Fulvestrant Teva 250 Mg
Sp.zn.sukls212132/2014 SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Fulvestrant Teva 250 mg
Injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje fulvestrantum 250 mg. Jeden ml roztoku obsahuje fulvestrantum50 mg.
Pomocné látky se známým účinkem
Jedna předplněná injekční stříkačka obsahuje: etanol 96% (alkohol) 500 mg benzylalkohol 500 mg benzyl - benzoat 750 mg čištěný ricinový olej do 5ml
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v předplněné injekční stříkačce
Čirý, bezbarvý až žlutý viskózní roztok. Parenterální roztoky je třeba před podáním vizuálně zkontrolovat na pevné částice nebo změnu barvy.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Fulvestrant Teva je indikován k léčbě postmenopauzálních žen s positivním estrogenovým receptorem, lokálně pokročilým nebo metastatickým karcinomem prsu při relapsu onemocnění v průběhu nebo po adjuvantní antiestrogenové terapii nebo progresi onemocnění při léčbě antiestrogeny.
4.2 Dávkování a způsob podání
Dávkování
Dospělé ženy (včetně starších žen)
Doporučená dávka je 500 mg jednou měsíčně s dodatečnou dávkou 500 mg podanou po dvou týdnech od zahajovací dávky.
Zvláštní populace
Porucha funkce ledvin
U pacientek s lehkou až středně těžkou poruchou funkce ledvin (clearance kreatininu > 30 ml/min) není třeba žádná úprava dávky. U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) nebyla bezpečnost a účinnost hodnocena, a proto je u těchto pacientů třeba postupovat s opatrností (viz bod 4.4).
Porucha funkce jater
U pacientek s lehkou až středně těžkou poruchou funkce jater není třeba žádná úprava dávky. Jelikož však může dojít k větší expozici fulvestrantu, je třeba u těchto pacientek používat Fulvestrant Teva s opatrností. U pacientek s těžkou poruchou funkce jater nejsou k dispozici žádné údaje (viz body 4.3, 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Fulvestrant Teva u dětí od narození do 18 let nebyla dosud stanovena. V současnosti dostupné údaje jsou uvedeny v bodech 5.1 a 5.2, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání
Fulvestrant Teva se podává jako dvě po sobě jdoucí 5ml injekce aplikované pomalou intramuskulární injekcí (1-2 minuty/injekce), po jedné injekci do každé hýždě.
Podrobné instrukce pro podání viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Těhotenství a kojení (viz bod 4.6).
Těžká porucha funkce jater (viz body 4.4 a 5.2).
4.4 Zvláštní upozornění a opatření pro použití
U pacientek s lehkou až středně těžkou poruchou funkce jater je třeba používat Fulvestrant Teva s opatrností (viz body 4.2, 4.3 a 5.2).
Fulvestrant Teva je třeba používat s opatrností při léčbě pacientek s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min).
Vzhledem k intramuskulárnímu způsobu podání je třeba používat Fulvestrant Teva s opatrností při léčbě pacientek s krvácivou diatézou, trombocytopenií nebo u pacientek léčených antikoagulancii.
U žen s pokročilým karcinomem prsu jsou často zaznamenávány tromboembolické příhody a byly pozorovány v klinických studiích s fulvestrantem (viz bod 4.8). Při předepisování přípravku Fulvestrant Teva rizikovým pacientkám je na to třeba brát ohled.
Zatím nejsou k dispozici dlouhodobé údaje o působení fulvestrantu na kosti. Vzhledem ke způsobu působení fulvestrantu existuje potenciální riziko vzniku osteoporózy.
Pediatrická populace
Fulvestrant Teva se nedoporučuje podávat dětem a dospívajícím, neboť nebyla stanovena bezpečnost a účinnost u této skupiny pacientů (viz bod 5.1).
Fulvestrant Teva obsahuje 96 % etanol (alkohol)
Tento léčivý přípravek obsahuje 10 % obj. etanolu (alkoholu), tj. do 1000 mg v jedné dávce, což odpovídá 20 ml piva nebo 8 ml vína. Je škodlivý pro osoby, které trpí alkoholismem. Je nutno vzít v úvahu u těhotných a kojících žen, dětí a vysoce rizikových skupin, jako jsou pacienti s jaterním onemocněním nebo epilepsií.
Fulvestrant Teva obsahuje benzylalkohol
Tento léčivý přípravek obsahuje benzylalkohol. Množství benzylalkoholu je 500 mg na 5 ml dávku (100 mg benzylalkoholu v 1 ml). Benzylalkohol může způsobit anafylaktické/anafylaktoidní reakce.
Fulvestrant Teva obsahuje čištěný ricinový olej
Tento léčivý přípravek obsahuje čištěný ricinový olej, který může způsobit těžké alergické reakce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Klinická interakční studie s midazolamem (substrát CYP3A4) prokázala, že fulvestrant neinhibuje CYP3A4. Klinická interakční studie s rifampicinem (induktor CYP3A4) a ketokonazolem (inhibitor CYP3A4) neprokázala žádné klinicky významné změny v clearance fulvestrantu. Pacientkám, kterým je podáván fulvestrant současně s inhibitory nebo s induktory CYP3A4, není nutné upravovat dávku.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Pacientky ve fertilním věku musí být poučeny, aby v průběhu léčby používaly účinnou antikoncepci. Těhotenství
Fulvestrant Teva je kontraindikován v těhotenství (viz bod 4.3). Bylo prokázáno, že fulvestrant po jednorázové nitrosvalové dávce u potkanů a králíků prostupuje placentou. Studie na zvířatech prokázaly reprodukční toxicitu včetně zvýšeného výskytu abnormalit a úmrtí plodu (viz bod 5.3). Dojde-li v průběhu podávání přípravku Fulvestrant Teva k otěhotnění, musí být pacientka informována o možném riziku pro plod a potenciálním riziku potratu.
Kojení
V průběhu léčby přípravkem Fulvestrant Teva musí být kojení přerušeno. Fulvestrant se u potkanů vylučuje do mateřského mléka. Není známo, zda se fulvestrant vylučuje do lidského mateřského mléka. Vzhledem k možnosti závažných nežádoucích účinků fulvestrantu pro kojence je kojení v průběhu léčby kontraindikováno (viz bod 4.3).
Fertilita
Účinek přípravku Fulvestrant Teva na fertilitu u člověka nebyl studován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Fulvestrant Teva nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. V průběhu léčby přípravkem Fulvestrant Teva však byla velmi často hlášena astenie, proto je třeba při řízení nebo obsluze strojů opatrnosti u pacientů, u nichž se tento nežádoucí účinek vyskytnul.
4.8 Nežádoucí účinky
Informace v tomto bodě shrnují všechny nežádoucí účinky z klinických studií, poregistračních studií nebo spontánních hlášení. Nejčastěji hlášenými nežádoucími účinky jsou reakce v místě vpichu, astenie, nauzea a zvýšené jaterní enzymy (ALT, AST, ALP).
Následující kategorie četností nežádoucích účinků byly vypočteny na základě analýzy souhrnných bezpečnostních údajů léčebné skupiny s fulvestrantem v dávce 500 mg ve studii CONFIRM (studie D6997C00002), FINDER 1 (studie D6997C00004), FINDER 2 (studie D6997C00006) a NEWEST (studie D6997C00003), které porovnávaly fulvestrant 500 mg a fulvestrant 250 mg. Četnosti uvedené v následující tabulce jsou založeny na všech hlášených nežádoucích účincích bez ohledu na hodnocení kauzality řešitelem.
Následující nežádoucí účinky jsou klasifikovány podle četnosti a třídy orgánových systémů (SOC). Četnosti výskytu jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1
Nežádoucí účinky
|
Nežádoucí účinky podle tříd orgánových systémů a frekvence | ||
|
Infekce a infestace |
Časté |
Infekce močových cest |
|
Poruchy krve a lymfatického systému |
Méně časté |
Snížení počtu krevních destiček |
|
Poruchy imunitního systému |
Časté |
Hypersensitivní reakce |
|
Poruchy metabolismu a výživy |
Časté | |
|
Poruchy nervového systému |
Časté | |
|
Cévní poruchy |
Časté |
Žilní tromboembolismus3, návaly horka |
|
Gastrointestinální poruchy |
Velmi časté | |
|
Časté | ||
|
Poruchy jater a žlučových cest |
Velmi časté |
Zvýšení jaterních enzymů (ALT, AST, Alp)3 |
|
Časté |
Zvýšená hladina bilirubinu3 | |
|
Méně časté |
Selhání jaterc, hepatitidac, zvýšená hladina GMT | |
|
Poruchy kůže a podkožní tkáně |
Časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté | |
|
Poruchy reprodukčního systému a prsu |
Méně časté |
Vaginální moniliáza, leukorhea, vaginální krvácení |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Astenie3, reakce v místě injekceb |
|
Méně časté |
Krvácení v místě aplikace, hematom v místě aplikace | |
a Zahrnuje nežádoucí účinky, u kterých nelze přesně určit podíl fulvestrantu v důsledku probíhajícího onemocnění.
b Termín reakce v místě aplikace nezahrnuje termíny krvácení v místě aplikace a hematom v místě aplikace. c Příhoda nebyla pozorována v hlavních klinických studiích (CONFIRM, FINDER 1, FINDER 2, NEWEST). Frekvence byla vypočtena za použití horního limitu pro 95% interval spolehlivosti pro bodový odhad. Ten je vypočten jako 3/560 (kde 560 je počet pacientů v hlavních klinických studiích), což odpovídá kategorii četnosti „méně časté“.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-utinek
4.9 Předávkování
U člověka nejsou žádné zkušenosti s předávkováním. Studie na zvířatech naznačují, že kromě účinků přímo nebo nepřímo souvisejících s antiestrogenním účinkem nebyly u vyšších dávek fulvestrantu prokázány žádné jiné účinky (viz bod 5.3). Dojde-li k předávkování, doporučuje se symptomatická podpůrná léčba.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: endokrinní léčba, antiestrogeny, ATC kód: L02BA03 Mechanismus účinku a farmakodynamické účinky
Fulvestrant je kompetitivní antagonista estrogenového receptoru (ER) s afinitou srovnatelnou s estradiolem. Fulvestrant blokuje trofický účinek estrogenů bez projevů částečného agonistického (tj. estrogenům podobného) účinku. Mechanismus účinku je spojen s down regulací hladin proteinu estrogenového receptoru. Klinické studie u postmenopausálních žen s primárním karcinomem prsu prokázaly, že fulvestrant významně snižuje hladinu proteinu ER v ER pozitivních nádorech ve srovnání s placebem. Docházelo také k významnému poklesu exprese progesteronového receptoru bez vnitřního agonistického estrogenního účinku. Bylo prokázáno, že fulvestrant 500 mg snižuje počet ER a proliferaci markeru Ki67 ve větší míře než fulvestrant 250 mg u postmenopauzálních žen s karcinomem prsu v neoadjuvantním uspořádání.
Klinická účinnost a bezpečnost u pokročilého karcinomu prsu
U 736 postmenopauzálních žen s pokročilým karcinomem prsu, které relabovaly při adjuvantní endokrinní léčbě nebo po této léčbě nebo progredovaly po endokrinní léčbě pokročilého onemocnění, byla provedena klinická studie fáze III. Ve studii bylo zařazeno 423 pacientek, které relabovaly nebo progredovaly při léčbě antiestrogeny (podskupina AE) a 313 pacientek, které relabovaly nebo progredovaly při léčbě inhibitory aromatázy (podskupina AI). Tato studie srovnávala účinnost a bezpečnost fulvestrantu 500 mg (n=362) s fulvestrantem 250 mg (n=374). Primárním cílovým parametrem bylo přežití bez progrese (PFS), klíčové sekundární parametry účinnosti zahrnovaly výskyt objektivní odpovědi (ORR), klinickou míru prospěšnosti (CBR) a celkové přežití (OS). Výsledky účinnosti ze studie CONFIRM jsou shrnuty v tabulce 2.
Tabulka 2 Souhrn výsledků primárních cílových parametrů účinnosti (PFS) a klíčové sekundární parametry účinnosti ze studie CONFIRM
|
Proměnná |
Druh odhadu; srovnání léčby |
Fulvestrant 500 mg (N=362) |
Fulvestrant 250 mg (N=374) |
Srovnání mezi skupinami (Fulvestrant 500 mg/Fulvestrant 250 mg) Poměr rizik 95% CI p-hodnota | ||
|
PFS |
K-M medián | |||||
|
V měsících; | ||||||
|
Poměr rizik | ||||||
|
Všichni pacienti |
6,5 |
5,5 |
0,80 |
0,68; 0,94 |
0,006 | |
|
-podskupina AE (n=423) |
8,6 |
5,8 |
0,76 |
0,62; 0,94 |
0,013 | |
|
-podskupina AI (n=313)a |
5,4 |
4,1 |
0,85 |
0,67; 1,08 |
0,195 | |
|
OSb |
K-M medián | |||||
|
v měsících; | ||||||
|
poměr rizik | ||||||
|
Všichni pacienti |
26,4 |
22,3 |
0,81 |
0,69; 0,96 |
0,016c | |
|
-podskupina AE (n=423) |
30,6 |
23,9 |
0,79 |
0,63; 0,99 |
0,038c | |
|
-podskupina AI (n=313)a |
24,1 |
20,8 |
0,86 |
0,67; 1,11 |
0,241c | |
|
Proměnná |
Druh |
Fulvestrant |
Fulvestrant |
Srovnání mezi |
skupinami | |
|
odhadu; |
500 mg |
250 mg |
(Fulvestrant 500 mg/Fulvestrant 250 mg) | |||
|
Srovnání |
(N=362) |
(N=374) |
Absolutní rozdíl |
95% CI | ||
|
léčby |
v % | |||||
|
ORRd |
% pacientů | |||||
|
s OR; | ||||||
|
absolutní | ||||||
|
rozdíl v % | ||||||
|
Všichni pacienti |
13,8 |
14,6 |
-0,8 |
-5,8; 6,3 | ||
|
-podskupina AE (n=296) |
18,1 |
19,1 |
-1,0 |
-8,2; 9,3 | ||
|
-podskupina AI (n=205)a |
7,3 |
8,3 |
-1,0 |
-5,5; 9,8 | ||
CBRe % pacientů
s CB;
absolutní rozdíl
v %
|
Všichni pacienti |
45,6 |
39,6 |
6,0 |
-1,1; 13,3 |
|
-podskupina AE (n=423) |
52,4 |
45,1 |
7,3 |
-2,2; 16,6 |
|
-podskupina AI (n=313)a |
36,2 |
32,3 |
3,9 |
-6,1; 15,2 |
a
Fulvestrant je indikován u pacientek, které relabovaly nebo progredovaly při léčbě antiestrogeny. Výsledky v podskupině AI jsou neprůkazné.
OS je uvedeno pro finální analýzu celkového přežití při 75% úplnosti dat.
Nominální hodnota p bez úpravy na opakované hodnoty mezi původní analýzou celkového přežití při 50% úplnosti dat a aktualizovanou analýzou celkového přežití při 75% úplnosti dat.
ORR byl hodnocen u pacientek, u kterých byla hodnotitelná odpověď při vstupu do studie (tj. ty s měřitelnou nemocí při vstupu do studie: 240 pacientek ve skupině fulvestrant 500 mg a 261 pacientek ve skupině fulvestrant 250 mg).
Pacientky s nejlepší objektivní odpovědí, částečnou odpovědí nebo stabilní nemocí po dobu>24 týdnů.
b
c d e
PFS: přežití bez progrese; ORR: výskyt objektivní odpovědi; OR: relativní riziko; CBR: klinická míra prospěšnosti; CB: klinická prospěšnost; OS: celkové přežití; K-M: Kaplan-Meier; CI: interval spolehlivosti, AI: inhibitor aromatázy, AE: antiestrogen.
Byly provedeny dvě klinické studie fáze III. Bylo do nich zařazeno celkem 851 postmenopauzálních žen s pokročilým karcinomem prsu, u kterých došlo k návratu onemocnění při nebo po adjuvantní endokrinní léčbě nebo k progresi po endokrinní léčbě pokročilého onemocnění. 77 % populace ve studii mělo estrogen receptor pozitivní karcinom prsu. V těchto studiích byla porovnávána bezpečnost a účinnost podávání fulvestrant 250 mg jednou měsíčně s denním podáváním 1 mg anastrozolu (inhibitor aromatázy). Celkově byl fulvestrant v dávce 250 mg měsíčně nejméně stejně účinný jako anastrozol, pokud jde o přežití bez progrese, výskyt objektivní odpovědi a doby přežití. Mezi oběma léčenými skupinami nebyly u primárních cílových parametrů studie žádné statisticky významné rozdíly. Primárním cílovým parametrem studie bylo přežití bez progrese. Kombinovaná analýza obou studií prokázala, že k progresi došlo u 83 % pacientek, které dostávaly fulvestrant, oproti 85 % pacientek, které užívaly anastrozol. Kombinovaná analýza obou studií ukázala, že poměr rizik fulvestrantu 250 mg oproti anastrozolu pro přežití bez progrese je 0,95 (95% CI 0,82 až 1,10). Výskyt objektivní odpovědi na fulvestrant 250 mg byl 19,2 % ve srovnání s 16,5 % na anastrozol. Střední doba přežití činila 27,4 měsíce u pacientek léčených fulvestrantem a 27,6 měsíce u pacientek léčených anastrozolem. Poměr rizik fulvestrantu 250 mg vs anastrozol pro dobu přežití byl 1,01 (95% CI 0,86 až 1,19).
Účinky na endometrium v menopauze
Předklinické údaje nenaznačují stimulační účinek fulvestrantu na endometrium u postmenopauzálních žen (viz bod 5.3). Dvoutýdenní studie u zdravých žen po menopauze léčených 20 mikrogramy ethinylestradiolu denně prokázala, že předchozí podávání fulvestrantu 250 mg vedlo k významně snížené stimulaci endometria v menopauze ve srovnání s předchozím podáváním placeba podle ultrazvukového měření tloušťky endometria.
Neoadjuvantní léčba po dobu až 16 týdnů u pacientek s karcinomem prsu léčených buďto fulvestrantem 500 mg nebo fulvestrantem 250 mg nevedla ke změně tloušťky endometria, což svědčí pro absenci agonistického účinku. Neexistuje důkaz pro nežádoucí účinky na děložní sliznici u sledovaných pacientek s karcinomem prsu. Nejsou k dispozici údaje týkající se morfologie endometria.
Ve dvou krátkodobých studiích (1 a 12 týdnů) u premenopauzálních žen s benigním gynekologickým onemocněním nebyly pozorovány žádné signifikantní změny tloušťky endometria (měřeno ultrazvukem) při srovnání skupin s fulvestrantem a placebem.
Účinky na kosti
Nejsou k dispozici dlouhodobé údaje o účinku fulvestrantu na kosti. Neoadjuvantní léčba po dobu až 16 týdnů u pacientek s karcinomem prsu léčených buďto fulvestrantem 500 mg nebo fulvestrantem 250 mg nevedla k významným změnám sérových markerů kostního metabolismu.
Pediatrická populace
Fulvestrant není indikován k použití u dětí. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s fulvestrantem u všech podskupin pediatrické populace s karcinomem prsu (informace o použití u dětí viz bod 4.2).
Otevřená studie fáze II zjišťovala bezpečnost, účinnost a farmakokinetiku fulvestrantu u 30 dívek ve věku 1 až 8 let s progresivní předčasnou pubertou ve spojení s McCune-Albrightovým syndromem (MAS). Pediatričtí pacienti dostávali dávku 4 mg/kg fulvestrantu intramuskulárně měsíčně. Tato 12měsíční studie hledala odpověď na řadu MAS cílových parametrů a prokázala snížení frekvence vaginálního krvácení a snížení rychlosti kostního zrání. Nejnižší koncentrace fulvestrantu v ustáleném stavu u dětí v této studii byly konzistentní s koncentracemi u dospělých (viz bod 5.2). V této malé studii nebyly zjištěny žádné nové bezpečnostní údaje, ale 5letá data ještě nejsou k dispozici.
5.2 Farmakokinetické vlastnosti
Absorpce
Po podání nitrosvalové injekce fulvestrantu s dlouhou dobou účinku se fulvestrant pomalu vstřebává a maximálních koncentrací v plazmě (Cmax) je dosaženo po přibližně 5 dnech. Po podání fulvestrantu 500 mg je dosaženo hladin na úrovni rovnovážné koncentrace nebo blízké rovnovážné koncentraci první měsíc po podání (průměr [CV]: AUC 475 [33,4 %] ng.dny/ml, Cmax 25,1 [35,3 %] ng/ml, Cmm 16,3 [25,9 %] ng/ml).
V ustáleném stavu se udržují koncentrace fulvestrantu v plazmě v relativně úzkém rozmezí s přibližně až 3násobným rozdílem mezi maximální a minimální koncentrací. Po nitrosvalovém podání je expozice v rozsahu dávek 50 až 500 mg zhruba úměrná dávce.
Distribuce
Fulvestrant podléhá rozsáhlé a rychlé distribuci. Velký zdánlivý distribuční objem v ustáleném stavu (Vdss) přibližně 3 až 5 l/kg signalizuje, že distribuce je většinou extravaskulární. Fulvestrant je ve velké míře (99 %) vázán na bílkoviny plazmy. Váže se hlavně na lipoproteinové frakce velmi nízké denzity (VLDL), nízké denzity (LDL) a vysoké denzity (HDL). Nebyly provedeny interakční studie kompetitivní vazby na bílkoviny. Role globulinu vázajícího pohlavní hormon (sex hormone-binding globulin, SHBG) nebyla stanovena.
Biotransformace
Metabolismus fulvestrantu nebyl dosud úplně zhodnocen, ale zahrnuje kombinaci celé řady možných biotransformačních cest, které odpovídají cestám endogenních steroidů. Identifikované metabolity (zahrnující metabolity 17-keton, sulfon, 3-sulfát, 3- a 17-glukuronid) jsou v antiestrogenových modelech buď méně účinné, nebo vykazují podobný účinek jako fulvestrant. Studie na preparátech z lidských jater a rekombinantních lidských enzymech ukazují, že na oxidaci fulvestrantu se podílí pouze CYP3A4 cytochromu P450; in vivo se však zdá, že převládají cesty, které nevyužívají cytochromu P450. Údaje in vitro naznačují, že fulvestrant neinhibuje izoenzymy CYP450.
Eliminace
Fulvestrant je vylučován především v metabolizované formě. Hlavní cestou exkrece je stolice, močí se vylučuje méně než 1 %. Fulvestrant má vysokou clearance, 11±1,7 ml/min/kg, což předpokládá vysoký hepatální extrakční poměr. Terminální poločas eliminace (t%) po nitrosvalové aplikaci se řídí rychlostí absorpce a odhaduje se na 50 dnů.
Zvláštní populace
V populační farmakokinetické analýze dat ze studií fáze III nebyly u fulvestrantu nalezeny žádné rozdíly ve farmakokinetickém profilu s ohledem na věk (rozmezí 33 až 89 let), tělesnou hmotnost (40-127 kg) nebo rasu.
Porucha funkce ledvin
Lehká až středně těžká porucha funkce ledvin neovlivnila v klinicky významném rozsahu farmakokinetiku fulvestrantu.
Porucha funkce jater
Farmakokinetika fulvestrantu byla sledována v klinickém hodnocení po jednorázovém podání u pacientů s lehkou až středně těžkou poruchou funkce jater (Child-Pugh syndrom třída A a B). Byla použita vysoká dávka krátkodoběji působícího nitrosvalově podávaného fulvestrantu. AUC u pacientů s poruchou funkce jater bylo 2,5krát vyšší než u zdravých jedinců. Předpokládá se, že toto zvýšení expozice po podání fulvestrantu bude dobře tolerováno. Pacienti s těžkou poruchou funkce jater (Child-Pugh syndrom třída C) nebyli hodnoceni.
Pediatrická populace
V klinické studii provedené u 30 dívek s progresivní předčasnou pubertou spojenou s McCune-Albrightovým syndromem (MAS) byla hodnocena farmakokinetika fulvestrantu (viz bod 5.1). Pacienti byli ve věku 1 až 8 let a byla jim podávána dávka 4 mg/kg fulvestrantu intramuskulárně každý měsíc. Geometrický průměr (směrodatná odchylka) ustálených minimálních koncentrací (Cmin, ss) a AUCss byly 4,2 (0,9) ng/ml, resp. 3680 (1020) ng.h/ml. Ačkoliv jsou získaná data omezená, zdá se, že minimální ustálené koncentrace fulvestrantu u dětí jsou konzistentní s koncentracemi u dospělých.
5.3 Předklinické údaje vztahující se k bezpečnosti
Akutní toxicita fulvestrantu je nízká.
Fulvestrant i jiné lékové formy fulvestrantu byly u všech zvířecích druhů ve studiích s opakovanými dávkami dobře snášeny. Místní reakce včetně myositidy a granulomu v místě injekce byly připsány vehikulu, avšak závažnost myositidy u králíků byla vyšší u fulvestrantu ve srovnání se srovnávací skupinou, které byl podán fyziologický roztok. Ve studiích toxicity s opakovanými intramuskulárními dávkami fulvestrantu u potkanů a psů byla antiestrogenní aktivita fulvestrantu odpovědná za většinu pozorovaných účinků, a to především na samičí reprodukční systém, ale také na další orgány, citlivé na hormony u obou pohlaví. U některých psů byla po chronickém podávání (12 měsíců) pozorována arteritida zahrnující více různých tkání.
Ve studiích na psech po perorálním i intravenózním podání byly zjištěny účinky na kardiovaskulární systém (mírná elevace S-T segmentu na EKG [po perorálním podání] a sinusová zástava u jednoho psa [intravenózní podání]). Tyto účinky se vyskytly při hladině fulvestrantu vyšší než u pacientů (Cmax > 15krát) a je pravděpodobné, že mají omezený klinický význam pro bezpečnost u člověka při podávání klinických dávek.
Fulvestrant nevykazoval žádný genotoxický potenciál.
Účinky fulvestrantu na reprodukci a embryo/fetální vývoj odpovídaly při dávkách podobných dávkám klinickým jeho antiestrogennímu účinku. U potkanů byl pozorován reverzibilní pokles plodnosti samic a přežití embryí, dystokie a zvýšený výskyt abnormalit plodu včetně tarsální flexury. Králičí samice po podání fulvestrantu neudržely březost. Bylo pozorováno zvýšení hmotnosti placenty a poimplantační ztráta plodů. U králíků došlo ke zvýšenému výskytu změn plodu (zpětný posun pánevního pletence a 27 presakrálních obratlů).
Dvouletá studie onkogenity na potkanech (nitrosvalové podání fulvestrantu) prokázala zvýšený výskyt benigních granulózních buněčných nádorů ovaria u potkaních samic při vysokých dávkách 10 mg na potkana/15 dnů a zvýšený výskyt nádorů testikulárních Leydigových buněk u samců. Ve dvouleté studii onkogenity u myší (každodenní perorální podání) byl zjištěn zvýšený výskyt pohlavně vázaných stromálních tumorů vaječníků (jak benigních, tak maligních) po podání dávek 150 a 500 mg/kg/den. Pro úroveň nulového účinku s ohledem na tyto nálezy byla systémová expozice (AUC) u laboratorních potkanů přibližně 1,5krát vyšší než očekávaná expozice u žen a 0,8krát vyšší než expozice u mužů a u myší přibližně 0,8krát vyšší než očekávaná expozice u žen či mužů. Tvorba těchto nádorů odpovídá farmakologicky navozeným endokrinním poruchám zpětnovazebné regulace hladin gonadotropinů vyvolaných antiestrogeny u zvířat s estrálním cyklem. Z tohoto důvodu nejsou tyto poznatky považovány za relevantní k použití fulvestrantu u žen po menopauze s pokročilým karcinomem prsu.
6.1 Seznam pomocných látek
Etanol (96%)
Benzylalkohol Benzyl-benzoát Čištěný ricinový olej
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a transportujte v chladničce (2 °C - 8 °C).
Teplotní výkyvy mimo rozmezí 2 °C - 8 °C by měly být omezeny. To zahrnuje vyloučení uchovávání při teplotách vyšších než 25 °C a nepřekračující 28 denní období při průměrné teplotě uchovávání přípravku do 25 °C (ale vyšší než 2 °C - 8 °C). Po teplotních výkyvech by měl být přípravek ihned vrácen do režimu uchovávání za doporučených podmínek (uchovávejte a transportujte v chladničce 2 °C - 8°C). Teplotní výkyvy mají kumulativní vliv na kvalitu přípravku a 28 denní období nesmí být překročeno v průběhu celé 2 leté doby použitelnosti přípravku Fulvestrant Teva (viz bod 6.3). Vystavení přípravku teplotám nižším než 2 °C nemá za následek poškození přípravku za předpokladu, že nebyl uchováván při teplotách nižších než -20 °C.
Uchovávejte předplněnou stříkačku v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení Předplněná injekční stříkačka se skládá z:
Jedna předplněná injekční stříkačka z čirého skla (třída 1) s polystyrenovým pístem, opatřená uzávěrem garantujícím neporušenost obalu, obsahující 5 ml injekčního roztoku přípravku Fulvestrant Teva.
Přiložena je také bezpečnostní jehla pro připojení k válci injekční stříkačky.
Nebo
Dvě předplněné injekční stříkačky z čirého skla (třída 1) s polystyrenovým pístem, opatřené uzávěremgarantujícím neporušenost obalu, každá obsahující 5 ml injekčního roztoku přípravku Fulvestrant Teva. Přiloženy jsou také bezpečnostní jehly pro připojení k válci injekční stříkačky.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro použití
Upozornění - bezpečnostní jehlu před použitím neautoklávujte.
Ruce musí zůstat po celou dobu použití a podání stále za jehlou.
Pro každou ze dvou injekčních stříkaček:
• Vyjměte skleněný válec injekční stříkačky z blistru a zkontrolujte, zda není poškozen.
• Zlomte pečeť čirého plastového krytu na spojce Luer injekční kryt sejměte společně s gumovým
víčkem (viz obrázek 1).
Obrázek 1
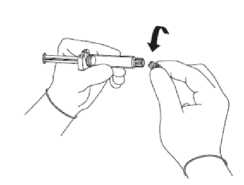
stříkačky
a
Sejměte vnější obal bezpečnostní jehly.
Připevněte bezpečnostní jehlu ke koncovce Luer (viz Obrázek 2).
• Otáčejte, až pevně dosedne.
Obrázek 2
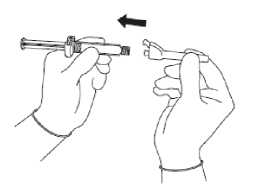
• Otáčejte, abyste jehlu zajistil(a) v koncovce Luer.
• Stáhněte chránítko jehly směrem dopředu tak, abyste nepoškodil(a) hrot jehly (viz obrázek 3).
Obrázek 3
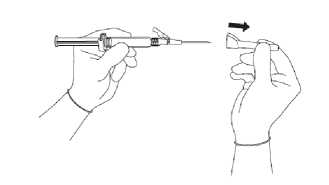
• Přeneste naplněnou injekční stříkačku na místo aplikace.
• Parenterální roztoky je třeba před podáním vizuálně zkontrolovat na pevné částice nebo změnu barvy.
• Vytlačte přebytečný plyn z injekční stříkačky.
• Podávejte jako pomalou nitrosvalovou injekci
(1-2 minuty/injekci) do hýžďového svalu. Pro komfort uživatele orientujte úkos jehly směrem k rameni páčky (viz Obrázek 4).
Obrázek 4

Po aplikaci jedním prstem ihned klepněte na rameno páčky, aby se aktivoval ochranný mechanismus (viz Obrázek 5).
UPOZORNĚNÍ: Vždy aktivujte směrem od sebe a jiných
Obrázek 5
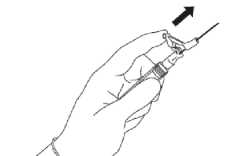
lidí. Zaslechnete kliknutí. Vizuálně se přesvědčte, že hrot jehly je zcela zakryt.
Likvidace
Předplněná injekční stříkačka je určena pouze pro jednorázové použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Teva Pharmaceuticals CR s.r.o Radlická 3185/1c,
Praha 150 00 Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
34/161/16-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13.4.2016 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
13.4.2016
11