Clinimix N17G35E
zastaralé informace, vyhledat novějšísp.zn. sukls37040/2011 sp.zn. sukls135726/2013
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
CLINIMIX N17G35E, infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Přípravek CLINIMIX N17G35E je dodáván ve dvoukomorových vacích: jedna komora obsahuje směs aminokyselin s elektrolyty, druhá komora obsahuje roztok glukózy s kalciem. Infuzní roztok aminokyselin obsahuje 15 L-aminokyselin (8 esenciálních aminokyselin), které jsou potřebné pro syntézu proteinů.
Poměr aminokyselin je následující:
Esenciální aminokyseliny/celkové aminokyseliny = 41,3%
Esenciální aminokyseliny /celkový obsah dusíku = 2,83 Aminokyseliny s rozvětveným řetězcem/celkové aminokyseliny = 19%
Kvantitativní složení přípravku CLINIMIX N17G35E je následující:
|
Léčivé látky: |
10% roztok aminokyselin s elektrolyty (g/l) |
35% roztok glukózy s kalciem (g/l) |
|
Leucinum |
7,30 | |
|
Phenylalaninum |
5,60 | |
|
Methioninum |
4,00 | |
|
Lysinum |
5,80 | |
|
(jako L-lysini hydrochloridum) |
(7,25) | |
|
Isoleucinum |
6,00 | |
|
Valinum |
5,80 | |
|
Histidinum |
4,80 | |
|
Threoninum |
4,20 | |
|
Tryptophanum |
1,80 | |
|
Alaninum |
20,70 | |
|
Argininum |
11,50 | |
|
Glycinum |
10,30 | |
|
Prolinum |
6,80 | |
|
Serinum |
5,00 | |
|
Tyrosinum |
0,40 | |
|
Natrii acetas trihydricus |
6,80 | |
|
Kalii hydrogenophosphas |
5,22 | |
|
Natrii chloridum |
1,17 | |
|
Magnesii chloridum hexahydricum |
1,02 | |
|
Glucosum monohydricum |
385 | |
|
(odp. Glucosum) |
(350) | |
|
Calcii chloridum dihydricum |
0,66 |
Úplný seznam pomocných látek viz bod 6.1.
Po smíchání obsahu obou komor je složení binární směsi všech dostupných velikostí balení následující:
|
N17G35E |
V 1 litru |
V 1,5 litru |
Ve 2 litrech |
|
Dusík (g) |
8,3 |
12,4 |
16,5 |
|
Aminokyseliny (g) |
50 |
75 |
100 |
|
Glukóza(g) |
175 |
263 |
350 |
|
Celková energetická hodnota (kcal) |
900 |
1350 |
1800 |
|
Energetická hodnota glukózy (kcal) |
700 |
1050 |
1400 |
|
Sodík (mmol) |
35 |
53 |
70 |
|
Draslík (mmol) |
30 |
45 |
60 |
|
Hořčík (mmol) |
2,5 |
3,8 |
5,0 |
|
Vápník (mmol) |
2,3 |
3,4 |
4,5 |
|
Acetáty (mmol) |
75 |
113 |
150 |
|
Chloridy (mmol) |
40 |
60 |
80 |
|
Fosfáty jako HPO 4-- (mmol) |
15 |
23 |
30 |
|
pH |
6 | ||
|
Osmolarita (mOsm/l) |
1625 |
3. LÉKOVÁ FORMA
Infuzní roztok.
• Popis přípravku před smícháním: roztoky aminokyselin a glukózy jsou čiré a bezbarvé nebo světle žluté
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Parenterální výživa v případech, ve kterých perorální nebo enterální výživa není možná, je nedostatečná nebo kontraindikovaná.
Pacientům na dlouhodobé parenterální výživě je možné přidat k přípravku CLINIMIX N17G35E emulzi lipidů k zajištění dostatečného přísunu energie a esenciálních mastných kyselin.
4.2 Dávkování a způsob podání
Dávkování
Dávkování se stanovuje podle metabolických potřeb, výdeje energie a klinického stavu pacienta.
U dospělých kolísá potřeba dusíku od 0,16 g/kg/den (přibližně 1 g aminokyselin/kg/den) do 0,35 g dusíku/kg/den (přibližně 2 g aminokyselin/kg/den).
U dětí se pohybuje potřeba mezi 0,35 g dusíku/kg/den (přibližně 2 g aminokyselin/kg/den) do 0,45 g dusíku/kg/den (přibližně 3 g aminokyselin/kg/den).
Potřeba energie se pohybuje od 25 kcal/kg/den do 40 kcal/kg/ den a je závislá na stavu výživy nemocného a stupni katabolismu.
Maximální denní dávky každé složky přípravku CLINIMIX N17G35 (t.j. aminokyselin nebo glukózy) mají být stanoveny podle individuální celkové nutriční potřeby a tolerance pacienta. Maximální rychlost infuze je 1,4 ml/kg/h, t.j. 85 až 100 ml/hod (u pacienta o hmotnosti 60 až 70 kg). Maximální denní dávka je 30 ml/kg, t.j. 1 800 až 2 100 ml (u pacienta o hmotnosti 60 až 70 kg).
Pediatrická populace Data nejsou k dispozici.
Způsob podání
Pouze k jednorázovému použití.
Doporučuje se po otevření obsah vaku okamžitě použít. Neuchovávejte k pozdějšímu podání infuze.
Přípravek podávejte až po protržení švů a smíchání obsahů obou komor.
Vzhled roztoku po smíchání: čirý a bezbarvý nebo světle žlutý roztok.
Návod na přípravu a zacházení s roztokem viz bod 6.6.
Při podání do periferie je nutno vzít v úvahu osmolaritu specifického infuzního roztoku. Roztoky nebo směsi s osmolaritou vyšší než 800 mOsm/l mají být podávány do centrální žíly (také viz bod 4.4).
Podle individuálních potřeb mohou být přidány vitamíny, stopové prvky a další komponenty (včetně lipidů), aby se předešlo rozvoji deficitu a komplikacím (viz bod 6.2).
V průběhu první hodiny by se měla rychlost infuze postupně zvyšovat.
Rychlost podání se má přizpůsobit dávkování, vlastnostem infuzního roztoku, celkovému požadovanému příjmu tekutin za 24 hodin a trvání infuze. Doba trvání infuze by měla být delší než 8 hodin.
Snížit riziko hypoglykémie po vysazení je možné postupným zpomalením rychlosti infuze v poslední hodině.
4.3 Kontraindikace
• Známá přecitlivělost na kteroukoli léčivou látku nebo na pomocné látky uvedené v bodu 6.1 nebo na složky obalu
• Poruchy metabolismu aminokyselin
• Těžká hyperglykémie
• Metabolická acidóza, zvýšená hladina laktátu
• CLINIMIX N17G35E s elektrolyty by neměl být používán u pacientů s hyperkalémií, hypernatrémií a u pacientů s patologicky zvýšenou plazmatickou koncentrací hořčíku, vápníku a/nebo fosforu
• Stejně jako u jiných infuzních roztoků obsahujících vápník je současná léčba přípravku CLINIMIX N17G35E s ceftriaxonem kontraindikována u novorozenců (ve věku nebo mladších 28 dní), i kdyby byly infuze vedeny odděleně (riziko fatální precipitace vápenaté soli ceftriaxonu v krevním oběhu novorozence). Současné podávání starším pacientům viz bod 4.5 a 6.2.
4.4 Zvláštní upozornění a opatření pro použití
UPOZORNĚNÍ
U přípravků řady CLINIMIX byly zaznamenány hypersenzitivní reakce/reakce na infuzi zahrnující hypotenzi, hypertenzi, periferní cyanózu, tachykardii, dyspnoi, zvracení, nauzeu, kopřivku, vyrážku, svědění, erytém, pocení, horečku a zimnici.
U jiných parenterálních nutričních přípravků byla zaznamenána anafylaxe.
Na počátku jakékoli intravenózní infuze je třeba pacienta klinicky sledovat. Při výskytu jakýchkoli abnormálních projevů a příznaků, např. hypersenzitivity nebo reakce na infuzi, je nutné infuzi okamžitě přerušit.
Roztoky obsahující glukózu podávejte s opatrností, pokud vůbec, u pacientů se známou alergií na obilí nebo obilné produkty.
U pacientů přijímajících parenterální výživu byly hlášeny pulmonální vaskulární precipitáty.
V některých případech došlo k úmrtí. Nadměrné doplňování vápníku a fosforu zvyšuje riziko tvorby precipitátů fosforečnanu vápenatého. Precipitáty byly zaznamenány i v roztoku bez fosfátové soli. Byly také hlášeny precipitáty na konečném filtru infuzního setu a suspektní in vivo tvorba precipitátů. Pokud se objeví pulmonální obtíže, infuze má být ihned zastavena a zahájeno vyhodnocení klinického stavu. Kromě kontroly roztoku se má také pravidelně provádět kontrola precipitátů u infuzního setu a katétru.
V důsledku použití intravenózních katétrů k podání parenterálních roztoků, špatné péče o katétry nebo u kontaminovaných roztoků se mohou objevit infekce a sepse.
Imunosuprese a další faktory jako hyperglykémie, malnutrice a /nebo základní onemocnění mohou pacienty predisponovat ke vzniku infekčních komplikací.
Rozpoznání časné infekce může usnadnit pečlivé symptomatické a laboratorní sledování horečky/třesavky, leukocytózy, technických komplikací v místě aplikace a hyperglykémie. Výskyt septických komplikací lze snížit zvýšeným důrazem na aseptickou techniku při umísťování katétru a jeho údržbě, a také při přípravě nutričního přípravku.
Realimentace vážně podvyživených pacientů může vést k realimentačnímu (refeeding) syndromu, při kterém dochází k intracelulárnímu přesunu draslíku, fosforu a hořčíku v důsledku počínajícího anabolismu pacienta. Může se rovněž rozvinout deficit thiaminu a retence tekutin. Důkladným sledováním a pomalým navyšování příjmu živin aniž by se podávalo nadbytečné množství (overfeeding) lze těmto komplikacím zabránit.
Hypertonické roztoky mohou při infuzi do periferní žíly vyvolat venózní iritaci. Výběr periferní nebo centrální žíly závisí na výsledné osmolaritě směsi. Obecně přijatý limit pro periferní infuzi je přibližně 800 mOsm/l, je však závislý na věku pacienta a jeho celkovém stavu a na stavu periferních žil.
Nepropojujte vaky do série, aby nedošlo ke vzduchové embolii způsobené případným reziduálním vzduchem v primárním vaku.
OPATŘENÍ
Před zahájením infuze musí být korigovány těžké poruchy rovnováhy vody a elektrolytů, těžké stavy hyperhydratace a těžké metabolické poruchy.
Není-li příjem výživy přizpůsoben požadavkům pacienta nebo není-li správně posouzena metabolická kapacita kterékoli z podávaných složek výživy, mohou nastat metabolické komplikace. Nežádoucí metabolické účinky se mohou objevit po nedostatečném nebo přebytečném podání živin nebo v důsledku nevhodného složení směsi vzhledem ke specifickým potřebám pacienta.
Pro správné monitorování průběhu infuze jsou zapotřebí časté klinické a laboratorní kontroly. Ty by měly zahrnovat iontogram a funkční testy jater a ledvin.
Intolerance glukózy je častá metabolická komplikace u pacientů v těžkém šoku. Během infuze se může objevit hyperglykémie, glykosurie a hyperosmolární syndrom. Proto by se měla pravidelně vyšetřovat hladina glukózy v krvi a v moči a u diabetiků by mělo být v případě potřeby upraveno dávkování inzulínu.
U pacientů s renální insuficiencí používejte přípravek s opatrností, zejména v případě hyperkalémie, protože existuje riziko rozvoje nebo zhoršení metabolické acidózy a hyperazotémie, pokud se neprovádí extrarenální odstraňování odpadních látek. U těchto pacientů je potřeba důsledně monitorovat stav tekutin a elektrolytů. V případě těžkého poškození ledvin by měly být přednostně podány speciálně navržené roztoky aminokyselin.
Opatrnosti je třeba při současném podávání přípravků CLINIMIX pacientům s adrenální insuficiencí.
Je třeba se vyvarovat oběhového přetížení, zejména u nemocných s plicním edémem, srdeční nedostatečností a srdečním selháním. Stav tekutin musí být pečlivě sledován.
U nemocných s preexistujícím onemocněním jater nebo jaterní insuficiencí kontrolujte kromě rutinních jaterních funkcí i možné příznaky hyperamonémie.
U některých pacientů na parenterální výživě se někdy mohou rozvinout hepatobiliární poruchy včetně cholestázy, jaterní steatózy, fibrózy a cirhózy, které mohou vyústit v selhání jater, podobně jako cholecystitida a cholelitiáza. Předpokládá se, že etiologie těchto poruch je multifaktoriální a může se u různých pacientů lišit. Pacienti s rozvojem laboratorních parametrů nebo jiných známek hepatobiliárních poruch by měli být včas vyšetřeni lékařem specializovaným na jaterní choroby, aby mohly být identifikovány možné kauzální a podpůrné faktory a provedeny možné terapeutické a profylaktické intervence.
U pacientů, kteří přijímají roztoky aminokyselin, se mohou objevit zvýšené hladiny amoniaku a hyperamonémie. U některých pacientů může tento stav indikovat vrozenou poruchu metabolismu aminokyselin (viz bod 4.3) nebo jaterní selhání.
Časté kontroly hladiny plazmatického amoniaku se mají provádět u novorozenců a kojenců. Těmito kontrolami lze zjistit hyperamonémii, která může indikovat vrozenou poruchu metabolismu aminokyselin. Podle závažnosti a etiologie může hyperamonémie vyžadovat okamžitou intervenci.
Příliš rychlá infuze aminokyselin může způsobit nevolnost, zvracení a zimnici. V těchto případech musí být infuze okamžitě zastavena.
Obecně by mělo být dávkování u starších pacientů stanoveno s opatrností, aby reflektovalo vyšší frekvenci výskytu snížených funkcí jater, ledvin nebo srdce a souběžná onemocnění nebo medikamentózní léčbu.
Pediatrická populace:
• U pediatrické populace nebyly provedeny žádné studie.
• Sledování hyperamonémie u pediatrických pacientů, viz výše.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Stejně jako u dalších infuzních roztoků obsahujících vápník je současná léčba přípravku CLINIMIX N17G35E s cefotriaxonem kontraindikována u novorozenců (ve věku nebo mladších 28 dní), i kdyby byly infuze vedeny odděleně (riziko fatální precipitace vápenaté soli ceftriaxonu v krevním oběhu novorozence) (viz bod 4.3).
U pacientů starších než 28 dnů (včetně dospělých), nesmí být ceftriaxon podáván stejnou infuzní linkou současně s intravenózními roztoky obsahující vápník, včetně přípravku CLINIMIX N17G35E. Pokud se použije stejná infuzní linka pro postupné podávání, musí se mezi jednotlivými aplikacemi linka důkladně propláchnut kompatibilní tekutinou.
Přípravek CLINIMIX N17G35E obsahuje draslík, proto má být podáván s opatrností pacientům, kteří se léčí látkami nebo přípravky způsobujícími hyperkalémii nebo zvyšujícími riziko hyperkalémie, jako jsou kalium šetřící diuretika (amilorid, spironolakton, triamteren), ACE inhibitory, antagonisté receptoru angiotensinu II nebo imunosupresiva takrolimus a cyklosporin.
4.6 Fertilita, těhotenství a kojení
Bezpečnost podávání přípravku CLINIMIX N17G35E ve fertilním věku, těhotenství a během kojení nebyla prokázána pro nedostatek klinických studií. O podání přípravku těhotným a kojícím ženám je třeba rozhodnout na základě posouzení možného rizika vzhledem k přínosu léčby.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie na schopnost řídit a obsluhovat stroje nebyly provedeny.
4.8 Nežádoucí účinky
Případné nežádoucí účinky mohou vzniknout v důsledku nesprávného použití: např. při předávkování nebo při příliš rychlé infuzi (viz též bod 4. a 4.9).
Postmarketingové nežádoucí účinky:
Následující nežádoucí účinky byly zaznamenány u přípravků CLINIMIX
během postmarketingového sledování a jsou řazeny podle MedDRA třídy orgánových
systémů (TOS) a upřednostňovaného termínu.
|
Třída orgánového systémů (TOS) |
Upřednostňovaný termín MedDRA |
Frekvencea |
|
Poruchy imunitního systému |
Přecitlivělost* |
Není známo |
a: Frekvence je definována jako velmi častá (>1/10); častá (>1/100 až <1/10); méně častá (>1/1000 až <1/100), vzácná (>1/10000 až <1/1000); velmi vzácná (<1/10000); není známo (z dostupných dat nelze určit)
*: Zahrnuje následující projevy: hypotenzi, hypertenzi, periferní cyanózu, tachykardii, dyspnoi, zvracení, nauzeu, kopřivku, vyrážku, svědění, erytém, pocení, horečku, zimnici
Další nežádoucí účinky popsané u přípravků pro parenterální výživu zahrnují:
• Anafylaxi
• Hyperglykémii, hyperamonémii, azotémii
• Pulmonální vaskulární precipitáty
• Selhání jater, cirhózu jater, fibrózu jater, cholestázu, steatózu jater, zvýšení krevního bilirubinu, zvýšení jatemích enzymů
• Cholecystitidu, cholelitiázu
• Tromboflebitidu v místě vpichu, žilní podráždění (flebitidu v místě vpichu, bolest, erytém, horkost, otok, induraci)
Glukózová intolerance je častou metabolickou komplikací u těžce stresovaných pacientů. Infuze přípravků může způsobit hyperglykémii, glykosurii a hyperosmolární koma.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu: Státní ústav pro kontrolu léčiv, Šrobárova 48,
100 41 Praha 10. Webové stránky: http: //www .sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Při nesprávném podávání přípravku (předávkování a/nebo překročení doporučené rychlosti podání) může dojít k hypervolémii, poruchám rovnováhy elektrolytů nebo acidóze, které mohou mít závažné nebo fatální důsledky. V těchto případech musí být infuze okamžitě přerušena. Podle klinického stavu může být indikovaná další intervence.
Při předávkování glukózou se může objevit hyperglykémie, glykosurie a hyperosmolární syndrom. Příliš rychlá infuze aminokyselin může vyvolat nevolnost, zvracení a třes. V těchto případech je třeba infuzi okamžitě přerušit (viz bod 4.4).
V některých závažných případech může být nutná hemodialýza, hemofiltrace nebo hemodiafiltrace.
Neexistuje specifické antidotum pro případ předávkování. Záchranné postupy by měly zahrnovat vhodná opatření zejména s ohledem na dýchací a kardiovaskulární systém.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Roztoky pro parenterální výživu, kombinace ATC kód: B05BA10
Jako intravenózní roztok pro parenterální výživu zajišťuje přípravek CLINIMIX N17G35E nutriční podporu pro udržení dusíkové a energetické rovnováhy, která může být narušena nedostatečnou výživou a traumatem. Roztoky přípravku CLINIMIX N17G35E představují biologicky dostupný zdroj dusíku (L-aminokyseliny), sacharidů (glukóza) a elektrolytů.
5.2 Farmakokinetické vlastnosti
Aminokyseliny, elektrolyty a glukóza obsažené v přípravku CLINIMIX N17G35E jsou distribuovány, metabolizovány a vylučovány stejným způsobem jako jednotlivě podávané intravenózní roztoky aminokyselin, glukózy a elektrolytů.
5.3 Předklinické údaje vztahující se k bezpečnosti
S přípravkem CLINIMIX N17G35E nebyly prováděny žádné preklinické studie.
Preklinické studie s roztoky aminokyselin a glukózy, které jsou obsaženy také v přípravku CLINIMIX, avšak v odlišném kvalitativním složení a koncentracích, neprokázaly specifickou toxicitu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Roztok aminokyselin: kyselina octová, voda na injekci Roztok glukózy: kyselina chlorovodíková, voda na injekci
6.2 Inkompatibility
Aditiva mohou být inkompatibilní, podrobné informace lze získat u výrobce.
Před přidáním jakýchkoli látek k infuznímu roztoku musí být ověřena jejich kompatibilita a stabilita směsi.
Roztok nesmí být podáván před, během nebo po transfuzi krve stejným infuzním zařízením, protože by mohlo dojít k pseudoaglutinaci.
Přípravek CLINIMIX N17G35E obsahuje ionty vápníku, které představují další riziko koagulace a tvorby precipitátů v citrátem antikoagulačně ošetřené krvi nebo jejích složkách.
• Stejně jako u jiných směsí parenterální výživy musí být zváženy poměry vápníku a fosforu. Přidání nadměrného množství vápníku a fosforu, zvláště ve formě minerálních solí, může vést k vytvoření precipitátů fosforečnanu vápenatého.
• Stejně jakou jiných infuzních roztoků obsahujících vápník je současná léčba přípravkem CLINIMIX N17G35E s ceftriaxonem kontraindikována u novorozenců (ve věku nebo mladších 28 dní), i kdyby byly vedeny oddělenou infuzní linkou (riziko fatální precipitace vápenaté soli ceftriaxonu v krevním oběhu novorozence).
• Pacientům starším 28 dnů (včetně dospělých) se nesmí ceftriaxon podávat současně s intravenózními roztoky obsahujícími vápník, včetně přípravku CLINIMIX N17G35E, stejnou infuzní linkou.
• Pokud se stejná infuzní linka použije k sekvenčnímu podání, musí být mezi jednotlivými infuzemi důkladně propláchnuta kompatibilním tekutinou.
6.3 Doba použitelnosti
• Dvoukomorové vaky v zevním přebalu mají dobu použitelnosti 2 roky.
• Po smíchání obsahu obou komor byla prokázána chemická a fyzikální stabilita 7 dní při 2 až 8°C a následně 48 hodin při teplotě do 25°C.
• Po přidání aditiv má být směs z mikrobiologického hlediska ihned použita. Není-li ihned použita, zodpovídá za dobu a podmínky uchovávání před použitím uživatel. Doba použitelnosti by neměla překročit 24 hodin při teplotě 2 až 8°C, pokud přidání aditiv neproběhlo za kontrolovaných a validovaných aseptických podmínek. Pokud je za výjimečných okolností požadována delší doba uchovávání, lze kontaktovat výrobce, protože pro přípravky uvedené v bodě 6.6.c jsou k dispozici údaje o stabilitě před použitím po dobu 7 dnů při 2-8°C a následně 48 hodin při teplotě do 25°C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25°C. Chraňte před mrazem.
Podmínky uchovávání přípravku viz bod 6.3.
Přípravky dodávané v průhledném přebalu uchovávejte ve vnějším kartonu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a velikost balení
Přípravek CLINIMIX N17G35E je dodáván ve dvoukomorových plastových vacích: jedna komora obsahuje směs aminokyselin s elektrolyty, druhá komora obsahuje roztok glukózy s kalciem.
Dvoukomorový vícevrstevný plastový vak (EP + SEBS/EVA/ mod EVA/PCCE), těsnící šev veden podélně, v laminátovém transparentním nebo Al přebalu, s absorbentem kyslíku. Sáček s absorbentem kyslíku musí být po sejmutí přebalu zlikvidován.
Vícevrstevný plastový vak je kompatibilní s lipidy.
Obě komory jsou od sebe odděleny svárem. Těsně před podáním se obsah obou komor smíchá stlačením nebo srolováním komor, čímž dojde k porušení sváru.
Velikost balení: 8 x 1000 ml
6 x 1500 ml 4 x 2000 ml
Objemy jednotlivých komor jsou následující:
|
Velikost vaku | |||
|
Komora |
1 l |
1,5 l |
2 l |
|
Roztok aminokyselin |
500 ml |
750 ml |
1000 ml |
|
Roztok glukózy |
500 ml |
750 ml |
1000 ml |
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Upozornění: Přípravek podávejte až po protržení těsnícího švu mezi komorami a promíchání jejich obsahu.
Aktivaci přípravku CLINIMIX lze provést v přebalu anebo po jeho sejmutí. a: Otevření přebalu:
• Přebal roztrhněte v místě zářezů.
• Používejte pouze čirý, bezbarvý nebo světle žlutý roztok v neporušeném obalu. b: Smíchání roztoků:
• Ujistěte se, že přípravek má pokojovou teplotu.
• Vak pevně uchopte za oba horní okraje.
• Zmáčkněte nebo srolujte tak, aby došlo k protržení těsnících švů (viz obr. 1).
• 2-3x obraťte a obsah dobře promíchejte.
• Vzhled roztoku po smíchání: čirý a bezbarvý nebo světle žlutý roztok.
c: Přidání aditiv k přípravku CLINIMIX N17G35E :(viz také bod 6.2):
Jak postupovat:
• Musí být dodrženy aseptické podmínky.
• Ujistěte se o stabilitě a kompatibilitě aditiv.
• Před přidáním aditiv aktivujte komory vaků.
• Připravte injekční vstup vaku.
• Propíchněte injekční vstup a přidejte aditiva stříkačkou s injekční jehlou nebo pomocí převodního setu.
• Důkladně promíchejte obsah vaku s aditivy.
• Zkontrolujte výsledný roztok, jeho zabarvení a přítomnost částic.
• Zkontrolujte, zda nedochází k úniku roztoku z vaku.
• Zajistěte, aby byly dodržovány požadavky na uchovávání aditiv.
Podobně jako u všech parenterálních roztoků, je třeba vždy před použitím aditiv ověřit jejich kompatibilitu. Je nutné důkladné a pečlivé aseptické smíchání všech přísad.
Varování: Aditiva mohou být přidávána po protržení švů mezi komorami (po smíchání obou roztoků). K přípravku CLINIMIX N17G35E lze přidávat:
• Emulze lipidů (např. ClinOleic) v množství 50 až 250 ml na 1000 ml přípravku CLINIMIX N17G35E
|
CLINIMIX |
CLINIMIX |
CLINIMIX | |
|
N17G35E |
N17G35E |
N17G35E | |
|
1 l+250 ml 20% |
1,5 l+500 ml 20% |
2 l+500 ml 20% | |
|
lipidů |
lipidů |
lipidů | |
|
Dusík (g) |
8,3 |
12,4 |
16,5 |
|
Aminokyseliny (g) |
50 |
75 |
100 |
|
Glukóza (g) |
175 |
263 |
350 |
|
Lipidy (g) |
50 |
100 |
100 |
|
Celková energetická hodnota (kcal) |
1400 |
2350 |
2800 |
|
Energetická hodnota glukózy (kcal) |
700 |
1050 |
1400 |
|
Energetická hodnota lipidů (kcal) |
500 |
1000 |
1000 |
|
Poměr glukóza / lipidy |
58/42 |
51/49 |
58/42 |
|
Sodík (mmol) |
35 |
53 |
70 |
|
Draslík (mmol) |
30 |
45 |
60 |
|
Hořčík (mmol) |
2,5 |
3,8 |
5,0 |
|
Vápník (mmol) |
2,3 |
3,4 |
4,5 |
|
Acetáty (mmol) |
70 |
113 |
150 |
|
Chloridy (mmol) |
40 |
60 |
80 |
|
Fosfáty jako HPO 4-- (mmol) |
15 |
23 |
30 |
|
pH |
6 |
6 |
6 |
|
Osmolarita (mOsm/l) |
1360 |
1290 |
1360 |
• Elektrolyty: na 1000 ml přípravku CLINIMIX N17G35E
|
Sodík |
Draslík |
Hořčík |
Vápník | |
|
Až do výsledné koncentrace |
80 mmol |
60 mmol |
5,6 mmol |
3,0 mmol |
|
• Stopové prvky |
: na 1000 ml přípravku CLINIMIX N17G35E | |||
|
Až do výsledné |
Měď |
10 pmol |
Zinek |
77 pmol |
|
koncentrace |
Chróm |
0,14 pmol |
Mangan |
2,5 pmol |
|
Fluor |
38 pmol |
Kobalt |
0,0125 pmol | |
|
Selen |
0,44 pmol |
Molybden |
0,13 pmol | |
|
Jód |
0,5 pmol |
v Železo |
10 pmol |
• Vitamíny: na 1000 ml přípravku CLINIMIX N17G35E
|
Až do výsledné koncentrace |
Vitamín A |
1750 IU |
Biotin |
35 pg |
|
Vitamín B6 |
2,27 mg |
Vitamín B1 |
1,76 mg | |
|
Vitamín D |
110 IU |
Kys. listová |
207 pg | |
|
Vitamín B12 |
3,0 pg |
Vitamín B2 |
2,07 mg | |
|
Vitamín E |
5,1 mg |
Vitamín C |
63 mg | |
|
Vitamín PP |
23 mg |
Vitamín B5 |
8,63 mg | |
|
Vitamín K |
75 pg |
Údaje o stabilitě po přidání jiných tukových emulzí a dalších aditiv k přípravku CLINIMIX N17G35E jsou k dispozici na vyžádání.
Pokud zpozorujete mírně krémovitou konzistenci, směs pečlivě promíchejte mírnými pohyby, abyste před podáním získali homogenní emulzi.
Přidání aditiv musí probíhat za aseptických podmínek.
Aditiva lze přidávat injekční stříkačkou nebo pomocí převodního setu.
• Přidání aditiva stříkačkou nebo převodním setem vybaveným jehlou:
- Připravte injekční vstup (samostatný vstup, viz obr. 2).
- Propíchněte jej a injikujte aditivum.
- Dobře promíchejte roztok s aditivem.
• Přidání aditiv převodním setem vybaveným hrotem:
- Prosím postupujte podle návodu k použití příslušného převodního setu pro lipidy.
- Nasaďte hrot převodního setu do vstupu pro přidání lipidů (nej delší vstup).
d: Příprava podání
• Zavěste vak.
• Z aplikačního vstupu odstraňte ochranný kryt (kratší z párových vstupů, viz obr. 2).
• Hrot infuzního setu pevně zapíchněte do aplikačního vstupu.
e: Podání
K jednorázovému použití.
Přípravek podávejte až po protržení těsnicích švů mezi komorami a promísení obsahu obou komor.
Částečně použité vaky dále neuchovávejte a veškeré příslušenství po použití zlikvidujte. Částečně použité vaky znovu nenapojujte.
Vaky nepropojujte do série, aby se předešlo vzduchové embolii způsobené možnou přítomností zbytkového vzduchu v primárním vaku.
Pro podávání parenterálních nutričních roztoků se doporučuje používat konečný filtr, je-li to možné.
Všechen nepoužitý přípravek nebo odpad má být zlikvidován v souladu s místními požadavky.
Obrázek 1
Stiskněte a rolujte vak přípravku CLINIMIX vertikálně

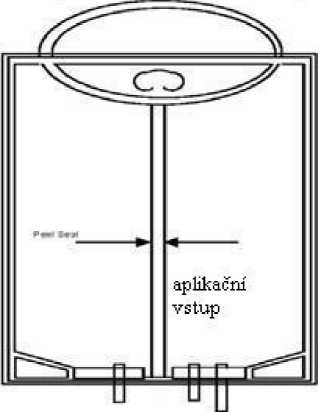
inj ekční vstup vstup pro převo dní s et
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter Healthcare Ltd.
Caxton Way, Thetford Velká Británie
8. REGISTRAČNÍ ČÍSLO
76/264/01-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
29.8.2001 / 11.3.2015
10. DATUM REVIZE TEXTU
11.3.2015
12