Cluvot 1250 Iu
Sp.zn. sukls260181/2012; sukls260182/2012
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Cluvot 250 IU, prášek a rozpouštědlo pro injekční/infuzní roztok. Cluvot 1250 IU, prášek a rozpouštědlo pro injekční/infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ"
Léčivá látka: Cluvot je purifikovaný koncentrát lidského plazmatického koagulačního faktoru XIII (FXIII). Je ve formě bílého prášku.
Jedna injekční lahvička obsahuje nominálně 250 nebo 1250 IU lidského plazmatického koagulačního faktoru XIII (Factor XIII coagulationis humanus).
Cluvot obsahuje přibližně 62,5 IU/ml (250 IU/4 ml a 1250 IU/20 ml) lidského plazmatického koagulačního faktoru XIII po rekonstituci v 4 ml, resp. 20 ml vody na injekci.
Specifická aktivita přípravku Cluvot je přibližně 6 - 10 IU/mg proteinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok. Bílý prášek a čiré, bezbarvé rozpouštědlo.
4. KLINICKÉ ÚDAJE 4.1. Terapeutické indik ace
Cluvot je indikován pro dospělé a pediatrické pacienty
• k profylaktické léčbě vrozeného nedostatku FXIII a
• k perioperační léčbě chirurgického krvácení u pacientů s vrozeným nedostatkem FXIII.
4.2 Dávkování a způsob podání
Dávkování
1 ml odpovídá přibližně 62,5 IU, respektive 100 IU odpovídá 1,6 ml.
Důležité:
Množství, které je potřeba podat, a frekvence podávání mají být vždy zaměřené na klinickou účinnost u individuálního případu.
Dávka
Dávkovací režim se upravuje individuálně podle tělesné hmotnosti, laboratorních hodnot a klinického stavu pacienta.
Rozpis dávkování při rutinní profylaxi
Úvodní dávka
• 40 mezinárodních jednotek (IU) na kg tělesné hmotnosti
• Rychlost injekce nemá překročit 4 ml za minutu.
Následné dávky
• Dávkování se řídí podle poslední minimální úrovně aktivity FXIII, s dávkováním jednou za 28 dní (každé 4 týdny), aby se minimální úroveň aktivity FXIII udržela přibližně na 5 až 20 %.
• Doporučená úprava dávkování o ±5 IU na kg se má opírat o minimální úroveň aktivity FXIII (jak je uvedeno v tabulce 1) a o klinický stav pacienta.
• Úprava dávkování se má opírat o specifický, citlivý test používaný ke stanovení hladin FXIII. Příklad úpravy dávky s použitím standardního testu aktivity Berichrom je popsán v níže uvedené tabulce 1.
Tabulka 1: Úprava dávky s použitím testu aktivity Berichrom
|
Minimální hladina aktivity faktoru XIII (v %) |
Změna dávkování |
|
Jedna minimální hladina < 5 % |
Zvýšení o 5 jednotek na kg |
|
Minimální hladina 5 % až 20 % |
Žádné změny. |
|
Dvě minimální hladiny > 20 % |
Snížení o 5 jednotek na kg |
|
Jedna minimální hladina > 25 % |
Snížení o 5 jednotek na kg |
Účinnost vyjádřená v jednotkách je stanovena pomocí testu aktivity Berichrom, citovaného podle aktuální normy International Standard for Blood Coagulation Factor XIII, Plasma [Mezinárodní norma pro krevní koagulační faktor XIII, plazma]. Proto se jedna jednotka rovná jedné mezinárodní jednotce (IU).
Předoperační profylaxe
Po podání poslední rutinní profylaktické dávky, pokud je operace naplánována:
• mezi 21. a 28. dnem po poslední dávce - pacientovi se podá plná profylaktická dávka bezprostředně před operací a následující profylaktická dávka o 28 dní později.
• mezi 8. až 21. dnem po poslední dávce - před operací lze podat doplňující dávku (plnou nebo částečnou). Dávka se řídí úrovní aktivity FXIII a klinickým stavem pacienta a upravuje se podle poločasu přípravku Cluvot.
• v průběhu 7 dní po poslední dávce - doplňující dávka nemusí být nutná.
Úpravy dávkování se mohou lišit od těchto doporučení a je třeba je individualizovat na základě úrovně aktivity FXIII a klinického stavu pacienta. Všichni pacienti musí být během operace i po operaci pečlivě sledováni.
Proto se doporučuje monitorovat zvýšení aktivity FXIII testem na FXIII. V případě velkého chirurgického zákroku a závažných krvácení je cílem dosáhnout téměř normálních hodnot (zdravé osoby: 70 % - 140 %).
Pediatrická populace
Dávkování a způsob podání u dětí a dospívajících je založeno na tělesné hmotnosti, a proto se celkově řídí stejnými pravidly jako u dospělých. Dávka a/nebo frekvence podávání se vždy u každého jedince řídí klinickou účinností a hladinami aktivity FXIII (Viz též body 5.1 a 5.2.).
Starší pacienti
Dávkování a způsob podání u starších pacientů (> 65 let) dosud nebylo dokumentováno klinickými studiemi.
Po rekonstituci je roztok čirý nebo slabě opalescentní. Přípravek se musí před podáním ohřát na pokojovou nebo tělesnou teplotu. Injekci nebo infuzi podávejte pomalu intravenózně rychlostí, kterou pacient vnímá jako příjemnou. Rychlost injekce nebo infuze by neměla překročit přibližně 4 ml za minutu.
Pozorujte bezprostřední reakce pacienta. Pokud se dostaví jakákoli reakce, která může souviset s podáváním přípravku Cluvot, je potřeba snížit rychlost infuze nebo infuzi zastavit podle toho, jak to vyžaduje klinický stav pacienta.
Pokyny k rekonstituci léčivého přípravku před podáním, viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
U pacientů se známou alergií na přípravek (s příznaky, jako generalizovaná kopřivka, vyrážka, pokles krevního tlaku, dušnost) lze podat profylakticky antihistaminika a kortikosteroidy.
Při podávání přípravku se mohou vyskytnout reakce z přecitlivělosti alergického typu. Pokud se objeví příznaky přecitlivělosti (jako kopřivka, generalizovaná kopřivka, svírání na hrudi, sípání, hypotenze a anafylaxe), musí se infuze přípravku Cluvot okamžitě přerušit. V případě šoku se musí postupovat podle aktuálních medicínských standardů pro protišokovou léčbu.
V případě čerstvé trombózy je nutná opatrnost, protože FXIII má fibrin-stabilizující účinek.
Imunogenicita
U pacientů, kterým byl podáván Cluvot, byl detekován vznik inhibičních protilátek proti FXIII. Proto by měli být pacienti monitorováni kvůli možnému vzniku inhibičních protilátek. Přítomnost inhibičních protilátek se může manifestovat jako nedostatečná odpověď na léčbu. Pokud se přes očekávání nedosáhne jistá úroveň aktivity plazmatického FXIII, nebo pokud se během aplikace profylaxe objeví intermenstruační krvácení, měla by se změřit koncentrace inhibičních protilátek FXIII.
Upozorněnípro pacienty na dietě s nízkým příjmem sodíku
Cluvot obsahuje 124,4 až 195,4 mg (5,41 až 8,50 mmol) sodíku v jedné dávce (40 IU/kg tělesné hmotnosti - při průměrné tělesné hmotnosti 70 kg), pokud je podána doporučená dávka (2800 IU = 44,8 ml). Tuto okolnost je třeba vzít v úvahu u pacientů, kteří jsou na dietě s kontrolovaným příjmem sodíku.
Virová bezpečnost
Standardní opatření zaměřená na předcházení infekcím vznikajícím jako důsledek použití léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují výběr dárců, kontrolu jednotlivých odběrů a plazmatických poolů na specifické markery infekce a začlenění efektivních výrobních postupů inaktivace/ odstranění virů. Přesto možnost přenosu infekčních agens při podávání léčivých přípravků, které byly vyrobeny z lidské krve nebo plazmy, nelze zcela vyloučit. To platí i o neznámých nebo objevujících se virech a jiných patogenech.
Uplatněná opatření jsou považována za účinná u obalených virů, jako jsou virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV), a u neobalených virů hepatitidy A a parvoviru B19.
Důrazně se doporučuje, aby se při každém podání přípravku Cluvot pacientovi zaznamenal název a číslo šarže přípravku, aby byla zachována vazba mezi pacientem a šarží přípravku.
U pacientů, kteří pravidelně/opakovaně dostávají deriváty lidské plazmy, by se měla uvážit možnost vhodného očkování (hepatitida A a B).
4.5 Inte rakce s jinými lé čivými přípravky a jiné formy inte rakce
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení Těhotenství
Omezené množství údajů o klinickém použití přípravku Cluvot během těhotenství neprokázalo žádné nežádoucí účinky na průběh těhotenství a prenatální nebo postnatální vývoj. O použití přípravku Cluvot během těhotenství lze uvažovat, pokud je to nutné.
Kojení
Údaje o vylučování přípravku Cluvot do lidského mateřského mléka nejsou k dispozici. Nicméně vzhledem k velikosti jeho molekuly je vylučování do mateřského mléka nepravděpodobné a vzhledem k jeho bílkovinné struktuře je absorpce nezměněných molekul kojencem také nepravděpodobná. Proto lze Cluvot používat během kojení.
Fertilita
Nejsou k dispozici žádné údaje týkající se účinku přípravku Cluvot na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie hodnotící účinky na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Následující nežádoucí účinky jsou založeny na zkušenostech po uvedení na trh.
Tabulkový přehled nežádoucích účinků
Tabulka uvedená níže je sestavena podle tříd orgánových systémů MedDRA. Frekvence byly hodnoceny podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až <1/10), méně časté (> 1/1 000 až <1/100), vzácné (> 1/10 000 až <1/1 000 ) a velmi vzácné (<1/10 000).
|
Standardní třídy orgánových systémů MedDRA |
Nežádoucí účinek |
Frekvence |
|
Poruchy imunitního systému |
Alergoidní-anafylaktoidní reakce (jako generalizovaná kopřivka, vyrážka, pokles krevního tlaku, dušnost) |
Vzácné |
|
Vývoj inhibitorů FXIII |
Velmi vzácné | |
|
Celkové poruchy a reakce v místě aplikace |
Vzestup tělesné teploty |
Vzácné |
V případě, že se objeví alergoidní-anafylaktoidní reakce, musí se okamžitě přerušit podávání přípravku Cluvot a zahájit vhodná léčba. Musí se dodržovat aktuální medicínské standardy pro protišokovou léčbu.
Pediatrická populace:
V klinických studiích se bezpečnostní profil u pediatrických pacientů neliší od profilu dospělých.
Informace o bezpečnosti týkající se přenosných agens viz bod 4.4.
4
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www. sukl. cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Nebyl hlášen žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika ATC kód: B02B D07
Faktor XIII spojuje aminoskupinu lysinu s glutaminem s využitím své enzymatické funkce (transamidázová aktivita), což vede k zesíťování mezi molekulami fibrinu. Zesíťování fibrinu a jeho stabilizace podporuje pronikání fibroblastů a hojení ran.
Pediatrická populace
V klinických studiích, které zahrnovaly subjekty < 18 let s vrozeným nedostatkem FXIII, bylo profylaktické podávání přípravku Cluvot každých 28 dnů úspěšné při udržování minimálních úrovní aktivity FXIII na přibližně 5-20 %.
5.2 Farmakokinetické vlastnosti
Distribuce
Přípravek se podává intravenózně; proto je okamžitě biologicky dostupný, čehož výsledkem je plazmatická koncentrace odpovídající aplikované dávce.
Eliminace
U pacientů s vrozeným nedostatkem FXIII byl stanoven biologický poločas přípravku Cluvot na 6,6 ± 2,29 dnů (střední hodnota ± SD). Cluvot se metabolizuje stejným způsobem, jako endogenní koagulační FXIII.
Přehled farmakokinetických parametrů (dospělá populace/18 let a starší) je uveden v následující tabulce:
|
Parametry |
Střední hodnota (min-max) |
|
AUC ss, 0-inf (jednotky»hod/ml) |
182,9 (133,5-300,2) |
|
Css, max (jednotky/ml)* |
0,9 (0,6-1,2) |
|
Css, min (jednotky/ml)* |
0,07 (0,0-0,16) |
|
Tmax (hod) |
1,2 (0,7-4,2) |
|
Poločas [dny] |
7,8 (3,1-11,02) |
|
CL [ml/hod/kg] |
0,22 (0,13-0,30) |
|
Vss [ml/kg] |
49,4 (31,65-62,91) |
|
MRT [dny] |
11.7 (5,7-17,02) |
AUC ss, (o-infi = plocha pod křivkou plazmatické koncentrace od času 0 do nekonečna v ustáleném stavu
* 100% aktivita odpovídá 1 jednotce/ml Css, max: maximální koncentrace v ustáleném stavu Css, min: minimální koncentrace v ustáleném stavu Tmax: doba k dosažení maximální koncentrace CL: clearance
Vss: distribuční objem v ustáleném stavu MRT = střední rezidenční čas
Pediatrická populace
Ze 188 unikátních subjektů v klinických studiích koncentrátu (lidského) faktoru XIII, bylo 117 subjektů < 18 let v době zařazení do studie (1 měsíc až < 2 roky, n = 17; 2 až < 12 let n = 62; 12 až < 16 let, n = 30; 17 až 18 let, n = 8). Ve farmakokinetické studii PK 2002, 5 ze 14 subjektů bylo ve věku od 2 do < 18 let (2 - 11 let, n = 3; 12 - 16 let, n = 2; 17 - 18 let, n = 0). Subjekty mladší než 16 let měly kratší poločas a rychlejší clearance (poločas: 5,7 ± 1,00 dnů; clearance: 0,291 ± 0,12 ml/hod/kg) ve srovnání s dospělými (poločas: 7,1 ± 2,74 dnů, clearance: 0,22 ± 0,07 ml/hod/kg).
Přípravek má u dětí ve srovnání s dospělými kratší poločas a rychlejší clearance.
Nicméně vzhledem k tomu, že u všech věkových skupin je dávkování individuálně určeno podle tělesné hmotnosti pacienta a upraveno podle požadované aktivity FXIII, není nutné žádné specifické dávkování s ohledem na věk.
5.3 Předklinické údaje vztahující se k bezpečnosti
Bílkoviny obsažené v přípravku Cluvot pocházejí z lidské plazmy a působí jako proteiny lidské plazmy.
Studie toxicity po jedné dávce a po opakovaném podávání u zvířat neodhalily toxický potenciál pro přípravek Cluvot.
Studie hodnotící vliv na reprodukci a embryofetální vývoj nebyly provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek: lidský albumin monohydrát glukosy chlorid sodný
hydroxid sodný (na úpravu pH)
Rozpouštědlo: voda na injekci
6.2 Inkompatibility
Cluvot se nesmí mísit s jinými léčivými přípravky, ředidly nebo rozpouštědly s výjimkou těch, které jsou uvedeny v bodě 6.6 a měl by být podáván oddělenou infuzní soupravou.
6.3 Doba použitelnosti
3 roky
Nepoužívejte po uplynutí doby použitelnosti, uvedené na krabičce a vnitřním obalu.
Chemická a fyzikální stabilita při použití byla prokázána po dobu 24 hodin při teplotě < 25 ° C. Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba uchovávání nemá přesáhnout 4 hodiny při pokojové teplotě. Rekonstituovaný roztok neochlazujte ani nezmrazujte.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C -8°C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvičky:
250 IU
Prášek: injekční lahvička z bezbarvého skla, uzavřená pryžovou zátkou (brombutylová guma), hliníkovým uzávěrem a plastovým krytem.
Rozpouštědlo (voda na injekci): injekční lahvička z bezbarvého skla
1250IU
Prášek: injekční lahvička z bezbarvého skla, uzavřená pryžovou zátkou (brombutylová guma), hliníkovým uzávěrem a plastovým krytem.
Rozpouštědlo (voda na injekci): injekční lahvička z bezbarvého skla
Velikost balení:
Balení s 250 IU 1 injekční lahvička s práškem 1 injekční lahvička s 4 ml vody na injekci 1 přepouštěcí adaptér s filtrem 20/20 (Mix2Vial)
Balení s 1250 IU 1 injekční lahvička s práškem 1 injekční lahvička s 20 ml vody na injekci 1 přepouštěcí adaptér s filtrem 20/20 (Mix2Vial)
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obecné pokyny
Roztok musí být čirý nebo lehce opalescentní. Po přefiltrování/natáhnutí (viz níže) se musí rekonstituovaný přípravek před podáním vizuálně zkontrolovat, zda neobsahuje částice a jak je zbarvený. Nepoužívejte viditelně zakalené roztoky nebo roztoky obsahující vločky nebo částice. Rekonstituce a natáhnutí musí být provedeno za aseptických podmínek.
Rekonstituce
Zahřejte rozpouštědlo na pokojovou teplotu. Ujistěte se, že byla odstraněna odklápěcí víčka na přípravku a rozpouštědle, zátky se ošetří aseptickým roztokem a nechají oschnout před otevřením balení Mix2Vial.
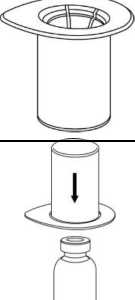
1
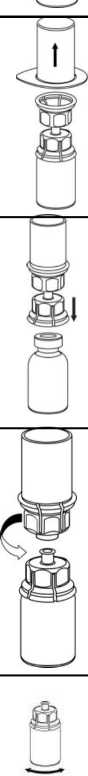
4
3
5
2
6
1. Sloupněte víčko balení Mix2Vial. Nevytahujte Mix2Vial z blistru!
2. Postavte injekční lahvičku s rozpouštědlem
na rovný a čistý povrch a pevně ji držte. Uchopte Mix2Vial společně s blistrem a zatlačte hrot modrého konce adaptéru rovně dolů skrz pryžovou zátku injekční lahvičky s rozpouštědlem.
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle vzhůru. Přesvědčte se, že jste vytáhl(a) je n blistrový obal a ne soupravu Mix2Vial.
4. Postavte injekční lahvičku s práškem na
rovný a pevný povrch. Obraťte injekční lahvičku s rozpouštědlem spolu s nasazenou soupravou Mix2Vial a zatlačte hrot průhledného konce adaptéru přímo dolů skrz pryžovou zátku injekční lahvičky s práškem. Rozpouštědlo se samo automaticky nasaje do injekční lahvičky s práškem.
5. Uchopte jednou rukou tu část soupravy Mix2Vial, kde je injekční lahvička s práškem a druhou rukou tu část, kde je injekční lahvička od rozpouštědla a odšroubujte je od sebe opatrně na dvě části. Injekční lahvičku od rozpouštědla
s připojeným modrým adaptérem soupravy Mix2Vial vyhoďte.
6. Jemně otáčejte injekční lahvičkou s práškem s připojeným průhledným adaptérem, dokud se prášek zcela nerozpustí. Netřepejte s ní.

7
7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Při svislé poloze injekční lahvičky s přípravkem připojte injekční stříkačku k nástavci Luer-Lock na Mix2Vial. Do injekční lahvičky s přípravkem injikujte vzduch.
|
P 8 |
8. Zatímco držíte píst injekční stříkačky stlačený, obraťte celý systém dnem vzhůru. Pomalým vytahováním pístu natáhněte roztok do injekční stříkačky. | |
|
i |
9 |
9. Po natažení roztoku do injekční stříkačky uchopte pevně válec injekční stříkačky (píst stále směřuje dolů) a odpojte průhledný adaptér soupravy Mix2Vial od injekční stříkačky. |
Je třeba dbát na to, aby se do stříkačky naplněné přípravkem nedostala krev, protože je zde riziko, že by krev mohla v stříkačce koagulovat a fibrinové sraženiny by byly podány pacientovi.
Rekonstituovaný roztok se má podávat samostatnou injekční/infuzní soupravou pomalou intravenózní injekcí rychlostí nepřesahující 4 ml za minutu.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH Emil-von-Behring-Strasse 76 35041 Marburg Německo
8. REGISTRAČNÍ ČÍSLO(A)
Cluvot 250 IU: 16/232/14-C Cluvot 1250 IU: 16/233/14-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11.6.2014
10. DATUM REVIZE TEXTU
11.6.2014
9