Iluvien 190 Mikrogramů Intravitreální Implantát V Aplikátoru
Sp. zn. sukls33003/2015 SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
ILUVIEN 190 mikrogramů intravitreální implantát v aplikátoru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden implantát obsahuje fluocinoloni acetonidum 190 mikrogramů.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Intravitreální implantát v aplikátoru.
Světle hnědý tyčinkový implantát velikosti asi 3,5 mm x 0,37 mm.
Aplikátor implantátu s jehlou velikosti 25 G.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek ILUVIEN je indikován k léčbě poškození zraku souvisejícím s chronickým diabetickým makulárním edémem, který nereaguje dostatečně na dostupnou léčbu (viz bod 5.1).
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je jeden implantát ILUVIEN do postiženého oka. Podání do obou očí současně se nedoporučuje (viz bod 4.4).
Jeden implantát ILUVIEN uvolňuje fluocinolon-acetonid po dobu až 36 měsíců. Další implantát je možné zavést po 12 měsících, pokud se u pacienta objeví zhoršení zraku nebo větší tloušťka sítnice v důsledku rekurence nebo zhoršení diabetického makulárního edému (viz bod 5.1).
Opakovaná léčba se nemá podávat, pokud potenciální přínosy nepřevyšují rizika.
Pouze pacienti, kteří nedostatečně reagovali na předchozí léčbu laserovou fotokoagulací nebo jinou dostupnou terapii diabetického makulárního edému, nemají být léčeni přípravkem ILUVIEN.
Pediatrická populace
Neexistuje žádné relevantní použití intravitreálně podaného fluocinolon-acetonidu u pediatrické populace s diabetickým makulárním edémem (DMO).
U starších pacientů nebo u pacientů s poruchou funkce ledvin nebo jater není nutná žádná úprava dávky.
Způsob podání
POUZE PRO INTRAVITREÁLNÍ PODÁNÍ.
Léčba přípravkem ILUVIEN je určena pouze pro podání do sklivce a má být podávána oftalmologem se zkušenostmi v aplikaci intravitreálních injekcí. Intravitreální injekční zákrok má být prováděn za kontrolovaných aseptických podmínek, které zahrnují použití sterilních rukavic, sterilních roušek a sterilního víčkového spekula (nebo ekvivalentního nástroje). Před injekcí implantátu má být podána adekvátní anestezie a širokospektrá antibiotika.
Postup injekce implantátu ILUVIEN je následující:
1. Na základě rozhodnutí ošetřujícího očního lékaře mohou být před operací podány antibiotické kapky.
2. Těsně před injekcí podejte lokální anestezii k místu injekce (doporučuje se dolní temporální kvadrant) jako jedna kapka následovaná buď vatovou štětičkou namočenou v anestetiku nebo jako subkonjunktivální podání adekvátní anestezie.
3. Aplikujte 2-3 kapky adekvátního lokálního antiseptika do dolního fornixu. Víčka mohou být omývána aplikátorem s vatovou štětičkou namočenou v adekvátním lokálním antiseptiku. Vložte sterilní víčkové spekulum. Nechte pacienta podívat nahoru a na místo injekce použijte vatovou štětičku namočenou v adekvátním antiseptiku. Před injekcí přípravku ILUVIEN ponechte lokální antiseptikum zaschnout po dobu 30-60 sekund.
4. Vnější část fóliového tvarovaného blistru nemá být považována za sterilní. Asistent (nesterilní) má odstranit tvarovaný blistr z obalu a zkontrolovat tvarovaný blistr a fólii, zda není přítomné poškození. Pokud je přítomné poškození, soupravu nepoužívejte.
Pokud je přijatelná, asistent by měl sloupnout fólii z tvarovaného blistru, aniž by se dotknul vnitřního povrchu.
5. Vizuálně zkontrolujte okénko na připraveném aplikátoru, abyste se ujistili, že je uvnitř lékový implantát.
6. Vyjměte aplikátor z tvarovaného blistru pomocí sterilních rukavic a dotýkejte se pouze sterilního povrchu a aplikátoru.
Ochranný kryt na jehle nemá být odstraněn, dokud se nebude přípravek ILUVIEN injikovat.
Před injekcí musí být hrot aplikátoru nad horizontální rovinou, aby bylo zajištěno, že je implantát v aplikátoru ve správné poloze.
7. Zavádění vyžaduje dva kroky, aby se snížilo množství vzduchu aplikovaného spolu
s implantátem. Před injekcí jehly do oka stiskněte tlačítko směrem dolů a posuňte ho k první zarážce (na zahnuté černé značky, podél tlačítkové lišty). U první zarážky uvolněte tlačítko a to se přesune do polohy nahoru („UP“). Pokud se v této poloze tlačítko nezvedne, nepokračujte s použitím této soupravy.
8. Optimální umístění implantátu je níže, než je disk optického nervu a vzadu za rovníkem oka. Změřte 4 milimetry inferotemporálně od limbu za pomocí kaliper.
9. Opatrně odstraňte ochranný kryt z jehly a zkontrolujte hrot, abyste se ujistili, že není ohnutý.
10. Jemně posuňte oční spojivku tak, aby po vytažení jehly nebyla místa vstupu jehly do spojivky a skléry ve stejné úrovni. Je třeba zabránit kontaktu mezi jehlou a okrajem víčka nebo řasami. Zasuňte jehlu do oka. Pro uvolnění implantátu, zatímco je tlačítko v poloze nahoru („up“), posuňte tlačítko dopředu až do konce tlačítkové lišty a vytáhněte jehlu. Poznámka: ujistěte se, že tlačítko dosáhne konec lišty před odstraněním jehly.
11. Odstraňte víčkové spekulum a proveďte nepřímou oftalmoskopii pro ověření umístění implantátu, dostatečné perfuze centrální sítnicové tepny a absenci jakýchkoli dalších komplikací. Deprese skléry může zlepšit vizualizaci implantátu. Vyšetření má zahrnovat kontrolu prokrvení hlavového optického nervu bezprostředně po injekci implantátu. Podle rozhodnutí očního lékaře se může provést bezprostřední měření nitroočního tlaku.
V návaznosti na zákrok by měli být pacienti sledování s ohledem na možné komplikace, jako je endoftalmitida, zvýšený nitrooční tlak, odchlípení sítnice a krvácení do sklivce nebo jeho odchlípení. Biomikroskopie s tonometrií mají být provedeny v době mezi dvěma a sedmi dny po injekci implantátu.
Poté se doporučuje sledování pacientů alespoň čtvrtletně s ohledem na potenciální komplikace kvůli prodloužení doby uvolňování fluocinolon-acetonidu přibližně o 36 měsíců (viz bod 4.4).
4.3 Kontraindikace
Intravitreální implantát s přípravkem ILUVIEN je kontraindikován v přítomnosti již existujícího glaukomu, případně aktivní nebo suspektní oční nebo periokulární infekce zahrnující většinu virových onemocnění rohovky a spojivky, včetně aktivní epiteliální herpes simplex keratitidy (dendritická keratitida), vakcinie, varicely, mykobakteriální infekce a mykotických onemocnění.
Přípravek ILUVIEN je kontraindikován u pacientů s hypersenzitivitou na léčivou látku nebo na kteroukoliv pomocnou látku přípravku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Intravitreální injekce byly dávány do souvislosti s endoftalmitidou, zvýšeným intraokulárním tlakem, odchlípením sítnice a krvácením do sklivce nebo jeho odchlípením. Pacienty je třeba poučit, že mají okamžitě hlásit jakékoliv příznaky, které poukazují na endoftalmitidu. Sledování pacienta během dvou až sedmi dnů po injekci může umožnit časnou identifikaci a léčbu oční infekce, zvýšeného nitroočního tlaku nebo dalších komplikací. Doporučuje se, aby se potom nitrooční tlak monitoroval minimálně jednou za čtvrt roku.
Použití intravitreálních kortikosteroidů může způsobovat kataraktu, zvýšení nitroočního tlaku, glaukom a může zvyšovat riziko sekundárních infekcí.
Bezpečnost a účinnost přípravku ILUVIEN podaného do obou očí současně nebyla hodnocena. Doporučuje se, aby se implantát nepodával současně při stejné návštěvě do obou očí. Souběžná léčba obou očí se nedoporučuje, dokud nebude známa systémová a oční odpověď pacienta na první implantát (viz bod 4.2).
Ve studiích FAME podstoupilo 80 % fakických subjektů léčených fluocinolon-acetonidem operaci katarakty (viz bod 4.8). Fakičtí pacienti mají být důkladně sledováni s ohledem na známky katarakty po léčbě.
Ve studiích FAME vyžadovalo 38 % pacientů léčených fluocinolon-acetonidem léčbu přípravky snižujícími nitrooční tlak (viz bod 4.8). Fluocinolon-acetonid se má používat s opatrností u pacientů s vysokým NOT a nitrooční tlak se musí důsledně monitorovat. V případě zvýšení NOT, který nereaguje na léky snižující NOT nebo postupy na snížení NOT, je možné implantát ILUVIEN odstranit pomocí vitrektomie.
Existují omezené zkušenosti s účinkem fluocinolon-acetonidu na oči po vitrektomii. Je pravděpodobné, že clearance léku by se urychlila po vitrektomii, neočekává se však, že by byly koncentrace v ustáleném stavu ovlivněny. To může zkrátit trvání účinku implantátu.
Ve studiích FAME bylo 24 % subjektů ve skupině se simulovanou léčbou, kteří byli léčeni kdykoliv buď antikoagulantem nebo antitrombocytární medikací, ve srovnání s 27 % subjektů léčených přípravkem ILUVIEN. U subjektů léčených přípravkem ILUVIEN souběžně nebo v průběhu 30 dnů od ukončení léčby pomocí antioagulancia nebo antitrombocytární léčby se objevila mírně zvýšená incidence spojivkového krvácení ve srovnání se subjekty léčenými se simulovanou léčbou (0,5 % simulovaná léčba a 2,7 % ILUVIEN). Jedinou další příhodou hlášenou s vyšší incidencí u subjektů léčených přípravkem ILUVIEN byla komplikace oční operace (0 % simulovaná léčba a 0,3 % léčených přípravkem ILUVIEN).
Existuje možnost, že by mohlo dojít k posunu implantátu do přední komory, zejména u pacientů s abnormalitami zadní kapsuly, jako j sou trhliny. To je třeba zvážit při vyšetřování pacientů, kteří si stěžují na poruchy zraku po léčbě.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Údaje o intravitreálním použití fluocinolon-acetonidu u těhotných žen nejsou k dispozici. Studie reprodukční toxicity intravitreálně podaného fluocinolon-acetonidu na zvířatech jsou nedostatečné (viz bod 5.3). I když není fluocinolon-acetonid detekovatelný v systémové cirkulaci po lokálním nitroočním ošetření, je fluocinolon potentní kortikoid a dokonce i velmi nízké hladiny systémové expozice mohou představovat určité riziko pro vývoj plodu. Použití přípravku ILUVIEN v těhotenství se z preventivních důvodů nedoporučuje.
Kojení
Systémově podávaný fluocinolon-acetonid je vylučován do mateřského mléka. I když se očekává, že systémová expozice u kojících žen po intravitreálním podání fluocinolon-acetonidu je velmi nízká, je nutno na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku rozhodnout, zda přerušit kojení nebo podávání přípravku ILUVIEN nezahájit.
Fertilita
Nejsou dostupné žádné údaje týkající se fertility. Nicméně účinky na plodnost u mužů a žen jsou nepravděpodobné, protože systémová expozice fluocinolon-acetonidu po intravitreálním podání je velmi nízká.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek ILUVIEN má malý vliv na schopnost řídit a používat stroje. U pacientů se může po podání přípravku ILUVIEN vyskytnout dočasné zhoršení zraku mají se vyhnout řízení nebo používání strojů, dokud to neustoupí.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Intravitreálně podaný fluocinolon-acetonid byl hodnocen u 768 subjektů (375 ve skupině 0,2 mikrogramů/den/ILUVIEN; 393 ve skupině 0,5 mikrogramů/den) s diabetickým makulárním edémem v rámci klinických studií FAME. Nejčastěji hlášené nežádoucí účinky léků zahrnovaly operaci katarakty, kataraktu a zvýšený nitrooční tlak.
Ve studiích fáze 3 vyžadovalo 38,4 % subjektů léčených přípravkem ILUVIEN léky na snižování nitroočního tlaku a 4,8 % vyžadovalo operaci na snižování nitroočního tlaku. Použití léků na snižování nitroočního tlaku bylo podobné u subjektů, kteří podstoupili dvě nebo více ošetření přípravkem ILUVIEN.
Během studií fáze 3 byly hlášeny dva případy endoftalmitidy u subjektů léčených přípravkem ILUVIEN. To představuje incidenci 0,2 % (2 případy rozdělené na 1022 injekcí).
Vzhledem k tomu, že většina subjektů v klinických studiích FAME dostala pouze jeden implantát (viz bod 5.1), nejsou dlouhodobé implikace retence nebiodegradabilního implantátu uvnitř oka známé.
V klinických studiích FAME ukazují 3leté údaje, že se příhody, jako je katarakta, zvýšený nitrooční tlak a mušky objevovaly pouze mírně častěji u subjektů, kteří dostávali 2 nebo více implantátů. To je považováno za funkci zvýšené expozice léku a nikoliv za účinek samotného implantátu.
V neklinických studiích se nevyskytly doklady pro zvýšení bezpečnostních rizik kromě změn čočky v oku králíka s 2-4 implantáty za 24 měsíců. Implantát je vyroben z polyimidu a je prakticky podobný haptice intraokulární čočky. Proto se očekává, že zůstane uvnitř oka inertní.
Tabulkový seznam nežádoucích účinků
Následující nežádoucí účinky byly považovány jako související s léčbou a jsou klasifikovány dle následující konvence: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000), velmi vzácné (< 1/10000). V rámci jednotlivých skupin frekvence jsou nežádoucí účinky řazeny dle klesající závažnosti.
|
Infekce a infestace |
Méně časté: Endoftalmitida |
|
Poruchy nervového systému |
Méně časté: Bolesti hlavy |
|
Poruchy oka |
Velmi časté: katarakta1, zvýšený nitrooční tlak2 Časté: glaukom3, bolesti oka4, krvácení do sklivce, krvácení do spojivky, rozmazané vidění5, snížená zraková ostrost, sklivcové plovoucí zákaly |
|
Méně časté: Retinální cévní okluze6, porucha optického nervu, makulopatie, atrofie optiku, vřed spojivky, neovaskularizace duhovky, retinální exsudáty, degenerace sklivce, odchlípení sklivce, zakalení zadní kapsuly, adheze duhovky, oční hyperemie, ztenčení sklery, výtok z oka, pruritus oka | |
|
Poranění, otravy a procedurální komplikace |
Méně časté: Extruze implantátu, implantát v rovině pohledu, komplikace zákroku, bolest při zákroku |
|
Chirurgické a léčebné postupy |
Velmi časté: operace katarakty Časté: trabekulektomie, operace glaukomu, vitrektomie, trabekuloplastika Méně časté: odstranění extrudovaného implantátu ze sklery |
|
Celkové poruchy a reakce v místě aplikace |
Méně časté: dislokace přístroje |
1 Zahrnuje MedDRA terminologii pro kataraktu (jinak nespecifikováno), subkapsulámí kataraktu, kortikální kataraktu, nukleární kataraktu a diabetickou kataraktu.
2 Zahrnuje MedDRA terminologii pro zvýšení nitroočního tlaku a oční hypertenzi.
3 Zahrnuje MedDRA terminologii pro glaukom, glaukom s otevřeným úhlem, hraniční glaukom, cupping optického nervu a zvýšení poměru jamky/disku optického nervu.
4 Zahrnuje MedDRA terminologii pro bolesti oka, podráždění oka a okulární diskomfort.
5 Zahrnuje MedDRA terminologii pro rozmazané vidění a poruchu zraku.
6 Zahrnuje MedDRA terminologii pro okluzi retinální žíly, okluzi retinální arterie a vaskulární retinální okluzi.
Popis vybraných nežádoucích účinků
Dlouhodobé použití kortikosteroidů může způsobovat kataraktu a zvýšení nitroočního tlaku. Frekvence výskytu uvedená níže reflektuje nálezy u všech pacientů ve studiích FAME. Pozorované frekvence u pacientů s chronickou DMO nebyl významně odlišný od výskytu v celkové populaci.
Incidence katarakty u fakických pacientů byla v klinických studiích fáze 3 asi 82 % u pacientů léčených přípravkem ILUVIEN a 50 % u pacientů se simulovanou léčbou. 80 % fakických pacientů léčených přípravkem ILUVIEN vyžadovalo operaci katarakty do 3. roku ve srovnání s 27 % pacientů se simulovanou léčbou s tím, že většina pacientů vyžadovala operaci do 21 měsíců. Zadní subkapsulární katarakta je nejčastější typ katarakty související s kortikosteroidy. Operace tohoto typu katarakty je obtížnější a může být spojena s vyšším rizikem chirurgických komplikací.
Ve studiích FAME byly pacienti s výchozím NOT > 21 mmHg vyloučeny. Incidence zvýšeného nitroočního tlaku byla 37 % a 38 % pacientů vyžadovalo léky na snižování nitroočního tlaku, přičemž polovina z nich vyžadovala minimálně dva léky pro kontrolu NOT. Použití léků na snižování nitroočního tlaku bylo podobné u pacientů, u kterých byla aplikována léčba dalším implantátem během studie. Kromě toho 5,6 % (21/375) pacientů, kteří dostali implantát, vyžadovalo chirurgickou nebo laserovou operaci pro kontrolu nitroočního tlaku (trabekuloplastika 5 (1,3 %), trabekulektomie 10 (2,7 %), endocykloablace 2 (0,5 %) a další chirurgické metody 6 (1,6 %)).
V podskupině pacientů s vyšším než středním nitroočním tlakem na začátku (> 15 mmHg), 47 % vyžadovalo přípravky na snižování nitroočního tlaku a podíl chirurgických a laserových zákroků se zvýšil na 7,1 %. V této podskupině bylo 5 pacientů (2,2 %) léčeno pomocí trabekuloplastiky, 7 (3,1 %) pomocí trabekulektomie, 2 (0,9 %) pomocí endocykloablace a 4 (1,8 %) pomocí další chirurgické metody pro léčbu glaukomu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www .sukl .cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Nebyl popsán žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: PROTIZÁNĚTLIVÁ LÉČIVA, kortikosteroidy, samotné ATC kód: S01BA15
Kortikosteroidy inhibují zánětlivou odpověď na různé vyvolávající látky. Inhibují otok, fibrinová depozita, dilataci kapilár, migraci leukocytů, kapilární proliferaci, proliferaci
fibroblastů, depozita kolagenu a vznik jizev související se zánětem.
Kortikosteroidy pravděpodobně působí prostřednictvím indukce inhibičních proteinů fosfolipázy A, souhrnně označovaných jako lipokortiny. Předpokládá se, že tyto proteiny kontrolují biosyntézu potentních mediátorů zánětu, jako jsou prostaglandiny a leukotrieny inhibicí uvolnění společných prekurzorů arachidonové kyseliny. Kyselina arachidonová je uvolňována z membránových fosfolipidů prostřednictvím fosfolipázy A2. Kortikosteroidy také prokazatelně redukují hladinu vaskulárního endotheliálního růstového faktoru, což je protein, který zvyšuje cévní permeabilitu a způsobuje otok.
Účinnost přípravku ILUVIEN byla hodnocena ve dvou randomizovaných, multicentrických, dvojitě zaslepených paralelních studiích zahrnujících pacienty s diabetickým makulárním edémem, kteří byli dříve léčeni laserovou fotokoagulací minimálně jednou, z nichž každá zahrnovala tři roky sledování. 74,4 % pacientů bylo léčeno 1 implantátem, 21,6 % bylo léčeno 2 implantáty, 3,5 % bylo léčeno 3 implantáty a 0,5 % 4 implantáty a 0 % > 4 implantáty). Primární cílový bod účinnosti v obou studiích byl podíl pacientů, jejichž zrak se zlepšil o 15 písmen nebo více po 24 měsících. U každé z těchto studií byl splněn primární cílový bod pro přípravek ILUVIEN (viz obrázek 1 pro integrované výsledky primárního cílového bodu účinnosti).
Procento subjektů se zlepšením o > 15 písmen oproti výchozímu stavu, integrované studie FAME
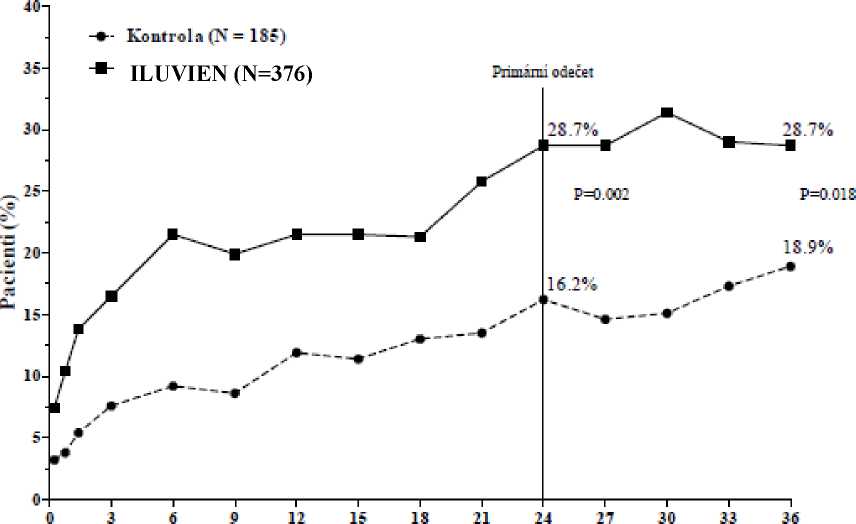
Měsíce
Když byla hodnocena účinnost jako funkce trvání choroby, pacienti s trváním DMO větším, než je medián (> 3 roky), měli významně příznivou reakci na přípravek ILUVIEN, zatímco pacienti s kratším trváním DMO nevykazovali další přínos proti kontrolní léčbě s ohledem na zlepšení zraku (Obrázek 2 a 3). Tyto údaje z podskupiny podporují indikaci v bodě 4.1 o použití u pacientů s chronickou DMO (tzn. trvání alespoň 3 roky).
Srovnání procenta pacientů se zlepšením > 15 písmen oproti výchozímu BCVA (nejlépe upravené ostrosti zraku) a průměrná změna od výchozí hodnoty překročení tloušťky centrálního bodu podle skupiny trvání DMO > 3 roky
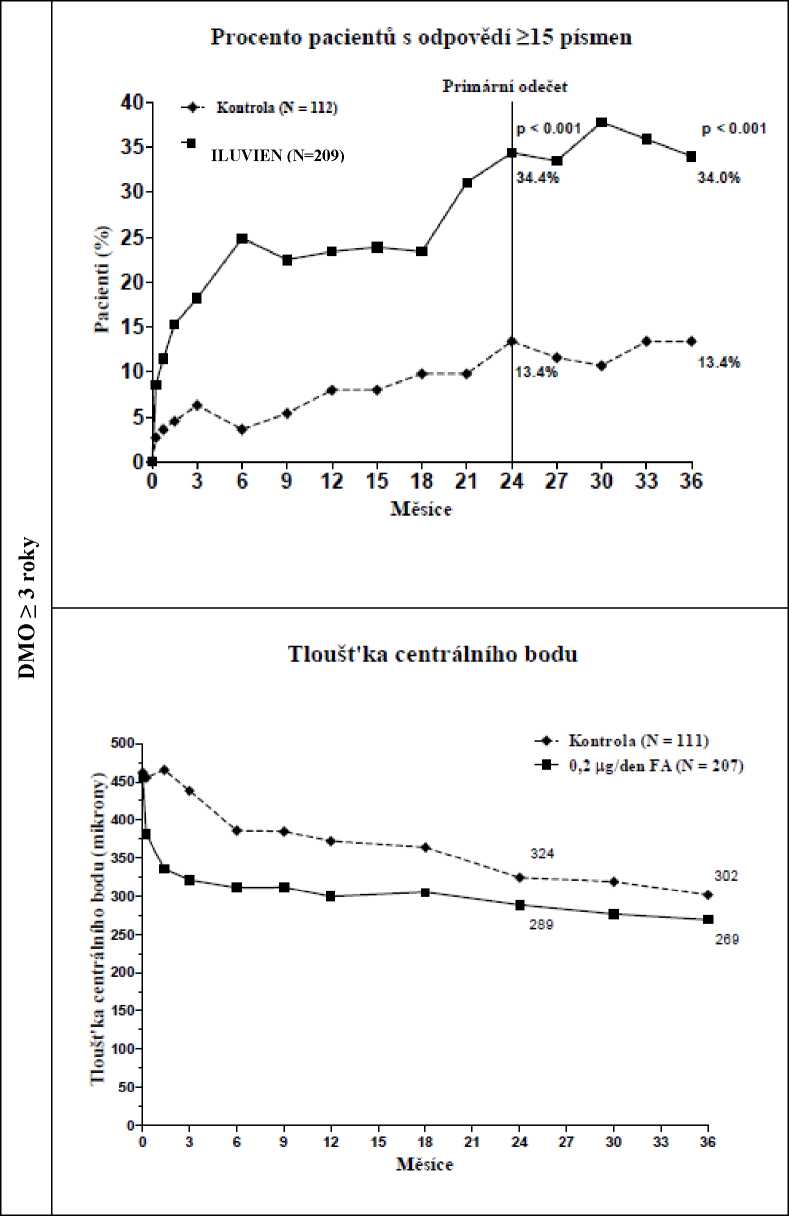
Obrázek 3: Srovnání průměrné změny od výchozí hodnoty překročení tloušťky centrálního
bodu a procento pacientů se zlepšením > 15 písmen od výchozího BCVA podle skupiny trvání DMO < 3 roky
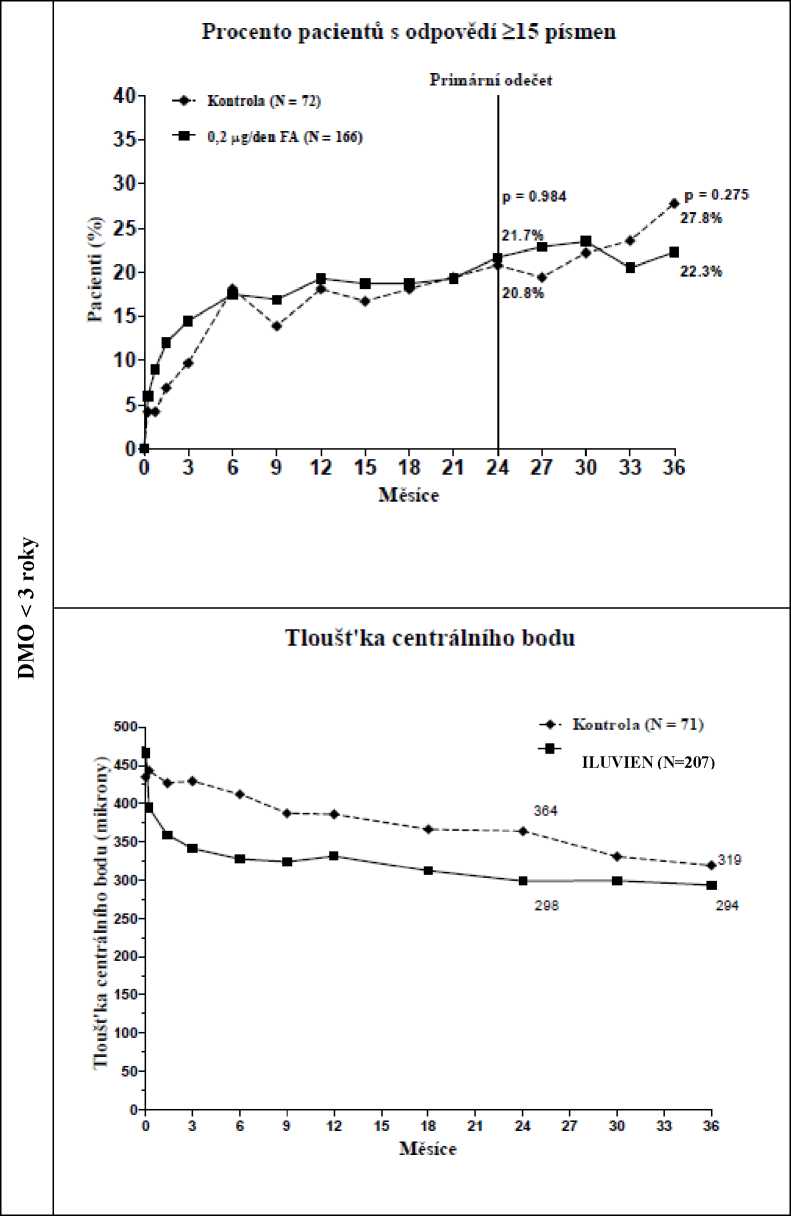
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s intravitreálně podávaným fluocinolon-acetonidem u všech podskupin pediatrické populace pro léčbu diabetického makulárního edému. Informace o použití u dětí viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Ve farmakokinetických studiích u člověka (C-01-06-002, studie FAMOUS) byly koncentrace fluocinolon-acetonidu v plazmě nižší, než dolní mez kvantifikace analýzy (100 pg/ml) ve všech časových bodech od dne 1 do měsíce 36. Maximální koncentrace fluocinolon-acetonidu ve sklivci byla pozorována v den 7 pro většinu pacientů. Koncentrace fluocinolon-acetonidu ve sklivci se snižovaly v průběhu prvních 3 - 6 měsíců a zůstaly prakticky stejné do 36. měsíce u subjektů, které nebyly léčené opakované. U pacientů, kteří byli léčeni opakovaně, se objevila druhá vrcholová koncentrace fluocinolon-acetonidu podobná té, která se vyskytla po úvodní dávce. Po opakované léčbě se koncentrace fluoxinolon-acetonidu ve sklivci vrátily na hladiny přibližně podobné hladinám pozorovaným v době prvního ošetření.
Obrázek 4: Hladiny FA ve sklivci člověka u pacientů, kteří dostali 1 implantát ILUVIEN
(studie FAMOUS)
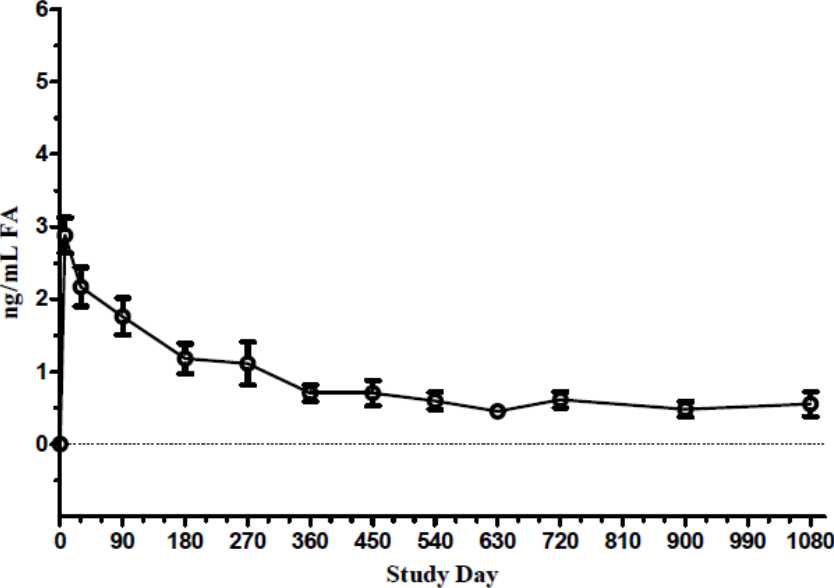
Den studie
5.3 Předklinické údaje vztahující se k bezpečnosti
Po systémovém podání byly zjištěny teratogenní účinky fluocinolon-acetonidu u myší a králíků. Nejsou dostupné žádné údaje o mutagenicitě, karcinogenicitě nebo vývojové toxicitě fluocinolon-acetonidu po intravitreálním podání. Nicméně intravitreálně podaný fluocinolon-acetonid nebyl detekovatelný systémově a proto se systémové účinky nepředpokládají.
Místní účinky (fokálně degenerativní léze postihující vlákna v zadní polární a zadní kortikální oblasti čočky) byly pozorovány u králíků při dávkách intravitreálního fluocinolon-acetonidu přesahujících klinicky používanou dávku. Místní účinky (fokální retinální jizvení) byly také pozorovány u králíků léčených placebem a fluocinolon-acetonidem v prostředku. Toto jizvení nebylo zjištěno klinicky u člověka a předpokládá se, že je důsledkem anatomických odlišností mezi králičím a lidským okem.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Polyvinylalkohol Polyimidová trubička Upravený dimetikon
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Po prvním otevření fólie okamžitě použijte.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C. Chraňte před chladem nebo mrazem.
Otevírejte uzavřený tvarovaný fóliový blistr až těsně před aplikací.
6.5 Druh obalu a obsah balení
Implantát se dodává v jednorázovém aplikátoru s jehlou velikosti 25 G. Jeden sterilní aplikátor obsahuje světle hnědý tyčinkový implantát délky 3,5 mm. Aplikátor je zabalen v tvarovaném fóliovém blistru.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Aplikátor zlikvidujte bezpečně v nádobě na biologicky nebezpečné ostré předměty.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Alimera Sciences Limited
Royal Pavilion Wellesley Road Aldershot Hampshire GU11 1PZ Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
64/016/15-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25.2.2015 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
13.10.2015