Azzalure 10 Speywood Jednotek/0,05 Ml
sp.zn.sukls10116/2015 a k sp.zn.sukls10112/2015
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
AZZALURE 10 Speywood jednotek/0,05ml, prášek pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Botulini toxinum typus A* 10 Speywood jednotek **/0,05ml naředěného roztoku Lahvička obsahuje 125 jednotek
*Komplex Clostridium botulinum toxin A s hemaglutininem
**Jedna Speywood jednotka (U) je definována jako střední letální peritoneální dávka u myší (LD50).
Speywood jednotky přípravku Azzalure jsou specifické pro tento přípravek a nejsou zaměnitelné s jinými přípravky obsahujícími botulotoxin.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro injekční roztok Bílý prášek
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Azzalure je indikován k dočasnému zlepšení vzhledu středně závažných až závažných glabelárních linií (svislé linie mezi obočím) viditelných při zamračení u dospělých pacientů do 65 let věku, jestliže závažnost těchto linií má důležitý psychologický dopad na pacienta.
4.2 Dávkování a způsob podání
Dávkování:
Jednotky botulotoxinu se liší v závislosti na léčivém přípravku. Speywood jednotky léčivého přípravku Azzalure jsou pro tento přípravek specifické a nejsou zaměnitelné s jednotkami v jiných přípravcích obsahujících botulotoxin.
Pediatrická populace:
Bezpečnost a účinnost přípravku Azzalure u jedinců mladších 18 let věku nebyla prokázána.
Způsob podání (viz bod 6.6):
Azzalure by měl být podán pouze lékařem, který má odpovídající kvalifikaci a zkušenosti s touto léčbou a který má požadované vybavení.
Po naředění se má Azzalure použít k léčbě jednoho pacienta během jednoho sezení.
Před injekcí je třeba přípravek rekonstituovat, instrukce pro rekonstituci jsou v bodě 6.6. Odstraňte veškerý make-up a desinfikujte kůži lokálním antiseptikem.
Intramuskulární injekce se vpíchne pravoúhle do kůže pomocí sterilní jehly o rozměru 29-30 gauge.
Instrukce pro podání:
Doporučená dávka je 50 Speywood jednotek (0,25 ml naředěného roztoku) přípravku Azzalure, rozdělená do 5 injekčních míst, 10 Speywood jednotek (0,05 ml naředěného roztoku) se podá intramuskulárně do každého z 5 míst: 2 injekce do každého musculus corrugator a jedna do musculus procerus poblíž nasofrontálního úhlu tak jak je ukázáno níže:
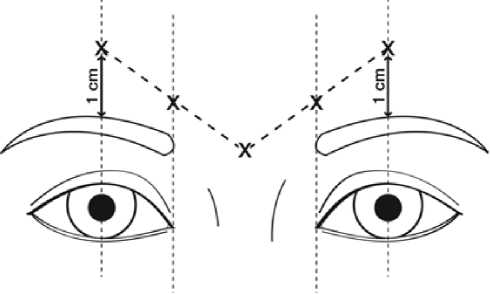
Anatomické orientační body mohou být snáze identifikovatelné, pokud se sledují a palpují při maximálním zamračení. Před podáním přípravku umístěte palec nebo ukazovák pevně pod hranu orbity, aby se předešlo extravazaci pod orbitální hranu. Během injekce by jehla měla mířit nahoru a mediálně. Aby se předešlo riziku ptózy, vyvarujte se podání blízko musculus levator palpebrae superioris, zvláště u pacientů s výraznějšími depresory obočí (depressor supercilii). Injekce do musculus corrugator musí být podány do střední části tohoto svalu, alespoň 1 cm nad hranu orbity.
Léčebný interval závisí na individuální odpovědi pacienta po vyhodnocení dosavadní léčby. V klinických studiích byl optimální účinek prokázán až do 4 měsíců po podání. Někteří pacienti odpovídali na léčbu ještě v 5. měsíci (viz bod 5.1). Léčebný interval nemá být kratší než 3 měsíce.
V případě selhání léčby nebo zmenšeného účinku po opakovaných podáních by se měla použít alternativní léčebná metoda. V případě selhání léčby po prvním léčebném sezení mohou být zváženy následující přístupy:
• Analýza příčin selhání, např. podání do nesprávných svalů, injekční technika, tvorba toxin-neutralizujících protilátek;
• Znovuvyhodnocení relevance léčby botulotoxinem A
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1;
- Při přítomnosti infekce v navrhovaných místech podání;
- Při myasthenia gravis, Lambert-Eatonově syndromu nebo amyotrofické laterální skleróze.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek Azzalure by měl být podáván s opatrností u pacientů s rizikem nebo s klinickými známkami poruchy neuromuskulárního přenosu. Tito pacienti mohou mít na agens, který je v přípravku Azzalure, zvýšenou citlivost, která může vést k nadměrné svalové slabosti.
Nežádoucí reakce pravděpodobně související s rozšířením účinku botulotoxinu do míst vzdálených od místa podání byly hlášeny velmi vzácně. Pacienti léčení terapeutickými dávkami mohou pocítit zvýšenou svalovou slabost.
Podání přípravku Azzalure se nedoporučuje u pacientů s anamnézou dysfagie a aspirace. Pacienti nebo pečovatelé by měli být poučeni, aby okamžitě vyhledali lékařskou pomoc v případě, že nastanou obtíže s polykáním, s řečí nebo s dýcháním.
Doporučená dávka a frekvence podání přípravku Azzalure nesmí být překročena.
Před podáním přípravku Azzalure je nezbytné vyšetřit anatomii obličeje pacienta. Obličejová asymetrie, ptóza, nadměrná dermatochalasie, zjizvení a jakékoli změny anatomie, jako výsledek předchozí chirurgické intervence, by měly být brány v úvahu.
Opatrnosti je třeba, jestliže se Azzalure používá v přítomnosti zánětu v navrženém místě podání nebo pokud cílový sval vykazuje nadměrnou slabost nebo atrofii.
Tak jako u všech intramuskulárních injekcí, léčba přípravkem Azzalure se nedoporučuje u pacientů, kteří mají prodlouženou dobu krvácivosti.
Injekce v častějším intervalu nebo ve vyšší dávce mohou zvýšit riziko tvorby protilátek proti botulotoxinu. Klinicky může tvorba neutralizujících protilátek snížit účinnost následné léčby.
Účinek podání různých botulotoxinů v průběhu léčby přípravkem Azzalure není znám a je nutné se mu vyhnout.
Přípravek Azzalure se může použít pouze pro léčbu jednoho pacienta během jednoho sezení. Zbytek nepoužitého přípravku se musí zlikvidovat tak, jak je uvedeno v bodě 6.6. Zvláštní opatrnosti je zapotřebí při přípravě a podání přípravku, jakož i při inaktivaci a likvidaci zbylého nepoužitého roztoku (viz bod 6.6).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Současná léčba přípravkem Azzalure a aminoglykosidy nebo jinými látkami interferujícími s neuromuskulárním přenosem (např. látky podobné kurare) by měla být podávána s opatrností, protože účinek botulotoxinu typu A může být umocněn.
Nebyly provedeny žádné interakční studie. Nebyly hlášeny žádné jiné interakce s klinickým významem.
4.6 Těhotenství a kojení Těhotenství
Přípravek Azzalure by se neměl používat v těhotenství, ledaže by to bylo nezbytně nutné. Neexistují žádné adekvátní údaje o podávání botulotoxinu typu A těhotným ženám.
Studie na zvířatech neprokázaly reprodukční toxicitu při vysokých dávkách (viz bod 5.3). Potenciální riziko pro člověka je neznámé.
Kojení
Není známo, zda je přípravek Azzalure vylučován do mateřského mléka. Použití přípravku Azzalure během kojení se nedoporučuje.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Existuje potenciální riziko vzniku místní svalové slabosti nebo poruch zraku spojených s použitím tohoto přípravku, jež může dočasně narušit schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Více než 1900 pacientů bylo vystaveno působení přípravku Azzalure v různých klinických studiích.
V pivotních klinických studiích bylo přes 1500 pacientů se středně závažnými až závažnými glabelárními liniemi léčeno doporučenou dávkou 50 jednotek v dvojitě slepých, placebem kontrolovaných a v dlouhodobých otevřených studiích.
V pivotních dvojitě slepých, placebem kontrolovaných studiích s jednou dávkou měly
22,3 % pacientů léčených doporučenou dávkou Azzalure (50 U) a 16,6 % pacientů léčených placebem reakci, která souvisela s léčbou, způsobem podání nebo s obojím.
V dlouhodobé otevřené studii fáze III, ve které podstoupili pacienti opakované cykly podání, mělo 26 % pacientů alespoň jednu související reakci po prvním podání. Incidence reakcí souvisejících s léčbou/způsobem podání klesala během opakovaných cyklů.
Nejčastěji se objevující související reakcí je bolest hlavy a reakce v místě vpichu. Obecně vzato se reakce související s léčbou/způsobem podání objevují během prvního týdne po aplikaci a jsou přechodné. Většina těchto hlášených reakcí byla mírná až středně závažná a byla reverzibilní.
Frekvence nežádoucích účinků je klasifikována následovně:
Velmi časté (>1/10); časté (>1/100 až < 1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000); není známo (nemůže být určena z dostupných dat).
|
Poruchy nervového systému |
Velmi časté Bolest hlavy Časté Lícní paréza (převážně paréza obočí) Méně časté Závratě |
|
Poruchy oka |
Časté Astenopie, ptóza, edém očního víčka, zvýšená lakrimace, suché oko, svalové záškuby (záškuby svalů okolo očí) Méně časté Zrakové poruchy, rozmazané vidění, diplopie, poruchy pohybu očí |
|
Poruchy kůže a podkožní tkáně |
Vzácné Kopřivka |
|
Celkové poruchy a reakce v místě |
Velmi časté |
|
aplikace |
Reakce v místě vpichu (např. zarudnutí, otok, podráždění, vyrážka, svědění, parestezie, bolest, nepříjemný pocit, bodání a modřiny) |
|
Poruchy imunitního systému |
Méně časté Přecitlivělost |
Velmi vzácně byly hlášeny nežádoucí účinky z rozšíření účinku toxinu z místa injekce do vzdálených míst (nadměrná svalová slabost, dysfagie, aspirační pneumonie, jež může být v některých případech fatální) (viz bod 4.4).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu: Státní ústav pro kontrolu léčiv, Šrobárova 48, 100 41 Praha 10. Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek.
4.9 Předávkování
Během klinických studií se neobjevily případy předávkování.
Po nadměrných dávkách botulotoxinu lze očekávat rozvoj neuromuskulární slabosti s řadou různých příznaků. Způsobí-li nadměrné dávky paralýzu respiračních svalů, může být zapotřebí respirační podpora. V případě předávkování by měl být pacient lékařsky sledován z hlediska příznaků nadměrné svalové slabosti nebo svalové paralýzy.
V případě potřeby by měla být zahájena symptomatická léčba.
Příznaky předávkování se nemusí objevit bezprostředně po podání.
Hospitalizace by měla být zvážena u pacientů se symptomy otravy botulotoxinem A (tj. kombinace svalové slabosti, ptózy, diplopie, poruchy polykání či řeči nebo paréza respiračních svalů).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: jiná periferně působící myorelaxancia,
ATC kód: M03AX01
Hlavním farmakodynamickým účinkem toxinu Clostridium botulinum typu A je chemická denervace léčeného svalu, jež má za následek měřitelný pokles sdruženého akčního potenciálu svalu, způsobující lokalizované snížení nebo paralýzu svalové aktivity.
Klinické údaje
Během klinického vývoje přípravku Azzalure bylo zařazeno do různých klinických studií více než 2 600 pacientů.
V klinických studiích bylo léčeno 1 907 pacientů se středně závažnými nebo závažnými glabelárními liniemi doporučenou dávkou 50 Speywood jednotek. 305 z nich bylo léčeno dávkou 50 U ve dvou pivotních, dvojitě slepých, placebem kontrolovaných studiích fáze III a 1 200 bylo léčeno dávkou 50 U v dlouhodobé otevřené studii fáze III s opakovanou dávkou. Zbývající pacienti byli léčeni v podpůrných studiích a ve studiích zkoumajících dávku.
Střední doba nástupu odpovědi byla 2 - 3 dny po léčbě s maximálním účinkem pozorovaným v den 30. V obou pivotních, placebem kontrolovaných studiích fáze III redukoval Azzalure signifikantně závažnost glabelárních linií až po dobu 4 měsíců.
V jedné ze dvou pivotních studií byl účinek signifikantní ještě po 5 měsících.
Třicet dní po injekci ukázalo vyhodnocení zkoušejících, že 90 % pacientů (273/305) odpovídalo na léčbu (neměli žádné nebo mírné glabelární linie při maximálním zamračení) v porovnání se 3 % pacientů (4/153) léčených placebem. Pět měsíců po injekci stále odpovídalo na léčbu 17 % pacientů (32/190) léčených Azzalure ve srovnání s 1 % pacientů (1/92) léčených placebem v dotyčné studii. Vlastní hodnocení pacientem po 30 dnech při maximálním zamračení dalo podíl odpovědi 82 % (251/305) pro léčené přípravkem Azzalure a 6 % (9/153) pro léčené placebem. Podíl pacientů, kteří dosáhli dvoustupňového zlepšení podle hodnocení zkoušejícího při maximálním zamračení, činil 77 % (79/103) v jedné pivotní studii fáze III, kde se toto hodnotilo.
Podskupina 177 pacientů měla středně závažné nebo závažné glabelární linie v klidu před léčbou. Hodnocení této populace provedené zkoušejícím 30 dní po léčbě ukázalo, že ve skupině pacientů léčených přípravkem Azzalure odpovídalo na léčbu 71 % pacientů (125/177) oproti 10 % pacientů (8/78) léčených placebem.
Dlouhodobá otevřená studie s opakovanou dávkou ukázala střední nástup odpovědi na léčbu 3 dny a odpověď se udržela po dobu opakovaných cyklů dávky. Podíl odpovědi na léčbu podle stanovení zkoušejícího v den 30 při maximálním zamračení se udržel po dobu opakovaných cyklů (pohyboval se mezi 80 % a 91 % během 5 cyklů). Podíl odpovídajících na léčbu v klidu během cyklů s opakovanou dávkou byl také konzistentní se studiemi s jednou dávkou; 56 % až 74 % pacientů léčených přípravkem Azzalure bylo považováno zkoušejícím za odpovídající v den 30 po léčbě.
Žádný z klinických cílů nezahrnoval objektivní hodnocení psychologického dopadu.
5.2 Farmakokinetické vlastnosti
Neočekává se, že by byl Azzalure po i.m. podání v doporučené dávce přítomen v periferní krvi v měřitelných hladinách. Z tohoto důvodu nebyly farmakokinetické studie s přípravkem Azzalure provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
V reprodukčních studiích na potkanech a králících bylo pozorováno několik případů závažné maternální toxicity spojené se ztrátou implantace při vysokých dávkách.
V dávkách odpovídajících 60 až 100-násobku doporučené dávky pro člověka (50 U) u králíků a potkanů nebyla pozorována žádná embryonální toxicita. Žádná teratogenní aktivita nebyla u těchto druhů pozorována. U potkanů byla fertilita samců a samic snížena v důsledku sníženého páření v důsledku svalové paralýzy při vysokých dávkách. Ve studiích chronické toxicity provedených na potkanech nebyly žádné náznaky systémové toxicity v dávkách odpovídajících 75-násobku doporučené dávky (50 U)
pro člověka, rozdělené stejnou měrou mezi pravý a levý m. gluteus maximus.
Studie akutní toxicity, chronické toxicity a lokální tolerance v místě podání neukázaly neobvyklé nežádoucí lokální nebo systémové účinky v klinicky relevantních dávkách.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Roztok lidského albuminu 200 g/l, monohydrát laktosy.
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
2 roky.
Naředěný roztok:
Chemická a fyzikální stabilita přípravku po otevření byla prokázána po dobu 24 hodin při teplotě 2 °C - 8 °C. Z mikrobiologického hlediska, pokud způsob otevření nevyloučí riziko mikrobiální kontaminace, má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Podmínky uchovávání naředěného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a velikost balení
125 Speywood jednotek prášku je v lahvičce (sklo typ I) se zátkou (halobutyl) a uzávěrem (aluminium).
Velikost balení: 1 nebo 2 lahvičky.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro použití, manipulaci a likvidaci by měly být striktně dodrženy.
Rekonstituce musí být provedena v souladu s pravidly správné praxe, zvláště se zřetelem na asepsi.
Azzalure musí být rekonstituován v injekčním roztoku chloridu sodného o koncentraci (0.9%).
Aby se získal čirý naředěný roztok o koncentraci 10 U/0,05 ml, je třeba natáhnout do injekční stříkačky požadované množství injekčního roztoku chloridu sodného o koncentraci 9 mg/ml (0,9%) dle tabulky ředění uvedené níže;
|
Množství rozpouštědla přidaného (0,9% roztok chloridu sodného) k lahvičce o 125 U |
Výsledná dávka (počet jednotek na 0,05 ml) |
|
0,63 ml |
10 U |
Přesné odměření 0,63 ml lze dosáhnout použitím 1ml stříkačky opatřené stupnicí s dělením po 0,1 ml a 0,01 ml.
DOPORUČENÍ PRO LIKVIDACI KONTAMINOVANÉHO MATERIÁLU
Bezprostředně po použití a před likvidací by měl být nepoužitý naředěný přípravek Azzalure (v lahvičce nebo stříkačce) inaktivován 2 ml roztoku chlornanu sodného v ředění 0,55 nebo 1 % (chloramin, chlorové vápno).
Použité lahvičky, stříkačky a materiál se nesmí vyprazdňovat a musí být odhozeny do patřičného kontejneru a zlikvidovány v souladu s místnímu požadavky.
DOPORUČENÍ PRO PŘÍPAD JAKÉKOLI UDÁLOSTI BĚHEM MANIPULACE S BOTULOTOXINEM
• Každý vysypaný/vylitý materiál se musí utřít: buď se použije absorpční materiál impregnovaný roztokem chlornanu sodného (chloramin, chlorové vápno)
v případě prášku, nebo suchý absorpční materiál v případě rekonstituovaného přípravku.
• Kontaminovaný povrch by se měl očistit s použitím absorpčního materiálu impregnovaného roztokem chlornanu sodného (chloramin, chlorové vápno) a poté vysušit.
• V případě rozbité lahvičky postupujte, jak je zmíněno výše, opatrným sebráním kousků rozbitého skla a setřením produktu tak, aby nedošlo k žádnému poranění kůže.
• Pokud dojde ke kontaktu přípravku s kůží, omyjte postiženou oblast roztokem chlornanu sodného (chloramin, chlorové vápno) a potom hojně opláchněte vodou.
• Pokud přijde přípravek do kontaktu s očima, vypláchněte je důkladně velkým množstvím vody nebo očním roztokem k výplachu očí.
• Přijde-li přípravek do kontaktu se zraněním, pořezanou nebo porušenou kůží, důkladně vypláchněte postižené místo velkým množstvím vody a učiňte vhodná lékařská opatření podle množství vstříknuté dávky.
Tyto instrukce pro použití, manipulaci a likvidaci by měly být striktně dodrženy.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Ipsen Biopharm Limited, Ash Road, Wrexham Industrial Estate, Wrexham, LL13 9UF, Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
63/120/10-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
10.2.2010/22.5.2014
10. DATUM REVIZE TEXTU
29.10.2015
10