Eprex 1000 Iu/0,1Ml
sp.zn. sukls111297/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
EPREX 200 IU/0,1 ml, injekční roztok EPREX 400 IU/0,1 ml, injekční roztok EPREX 1 000 IU/0,1 ml, injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
|
INJEKČNÍ STŘÍKAČKY |
LÉČIVÁ LÁTKA |
|
KONCENTRACE/ OBJEM |
Epoetinum alfa * |
|
1000 IU/0,5 ml |
8,4 pg/0,5 ml |
|
2000 IU/0,5 ml |
16,8 pg/0,5 ml |
|
3000 IU/0,3 ml |
25,2 pg/0,3 ml |
|
4000 IU/0,4 ml |
33,6 pg/0,4 ml |
|
5000 IU/0,5 ml |
42,0 pg/0,5 ml |
|
6000 IU/0,6 ml |
50,4 pg/0,6 ml |
|
8000 IU/0,8 ml |
67,2 pg/0,8 ml |
|
10 000 IU/ 1,0 ml |
84,0 pg/1,0 ml |
* Epoetinum alfa produkovaný v ovariálních buňkách křečíka čínského (Chinese Hamster Ovary, CHO) s použitím technologie rekombinantní DNA.
Tento přípravek obsahuje méně než 1 mmol sodných iontů (23 mg) na jednu dávku, dá se tedy považovat za „sodíku prostý“.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok k intravenózní a subkutánní aplikaci v předplněných injekčních stříkačkách.
Čirý bezbarvý roztok bez viditelných mechanických částic.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
EPREX je indikován k léčbě symptomatické anemie spojené s chronickým renálním selháním (CRF):
• u dospělých a pediatrických pacientů ve věku 1 až 18 let na hemodialýze a u dospělých pacientů na peritoneální dialýze.
• u dospělých pacientů s renální insuficiencí, kteří nejsou dosud dialyzováni, k léčbě těžké anemie renálního původu s vyjádřenými klinickými příznaky.
EPREX je indikován u dospělých pacientů podstupujících chemoterapii solidních tumorů, maligního lymfomu nebo mnohočetného myelomu, v případě rizika transfuze vyplývajícího z celkového stavu pacienta (např. stav kardiovaskulárního systému, pre-existující anemie při zahájení chemoterapie), k léčbě anemie nebo ke snížení potřeby transfuze.
EPREX je indikován u dospělých pacientů v programu předoperačního autologního odběru ke zvýšení výtěžku autologní krve.
Léčba je vhodná pouze u pacientů se středně závažnou anemií (koncentrace hemoglobinu 10 až 13 g/dl [6,2 až 8,1 mmol/l], bez deficitu železa) v případě, že nelze využít postupů pro úsporu krve nebo tyto postupy nepostačují při plánovaném velkém chirurgickém výkonu náročném na potřebu velkého objemu krve (4 nebo více krevních jednotek u žen a 5 nebo více u mužů).
EPREX je indikován u dospělých pacientů bez deficitu železa ke snížení počtu alogenních krevních transfuzí u plánovaných velkých ortopedických operací s vysokým rizikem komplikací spojených s transfuzí. Použití by mělo být omezeno na pacienty se středně závažnou anemií (koncentrace hemoglobinu 10 až 13 g/dl), kteří nejsou zařazeni do programu předoperačního autologního odběru a při očekávané krevní ztrátě středního rozsahu (900-1800 ml).
4.2 Dávkování a způsob podání
Dávkování
Před zahájením léčby epoetinem alfa a před rozhodnutím o zvýšení dávky by měly být vyloučeny a případně léčeny všechny ostatní příčiny anemie (deficit železa, kyseliny listové nebo vitaminu BJ2, intoxikace hliníkem, infekce nebo zánět, ztráta krve, hemolýza a fibróza kostní dřeně jakéhokoli původu). Pro zajištění optimální odezvy na léčbu epoetinem alfa by měla být před začátkem léčby zajištěna adekvátní zásoba železa a v případě nutnosti podána suplementace železa (viz bod 4.4).
Léčba symptomatické anemie u dospělých s chronickým renálním selháním
Příznaky a následky anemie se mohou lišit v závislosti na věku a pohlaví pacienta a dalších současně probíhajících onemocněních. Z tohoto důvodu je zapotřebí, aby lékař individuálně u každého pacienta zhodnotil klinický stav a průběh onemocnění.
Doporučené požadované rozmezí koncentrace hemoglobinu je 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l). EPREX se podává za účelem zvýšení koncentrace hemoglobinu nejvýše na 12 g/dl (7,5 mmol/l). Koncentrace hemoglobinu se nemá zvyšovat o více než 2 g/dl (1,25 mmol/l) během čtyř týdnů. Pokud k tomu dojde, je nutné upravit dávku, jak je dále uvedeno.
Vzhledem k variabilitě mezi jednotlivými pacienty mohou koncentrace hemoglobinu u jednotlivých pacientů být nižší nebo vyšší, než je požadovaný limit. Rozdíly v koncentraci hemoglobinu je nutné korigovat pomocí úpravy dávky tak, aby cílové rozmezí koncentrace hemoglobinu činilo 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l).
Je zapotřebí zabránit tomu, aby koncentrace hemoglobinu byla trvale vyšší než 12 g/dl (7,5 mmol/l). Pokud se koncentrace hemoglobinu zvyšuje více než o 2 g/dl (1,25 mmol/l) za měsíc nebo pokud je koncentrace trvale vyšší než 12 g/dl (7,5 mmol/l), musí být dávka přípravku EPREX snížena o 25 %. Pokud koncentrace hemoglobinu přesáhne 13 g/dl (8,1 mmol/l), léčba přípravkem EPREX musí být přerušena až do poklesu pod 12 g/dl (7,5 mmol/l) a znovu zahájena dávkou sníženou o 25 % oproti původní dávce.
Pacienti musí být pečlivě monitorováni, aby bylo zaručeno, že dostávají nejnižší možnou dávku přípravku EPREX, která zajišťuje odpovídající kontrolu anemie a jejích příznaků.
Léčba přípravkem EPREX se dělí do dvou fází - korekční fáze a udržovací fáze.
Dospělí hemodialyzovaní pacienti
U hemodialyzovaných pacientů se snadno dostupným intravenózním přístupem je přednostní intravenózní podání.
Korekční _ fáze
Počáteční dávka je 50 IU/kg 3x týdně.
V případě nutnosti zvyšte nebo snižte dávku o 25 IU/kg (3krát týdně), dokud nebude dosaženo požadovaného rozmezí koncentrace hemoglobinu 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l) (je třeba provádět v jednotlivých krocích s odstupem alespoň čtyř týdnů).
Udržovací fáze
Doporučená celková týdenní dávka se pohybuje v rozmezí 75 IU/kg až 300 IU/kg.
Je třeba provést náležitou úpravu dávky tak, aby se koncentrace hemoglobinu udržely v požadovaném rozmezí 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l).
Léčba pacientů s velmi nízkými počátečními koncentracemi hemoglobinu (< 6 g/dl nebo < 3,75 mmol/l) může vyžadovat vyšší udržovací dávky, než u pacientů s méně závažnou počáteční anemií (> 8 g/dl nebo > 5 mmol/l).
Dospělí pacienti s renální insuficiencí, kteří nejsou dosud dialyzováni
V případě, že intravenózní přístup není snadno dostupný, může být přípravek EPREX podáván subkutánně.
Korekční fáze
Počáteční dávka je 50 IU/kg 3x týdně, v případě nutnosti se dávka zvyšuje o 25 IU/kg (3krát týdně) až do dosažení léčebného cíle (je třeba provádět v jednotlivých krocích s odstupem alespoň čtyř týdnů).
Udržovací fáze
V udržovací fázi může být přípravek EPREX podáván buď 3krát týdně nebo v případě subkutánního podání jednou týdně nebo jednou za 2 týdny.
Vhodná úprava dávkování a dávkovacího intervalu by měla směřovat k udržení koncentrace hemoglobinu na požadované úrovni: hladina hemoglobinu v rozmezí 10 až 12 g/dl (6,2 až 7,5 mmol/l). Prodloužený dávkovací interval může vyžadovat zvýšení dávky.
Maximální dávka nemá překročit 150 IU/kg 3krát týdně, 240 IU/kg (až do maxima 20 000 IU) jednou týdně, nebo 480 IU/kg (až do maxima 40 000 IU) jednou za 2 týdny.
Dospělí pacienti léčení peritoneální dialýzou
V případě, že intravenózní přístup není snadno dostupný, může být přípravek EPREX podáván subkutánně.
Korekční fáze
Počáteční dávka je 50 IU/kg 2x týdně.
Udržovací fáze
Doporučená udržovací dávka je 25 IU/kg až 50 IU/kg 2x týdně ve dvou stejných injekcích.
Je třeba provést náležitou úpravu dávky tak, aby se hodnoty hemoglobinu udržely na požadované hladině 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l).
Léčba dospělých pacientů s anemií v důsledku chemoterapie
Příznaky a následky anemie se mohou lišit v závislosti na věku a pohlaví pacienta a jeho celkovém zdravotním stavu. Z tohoto důvodu je zapotřebí, aby lékař individuálně u každého pacienta zhodnotil klinický stav a průběh onemocnění.
Přípravek EPREX se podává pacientům s anemií (koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l). Počáteční dávka je 150 IU/kg subkutánně, 3x týdně.
Alternativně lze přípravek EPREX podat v počáteční dávce 450 IU/kg subkutánně jednou týdně.
Je třeba provést náležitou úpravu dávky tak, aby se koncentrace hemoglobinu udržely v požadovaném rozmezí 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l).
Vzhledem k variabilitě mezi jednotlivými pacienty mohou koncentrace hemoglobinu u jednotlivých pacientů být nižší nebo vyšší, než je požadované rozmezí koncentrace hemoglobinu. Rozdíly v koncentraci hemoglobinu je nutné korigovat pomocí úpravy dávky tak, aby požadované rozmezí koncentrace hemoglobinu činilo 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l). Je zapotřebí zabránit tomu, aby koncentrace hemoglobinu byla trvale vyšší než 12 g/dl (7,5 mmol/l). Pokyny pro úpravu dávky v případě, že koncentrace hemoglobinu překročí 12 g/dl (7,5 mmol/l), jsou uvedeny níže.
Pokud se po čtyřech týdnech léčby zvýší koncentrace hemoglobinu nejméně o 1 g/dl (0,62 mmol/l) nebo se zvýší počet retikulocytů o > 40 000 buněk/pl nad výchozí úroveň, může být zachováno dávkování 150 IU/kg 3x týdně nebo 450 IU/kg jednou týdně.
Pokud se koncentrace hemoglobinu zvýší o < 1 g/dl (< 0,62 mmol/l) a počet retikulocytů vzroste o
< 40 000 buněk/pl nad výchozí úroveň, dávka se zvýší na 300 IU/kg 3x týdně. Jestliže se po dalších čtyřech týdnech léčby při dávkování 300 IU/kg koncentrace hemoglobinu zvýší o > 1 g/dl
(> 0,62 mmol/l) nebo se zvýší počet retikulocytů o > 40 000 buněk/pl nad výchozí úroveň, může být zachováno dávkování 300 IU/kg 3x týdně.
Pokud se koncentrace hemoglobinu zvýší o < 1 g/dl (< 0,62 mmol/l) a počet retikulocytů vzroste o
< 40 000 buněk/pl nad výchozí úroveň, je odpověď nepravděpodobná a léčba by měla být ukončena.
Úprava dávkování k udržení koncentrací hemoglobinu v rozmezí 10 g/dl až 12 g/dl
Pokud stoupá koncentrace hemoglobinu o více než 2 g/dl (1,25 mmol/l) za měsíc, nebo koncentrace hemoglobinu přesáhne hodnotu 12 g/dl (7,5 mmol/l), je třeba redukovat dávku přípravku EPREX o 25 až 50 %.
Pokud koncentrace hemoglobinu přesáhne 13 g/dl (8,1 mmol/l), léčba se přeruší, dokud koncentrace neklesne pod 12 g/dl (7,5 mmol/l) a poté se obnoví léčba přípravkem EPREX dávkou sníženou o 25 % oproti původní dávce.
Doporučované dávkovací schéma je uvedeno v diagramu:
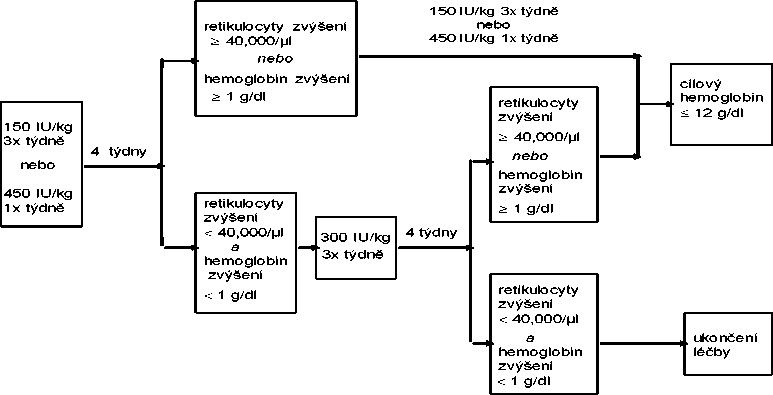
Pacienti musí být pečlivě monitorováni, aby bylo zaručeno, že dostávají nejnižší možnou dávku přípravku stimulujícího erytropoézu (ESA), která zajišťuje odpovídající kontrolu příznaků anemie.
Léčba přípravkem EPREX by měla pokračovat jeden měsíc po ukončení chemoterapie.
Léčba dospělých pacientů v programu předoperačního autologního odběru
Pacientům s mírnou anemií (hematokrit 33 až 39 %) a s potřebou odběru >4 jednotek krve by měl být podáván přípravek EPREX v dávce 600 IU/kg nitrožilně 2x týdně po tři týdny před chirurgickým výkonem. EPREX se má podávat po dokončení odběru.
Léčba dospělých pacientů s plánovanou velkou ortopedickou operací
Doporučovaná dávka přípravku EPREX je 600 IU/kg podávaná subkutánně 1x týdně, po dobu tří týdnů před operačním výkonem (ve dnech -21, -14 a -7), a v den výkonu.
V případech, kdy je z lékařského hlediska nutné zkrátit čas před výkonem na méně než tři týdny, by měl být přípravek EPREX podáván subkutánně v dávce 300 IU/kg jednou denně po dobu 10 po sobě jdoucích dnů před výkonem, v den operace a po dobu čtyř dnů bezprostředně po operaci.
Pokud v předoperačním období dosáhne koncentrace hemoglobinu hodnoty 15 g/dl nebo vyšší, podávání přípravku EPREX by mělo být ukončeno a další dávka by neměla být podána.
Pediatrická populace
Léčba symptomatické anemie u pacientů s chronickým renálním selháním na hemodialýze
Příznaky a následky anemie se mohou lišit v závislosti na věku a pohlaví pacienta a dalších současně probíhajících onemocněních. Z tohoto důvodu je zapotřebí, aby lékař individuálně u každého pacienta zhodnotil klinický stav a průběh onemocnění.
Doporučené rozmezí koncentrace hemoglobinu u pediatrických pacientů je 9,5 g/dl až 11 g/dl (5,9 až 6,8 mmol/l). EPREX se podává za účelem zvýšení koncentrace hemoglobinu nejvýše na 11 g/dl (6,8 mmol/l). Koncentrace hemoglobinu se nemá zvyšovat o více než 2 g/dl (1,25 mmol/l) během čtyř týdnů. Pokud k tomu dojde, je nutné upravit dávku, jak je dále uvedeno.
Pacienti musí být pečlivě monitorováni, aby bylo zaručeno, že dostávají nejnižší možnou dávku přípravku EPREX, která zajišťuje odpovídající kontrolu anemie a jejích příznaků.
Léčba přípravkem EPREX se dělí do dvou fází - korekční fáze a udržovací fáze.
U hemodialyzovaných pediatrických pacientů se snadno dostupným intravenózním přístupem je přednostní intravenózní podání.
Korekční _ fáze
Počáteční dávka je 50 IU/kg intravenózně, 3x týdně.
V případě nutnosti zvyšte nebo snižte dávku o 25 IU/kg (3krát týdně), dokud nebude dosaženo požadovaného rozmezí koncentrace hemoglobinu 9,5 g/dl až 11 g/dl (5,9 až 6,8 mmol/l) (je třeba provádět v jednotlivých krocích s odstupem alespoň čtyř týdnů).
Je třeba provést náležitou úpravu dávky tak, aby se koncentrace hemoglobinu udržely v požadovaném rozmezí 9,5 g/dl až 11 g/dl (5,9 až 6,8 mmol/l).
Obecně lze říci, že dětem s hmotností nižší než 30 kg je zapotřebí podávat vyšší udržovací dávky než dětem s hmotností nad 30 kg a dospělým.
Léčba pediatrických pacientů s velmi nízkými počátečními koncentracemi hemoglobinu (< 6,8 g/dl nebo < 4,25 mmol/l) může vyžadovat vyšší udržovací dávky než u pacientů s vyšší počáteční koncentrací hemoglobinu (Hb > 6,8 g/dl nebo > 4,25 mmol/l).
Léčba pediatrických pacientů s anemií v důsledku chemoterapie
Bezpečnost a účinnost přípravku EPREX u pediatrických pacientů, kteří podstupují chemoterapii, nebyla stanovena.
Léčba pediatrických pacientů v programu předoperačního autologního odběru
Bezpečnost a účinnost přípravku EPREX u pediatrických pacientů nebyla stanovena. Nejsou dostupné žádné údaje.
Léčba pediatrických pacientů s plánovanou velkou ortopedickou operací
Bezpečnost a účinnost přípravku EPREX u pediatrických pacientů nebyla stanovena.
Nejsou dostupné žádné údaje.
Způsob podání
Opatření, která je třeba provést před zacházením nebo podáním léčivého přípravku,
Před použitím nechte injekční stříkačku EPREX stát, dokud nedosáhne pokojové teploty. To obvykle trvá 15 až 30 minut.
Léčba symptomatické anemie u dospělých pacientů s chronickým selháním ledvin
U pacientů s chronickým selháním ledvin, u nichž je běžně k dispozici intravenózní přístup (hemodialyzovaní pacienti) je upřednostňováno podání přípravku EPREX intravenózní cestou.
V případě, že intravenózní přístup není snadno dostupný (pacienti dosud nepodstupující dialýzu a pacienti léčení peritoneální dialýzou), může být přípravek EPREX podáván subkutánní injekcí.
Léčba dospělých pacientů s anemií v důsledku chemoterapie
Přípravek EPREX se má podávat subkutánní injekcí.
Léčba dospělých pacientů v programu předoperačního autologního odběru Přípravek EPREX se má podávat intravenózní cestou.
Léčba dospělých pacientů s plánovanou velkou ortopedickou operací Přípravek EPREX se má podávat subkutánní injekcí.
Léčba symptomatické anemie u pediatrických pacientů s chronickým renálním selháním na hemodialýze
U pediatrických pacientů s chronickým selháním ledvin, u nichž je běžně k dispozici intravenózní přístup (hemodialyzovaní pacienti) je upřednostňováno podání přípravku EPREX intravenózní cestou.
Intravenózní podání
Aplikace by měla trvat nejméně 1-5 minut, v závislosti na celkové dávce. U hemodialyzovaných pacientů může být podána bolusová injekce v průběhu dialýzy prostřednictvím vhodného žilního portu na dialyzační lince. Injekce může být rovněž podána na konci dialýzy kanylou dialyzační soupravy, s následnou aplikací 10 ml izotonického roztoku chloridu sodného za účelem propláchnutí uzávěru a zajištění bezpečného průniku léčivé látky do systémové cirkulace.
U pacientů reagujících na léčbu „chřipkovými příznaky“ je upřednostňována pomalejší rychlost aplikace (viz bod 4.8).
Nepodávejte přípravek EPREX intravenózní infuzí nebo ve spojení s roztoky jiných léků.
Subkutánní podání
Maximální objem aplikovaný do jednoho místa by neměl přesáhnout 1 ml. Při nutnosti podání většího objemu je zapotřebí injekci aplikovat do více míst.
Injekce se aplikují pod kůži končetin nebo přední stěny břišní.
V případech, kdy lékař rozhodne, že je možné, aby pacient nebo jeho pečovatel aplikoval subkutánně injekci přípravku EPREX samostatně, je nutné poskytnout pacientovi nebo pečovateli přesné instrukce týkající se správné dávky a způsobu podání přípravku.
Stejně jako u ostatních injekčně aplikovaných přípravků zkontrolujte, zda nejsou v injekčním roztoku přítomny částice nebo zda nedošlo ke změně jeho barvy.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1..
Pacienti, u nichž došlo v důsledku léčby kterýmkoli erytropoetinem k rozvoji čisté aplazie červené krevní řady, by neměli být léčeni přípravkem EPREX ani jakýmkoli jiným erytropoetinem (viz bod
4.4 - Čistá aplazie červené krevní řady).
Nezvládnutelná hypertenze.
U pacientů suplementovaných přípravkem EPREX musí být respektovány všechny kontraindikace spojené s programem předoperačního autologního odběru.
Použití přípravku EPREX u pacientů připravovaných na plánovanou velkou ortopedickou operaci a nezařazených do programu předoperačního autologního odběru je kontraindikováno u pacientů se závažným onemocněním koronárních arterií, periferních arterií, karotid, nebo s cerebrovaskulárním onemocněním, včetně pacientů, kteří nedávno prodělali infarkt myokardu nebo cévní mozkovou příhodu.
Chirurgičtí pacienti, kteří se z jakéhokoli důvodu nemohou podrobit antitrombotické profylaxi.
4.4 Zvláštní upozornění a opatření pro použití
Všeobecná
U všech pacientů léčených epoetinem alfa by měl být pečlivě sledován, a v případě nutnosti korigován krevní tlak. Epoetin alfa by měl být používán s opatrností v případě neléčené, neadekvátně léčené nebo obtížně zvládnutelné hypertenze. Antihypertenzní léčbu může být zapotřebí doplnit nebo zvýšit. Není-li kontrola krevního tlaku možná, měla by být léčba epoetinem alfa ukončena.
Hypertenzní krize s encefalopatií a záchvaty vyžadující okamžitý zásah lékaře a intenzivní péči se vyskytly při léčbě epoetinem alfa i u pacientů, kteří měli před zahájením léčby normální nebo nízký krevní tlak. Zvláštní pozornost je nutno věnovat náhlým bodavým bolestem hlavy připomínajícím migrénu, které mohou být varovným signálem (viz bod 4.8).
Epoetin alfa by měl být podáván s opatrností pacientům s epilepsií, s výskytem záchvatů v anamnéze, nebo se zdravotními stavy spojenými s predispozicí k záchvatové aktivitě, jako jsou infekce CNS a mozkové metastázy.
Epoetin alfa by měl být používán s opatrností u pacientů s chronickým selháním jater. Bezpečnost epoetinu alfa u pacientů s poruchou funkce jater nebyla stanovena.
U pacientů užívajících ESA byl pozorován zvýšený výskyt trombotických cévních příhod (TVE) (viz bod 4.8). Mezi TVE patří žilní a arteriální trombózy a embolie (včetně příhod s fatálními následky), jako je hluboká žilní trombóza, plicní embolie, retinální trombóza a infarkt myokardu. Dále byly hlášeny cerebrovaskulární příhody (včetně mozkového infarktu, mozkového krvácení a tranzitorní ischemické ataky).
Výše zmíněné riziko TVE je třeba pečlivě zvážit proti výhodám, které by měly vyplynout z léčby epoetinem alfa, zejména u pacientů s preexistujícími rizikovými faktory pro TVE, včetně obezity a předchozího výskytu TVE (např. hluboká žilní trombóza, plicní embolie, a cévní mozková příhoda).
U všech pacientů je zapotřebí pečlivě sledovat koncentrace hemoglobinu z důvodu potenciálního zvýšeného rizika tromboembolických příhod a fatálních důsledků, ke kterým může dojít u pacientů, u nichž během léčby hladina hemoglobinu přesáhne horní hranici rozmezí koncentrace pro indikaci epoetinu alfa.
V průběhu léčby epoetinem alfa lze někdy pozorovat mírný, na dávce závislý vzestup počtu trombocytů v rámci normálních hodnot. Tento vzestup v průběhu další léčby odezní. Kromě toho byly hlášeny i případy trombocytemie nad normální hodnoty. Během prvních 8 týdnů léčby se doporučuje počet trombocytů pravidelně sledovat.
Před zahájením léčby epoetinem alfa a před rozhodnutím o zvýšení dávky by měly být vyloučeny a případně léčeny všechny ostatní příčiny anemie (deficit železa, kyseliny listové nebo vitaminu B12, intoxikace hliníkem, infekce nebo zánět, ztráta krve, hemolýza a fibróza kostní dřeně jakéhokoli původu). Ve většině případů klesá souběžně se vzestupem hematokritu sérový feritin. Pro zajištění optimální odezvy na léčbu epoetinem alfa by měla být před začátkem léčby zajištěna adekvátní zásoba železa a v případě nutnosti podána suplementace železa (viz bod 4.2).
• U pacientů s chronickým selháním ledvin se doporučuje suplementace železa (elementární železo 200 až 300 mg/den perorálně pro dospělé a 100 až 200 mg/den perorálně pro pediatrické pacienty), pokud je sérová koncentrace feritinu nižší než 100 ng/ ml.
• U onkologických pacientů se doporučuje suplementace železa (elementární železo 200 až 300 mg/den perorálně), pokud je saturace transferinu nižší než 20 %.
• U pacientů v programu předoperačního autologního odběru by měla být podávána suplementace železa (elementární železo 200 mg/den perorálně) během několika týdnů před autologním odběrem k zajištění vysokých zásob železa před zahájením léčby epoetinem alfa, a během celého průběhu léčby epoetinem alfa.
• U pacientů s plánovanou velkou ortopedickou operací by měla být podávána suplementace železa (elementární železo 200 mg/den perorálně) během celého průběhu léčby epoetinem alfa. Je-li to možné, suplementace železa by měla být zahájena před počátkem léčby epoetinem alfa k zajištění dostatečných zásob železa.
U všech pacientů s nádorovým onemocněním by měly být před zvýšením dávky pečlivě posouzeny všechny aditivní faktory anemie.
U pacientů léčených epoetinem alfa byl velmi vzácně pozorován vývoj nebo exacerbace porfyrie. Epoetin alfa by měl být podáván s opatrností pacientům s porfyrií.
Pro zlepšení dohledatelnosti látek stimulujících erytropoézu (ESA) je nutno naprosto přesně zaevidovat název předepsané ESA látky do záznamů o pacientovi.
Pacienti mají být převedeni z léčby jednou ESA na jinou pouze pod náležitým dohledem.
Čistá aplazie červené krevní řady
Po měsících až letech léčby subkutánním epoetinem byla hlášena čistá aplazie červené krevní řady zprostředkovaná tvorbou protilátek (PRCA), zejména u pacientů s chronickým selháním ledvin. Případy výskytu byly hlášeny rovněž u pacientů s hepatitidou C léčených interferonem a ribavirinen, kteří současně užívali ESA. Epoetin alfa není schválen k léčbě anemie asociované s hepatitidou C.
U pacientů s náhlou ztrátou účinnosti definovanou poklesem hemoglobinu (o 1 až 2 g/dl za měsíc) se zvýšenou potřebou transfuzí by měl být stanoven počet retikulocytů a prověřeny typické příčiny ztráty účinnosti (např. deficit železa, kyseliny listové nebo vitaminu B12, aluminiová intoxikace, infekce nebo zánět, krevní ztráta, hemolýza a fibróza kostí dřeně jakéhokoli původu).
Paradoxní pokles hemoglobinu a rozvoj závažné anemie související s nízkým počtem retikulocytů musí být podnětem k ukončení léčby epoetinem alfa a k provedení testu na anti-erythropoetinové protilátky. Ke stanovení diagnózy PRCA je rovněž třeba zvážit vyšetření kostní dřeně.
Vzhledem k riziku zkřížené reakce nesmí být zahájena žádná léčba jinými ESA.
Léčba symptomatické anemie u dospělých a pediatrických pacientů s chronickým renálním selháním
U pacientů s chronickým selháním ledvin léčených epoetinem alfa by měla být pravidelně měřena hladina hemoglobinu, až do dosažení stabilní úrovně, a poté v pravidelných intervalech.
K minimalizaci rizika zhoršení hypertenze u pacientů s chronickým renálním selháním by měla koncentrace hemoglobinu narůstat přibližně o 1 g/dl (0,62 mmol/l) za měsíc a nepřesáhnout vzestup o 2 g/dl (1,25 mmol/l) za měsíc.
U pacientů s chronickým renálním selháním nemá udržovací koncentrace hemoglobinu přesáhnout horní hranici rozmezí koncentrace hemoglobinu dle doporučení v bodu 4.2. V klinických studiích bylo při podávání ESA za účelem dosažení koncentrace hemoglobinu vyšší než 12 g/dl (7,5 mmol/l) pozorováno zvýšené riziko úmrtí a závažných kardiovaskulárních příhod.
Kontrolované klinické studie neprokázaly významný přínos podávání erytropoetinů, pokud se koncentrace hemoglobinu zvýší nad úroveň potřebnou ke kontrole příznaků anemie a zamezení potřeby krevní transfuze.
Pacienti s chronickým renálním selháním léčení epoetinem alfa subkutánní cestou by měli být pravidelně monitorováni z hlediska ztráty účinnosti, definované absencí nebo sníženou odpovědí na léčbu epoetinem alfa u pacientů, kteří dříve na tuto léčbu reagovali. Tento stav je charakterizován trvalým poklesem hemoglobinu navzdory zvýšenému dávkování epoetinu alfa.
Někteří pacienti s prodlouženými dávkovacími intervaly (interval mezi dávkami delší než 1 týden) epoetinu alfa nemusí být schopni udržet adekvátní hladiny hemoglobinu (viz bod 5.1), a mohou potřebovat zvýšení dávky epoetinu alfa. Hodnoty hemoglobinu mají být pravidelně sledovány.
U pacientů podstupujících hemodialýzu může dojít k trombotizaci arteriovenózní spojky, zejména u pacientů s tendencí k hypotenzi nebo tam, kde již dříve nastaly komplikace v této oblasti (např.
stenózy, aneuryzmata atd.). U těchto pacientů se doporučuje včasná revize spojky a antitrombotická profylaxe např. podáním kyseliny acetylsalicylové.
V ojedinělých případech byla pozorována hyperkalemie, ačkoli příčina nebyla zjištěna. U pacientů
s chronickým renálním selháním je nutno pečlivě monitorovat koncentrace sérových elektrolytů. Při zvýšené nebo stoupající sérové koncentraci draslíku je zapotřebí posoudit přerušení aplikace epoetinu alfa až do korekce sérových hodnot draslíku a kromě toho zvolit příslušnou léčbu hyperkalemie .
V důsledku vzestupu hematokritu je během dialýzy u pacientů léčených epoetinem alfa často nutné zvýšení dávek heparinu. Při nedostatečné heparinizaci může nastat okluze dialyzačního systému.
Na základě dosud dostupných údajů nezvyšuje epoetin alfa u pacientů v predialýze rychlost progrese renální insuficience.
Léčba pacientů s anemií v důsledku chemoterapie:
U pacientů s nádorovým onemocněním léčených epoetinem alfa by měla být pravidelně měřena hladina hemoglobinu, až do dosažení stabilní úrovně, a poté v pravidelných intervalech.
Epoetiny jsou růstovými faktory primárně stimulujícími tvorbu červených krvinek. Erytropoetinové receptory mohou být exprimovány na povrchu některých maligních buněčných linií. Existuje možnost, jako u všech růstových faktorů, že epoetiny mohou stimulovat růst některých nádorů. V několika kontrolovaných studiích nevykázaly epoetiny u pacientů s anemií spojenou s nádorovým onemocněním vliv na zlepšení celkového přežití nebo snížení rizika progrese nádoru.
V kontrolovaných studiích s epoetinem alfa a dalšími ESA bylo pozorováno:
• snížení lokoregionální kontroly u pacientů s nádorem v pokročilém stádiu v oblasti hlavy nebo krku léčených radioterapií s cílovou koncentrací hemoglobinu vyšší než 14 g/dl (8,7 mmol/l),
• zkrácení celkového přežití a zvýšení počtu úmrtí v souvislosti s progresí choroby po 4 měsících u pacientek s metastazujícícím karcinomem prsu léčených chemoterapií s cílovou koncentrací hemoglobinu 12 až 14 g/dl (7,5 až 8,7 mmol/l),
• zvýšené riziko úmrtí při cílové koncentraci hemoglobinu 12 g/dl (7,5 mmol/l) u pacientů s aktivním maligním onemocněním, kteří nedostávali ani chemoterapii ani radioterapii. ESA nejsou indikovány pro použití u této skupiny pacientů.
Vzhledem k výše uvedenému by měla být v některých klinických situacích u anemických pacientů s nádorovým onemocněním upřednostňována krevní transfuze. Rozhodnutí podat rekombinantní erytropoetin by mělo být založeno na posouzení poměru mezi rizikem a prospěchem léčby s přihlédnutím na jednotlivého pacienta a jeho specifickému klinickému kontextu. Hodnotící faktory, které je třeba vzít v úvahu, jsou typ a stádium nádoru, stupeň anemie, předpokládaná doba přežití, prostředí, ve kterém je pacient léčen a pacientovo právo volby (viz bod 5.1).
U pacientů s nádorovým onemocněním léčených chemoterapií je zapotřebí při rozhodování o vhodnosti léčby epoetinem alfa počítat se zpožděním přibližně 2 až 3 týdny mezi podáním látky stimulující erytropoézu a výskytem erytropoetinem indukovaných červených krvinek (pacient je vystaven riziku, že dostane transfuzi).
Chirurgičtí pacienti v programu předoperačního autologního odběru
U pacientů léčených epoetinem alfa je nutné respektovat všechna varování a zvláštní opatření spojená s předoperačním autologním převodem se zvláštním důrazem na rutinní doplnění objemu.
Pacienti s plánovanou velkou ortopedickou operací
V perioperačním období je třeba vždy uplatňovat správnou praxi pro zacházení s krví.
Pacienti zařazení do programu plánovaných velkých ortopedických operací musí být vzhledem k možnosti rozvoje trombotických a vaskulárních příhod u operovaných pacientů, zvláště při výskytu kardiovaskulárního onemocnění, zajištěni adekvátní antitrombotickou profylaxí. Dále je zapotřebí věnovat zvýšenou pozornost pacientům s predispozicí k rozvoji hluboké žilní trombózy. U pacientů s výchozí koncentrací hemoglobinu > 13 g/dl nelze vyloučit zvýšené riziko pooperačních trombotických a vaskulárních příhod ve spojitosti s léčbou epoetinem alfa. Z těchto důvodů by epoetin alfa neměl být aplikován u pacientů s výchozí koncentrací hemoglobinu > 13 g/dl.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Údaje svědčící pro alteraci metabolismu jiných léků při podávání epoetinu alfa nebyly zjištěny. Léky snižující tvorbu červených krvinek mohou snížit odpověď na epoetin alfa.
Není vyloučena potenciální interakce s cyklosporinem vzhledem k jeho vazbě na erytrocyty. Podává-li se epoetin alfa současně s cyklosporinem, je zapotřebí monitorovat koncentrace cyklosporinu v krvi a v případě nutnosti upravit dávkování cyklosporinu úměrně vzestupu hematokritu.
Neexistují údaje svědčící o interakci mezi epoetinem alfa a G-CSF nebo GM-CSF v návaznosti na hematologickou diferenciaci nebo proliferaci nádorových buněk bioptických vzorků in vitro.
U dospělých pacientek s metastazujícícím karcinomem prsu nemělo subkutánní podání 40 000 IU/ml epoetinu alfa s trastuzumabem v dávce 6 mg/kg žádný účinek na farmakokinetiku trastuzumabu.
4.6 Fertilita, těhotenství a kojení
Adekvátní kontrolované studie u těhotných žen nejsou k dispozici. Studie se zvířaty prokázaly reprodukční toxicitu (viz bod 5.3). Epoetin alfa by proto měl být podáván v těhotenství pouze tehdy, pokud potenciální přínos léčby převýší možné riziko pro plod. Použití epoetinu alfa u těhotných pacientek zařazených do programu předoperačního autologního odběru se nedoporučuje.
Kojení
Není známo, zda exogenní epoetin alfa prostupuje do mateřského mléka. Epoetin alfa by měl být u kojících žen užíván s opatrností. Rozhodnutí o tom, zda pokračovat či ukončit kojení, nebo zda pokračovat či ukončit léčbu epoetinem alfa, by mělo být učiněno na základě posouzení přínosu kojení pro dítě a přínosu léčby epoetinem alfa pro ženu.
Použití epoetinu alfa u kojících pacientek zařazených do programu předoperačního autologního odběru se nedoporučuje.
Fertilita
Nebyly provedeny žádné studie hodnotící potenciální účinek epoetinu alfa na fertilitu u mužů či žen.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie hodnotící účinky na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Nejčastějším nežádoucím účinkem během léčby epoetinem alfa je na dávce závislé zvýšení krevního tlaku nebo zhoršení již existující hypertenze. Je nutné provádět monitorování krevního tlaku, zejména na začátku léčby (viz bod 4.4).
Nejčastěji se vyskytujícími nežádoucími účinky, pozorovanými v klinických hodnoceních s epoetinem alfa, jsou průjem, nauzea, zvracení, pyrexie a bolest hlavy. Dále se může vyskytnout onemocnění podobné chřipce, zejména na počátku léčby.
Ve studiích s prodlouženým intervalem dávkování u dospělých pacientů s renální nedostatečností, kteří dosud nepodstoupili dialýzu, byly hlášeny případy kongesce dýchacích cest, včetně kongesce horních cest dýchacích, nosní kongesce a nasofaryngitidy.
U pacientů užívajících ESA byl hlášen zvýšený výskyt trombotických cévních příhod (TVE) (viz bod 4.4).
Přehled nežádoucích účinků v tabulce
Z celkového počtu 3 262 subjektů ve 23 randomizovaných, dvojitě zaslepených, placebem nebo standardní léčbou kontrolovaných studiích, byl vyhodnocen celkový bezpečnostní profil přípravku EPREX u 1 992 anemických pacientů. Celkem bylo do analýzy zahrnuto 228 subjektů s CRF (chronickým selháním ledvin) léčených epoetinem alfa ve 4 studiích zahrnujících pacienty s chronickým selháním ledvin (2 studie zahrnující pacienty před dialýzou [N = 131 léčených subjektů s CRF] a 2 studie zahrnující dialyzované pacienty [N = 97 léčených subjektů s CRF]; 1 404 léčených subjektů s onkologickým onemocněním v 16 studiích hodnotících anemii indukovanou chemoterapií, 147 léčených subjektů ve 2 studiích hodnotících pacienty v programu autologního dárcovství krve; a 213 léčených subjektů v 1 studii hodnotící pacienty v perioperačním období. Nežádoucí účinky hlášené u > 1 % subjektů léčených epoetinem alfa v těchto studiích jsou uvedeny v následující tabulce:
Přibližná četnost: velmi časté (> 1/10), časté (> 1/100 až <1/10), méně časté (> 1/1 000 až <1/100), vzácné (> 1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (nelze určit z dostupných údajů).
|
Třídy orgánových systémů |
Četnost | |||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo | |
|
Poruchy krve a lymfatického systému |
Čistá aplazie červené krevní řady způsobená tvorbou protilátek proti erytropoetinu 14, Trombocytemie 1 | |||||
|
Poruchy metabolismu a výživy |
Hyperkalemie2 | |||||
|
Poruchy imunitního systému |
Anafylaktická reakce4, Hypersenzitivita 4 | |||||
|
Poruchy nervového systému |
Konvulze | |||||
|
Cévní poruchy |
Žilní a arteriální trombóza3, Hypertenze |
Hypertenzní krize4 | ||||
|
Respirační, |
Kongesce | |||||
|
Třídy orgánových systémů |
Četnost | |||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo | |
|
hrudní a mediastinální poruchy |
respiračního traktu | |||||
|
Gastrointestin ální poruchy | ||||||
|
Poruchy kůže a podkožní tkáně |
Angioneurotický edém4, Kopřivka4 | |||||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie, Bolest kostí, Myalgie, Bolest končetin | |||||
|
Vrozené, familiární a genetické vady |
Porfyrie4 | |||||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Zimnice, Onemocnění podobné chřipce, Reakce v místě vpichu, Periferní edém |
Neúčinnost léku4 | |||
1 Identifikované během postmarketingového sledování a na základě kategorií četnosti předpokládaných z míry spontánních hlášení
2 Časté u dialýzy
3 Zahrnuje arteriální a venózní, fatální a nefatální příhody, jako jsou: hluboká žilní trombóza, plicní embolie, trombóza sítnice, arteriální trombóza (včetně infarktu myokardu), cévní mozková příhoda (včetně trombotické cévní mozkové příhody a krvácení do mozku), tranzitorní ischemická ataka, trombóza shuntu (včetně dialyzačních zařízení) a trombóza v rámci arteriovenózního shuntu při aneuryzmatu
4 Viz podčást níže a/nebo bod 4.4.
Popis vybraných nežádoucích účinků
Byly hlášeny hypersenzitivní reakce, včetně případů vyrážky (včetně kopřivky), anafylaktické reakce a angioedému (viz bod 4.4).
Hypertenzní krize s encefalopatií a záchvaty vyžadující okamžitý zásah lékaře a intenzivní péči se vyskytly při léčbě epoetinem alfa i u pacientů, kteří měli před zahájením léčby normální nebo nízký krevní tlak. Zvláštní pozornost je nutno věnovat náhlým bodavým bolestem hlavy připomínajícím migrénu, které mohou být varovným signálem (viz bod 4.4).
Po několika měsících až letech léčby přípravkem EPREX byla velmi vzácně hlášena u <1/10 000 případů na pacientorok protilátkami zprostředkovaná čistá aplazie červené krevní řady (viz bod 4.4).
Pediatrická populace s chronickým selháním ledvin na hemodialýze
Expozice pediatrických pacientů s chronickým selháním ledvin na hemodialýze v klinických studiích a po uvedení přípravku na trh je omezená. U této populace nebyly hlášeny žádné specifické nežádoucí účinky, které nejsou uvedeny v tabulce výše, ani žádné nežádoucí účinky, které by neodpovídaly základnímu onemocnění.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Terapeutická šíře epoetinu alfa je značná. Předávkování epoetinem alfa se může projevit příznaky, které odpovídají vystupňovaným farmakologickým účinkům hormonu. Excesivní vzestup koncentrace hemoglobinu může vyústit v nutnost provedení flebotomie a v případě potřeby i dalších podpůrných opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antianemika, ATC kód: B03XA01. Mechanismus účinku
Erytropoetin (EPO) je glykoproteinový hormon produkovaný primárně v ledvinách v reakci na hypoxii, který působí jako klíčový regulátor tvorby červených krvinek (RBC). EPO je zapojen do všech fází vývoje buněk erytroidní řady, a působí zejména na úrovni erytroidních prekurzorů. Po navázání EPO na příslušný receptor buněčného povrchu se aktivují signální dráhy, které interferují s apoptózou a stimulují proliferaci erytroidních buněk. Rekombinantní lidský EPO (epoetin alfa) exprimovaný v ovariálních buňkách křečíka čínského má shodnou sekvenci 165 aminokyselin s EPO z lidské moči; obě formy jsou nerozlišitelné pomocí funkčních testů. Zdánlivá molekulová hmotnost erytropoetinu je 32 000 až 40 000 daltonů.
Erytropoetin je růstový faktor, který primárně stimuluje produkci červených krvinek. Receptory pro erytropoetin mohou být exprimovány na povrchu různých nádorových buněk.
Farmakodynamické účinky
Zdraví dobrovolníci
Po jednorázových dávkách epoetinu alfa (20 000 až 160 000 IU subkutánně) byla pozorována na dávce závislá odpověď u vyšetřovaných farmakodynamických markerů, včetně: retikulocytů, červených krvinek a hemoglobinu. U změn v procentuálním zastoupení retikulocytů byl pozorován definovaný profil závislosti koncentrace na čase s maximem a návratem k výchozímu stavu. Méně definovaný profil byl pozorován u červených krvinek a hemoglobinu. Obecně se všechny farmakodynamické markery zvyšovaly lineárně s dávkou a dosáhly maximální odpovědi při nejvyšších hladinách dávek.
Další farmakodynamické studie hodnotily dávku 40 000 IU jednou týdně oproti dávce 150 IU / kg 3krát týdně. I přes rozdíly v profilech závislosti koncentrace na čase byla farmakodynamická odezva (měřená změnami v procentuálním podílu retikulocytů, hemoglobinu a celkového počtu červených krvinek) podobná mezi oběma uvedenými režimy. Další studie srovnávala režim 40 000 IU epoetinu alfa jednou týdně s dávkami jednou za 2 týdny v rozmezí od 80 000 do 120 000 IU subkutánně. Na základě výsledků těchto farmakodynamických studií na zdravých dobrovolnících se celkově zdá, že dávkovací schéma 40 000 IU jednou týdně je účinnější z hlediska tvorby červených krvinek než režimy jednou za 2 týdny, i přes pozorovanou podobnost v tvorbě retikulocytů mezi režimem jednou týdně a jednou za 2 týdny.
Chronické selhání ledvin
Bylo prokázáno, že epoetin alfa stimuluje tvorbu červených krvinek u anemických pacientů s chronickým selháním ledvin, včetně dialyzovaných pacientů a pacientů s predialýzou. Prvním důkazem o léčebné odpovědi na epoetin alfa je zvýšení počtu retikulocytů během 10 dnů, následované zvýšením počtu červených krvinek, hemoglobinu a hematokritu, obvykle v rozmezí 2 až 6 týdnů. Odpověď hemoglobinu se liší mezi pacienty a může být ovlivněna zásobami železa a přítomností souběžně se vyskytujících zdravotních problémů.
Anemie v důsledku chemoterapie
Bylo prokázáno, že epoetin alfa podávaný 3krát týdně nebo jednou týdně zvyšuje hladinu hemoglobinu a snižuje potřebu transfuze po prvním měsíci léčby u anemických pacientů s nádorovým onemocněním, kteří podstupují chemoterapii.
Ve studii srovnávající dávkovací režimy 150 IU/kg, 3krát týdně a 40 000 IU jednou týdně u zdravých subjektů a u anemických pacientů s nádorovým onemocněním byly zjištěny podobné časové profily změn procentuálního zastoupení retikulocytů, hemoglobinu a celkového počtu červených krvinek mezi oběma dávkovacími režimy jak u zdravých subjektů, tak i u anemických pacientů s nádorovým onemocněním. AUC příslušných farmakodynamických parametrů byly podobné mezi dávkovacími režimy 150 IU/kg 3krát týdně resp. 40 000 IU jednou týdně u zdravých subjektů i u anemických pacientů s nádorovým onemocněním.
Dospělí chirurgičtí pacienti v programu předoperačního autologního odběru
Bylo prokázáno, že epoetin alfa stimuluje tvorbu červených krvinek za účelem zvýšení odběru autologní krve a omezení poklesu hemoglobinu u dospělých pacientů s plánovanou velkou operací, u kterých se nepředpokládá odběr v rozsahu kompletní perioperační potřeby krve. Nejvýraznější účinky byly pozorovány u pacientů s nízkou hladinou hemoglobinu (< 13 g/dl).
Léčba dospělých pacientů s plánovanou velkou ortopedickou operací
U pacientů s plánovanou velkou ortopedickou operací a s hladinou hemoglobinu před léčbou >10 až < 13 g/dl bylo prokázáno, že epoetin alfa snižuje riziko alogenní transfuze a urychluje regenerace erytroidních buněk (zvýšená hladina hemoglobinu, hematokritu a zvýšený počet retikulocytů).
Klinická účinnost a bezpečnost
Chronické selhání ledvin
Epoetin alfa byl zkoumán v klinických studiích u dospělých anemických pacientů s CRF, včetně hemodialyzovaných pacientů a pacientů, kteří dosud nepodstoupili hemodialýzu, z hlediska léčby anemie a udržení hematokritu v cílovém rozmezí koncentrace 30 až 36 %.
V klinických hodnoceních s počátečními dávkami 50 až 150 IU/kg 3krát týdně přibližně 95 % všech pacientů odpovědělo klinicky významným zvýšením hematokritu. Po cca dvou měsících léčby byli prakticky všichni pacienti nezávislí na transfuzi. Po dosažení cílové hodnoty hematokritu byla udržovací dávka individuálně přizpůsobena u každého pacienta.
Ve třech nejrozsáhlejších klinických studiích provedených u dospělých pacientů na dialýze činil medián udržovací dávky nutné k udržení hematokritu v rozmezí 30 až 36 % přibližně 75 IU/kg 3krát týdně.
Ve dvojitě zaslepené, placebem kontrolované, multicentrické studii hodnotící kvalitu života u pacientů s CRF na hemodialýze bylo prokázáno klinicky a statisticky významné zlepšení u pacientů léčených epoetinem alfa ve srovnání s placebovou skupinou z hlediska parametrů únavy, fyzických příznaků, vztahů a deprese (dotazník pro onemocnění ledvin Kidney Disease Questionnaire) po šesti měsících léčby. Pacienti ze skupiny léčené epoetinem alfa byli dále zařazeni do otevřené prodloužené studie, v níž bylo prokázáno zlepšení kvality života, které se udrželo po dobu dalších 12 měsíců.
Dospělí pacienti s renální insuficiencí, kteří nejsou dosud dialyzováni
V klinických hodnoceních prováděných u pacientů s CRF, kteří nebyli léčeni dialýzou, ale byli léčeni epoetinem alfa, byla průměrná doba trvání léčby téměř pět měsíců. Tito pacienti reagovali na terapii epoetinem alfa podobným způsobem, jaký byl pozorován u pacientů na dialýze. U pacientů s CRF neléčených dialýzou bylo prokázáno na dávce závislé a trvalé zvýšení hematokritu po podání epoetinu alfa buď intravenózní, nebo subkutánní cestou. Nezávisle na způsobu podání epoetinu alfa byly zaznamenány podobné míry nárůstu hematokritu. U dávek epoetinu alfa 75 až 150 IU/kg týdně bylo prokázáno udržení hematokritu v rozmezí 36 až 38 % po dobu až šesti měsíců.
Ve dvou studiích s prodlouženým dávkovacím intervalem přípravku EPREX (aplikace 3krát týdně, jednou týdně, jednou za 2 týdny a jednou za 4 týdny) se při prodloužení intervalů mezi jednotlivými dávkami u některých pacientů neudržely adekvátní hladiny hemoglobinu a pacienti dosáhli protokolem definovaná kritéria pro vystoupení ze studie (0% u skupiny s aplikací jednou týdně, 3,7% u skupiny s aplikací jednou za 2 týdny a 3,3% u skupiny s aplikací jednou za 4 týdny).
V randomizované prospektivní studii (CHOIR) bylo hodnoceno 1 432 anemických pacientů s chronickým selháním ledvin, kteří nepodstupovali dialýzu. Pacienti byli přiřazeni k léčbě epoetinem alfa s cílem udržet hladinu hemoglobinu 13,5 g /dl (vyšší než doporučená koncentrace hemoglobinu) nebo 11,3 g/dl. Závažné kardiovaskulární příhody (úmrtí, infarkt myokardu, cévní mozková příhoda nebo hospitalizace z důvodu srdečního selhání) se vyskytly u 125 (18 %) ze 715 pacientů ve skupině s vyšší cílovou hladinou hemoglobinu oproti 97 (14 %) ze 717 pacientů ve skupině s nižší hladinou hemoglobinu (poměr rizik [HR] 1,3; 95% CI: 1,0, 1,7, p = 0,03).
Léčba pacientů s anemií v důsledku chemoterapie
Epoetin alfa byl studován v klinických hodnoceních u anemických dospělých onkologických pacientů s lymfoidními a solidními nádory, a u pacientů na různých chemoterapeutických režimech, včetně režimů obsahujících platinu a režimů bez platiny. V těchto studiích bylo prokázáno, že epoetin alfa podávaný 3krát týdně a jednou týdně zvyšuje koncentrace hemoglobinu a snižuje potřebu transfuze po prvním měsíci léčby u anemických pacientů s nádorovým onemocněním. V některých studiích byla po dvojitě zaslepené fázi realizována otevřená fáze, během níž všichni pacienti dostávali epoetin alfa a byla pozorována doba udržení účinku.
Dostupné důkazy naznačují, že pacienti s hematologickými malignitami resp. solidními nádory reagují na léčbu epoetinem alfa ekvivalentně, stejně jako pacienti s nádorovou infiltrací resp. bez infiltrace kostní dřeně. V chemoterapeutických studiích byla prokázána srovnatelná intenzita chemoterapie ve skupině s epoetinem alfa a v placebové skupině na základě obdobné plochy pod časovou křivkou neutrofilů u pacientů léčených epoetinem alfa a pacientů léčených placebem, stejně jako na základě podobného poměru pacientů ve skupině léčené epoetinem alfa a skupině užívající placebo, kde absolutní počet neutrofilů klesl pod 1000 resp. 500 buněk/pl.
V prospektivní, randomizované, dvojitě zaslepené, placebem kontrolované studii se zařazenými 375 anemickými pacienty s různými nemyeloidními malignitami, léčenými chemoterapií bez použití derivátů platiny, bylo prokázáno signifikantní snížení průvodních příznaků anemie (např. únavy, snížené energie a snížené aktivity). Údaje byly hodnoceny dle těchto klasifikací: „Functional Assessment of Cancer Therapy-Aneamia (FACT-An) obecná klasifikace, FACT-An klasifikace hodnotící únavu a Cancer Linar Analogue Scale (CLAS). Výsledky dvou dalších, méně rozsáhlých, randomizovaných, placebem kontrolovaných studií neprokázaly signifikantní zvýšení parametrů kvality života dle klasifikace EORTC-QLQ-C30 nebo CLAS.
Přežití a progrese nádoru byly sledovány v pěti velkých kontrolovaných studiích celkově zahrnujících 2 833 pacientů, z nichž čtyři studie byly dvojitě zaslepené, placebem kontrolované a jedna studie byla otevřená. Do studií byli zařazeni jak pacienti s nádorovým onemocněním léčení chemoterapií (dvě studie), tak i pacienti, u kterých není léčba látkami stimulujícími erytropoézu (ESA) indikována: anemie u pacientů s nádorovým onemocněním, kteří nejsou léčeni chemoterapií, a pacienti s nádory hlavy a krku léčení radioterapií. Cílová koncentrace hemoglobinu byla > 13 g/dl, ve zbývajících třech studiích byla 12 - 14 g/dl. V otevřené studii nebyl zjištěn rozdíl v celkovém přežití mezi pacienty léčenými rekombinantním lidským erytropoetinem a kontrolní skupinou. Ve čtyřech placebem kontrolovaných studiích se poměr rizika pro celkové přežití pohyboval v rozmezí 1,25 a 2,47 ve prospěch kontrolní skupiny. V těchto studiích došlo, v porovnání s kontrolní skupinou, ke konzistentnímu neobjasněnému statisticky významnému zvýšení mortality u pacientů s anemií spojenou s různými běžnými nádory, kteří dostávali rekombinantní lidský erytropoetin. Výsledné celkové přežití v těchto studiích nemůže být uspokojivě vysvětleno rozdíly ve výskytu trombózy a souvisejících komplikací mezi skupinou s rekombinantním lidským erytropoetinem a kontrolní skupinou.
Byla rovněž provedena analýza údajů z 53 kontrolovaných klinických hodnocení s několika epoetiny u více než 13 900 onkologických pacientů (chemoterapie, radioterapie, chemoradioterapie nebo neléčení). Meta-analýza údajů celkového přežití vygenerovala odhad míry rizika přežití na 1,06 ve prospěch kontrolních skupin (95 % CI: 1,00, 1,12; celkem 53 studií a 13 933 pacientů) a u onkologických pacientů léčených chemoterapií byla míra celkového rizika1,04 (95 % CI: 0,97, 1,11; celkem 38 studií a 10 441 pacientů). Meta-analýzy rovněž ukázaly konzistentní a významné zvýšení relativního rizika tromboembolických příhod u onkologických pacientů léčených rekombinantním lidským erythropoetinem (viz bod 4.4).
Program autologního odběru
Účinek epoetinu alfa z hlediska usnadnění odběru autologní krve u pacientů s nízkým hematokritem (< 39% a bez anemie v důsledku nedostatku železa) s naplánovanou velkou ortopedickou operací byl hodnocen v dvojitě zaslepené, placebem kontrolované studii zahrnující 204 pacientů, a v jednoduše zaslepené placebem kontrolované studii zahrnující 55 pacientů.
Ve dvojitě zaslepené studii byli pacienti léčeni epoetinem alfa 600 IU/kg nebo placebem intravenózně jednou denně každé 3-4 dny po dobu 3 týdnů (celkem 6 dávek). V průměru se u pacientů léčených epoetinem alfa podařilo získat signifikantně více jednotek krve (4,5 jednotek) než u pacientů užívajících placebo (3,0 jednotky).
V jednoduše zaslepené studii byli pacienti léčeni epoetinem alfa 300 IU/kg nebo 600 IU/kg nebo placebem intravenózně jednou denně každé 3-4 dny po dobu 3 týdnů (celkem 6 dávek). U pacientů léčených epoetinem alfa se rovněž podařilo získat signifikantně více jednotek krve (epoetin alfa 300 IU / kg = 4,4 jednotek; epoetin alfa 600 IU / kg = 4,7 jednotek) než u pacientů léčených placebem (2,9 jednotek).
Léčba epoetinem alfa snížila riziko expozice alogenní krvi o 50 % ve srovnání s pacienty, kteří nedostávali epoetin alfa.
Plánovaná velká ortopedická operace
Účinek epoetinu alfa (300 IU/kg nebo 100 IU/kg) na expozici alogenní krevní transfuzi byl hodnocen v placebem kontrolovaném, dvojitě zaslepeném klinickém hodnocení u dospělých pacientů bez deficitu železa s plánovanou velkou ortopedickou operací kyčle nebo kolenního kloubu. Epoetin alfa byl podáván subkutánně po dobu 10 dnů před operací, v den operace a po dobu čtyř dnů po operaci. Pacienti byli stratifikováni podle výchozí hodnoty hemoglobinu (< 10 g/dl, > 10 až < 13 g/dl a > 13 g/dl).
Epoetin alfa 300 IU/kg významně snížil riziko alogenní transfuze u pacientů s hladinou hemoglobinu před léčbou > 10 až < 13 g/dl . Transfuze byla zapotřebí u 16 % pacientů léčených epoetinem alfa 300 IU/kg, u 23 % pacientů léčených epoetinem alfa 100 IU/kg a u 45 % pacientů léčených placebem.
Otevřená studie s paralelními skupinami zahrnující dospělé pacienty bez deficitu železa s hladinou hemoglobinu před léčbou > 10 až < 13 g/dl, u nichž byla naplánována velká ortopedická operace kyčelního nebo kolenního kloubu, srovnávala režim epoetinu alfa 300 IU/kg subkutánně jednou denně po dobu 10 dní před operací, v den operace a po dobu čtyř dnů po operaci s režimem epoetinu alfa 600 IU/kg podkožně jednou týdně po dobu 3 týdnů před operací a v den operace.
Ve skupině užívající epoetin alfa v dávce 600 IU/kg jednou týdně bylo pozorováno dvojnásobné zvýšení hladiny hemoglobinu (1,44 g/dl) za dobu od začátku předléčení do doby operace než ve skupině užívající 300 IU/kg denně (0,73 g/dl). Po celé pooperační období byly průměrné hladiny hemoglobinu v obou léčebných skupinách obdobné.
Erytropoetická odpověď na léčbu pozorovaná v obou léčebných ramenech vedla k obdobné četnosti transfuzí (16 % ve skupině 600 IU/kg jednou týdně a 20 % ve skupině 300 IU/kg jednou denně).
Pediatrická populace
Chronické selhání ledvin
Epoetin alfa byl hodnocen v otevřené, nerandomizované, 52týdenní klinické studii s otevřeným dávkovým rozmezím u dětských pacientů s CRF léčených hemodialýzou. Medián věku pacientů zařazených do studie činil 11,6 roků (rozmezí 0,5-20,1 roků).
Epoetin alfa byl podáván v dávce 75 IU/kg/týden intravenózně ve 2 nebo 3 dílčích dávkách po dialýze, titrován po 75 IU/kg/týden v intervalu 4 týdnů (až do maxima 300 IU/kg/týden) s cílem dosáhnout zvýšení hladiny hemoglobinu o 1 g/dl/měsíc. Požadované rozmezí koncentrace hemoglobinu činilo 9,6 až 11,2 g/dl. Cílové hladiny hemoglobinu dosáhlo 81 % pacientů. Medián doby potřebné k dosažení cílové hladiny byl 11 týdnů a medián dávky při dosažení cíle činil 150 IU/kg/týden. Ze všech pacientů, kteří dosáhli tohoto cíle, užívalo 90 % dávkovací režim 3krát týdně.
Po 52 týdnech zůstalo ve studii 57 % pacientů, kteří užívali dávku o mediánu 200 IU/kg/týden.
5.2 Farmakokinetické vlastnosti
Po subkutánní injekci dosahují sérové hladiny epoetinu alfa svého maxima v době 12 až 18 hodin po podání dávky. Po opakovaném podávání dávky 600 IU/kg subkutánně jednou týdně nedocházelo k akumulaci.
Absolutní biologická dostupnost po subkutánní injekcí epoetinu alfa u zdravých dobrovolníků je přibližně 20 %.
Distribuce
Průměrný distribuční objem u zdravých subjektů činil 49,3 ml/kg po intravenózních dávkách 50 a 100 IU/kg. Po intravenózním podání epoetinu alfa u pacientů s chronickým selháním ledvin se distribuční objem pohyboval v rozmezí 57 až 107 ml/kg po podání jedné dávky (12 IU/kg) resp. 42 až 64 ml/kg po opakovaném podávání (48 až 192 IU/kg). To znamená, že distribuční objem je nepatrně vyšší než plazmatický objem.
Eliminace
Biologický poločas epoetinu alfa po opakovaném intravenózním podání činí u zdravých osob přibližně 4 hodiny. V případě podkožního podání u zdravých subjektů činí poločas přibližně 24 hodin.
Průměrná hodnota CL/F pro dávky 150 IU/kg 3krát týdně a 40 000 IU jednou týdně u zdravých subjektů činila 31,2 resp. 12,6 ml/h/kg. Průměrná hodnota CL/F pro režimy 150 IU/kg, 3krát týdně a 40 000 IU jednou týdně u anemických pacientů s nádorovým onemocněním činila 45,8 resp. 11,3 ml/h/kg. U většiny anemických pacientů s nádorovým onemocněním léčených cyklicky chemoterapií byla hodnota CL/F nižší po subkutánní dávce 40 000 IU jednou týdně a 150 IU/kg 3krát týdně v porovnání s hodnotami u zdravých subjektů.
Linearita/Nelinearita
Po intravenózním podání 150 a 300 IU/kg 3krát týdně bylo u zdravých jedinců pozorováno zvýšení sérové koncentrace epoetinu alfa úměrné velikosti dávky. Subkutánní podání jednorázové dávky 300 až 2 400 IU/kg epoetinu alfa vedlo k lineárnímu vztahu mezi průměrnou hodnotou Cmax a dávkou a mezi průměrnou hodnotou AUC a dávkou. U zdravých jedinců byl zaznamenán inverzní vztah mezi zdánlivou clearance a dávkou.
Ve studiích hodnotících prodloužení dávkovacího intervalu (40 000 IU jednou týdně a 80 000, 100 000 a 120 000 IU jednou za 2 týdny) byl pozorován lineární vztah mezi průměrnou hodnotou Cmax a dávkou, který však nebyl úměrný dávce, a mezi průměrem AUC a dávkou v rovnovážném stavu.
Farmakokinetické/farmakodynamické vztahy
Epoetin alfa vykazuje na dávce závislý účinek na hematologické parametry, který je nezávislý na cestě podání.
Pediatrická populace
U pediatrických pacientů s chronickým selháním ledvin byl po opakovaném intravenózním podání epoetinu alfa hlášen biologický poločas přibližně 6,2 až 8,7 hodin. Farmakokinetický profil epoetinu alfa u dětí a dospívajících se zdá být podobný jako u dospělých.
U pacientů s chronickým selháním ledvin je biologický poločas po intravenózním podání epoetinu alfa mírně prodloužen, přibližně na 5 hodin, ve srovnání se zdravými jedinci.
5.3 Předklinické údaje vztahující se k bezpečnosti
V toxikologických studiích s opakovaným podáním dávky u psů a laboratorních potkanů, avšak nikoli u opic, bylo podání epoetinu alfa spojeno se subklinickou fibrózou kostní dřeně. Fibróza kostní dřeně je známá komplikace chronického renálního selhání u lidí a může být spojována se sekundárním hyperparathyreoidismem nebo dosud neznámými faktory. Ve studii s hemodialyzovanými pacienty léčenými epoetinem alfa po dobu tři roky nedošlo ke zvýšení incidence fibrózy kostní dřeně v porovnání se srovnatelnou kontrolní skupinou dialyzovaných pacientů, kteří nebyli léčeni epoetinem alfa.
Epoetin alfa neindukoval genové mutace u bakterií (Amesův test), chromozomové aberace v savčích buňkách, žádná poškození v mikronukleu myší ani genové mutace na lokus HGPRT.
Dlouhodobé studie karcinogenity nebyly prováděny. V literatuře existují nejednoznačné zprávy založené na sledování lidských nádorových buněk in vitro, které naznačují, že erytropoetiny mohou hrát roli v proliferaci tumorů. Tyto zprávy však mají nejistý klinický význam.
V buněčných kulturách odvozených z buněk lidské kostní dřeně epoetin alfa stimuluje specificky erytropoézu a neovlivňuje tvorbu bílých krvinek. Cytotoxické účinky epoetinu alfa na buňky kostní dřeně nebyly zaznamenány.
Ve studiích se zvířaty bylo prokázáno, že epoetin alfa snižuje fetální hmotnost, prodlužuje osifikaci a zvyšuje fetální mortalitu při podání v týdenních dávkách, které dvacetinásobně převyšují doporučenou týdenní dávku určenou pacientovi. Tyto změny jsou interpretovány jako sekundárně vzniklé v důsledku sníženého nárůstu maternální tělesné hmotnosti a jejich význam pro člověka není znám, vzhledem k hladině terapeutických dávek.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
polysorbát 80 glycin
voda na injekci
dihydrát dihydrogenfosforečnanu sodného dihydrát hydrogenfosforečnanu sodného chlorid sodný
6.2 Inkompatibility
Vzhledem k absenci studií kompatibility nelze tento přípravek podávat společně s jinými léčivými přípravky.
6.3 Doba použitelnosti
18 měsíců
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2-8 °C). Toto teplotní rozmezí musí být přísně dodržováno až do podání pacientovi. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem. Chraňte před mrazem. Před použitím neprotřepávejte!
Pokud je přípravek používán v rámci ambulantní péče, může být vyjmut z chladničky a uchováván bez možnosti navrácení do chladničky, při teplotě do 25 °C nejdéle 3 dny. Pokud léčivý přípravek nebude použit do konce tohoto období, musí být zlikvidován.
6.5 Druh obalu a obsah balení
Předplněná jednorázová skleněná stříkačka s plastovým pístem (opatřeným teflonovým uzávěrem) a s ocelovou jehlou chráněnou pevným krytem (kaučuk pokrytý polypropylenovou vrstvou) a bezpečnostním nástavcem PROTECS (polykarbonát) připojeným ke stříkačce, velikost balení po 6 kusech.
EPREX 200 IU/0,1 ml EPREX 400 IU/0,1 ml EPREX 1 000 IU/0,1 ml
|
6x |
1 000 |
IU/0,5 |
ml |
|
6x |
2 000 |
IU/0,5 |
ml |
|
6x |
3 000 |
IU/0,3 |
ml |
|
6x |
4 000 |
IU/0,4 |
ml |
|
6x |
5 000 |
IU/0,5 |
ml |
|
6x |
6 000 |
IU/0,6 |
ml |
|
6x |
8 000 |
IU/0,8 |
ml |
|
6x |
10 000 IU/1, |
0 ml | |
Velikost balení: Injekční stříkačky:
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nepodávejte intravenózní infuzí nebo spolu s roztoky jiných léků.
Před použitím ponechte předplněné injekční stříkačky stát do dosažení pokojové teploty. To obvykle trvá 15 až 30 minut.
Přípravek by neměl být použit, a měl by být zlikvidován
• pokud je porušen uzávěr,
• pokud je roztok zabarven nebo obsahuje viditelné částice,
• pokud víte, že roztok byl, nebo se domníváte, že mohl být nedopatřením zmražen,
• při poruše chladničky.
Přípravek je určen k jednorázovému použití. Z každé injekční stříkačky použijte vždy pouze jednu dávku přípravku EPREX, před použitím odstraňte přebytečný roztok ze stříkačky. Viz kapitola 3. Jak se přípravek EPREX používá (Pokyny, jak samostatně podávat injekci přípravku EPREX) příbalové informace.
Předplněné injekční stříkačky jsou opatřeny bezpečnostním nástavcem PROTECS, který pomáhá předejít poranění způsobenému použitou jehlou. Příbalová informace obsahuje podrobný návod k zacházení s předplněnými injekčními stříkačkami opatřenými bezpečnostním nástavcem PROTECS.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag s.r.o., Praha, Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
EPREX 200 IU/0,1 ml : 12/161/89-A/C EPREX 400 IU/0,1 ml : 12/161/89-B/C EPREX 1 000 IU/0,1 ml : 12/161/89-C/C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19.6.1989
Datum posledního prodloužení registrace: 23.2.2011
10. DATUM REVIZE TEXTU
5.3.2015
22/22