže na N-terminální aktivní místa 20S proteazomu obsahující threonin, což je proteolytická jádrová částice uvnitř 26S proteazomu. Carfilzomib prokázal in vitro antiproliferativní a proapoptotickou aktivitu u solidních a hematologických nádorových buněk. U zvířat carfilzomib inhiboval aktivitu proteazomu v krvi a tkáni a oddálil růst nádoru u modelů s mnohočetným myelomem, hematologickými a solidními nádory.
12.2 Farmakodynamika
Intravenózní podání carfilzomibu mělo za následek supresi chymotrypsinu podobné aktivity proteazomu, když se měřila v krvi za 1 hodinu po první dávce. První den prvního cyklu se inhibice proteazomu v mononukleárních buňkách periferní krve pohybovala v rozmezí 79 - 89 % při dávce 15 mg/m2 a v rozmezí 82 - 83 % při dávce 20 mg/m2. Podání carfilzomibu v dávce 20 mg/m2dále vedlo k inhibici podjednotek LMP2 a MECL1 imunoproteazomu v rozmezí 26 - 32 %, resp. 41 - 49 %. Inhibice proteazomu se udržela po dobu > 48 hodin po první dávce carfilzomibu v každém týdnu podání.
12.3 Farmakokinetika
Absorpce: Maximální koncentrace (Cmax) po jednorázové intravenózní dávce 27 mg/m2 byla 4232 ng/ml a plocha pod křivkou plazmatických koncentrací (AUC) byla 379 ng»hod./ml. Po opakovaných dávkách carfilzomibu 15 a 20 mg/m2 byly AUC a poločas obdobné 1. a 15. nebo 16. den prvního cyklu, což naznačuje, že nedochází k systémové akumulaci carfilzomibu. Při dávkách mezi 20 a 36 mg/m2 byl pozorován vzestup expozice závislý na dávce.
Distribuce: Průměrný distribuční objem v ustáleném stavu dávky 20 mg/m2 carfilzomibu byl 28 I. Při testování in vitro byla vazba carfilzomibu na plazmatické bílkoviny člověka průměrně 97 % v rozmezí koncentrací 0,4 - 4 pmol.
Metabolismus: Carfilzomib se rychle a výrazně metabolizuje. Hlavní metabolity naměřené v lidské plazmě a moči a vytvořené in vitro lidskými hepatocyty, byly bílkovinné fragmenty a diol carfilzomibu, což ukazuje na to, že hlavními metabolickými cestami jsou štěpení peptidázy a hydrolýza epoxidu. V celkovém metabolismu carfilzomibu hrají mechanismy zprostředkované cytochromem P450 pouze malou roli. Metabolity nemají žádnou známou biologickou aktivitu.
Eliminace: Po intravenózním podání dávek > 15 mg/m2 byl carfilzomib rychle odstraněn ze systémového oběhu s poločasem < 1 hodina 1. den prvního cyklu. Systémová clearance se pohybovala mezi 151 - 263 l/hod. a předčila průtok krve v játrech, což naznačuje, že carfilzomib se z větší části vylučuje extrahepatálně. Dráhy eliminace carfilzomibu u člověka dosud nebyly určeny.
Věk: Analýza populačních farmakokinetických dat po první dávce v prvním cyklu (1. den) u 154 pacientů, kterým byla intravenózně podána dávka 20 mg/m2, neprokázala klinicky významný rozdíl v expozici mezi pacienty mladšími 65 let a pacienty ve věku 65 let a více.
Pohlaví: V populační farmakokinetické studii byly průměrné hodnoty AUC a Cmax normalizované na dávku srovnatelné u mužů i žen.
Porucha funkce jater: U pacientů s poruchou funkce jater nebyly provedeny žádné farmakokinetické studie s KYPROLISEM [viz Zvláštní upozornění a opatření pro použití (5.7)].
Porucha funkce ledvin: Byla provedena farmakokinetické studie u 43 pacientů s mnohočetným myelomem, kteří měli různý stupeň poruchy funkce ledvin a kteří byli klasifikováni podle jejich clearance kreatininu (CLcr) do těchto skupin: normální funkce (CLcr > 80 ml/min., n = 8), mírná porucha (CLcr 50 - 80 ml/min., n = 12), středně těžká porucha (CLcr 30 - 49 ml/min., n = 8), těžká porucha (CLcr < 30 ml/min., n = 7) a chronická dialýza (n = 8). KYPROLIS se podával intravenózně po dobu 2 až 10 minut dva po sobě jdoucí dny, Ix týdně po dobu tří týdnů (1., 2., 8., 9., 15. a 16. den), poté následovala přestávka trvající 12 dní. Celý cyklus tak trval 28 dní. Pacienti dostali úvodní dávku 15 mg/m2, která mohla být počínaje 2. cyklem zvýšena na 20 mg/m2, pokud dávku 15 mg/m2 v prvním cyklu dobře snášeli. V této studii neměl stav funkce ledvin žádný vliv na odstranění nebo expozici carfilzomibu po jednorázovém nebo opakovaném podání dávky léku [viz Použití u speciálních populací pacientů (8.6)].
Cytochrom P450: Ve studii in vitro, ve které byly použity lidské jaterní mikrosomy, vykázal carfilzomib mírný a na čase závislý inhibiční efekt na lidský cytochrom CYP3A4/5. Studie in vitro naznačily, že carfilzomib neindukuje CYP1A2 a CYP3A4 v kultuře čerstvých lidských hepatocytů. V celkovém metabolismu carfilzomibu hrají mechanismy zprostředkované cytochromem P450 pouze malou úlohu. Klinická studie u 17 pacientů užívajících perorálně midazolam za účelem sledování CYP3A prokázala, že farmakokinetika midazolamu nebyla ovlivněna současným podáváním carfilzomibu. Nepředpokládá se, že by KYPROLIS inhiboval aktivitu CYP3A4/5, případně ovlivňoval expozici vůči substrátům CYP3A4/5.
P-glykoprotein: Carfilzomib je substrát P-glykoproteinu (P-gp) a vykázal pouze marginální inhibiční účinky na P-gp v jednovrstevné kultuře Caco-2 buněk. Vzhledem ktomu, že KYPROLIS se podává intravenózně a ve velké míře se metabolizuje, není pravděpodobné, že by byl ovlivňován inhibitory nebo induktory P-gp.
13. NEKLINICKÁ TOXIKOLOGIE
13.1 Karcinogeneze, mutageneze a postižení fertility
Studie karcinogenity s carfilzomibem nebyly prováděny.
Carfilzomib byl klastogenní v in vitro testu chromozomálních aberací na lymfocytech periferní krve. Carfilzomib nebyl mutagenní v (Amesově) in vitro testu bakteriální revezní mutace a nebyl klastogenní v in vivo analýze mikrojader kostní dřeně myší.
Studie fertility s carfilzomibem se neprováděly. Nebyly zaznamenány žádné účinky na reprodukční tkáně během 28denních studií toxicity s podáním opakované dávky potkanům a opicím nebo ve studiích chronické toxicity trvajících 6 měsíců u potkanů a 9 měsíců u opic.
13.2 Toxikologie a/anebo farmakologie u zvířat
U opic, kterým byl podán jednorázový bolus intravenózní dávky 3 mg/kg carfilzomibu (zhruba l,3násobek doporučené dávky 27 mg/m2 u člověka na základě plochy tělesného povrchu), byla pozorována hypotenze, zvýšená srdeční frekvence a zvýšené hladiny troponinu-T v séru. Opakované intravenózní bolusy carfilzomibu v dávce > 2 mg/kg/dávka potkanům a v dávce 2 mg/kg/dávka opicím v podobném dávkovacím schématu jako při klinickém použití způsobily mortalitu vyvolanou toxicitou na kardiovaskulární systém (srdeční selhání, srdeční fibróza, hromadění perikardiální tekutiny, srdeční krvácení/degenerace), gastrointestinální systém (nekróza/krvácení), renální systém (glomerulonefropatie, tubulámí nekróza, dysfunkce) a plicní systém (krvácení/zánět). Dávka 2 mg/kg/dávka u potkanů představuje přibližně polovinu doporučené dávky u člověka 27 mg/m2 na základě plochy tělesného povrchu. Dávka2 mg/kg/dávka u opic je přibližně stejná jako je doporučená dávka u člověka na základě plochy tělesného povrchu.
14. KLINICKÉ STUDIE
14.1 Studie u relapsu mnohočetného myelomu
Bezpečnost a účinnost přípravku KYPROLIS byla hodnocena v multicentrické jednoramenné klinické studii. Do studie bylo zařazeno 266 pacientů s relapsem mnohočetného myelomu, kteří předtím dostali nejméně 2 léčby (včetně bortezomibu a thalidomidu a/anebo lenalidomidu). Pacienti byli zařazeni do studie, pokud měli < 25% odpověď na poslední léčbu nebo došlo k progresi choroby během 60 dní od poslední léčby. Pacienti byli ze studie vyřazeni z těchto důvodů: hladina celkového bilirubinu byla > 2x vyšší než horní hranice normálních hodnot, clearance kreatininu byla pod 30 ml/min, městnavé srdeční selhání třídy III nebo IV podle klasifikace New York Heart Association, symptomatická ischemie myokardu, infarkt myokardu v posledních 6 měsících, periferní neuropatie 3. nebo 4. stupně nebo periferní neuropatie 2. stupně s bolestmi, aktivní infekce vyžadující léčbu a pleurální výpotek.
KYPROLIS byl podáván intravenózně 2 až 10 minut dva po sobě jdoucí dny každý týden po dobu 3 týdnů a poté následovala 12denní přestávka (28denní léčebný cyklus), a to až do progrese onemocnění, do nepřijatelné toxicity nebo maximálně 12 cyklů. Každá dávka byla 20 mg/m2 v prvním cyklu a 27 mg/m2 v následujících cyklech. Ke snížení výskytu a závažnosti horečky, svalové ztuhlosti, třesavky, dušnosti, myalgií a artralgií byl pacientům podán dexamethason 4 mg per os nebo v intravenózní infuzi před všemi dávkami přípravku KYPROLIS v prvním cyklu a před všemi dávkami přípravku KYPROLIS v prvním cyklu se zvýšením dávky (na 27 mg/m2). Premedikace dexamethasonem (4 mg per os nebo intravenózně) byla zopakována, pokud se v následujících cyklech opět objevily výše uvedené symptomy.
Výchozí charakteristické vlastnosti pacientů a nemoci jsou shrnuty v tabulce č. 5.
Tab. 5 Demografické parametry a vstupní charakteristika onemocnění
|
Charakteristická vlastnost |
Počet pacientů (%) |
|
Charakteristické vlastnosti pacientů Zařazení pacienti Střední věk, roky (rozptyl) Věková skupina, < 65 / > 65 (roky) Pohlaví (muži / ženy) Rasa (běloši / černoši / Asiaté / jiní) |
266 (100) 63,0 (37; 87) 146 (54,9) / 120 (45,1) 155 (58,3) / 111 (41,7) 190 (71,4) / 53 (19,9) / 6(2,3) / 17 (6,4) |
|
Charakteristické vlastnosti nemoci Počet předchozích režimů (medián) Předcházející transplantace Refrakterní stav na poslední léčbub |
5a 198 (74,4) |
|
Refrakterní pacienti: Progrese během poslední léčby Refrakterní pacienti: Progrese během 60 dní po dokončení poslední léčby Refrakterní pacienti: < 25% odpověď na léčbu Pacienti s relapsem: Progrese po 60 dnech po léčbě |
198 (74,4) 38 (14,3) 16 (6,0) 14 (5,3) |
|
Roky od diagnózy choroby, medián (rozptyl) Postižení plazmatických buněk (< 50 % / > 50 % / neznámé nebo chybí) |
5,35 (0,5; 22,3) 143 (53,8) / 106 (39,8) / 17 (6,4) |
|
Cytogenetika nebo FISH analýzy Normální / Příznivá Špatná prognóza Neznámá / Nevyšetřena |
159 (59,8) 75 (28,2) 32 (12,0) |
|
Beta-2 mikroglubuiin v séru Medián (rozptyl) Clearance kreatininu < 30 (ml/min) |
4,3 (0,4; 20,5) 6(2,3) |
|
3 Rozptyl: 1, 20 |
b Kategorie refrakterního stavu jsou odvozené podle pragmatického stavu s použitím dostupných laboratorních dat Medián počtu zahájených cyklů byl 4.
Primárním cílem byla celková míra odpovědí (ORR), která byla stanovená nezávislou hodnotící komisí s použitím kritérií Mezinárodní myelomové pracovní skupiny. ORR (zcela kompletní odpověď [sCR] + kompletní odpověď [CR] + velmi dobrá částečná odpověď [VGPR]+ částečná dopověď [PR]) byla 22,9 % (95% Cl: 18,0; 28,5) (N = 266) (viz tabulka č. 6). Medián trvání odpovědi (DOR) byl 7,8 měsíců (95% Cl: 5,6; 9,2).
Tab. 6 Kategorie odpovědí
|
Charakteristická vlastnost |
Pacienti v klinické studii |
|
n (%) | |
|
Počet pacientů (%) Kategorie odpovědi3 |
266 (100) |
|
Kompletní odpověď Velmi dobrá částečná odpověď Částečná odpověď Celková odpověď 95% Clb |
1 (0,4) 13 (4,9) 47 (17,7) 61 (22,9) (18,0; 28,5) |
|
3 Podle vyhodnocení nezávislé hodnotící komise b Přesný interval spolehlivosti |
16. JAK SE LÉK DODÁVÁ, UCHOVÁVÁNÍ LÉKU A ZACHÁZENÍ S NÍM 16.1 Jak se lék dodává
KYPROLIS (carfilzomib) pro injekce se dodává jako jednotlivě balená lahvička na jedno použití obsahující 60 mg carfilzomibu jako bílý až bělavý lyofilizovaný koláč nebo prášek.
• NDC 76075-101-01, 60 mg carfilzomibu v lahvičce
16.2 Uchovávání léku a zacházení s ním
Neotevřené lahvičky se uchovávají v lednici (při teplotě 2°C - 8°C; 36°F až 46°F). Lék uchovávejte v původním obalu, aby byl chráněn před světlem.
17. INFORMACE O PORADENSKÉ SLUŽBĚ PRO PACIENTY
Před léčbou přípravkem KYPROLIS má lékař probrat s pacientem tyto náležitosti:
Poučit pacienty, aby kontaktovali svého lékaře v případě rozvoje některého z následujících projevů: horečka, třesavka, svalová ztuhlost, bolest na hrudi, kašel nebo otoky nohou nebo dolních končetin.
Informovat pacienty, že KYPROLIS může způsobovat únavu, závratě, mdloby, případně pokles krevního tlaku. Doporučit pacientům, aby v případě výskytu některého z těchto projevů neřídili motorové vozidlo a neobsluhovali stroje.
Informovat pacienty, že během léčby přípravkem KYPROLIS se mohou vyskytnout dechové potíže (dusnost). Dechové potíže se nejčastěji vyskytnou během jednoho dne po podání léku. Je třeba poučit pacienty, aby v případě dechových potíží kontaktovali svého lékaře.
Poradit pacientům, aby se vyvarovali dehydratace, jelikož u pacientů léčených přípravkem KYPROLIS se může vyskytnout zvracení, případně průjem. Poučit pacienty, aby v případě výskytu zvracení, závratí nebo signálů mdlob vyhledali lékařskou pomoc.
Poučit pacientky v reprodukčním věku, aby používaly účinnou antikoncepci k zabránění otěhotnění během léčby přípravkem KYPROLIS. Poradit pacientce, aby se v případě otěhotnění během léčby neprodleně obrátila na svého lékaře. Poučit pacientky, aby v těhotenství a období kojení neužívaly KYPROLIS. Jestliže si pacientka bude přát po skončení léčby opět kojit, je třeba ji doporučit, aby správné načasování probrala se svým lékařem.
Doporučit pacientům, aby probrali se svým lékařem všechny léky, které užívají před zahájením léčby přípravkem KYPROLIS nebo před zahájením užívání jakéhokoliv nového léku během léčby přípravkem KYPROLIS.
Vyráběno pro:
Onyx Pharmaceuticals, lne.
249 East Grand Avenue South San Francisco, CA 94080
Čísla US patentů: 7,232,818; 7,417,042; 7,491,704; 7,737,112
*


'

'

Kapitoly nebo podkapitoly vynechané v úplné informaci pro preskripci nejsou v tomto seznamu uvedeny.
DÁVKOVACÍ FORMY A SÍLY
KYPROLIS lahvička pro jedno použití obsahuje 60 mg carfilzomibu jako sterilní bílý až bělavý lyofilizovaný koláč nebo prášek.