Factor Vii Baxalta 600 Iu
sp.zn. sukls135433/2016 a sp.zn. sukls135284/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
FACTOR VII BAXALTA 600 IU Prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Přípravek FACTOR VII BAXALTA je dodáván ve formě lyofilizovaného prášku a obsahuje factor VII coagulationis humanus 600 IU* v jedné injekční lahvičce.
Po rekonstituci v 10 ml vody na injekci obsahuje přípravek přibližně 60 IU/ml (600 IU/10ml) factor VII coagulationis humanus.
Aktivita je stanovena za použití chromogenního testu podle Evropského lékopisu (European Pharmacopoeia). Specifická aktivita přípravku FACTOR VII BAXALTA je > 2 IU faktoru VII/mg proteinu.
Rekonstituovaný přípravek obsahuje < 20 IU FII/100 IU FVII, <15 IU FIX/100 IU FVII a < 35 IU FX/100 IU FVII.
Vyrobeno z plazmy lidských dárců.
Pomocné látky se známým účinkem:
Sodná sůl heparinu (max. 0,5 IU/IU FVII)
Jedna injekční lahvička přípravku obsahuje 1,56 - 1,9 mmol sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok
Bílý nebo lehce zbarvený prášek nebo drobivá hmota. Roztok má po rekonstituci pH 6,5 - 7,5 a osmolalitu > 240 mosmol/kg.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek FACTOR VII BAXALTA je indikován k:
Léčba a profylaxe krvácivých poruch způsobených nedostatkem faktoru VII nebo spojených s nedostatkem faktoru VII:
• Akutní krvácení a profylaxe peroperačního krvácení v případech vrozeného nedostatku faktoru VII (hypo- nebo aprokonvertinémie).
• Akutní krvácení a profylaxe peroperačního krvácení v případech získaného nedostatku faktoru VII:
- perorální antikoagulační léčba
- nedostatek vitaminu K (poruchy vstřebávání, dlouhodobá parenterální výživa atd.)
- poškození jatemího parenchymu (hepatitida, jatemí cirhóza, těžké toxické poškození jater
atd.)
Tento přípravek neobsahuje aktivovaný faktor VIIa a nemá být použit u hemofilických pacientů s inhibitorem.
4.2 Dávkování a způsob podání
Léčba by měla být zahájena pod dohledem lékaře, který má zkušenosti se substituční léčbou koagulačními faktory.
Dávkování
Vzhledem ke vzácnosti výskytu onemocnění jsou k dispozici pouze omezené údaje o klinickém použití přípravků s faktorem VII. Z tohoto důvodu mohou být dána pouze obecná doporučení dávkování. Požadavky na individuální dávkování mohou být identifikovány pouze na základě pravidelného stanovení obsahu faktoru VII v plazmě a soustavného sledování klinického stavu pacienta.
Dávkování a doba substituční léčby závisí na závažnosti nedostatku faktoru VII, na místě a rozsahu krvácení a na klinickém stavu pacienta. Vztah mezi individuálními zbytkovými hladinami faktoru VII a klinickou tendencí ke krvácení je méně pevný než u klasické hemofilie.
Počet podaných jednotek faktoru VII se vyjadřuje v mezinárodních jednotkách (IU), které jsou vztaženy k současnému WHO standardu pro přípravky faktoru VII. Aktivita faktoru VII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě) nebo v mezinárodních jednotkách (ve vztahu k mezinárodnímu standardu pro faktor VII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VII se rovná množství faktoru VII v 1 ml normální lidské plazmy. Výpočet potřebné dávky faktoru VII, jak je níže specifikován, je založen na empirickém zjištění, že 1 mezinárodní jednotka (IU) faktoru VII na kg tělesné hmotnosti zvyšuje aktivitu faktoru VII v plazmě přibližně o 1,9 % (0,019 IU/ml) normální aktivity.
Potřebná dávka se určuje pomocí následujícího vzorce:
„Potřebný počet jednotek = tělesná hmotnost (kg) x požadované zvýšení faktoru VII (IU/ml) x 53* {převrácená hodnota pozorovaného zotavení (ml/kg)}“
*(1/0,019 = 52,6)
Pokud je známo individuální zotavení, má být pro výpočet použita tato hodnota.
Množství, které má být podáno, a četnost podání se musí vždy řídit podle klinické účinnosti v konkrétním případě. Toto je zvláště důležité při léčbě nedostatku faktoru VII, kdy individuální tendence ke krvácení není v přesném vztahu k aktivitě faktoru VII v plazmě naměřeném při laboratorních testech. Dávkovací intervaly musí být přizpůsobeny krátkému poločasu faktoru VII v krevním oběhu, který je přibližně 3 až 5 hodin. Pokud je přípravek FACTOR VII BAXALTA podáván přerušovanými injekcemi/infuzemi, jsou často adekvátní dávkovací intervaly 6 - 8 hodin. Při léčbě nedostatku faktoru VII jsou obvykle, ve vztahu k aktivitě v normální plazmě, požadovány nižší hladiny nedostatkového faktoru než u klasické hemofilie (hemofilie A a B). Tabulka níže dává návod pro podání přerušovanými injekcemi/infuzemi na základě omezené klinické zkušenosti. Medicínské potvrzení na základě zkoušek klinické účinosti neexistuje.
|
Stupeň krvácení / typ chirurgického zákroku |
Požadovaná hladina faktoru VII IU/ml |
Frekvence dávek (hodiny)/ doba léčby (dny) |
|
Menší krvácení |
0,10 - 0,20 |
Jedna dávka |
|
Těžké krvácení |
0,25 - 0,40 (trough-peak) |
Po dobu 8 až 10 dnů nebo do úplného zhojení rány |
|
Menší chrurgické výkony |
0,20 - 0,30 |
Jedna dávka před chirurgickým výkonem nebo pokud se očekává výraznější riziko krvácení do úplného zhojení rány |
|
Větší chirurgické výkony |
Před operací >0,50, poté 0,25 - 0,45 (trough-peak) |
Po dobu 8 až 10 dní nebo do úplného zhojení rány |
**Na základě klinického rozhodnutí u individuálního případu by mohly být dostatečné nižší dávky ke konci léčby, když je dosaženo odpovídající hemostáze.
Není dostatek údajů pro doporučení podávání přípravku FACTOR VII BAXALTA dětem do 6 let. Způsob podání
Rozpusťte přípravek podle návodu v bodě 6.6. Přípravek má být aplikován intravenózně. Roztok aplikujte pomalu (ne rychleji než 2 ml za 1 minutu).
4.3 Kontraindikace
Přípravek FACTOR VII BAXALTA nesmí být použit u pacientů s:
• Přecitlivělostí na léčivou látku nebo na některou pomocnou látku.
• Vysokým rizikem tromboembolie nebo diseminované intravaskulární koagulace (viz bod 4.4) nebo hyperfibrinolýzou bez předchozího zvládnutí základní konzumpční poruchy
• Známou alergií na heparin nebo heparinem navozenou trombocytopenií v anamnéze
4.4 Zvláštní upozornění a opatření pro použití VAROVÁNÍ
U přípravků obsahujích faktor VII, byly hlášeny hypersensitivní reakce včetně anafylaktických reakcí. Hypersensitivní reakce byly hlášeny také s přípravkem FACTOR VII BAXALTA (viz bod 4.8).
Pokud se objeví alergické reakce nebo reakce anafylaktického typu, má být používání přípravku okamžitě ukončeno. V případě šoku mají být dodrženy lékařské standardy pro léčbu šoku. Pacienti a/nebo jejich ošetřovatelé by měli být informováni o časných příznacích alergických reakcí. Pacienti musí být poučeni, aby ihned přerušili používání přípravku a kontaktovali svého lékaře, jestliže se tyto příznaky objeví.
Jako koncentrát faktoru VII odvozený z plazmy obsahuje tento přípravek další lidské proteiny.
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a zařazení účinných výrobních kroků, při nichž jsou inaktivovány/odstraněny viry. Přesto nemůže být při podávání léčiv vyráběných z lidské krve nebo plazmy zcela vyloučena možnost přenosu infekce. To platí i pro jakékoli neznámé nebo vznikající viry a jiné patogeny.
Přijatá opatření jsou považována za účinná u tzv. obalených virů, například u viru lidské imunitní nedostatečnosti (HIV), viru hepatitidy B (HBV), viru hepatitidy C (HCV), a u neobaleného viru hepatitidy A.
Omezený účinek mohou mít tato opatření u neobalených virů jako je parvovirus B19.
Infekce parvovirem B19 může být závažná u těhotných žen (infekce fetu) a u jedinců s imunodeficitem nebo zvýšenou erytropoézou (např. hemolytická anémie) (viz bod 4.6).
U pacientů, kteří pravidelně/opakovaně dostávají faktor VII z lidské plazmy, je třeba zvážit vhodnou vakcinaci (hepatitida A a B).
Při každé aplikaci přípravku FACTOR VII BAXALTA důrazně doporučujeme zaznamenat název a číslo šarže přípravku (FACTOR VII BAXALTA), aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže přípravku.
U pacientů léčených přípravkem obsahujícím lidský koagulační faktoru VII je potenciální riziko tromboembolie a diseminované intravaskulární koagulace. Při použití přípravku FACTOR VII BAXALTA byly hlášeny mozková žilní trombóza, hluboká žilní trombóza a tromboflebitida. Pacienti dostávající lidský koagulační faktor VII musí být pečlivě monitorováni na všechny příznaky nebo symptomy intravaskulární koagulace nebo tromboembolie. Vzhledem k možnému riziku tromboembolických komplikací musí být při podávání koncentrátů lidského koagulačního faktoru VII zvýšená opatrnost u pacientů s anamnézou koronárního onemocnění, pacientů s jaterními chorobami, u pooperačních stavů, u novorozenců či u pacientů s rizikem tromboembolických projevů nebo diseminované intravaskulární koagulace. Ve všech těchto situacích je nutno zvážit možný pozitivní účinek léčby přípravkem FACTOR VII BAXALTA oproti riziku těchto komplikací.
Substituční léčba lidským faktorem VII (včetně přípravku FACTOR VII BAXALTA) může vést k tvorbě cirkulujících protilátek inhibujících faktor VII. Pokud se tyto inhibitory objeví, projeví se to jako nedostatečná klinická odpověď.
UPOZORNĚNÍ
Vypočtené množství obsahu sodíku v přípravku FACTOR VII BAXALTA je 1,56 - 1,90 mmol na injekční lahvičku. To má být vzato v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou známy žádné interakce přípravků lidského koagulačního faktoru VII s jinými léčivými přípravky.
Interference s laboratorními testy:
Při vyšetřování hemokoagulace v testech, jež jsou citlivé na heparin, u pacientů, kteří dostávají vysoké dávky přípravku FACTOR VII BAXALTA, se musí vzít v úvahu obsah heparinu podaného s přípravkem.
Pokud je to nutné, je možno účinek heparinu neutralizovat přidáním protaminu k vyšetřovanému vzorku.
4.6 Těhotenství a kojení
Bezpečnost humánního koagulačního faktoru VII při používání u těhotných žen nebyla v kontrolovaných klinických studiích u člověka stanovena.
Experimentální studie na zvířatech nejsou ke stanovení bezpečnosti s ohledem na reprodukci, vývoj embrya a plodu, průběh těhotenství a peri- a postnatální vývoj vhodné. Při předepisování koncentrátu faktoru VII těhotným ženám je třeba opatrnosti.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly pozorovány žádné účiny na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Nežádoucí účinky přípravku FACTOR VII BAXALTA na základě hlášení z klinických studií a z poregistračního použití.
Četnost nežádoucích účinků je hodnocena podle následující stupnice: velmi časté (> 1/10), časté (> 1/100 - < 1/10), méně časté (> 1/1 000 - < 1/100), vzácné (>1/10 000 - <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
Následující nežádoucí účinky byly identifikovány při klinických studiích s 57 pediatrickými a dospělými pacienty s vrozeným nedostatkem faktoru VII. Pacientům byl podán koncentrát faktoru VII pro kontrolu akutních krvácivých epizod, pokrytí chirurgického výkonu a pro dlouhodobou profylaxi krvácivých epizod. V průběhu této studie bylo 8234 dnů expozice koncentrátu faktoru VII.
Obsah tabulky níže zahrnuje nežádoucí účinky hlášené z klinických studií i z poregistračního použití seřazené podle tříd orgánových systémů MedDRA (TOS), dále podle upřednostňovaných termínů v pořadí závažnosti, pokud je to možné:
|
Třídy orgánových systémů (TOS) |
Nežádoucí účinek (Upřednostňovaný termín MedDRA) |
Četnost |
|
PORUCHY KRVE A LYMFATICKÉHO SYSTÉMU |
Inhibitor faktoru VII (pozitivní protilátky faktoru VII) |
Není známo |
|
PORUCHY IMUNITNÍHO SYSTÉMU |
Hypersensitivní reakce3 |
Není známo |
|
Anafylaktická reakce |
Není známo | |
|
CÉVNÍ PORUCHY |
Zrudnutí |
Častéa |
|
Hluboká žilní trombóza |
Není známo | |
|
Povrchová tromboflebitida |
Není známo | |
|
PORUCHY KŮŽE A PODKOŽNÍ TKÁNĚ |
Častéa | |
|
Není známo | ||
|
CELKOVÉ PORUCHY A REAKCE V MÍSTĚ APLIKACE |
Pyrexie |
Častéa |
aSeznam symptomů hypersenzitivních reakcí může zahrnovat závrať, dysestézii, bolest hlavy, hypotenzi, bronchospasmus, dušnost, nevolnost, návaly horka a zvracení.
Následující další nežádoucí účinky jsou stanoveny na základě poregistračního použití přípravků stejné třídy:
- Alergické reakce nebo reakce anafylaktického typu:
Tyto reakce jsou pozorovány vzácně.
Byla hlášena kopřivka a zvracení.
- Celkové poruchy a reakce v místě aplikace:
Vzácně je pozorována zvýšená tělesná teplota.
- Cévní poruchy:
Po podání lidského koagulačního faktoru VII se mohou vzácně objevit tromboembolické epizody (viz bod 4.4).
Byly hlášeny mrtvice, infarkt myokardu, arteriální trombóza, plicní embolie a diseminované intravaskulámí koagulace.
Bezpečnostní informace týkající se přenosu infekce viz bod 4.4.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu: Státní ústav pro kontrolu léčiv, Šrobárova 48,
100 41 Praha 10, webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek.
4.9 Předávkování
Podávání vysokých dávek přípravků obsahujících faktor VII (přípravky protrombinového komplexu) bylo spojeno s případy infarktu myokardu, diseminované intravaskulární koagulace, žilní trombózy a plicní embolie. Při předávkování je tedy vyšší pravděpodobnost rozvoje tromboembolických komplikací.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Koagulační faktory: koagulační faktor VII ATC kód: B02BD05
Koagulační faktor VII je jedním z faktorů krevního srážení závislých na vitaminu K přítomných v normální lidské plazmě. Je to jednořetězcový glykoprotein s molekulovou hmotností přibližně 50 000 daltonů. Faktor VII je zymogen aktivní serinové proteázy faktoru VIIa, kterým je iniciována zevní cesta krevního srážení. Komplex tkáňový faktor-faktor VIIa aktivuje koagulační faktory IX a X, přičemž se vytváří faktory IXa a Xa. S další propagací koagulační kaskády se generuje trombin, fibrinogen se přeměňuje na fibrin a vytváří se sraženina. Normální generování thrombinu je také velmi důležité pro funkci destiček jako součást primární hemostázy. Vrozený deficit faktoru VII je autosomálně recesivně dědičné onemocnění. Podávání koncentrátu lidského koagulačního faktoru VII vede ke zvýšení plazmatických hladin faktoru VII a může dočasně upravit poruchu koagulace u pacientů s nedostatkem faktoru VII.
5.2 Farmakokinetické vlastnosti
Po intravenózním podání přípravku FACTOR VII BAXALTA je odhadovaná in vivo recovery přibližně 60 - 100% s průměrným poločasem asi 3 až 5 hodin. Níže uvedená tabulka shrnuje výsledky farmakokinetické studie na téma “Recovery and half life of vapor heated factor VII Concentrate”1, která poskytuje následující farmakokinetické údaje o zotavení (Incremental Recovery, IR), ploše pod křivkou (AUC), průměrné době setrvání v těle (Mean Residence Time, MRT), clearance (C1), distribučním objemu v ustáleném stavu (volume of distribution of steady state, VSS), poločase (HL) jak pro počáteční fázi (HL1) tak i o poločase eliminačním (HL2).
Výsledky:
|
IR (IU/dl na IU/kg) |
AUC (IU x h/ml) |
MRT (h) |
C1 (ml/h) |
Vss (ml) |
HL (h) |
HL1 (h) |
HL2 (h) | |
|
Min |
1,6 |
1,9 |
3,8 |
100 |
503 |
2,7 |
0,21 |
2,5 |
|
Q1 |
1,7 |
3,9 |
5,5 |
206 |
1345 |
3,8 |
0,68 |
2,7 |
|
Median |
1,9 |
4,3 |
6,9 |
326 |
1893 |
4,8 |
1,19 |
3,1 |
|
Q3 |
3,0 |
7,2 |
7,4 |
396 |
3377 |
5,1 |
1,87 |
5,3 |
|
Max |
3,4 |
9,8 |
15,1 |
531 |
6410 |
10,5 |
2,79 |
10,8 |
5.3 Předklinické údaje vztahující se k bezpečnosti
Lidský koagulační faktor VII (obsažený v přípravku FACTOR VII BAXALTA) je normální složkou lidské plazmy a účinkuje jako endogenní faktor VII.
Testování toxicity jedné dávky není relevantní, protože vyšší dávky vedou k oběhovému přetížení. Testování toxicity opakovaných dávek u zvířat je neproveditelné vzhledem k interferenci s vytvářenými protilátkami proti heterolognímu proteinu.
Protože klinická zkušenost nenasvědčuje žádným karcinogenním či mutagenním účinkům lidského koagulačního faktoru VII, nejsou považovány experimentální studie, zejména u heterologních druhů, za nezbytné.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Dihydrát citronanu sodného Chlorid sodný
Sodná sůl heparinu < 0,5 IU/IU FVII
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky včetně heparinu. Doporučuje se propláchnout žilní vstup fyziologickým roztokem před a po infuzi přípravku FACTOR VII BAXALTA.
Vzhledem k tomu, že může dojít k selhání léčby v důsledku adsorpce lidského koagulačního faktoru VII na vnitřní povrchy aplikačního zařízení, smí být použito pouze přiložené příslušenství pro rekonstituci a aplikaci.
6.3 Doba použitelnosti
Lyofilizovaný FACTOR VII BAXALTA má dobu použitelnosti 3 roky.
Po rozpuštění musí být FACTOR VII BAXALTA ihned použit, protože přípravek neobsahuje konzervační látky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě +2°C až +8°C (v chladničce).
Chraňte před mrazem. Uchovávejte vnitřní obal v krabičce, aby byl přípravek chráněn před světlem. Nepoužívejte po uplynutí doby použitelnosti uvedené na obalu.
6.5 Druh obalu a obsah balení
FACTOR VII BAXALTA je dodáván v injekční lahvičce s lyofilizovaným práškem 600 IU, který se má rozpustit v 10 ml vody na injekci.
Přípravek FACTOR VII BAXALTA je dodáván v injekčních lahvičkách ze skla typu II opatřených pryžovou zátkou, kovovým uzávěrem a plastikovým krytem (s obsahem jedné dávky). Rozpouštědlo se dodává v injekčních lahvičkách ze skla typu I opatřených pryžovou zátkou, kovovým uzávěrem a plastikovým krytem (s obsahem pro jednu dávku).
Každé balení obsahuje také set pro rekonstituci a aplikaci, který se skládá z:
1 injekční stříkačky k jednorázovému použití, 1 jehly k jednorázovému použití, 1 převodní jehly, 1 filtrační jehly, 1 zavzdušňovací jehly a 1 infuzního setu s křidélky.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rekonstituce přípravku FACTOR VII BAXALTA se provádí bezprostředně před podáním. Měl by se použít pouze infuzní set dodávaný s přípravkem. Roztok musí být čirý nebo lehce opalescentní. Po rekonstituci je třeba přípravek prohlédnout s ohledem na přítomnost částic a změnu barvy před podáním. Nepoužívejte roztoky, které jsou zakalené nebo obsahují usazeniny. Po rozpuštění by měl být přípravek použit okamžitě. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce lyofilizátu
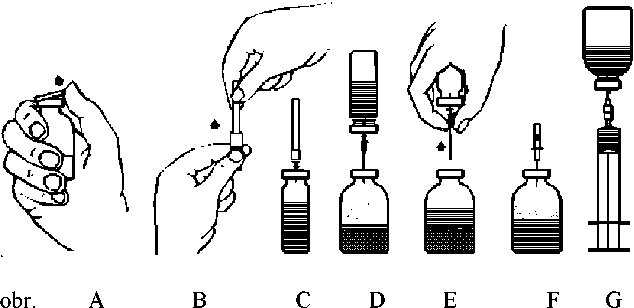
1. Ještě uzavřenou injekční lahvičku obsahující rozpouštědlo zahřejte na pokojovou teplotu (max. 37°C).
2. Odstraňte víčko z injekční lahvičky s koncentrátem a z injekční lahvičky s rozpouštědlem (obr. A) a proveďte dezinfekci pryžových zátek na obou injekčních lahvičkách.
3. Pootočením a povytažením sejměte ochranný kryt z jednoho konce přiložené převodní jehly (obr. B). Touto odkrytou jehlou propíchněte pryžovou zátku injekční lahvičky s rozpouštědlem (obr. C).
4. Sejměte ochranný kryt z druhého konce převodní jehly. Dbejte přitom, abyste se odkrytého konce nedotkli.
5. Injekční lahvičku s rozpouštědlem obraťte vzhůru dnem nad injekční lahvičkou s koncentrátem a volným koncem převodní jehly propíchněte pryžovou zátku injekční lahvičky s koncentrátem (obr. D). Rozpouštědlo je vtaženo vakuem do lahvičky s koncentrátem.
6. Injekční lahvičky odpojte vytažením jehly z lahvičky s koncentrátem (obr. E). Rozpouštění urychlete protřepáním nebo kroužením lahvičkou s koncentrátem.
7. Po úplném rozpuštění koncentrátu zapíchněte do injekční lahvičky přiloženou zavzdušňovací jehlu (obr. F) a veškerá pěna opadne. Odstraňte zavzdušňovací jehlu.
Injekce
1. Pootočením a povytažením sejměte ochranný kryt z přiložené „filtrační jehly“ a jehlu napojte na sterilní injekční stříkačku k jednorázovému použití. Do injekční stříkačky natáhněte roztok (Obr.
G).
2. Odpojte filtrační jehlu od injekční stříkačky a roztok podávejte pomalu intravenózně (maximální rychlost injekce: 2 ml/min.) pomocí přiloženého infuzního setu s křidélky (nebo pomocí přiložené jehly k jednorázovému použití).
3. Při domácí léčbě zajistěte, aby byly použité jehly a injekční stříkačky vloženy zpět do krabičky soupravy pro rozpuštění a aby tato krabička byla vrácena zpět do hemofilického centra.
Infuze
Pokud se podává v infuzi, je třeba použít infuzní soupravu k jednorázovému použití s odpovídajícím filtrem.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Do 30.11.2016
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
Od 1.12.2016
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO
75/118/81-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19.3.1981
Datum posledního prodloužení registrace: 11.5.2016
10. DATUM REVIZE TEXTU
3.8.2016
9/9
References:
Rivard GE, Kovac I., Kunschak M.. Thoene P., and the Factor VII Study Group. Clinical Study of Recovery and Half Life of Vapor-Heated Factor VII Concentrate. Transfusion 1994, 34 (11): 975-979
W. Engl. Statistical Report for the Clinical Study of Recovery and Half Life of Factor VII Concentrate (Human) IMMUNO, Vapor Heated dated August 25, 2006