Anbinex
sp.zn. sukls43174/2010
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Anbinex
50 IU/ml, prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Léčivá látka : Antithrombinum III
Anbinex se dodává jako lyofilizovaný prášek, který obsahuje nominálně antithrombinum získaný z lidské plazmy 500 IU nebo 1000 IU v jedné injekční lahvičce.
Přípravek obsahuje přibližně 500 IU/10 ml nebo 1000 IU/20 ml antithrombinum získaný z lidské plasmy, když je rekonstituován s 10 ml nebo 20 ml sterilní vody na injekci.
Účinnost (IU) se určuje pomocí chromogenního stanovení podle Evropského lékopisu. Specifická aktivita Anbinex je alespoň 5 IU/mg proteinu.
Anbinex 500 IU obsahuje 1,45 mmol (33,35 mg) sodíku na 10 ml (injekční stříkačka).
Anbinex 1000 IU obsahuje 2,90 mmol (66,7 mg) sodíku na 20 ml (injekční stříkačka).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok
Injekční lahvička obsahuje prášek pro injekční roztok: bílá, hygroskopická pevná látka nebo prášek.
Injekční stříkačka obsahuje rozpouštědlo: bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
- Vrozený nedostatek antitrombinu:
a) Profylaxe hluboké žilní trombózy a tromboembolismu v klinicky rizikové situaci (především při operaci nebo v období okolo porodu), v kombinaci s heparinem, pokud je indikován
b) Prevence rozvoje hluboké žilní trombózy nebo tromboembolismu v kombinaci s heparinem, pokud je indikován
- Získaný nedostatek antitrombinu
4.2. Dávkování a způsob podání
Léčbu je nutné zahájit pod dohledem lékaře, který má zkušenosti s léčbou pacientů s deficitem
antitrombinu.
Při léčbě vrozeného deficitu antitrombinu je nutné dávkování a trvání léčby individuálně upravit pro jednotlivé pacienty. Při tom je nutné brát v úvahu rodinnou anamnézu tromboembolických příhod, současné faktory klinických rizik a laboratorní hodnocení.
Dávkování a doba substituční terapie získaného deficitu závisí na hladině antitrombinu v plazmě, na rychlosti odbourávání, na výchozím onemocnění a na vážnosti klinického stavu. Množství, jaké se má podávat a frekvence aplikace by vždy měla být založena na klinické účinnosti a na laboratorním posouzení každého případu individuálně.
Počet podávaných jednotek antitrombinu je vyjádřen v mezinárodních jednotkách (IU), což odpovídá současnému standardu Světové zdravotnické organizace pro antitrombin. Aktivita antitrombinu v plazmě je vyjádřena buď v procentech (vzhledem k normální lidské plazmě), nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro obsah antitrombinu v plazmě).
Jedna mezinárodní jednotka (IU) aktivity antitrombinu je ekvivalentní tomuto množství antitrombinu v jednom ml normální lidské plazmy. Výpočet požadované dávky antitrombinu je založen na empirickém zjištění, že jedna mezinárodní jednotka (IU) antitrombinu na 1 kg tělesné hmotnosti zvýší antitrombinovou aktivitu plazmy přibližně o 1,1 % až 1,6 %.
Počáteční dávka se určí pomocí následujícího vzorce:
Požadovaný počet jednotek = tělesná hmotnost (kg) x (100 - bazální antitrombinová aktivita [%]) x 0,8
Počáteční dávka, pokud jde o aktivitu antitrombinu, závisí na klinické situaci. Při potvrzení vhodnosti substituce antitrombinu, by dávkování mělo být dostatečné k dosažení cílové aktivity antitrombinu a k udržení její účinné úrovně. Dávkování je nutno upravit na základě laboratorních měření aktivity antitrombinu, která by se měla provádět alespoň dvakrát denně dokud nebude pacient ve stabilizovaném stavu. Potom postačí jednou denně, nejlépe těsně před další infuzí. O úpravě dávkování by se mělo uvažovat jak na základě příznaků zvýšené přeměny antitrombinu, zjištěných na základě laboratorních testů, tak i na základě klinického průběhu. Úroveň hladiny antitrombinu musí být udržována vyšší než 80% po dobu trvání léčby, pokud klinické údaje neindikují jinou účinnou hladinu.
Počáteční dávka u vrozeného deficitu je 30 - 50 IU/kg.
Dále by se mělo dávkování a frekvence podávání stejně jako trvání terapie přizpůsobit biologickým datům a klinické situaci.
Pediatrická populace
Anbinex se nedoporučuje podávat dětem do 6 let vzhledem k nedostatku údajů o bezpečnosti a účinnosti.
Způsob podání
Nařeďte přípravek tak, jak je popsáno v bodu 6.6. Přípravek se podává intravenózně.
Rychlost podání nesmí překročit 0,08 ml/kg/min
4.3. Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku.
Zvláštní upozornění o pomocných látkách jsou uvedeny v bodě 4.4.
4.4. Zvláštní upozornění a opatření pro použití
Mohou nastat projevy přecitlivělosti alergického typu, podobně jako při intravenózní aplikaci jakéhokoli bílkovinného produktu. Pacienti musí být pozorně sledováni a pečlivě pozorováni, aby byly zjištěny jakékoli symptomy, projevující se v období podávání infuze. Pacienti by měli být informováni o počátečních příznacích reakce přecitlivělosti včetně kopřivky postihující celé tělo, napětí na hrudi, dušnosti, nízkého krevního tlaku a anafylaxe. Jestliže se tyto symptomy projeví, měli by se pacienti obrátit na svého lékaře.
V případě šoku by mělo být poskytnuto standardní lékařské ošetření.
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a účinné výrobní kroky, při nichž jsou inaktivovány nebo odstraněny viry. Přes všechna tato opatření při přípravě léků vyráběných z lidské krve nebo plazmy nelze možnost přenosu infekčních agens zcela vyloučit. To platí i pro jakékoli neznámé nebo nově vznikající viry a jiné patogeny.
Provedená opatření jsou považována za efektivní pro viry s obalem, jakým jsou HIV, HBV a HCV, a pro viry HAV bez obalu. Přijatá opatření mohou být účinná jen omezeně v případě virů bez obalu jakým je parvovirus B19. Infekce způsobená parvovirem B19 může být závážná pro těhotné ženy (fetální infekce) a pro pacienty s immunodeficitem, nebo se zvýšenou erytropoézou (např. hemolytická anemie).
Důrazně se doporučuje, aby se vždy, když bude pacientovi Anbinex podán, zaznamenal název a číslo šarže výrobku, aby bylo možno vysledovat souvislost mezi pacientem a výrobní šarží.
Doporučuje se očkování proti hepatitidě A a B u pacientů léčených antitrombinem z lidské plazmy.
Klinické a biologické sledování situace při současném podání antitrombinu s heparinem:
- aby bylo možno stanovit dávkování heparinu a zabránit nadměrnému snížení srážlivosti, měly by se pravidelně, v krátkých intervalech, provádět kontroly úrovně koagulace (APPT a vhodná anti-FXa aktivita), především v prvních minutách / hodinách po aplikaci antitrombinu.
- každý den je třeba měřit hladinu antitrombinu, aby mohla být stanovena individuální dávka, protože existuje riziko snížení hladiny antitrombinu v důsledku déle trvající léčby nefrakcionovaným heparinem.
Anbinex 500 IU obsahuje 1,45 mmol (33,35 mg) sodíku na 10 ml (injekční stříkačka).
Anbinex 1000 IU obsahuje 2,90 mmol (66,7 mg) sodíku na 20 ml (injekční stříkačka).
Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Heparin: substituce antitrombinu v průběhu podávání heparinu v terapeutických dávkách zvyšuje riziko krvácení. Účinek antitrombinu silně roste vlivem heparinu. Poločas rozpadu antitrombinu se může silně zkrátit při současné aplikaci heparinu v důsledku zrychlené přeměny antitrombinu. Proto je nutno klinicky i biologicky monitorovat současné podávání heparinu a antitrombinu u těch pacientů, kteří mají zvýšené riziko krvácení.
4.6. Těhotenství a kojení
Zkušenosti týkající se bezpečnosti produktů obsahujících lidský antitrombin pro těhotné ženy jsou omezené.
Anbinex by se měl podávat těhotným a kojícím ženám s deficitem antitrombinu pouze v jasně indikovaných případech, s přihlédnutím k tomu, že těhotenství přináší zvýšené riziko tromboembolických událostí u těchto pacientek.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Anbinex nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8. Nežádoucí účinky
Při podávání přípravku byly vzácně pozorované příznaky přecitlivělosti organismu nebo alergických reakcí (angioedém, bolest v místě zavedení infuze, zimnice, zčervenání, kopřivka, bolest hlavy, vyrážka, hypotenze, letargie, nausea, neklid, tachykardie, tlak na hrudi, mravenčení, zvracení, těžké dýchání), které se mohou v některých případech rozvinout do podoby těžké anafylaxe (včetně šoku).
Vzácně se vyskytla zvýšená teplota.
Informace o virové bezpečnosti viz bod 4.4.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9. Předávkování
Nebyl hlášen žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI 5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antikoagulancia, antitrombotica: ATC kód: B01AB02.
Antitrombin, glykoprotein s molekulovou hmotností 58 kD, který obsahuje 432 aminokyselin, patří do skupiny serpinů (inhibitorů serin proteázy). Je jedním z nejdůležitějších přirozených inhibitorů koagulace krve. Nejsilněji inhibované faktory jsou trombin a faktor Xa, ale i faktory kontaktní aktivace, faktory vnitřního systému a komplex faktor VIIa/tkáňový faktor. Aktivita antitrombinu je výrazně zvýšena heparinem a antikoagulační účinky heparinu jsou závislé na přítomnosti antitrombinu.
Antitrombin obsahuje dvě funkčně důležité domény. První obsahuje reakční centrum a poskytuje místo štěpení pro proteinázy, jako je trombin, což je předpoklad vytvoření stabilního komplexu proteináza-inhibitor. Druhé je doména vážící glykosaminoglykany zodpovídající za interakci s heparinem a příbuznými látkami, která zrychluje inhibici trombinu. Komplexy inhibitor-koagulační enzym jsou odbourávány retikulo-endoteliálním systémem.
Aktivita antitrombinu u dospělých je 80 - 120% a úroveň u novorozenců činí asi 40 - 60% .
5.2. Farmakokinetické vlastnosti
Farmakokinetické studie antitrombinu ukázaly biologický poločas přibližně 3 dny. Při současné léčbě heparinem může být snížen na přibližně 1,5 dne. Ke snížení na hodiny může dojít v případě jeho velké spotřeby.
Výsledky získané nezávislým typem analýzy při klinických zkouškách přípravku Anbinex u pacientů s vrozeným deficitem antitrombinu byly:
- Recovery 1,3 ± 0,2 v rozmezí 1,1 až 1,6% - stanoveno funkční analýzou
- Plocha pod křivkou (AUC) = 66.461 ± 15.445 IU h/l.
- Konečný poločas = 98,1 ± 45,0 h based - stanoveno funkční analýzou
- Střední pobytový čas (MRT) = 121,7 ± 52,1 h.
- Clearance = 0,931 ± 0,214 ml/h/kg.
5.3 Předklinické údaje vztahující se k bezpečnosti
Antitrombin je normální složkou lidské plazmy.
Testování toxicity jedné dávky je málo spolehlivé a neumožňuje odhadnout toxickou nebo smrtelnou dávku.
Opakované testování toxicity na zvířatech je neproveditelné vzhledem k tvorbě protilátek.
Na zvířecích modelech nebyly popsány žádné příznaky akutní toxicity.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
Mannitol Chlorid sodný Dihydrát natrium-citrátu Voda na injekci (rozpouštědlo)
6.2. Inkompatibility
Tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
Vzhledem k tomu, že může dojít k selhání léčby v důsledku adsorbce liského antitrombinu na vnitřní povrchy aplikačního zařízení, smí být použito pouze přiložené příslušenství pro rekonstituci a aplikaci.
6.3. Doba použitelnosti
Doba použitelnosti neotevřeného přípravku jsou 3 roky.
Chemická a fyzikální stabilita přípravku po rekonstituci byla prokázána po dobu 12 hod při teplotě 25 °C. Z mikrobiologického hlediska by měl být přípravek použít okamžitě. Pokud není použit okamžitě, je doba a podmínky uchovávání přípravku po rekonstituci před použitím na zodpovědnosti uživatele a neměla by normálně být delší než 24 hodin při teplotě 2 - 8 °C, pokud nebyla rekonstituce provedena za kontrolovaných a validovaných aseptických podmínek.
6.4. Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Chraňte před mrazem.
6.5. Druh obalu a velikost balení
Anbinex, 500 IU prášek a rozpouštědlo pro injekční roztok (10 ml vody na injekci)
Anbinex, 1000 IU prášek a rozpouštědlo pro injekční roztok (20 ml vody na injekci)
Anbinex je dodáván v injekční lahvičce z bezbarvého skla (třídy II) obsahující 500 IU nebo 1000 IU lidského anthitrombinum III (prášek), lahvička je uzavřená zátkou z bromobutylové pryže. Rozpouštědlo je dodáváno v předplněné injekční stříkačce (sklo třídy I) se zátkou z bromobutylové pryže, obsahující 10 ml (500 IU) nebo 20 ml (1000 IU) vody na injekci .
Příslušenství dodávané s přípravkem Anbinex sloužící k rekonstituci: adaptér lahvičky a mikrofiltr.
Jedno balení obsahuje: 1 lahvičku s lyofilizátem, 1 předplněnou injekční stříkačku s vodou na injekci a příslušenstvím pro rekonstituci.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
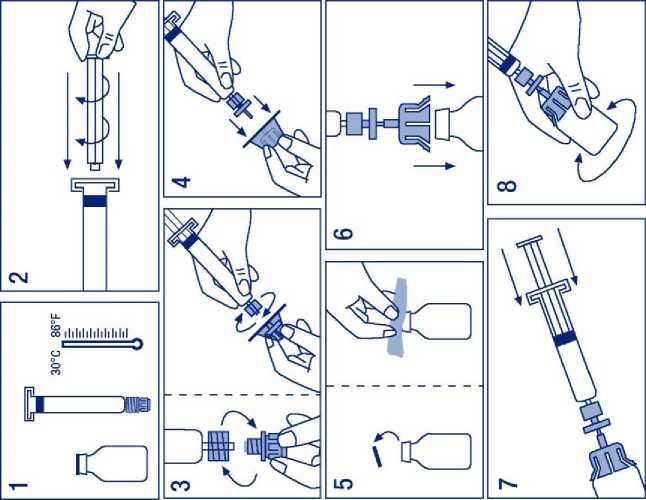
Příprava roztoku:
1. Injekční lahvičku s práškem a injekční stříkačku zahřejte maximálně na 30 °C.
2. Na injekční stříkačku s rozpouštědlem nasaďte píst.
3. Z balení vyjměte filtr. Z konce injekční stříkačky sejměte krytku a nasaďte filtr.
4. Z balení vyjměte adaptér injekční lahvičky a nasaďte ho na filtr se stříkačkou.
5. Z injekční lahvičky s práškem sejměte klobouček a zátku injekční lahvičky otřete přiloženým antiseptickým tampónem.
6. Zátku injekční lahvičky propíchněte jehlou adaptéru.
7. Roztok z injekční stříkačky přeneste kompletně do lahvičky s práškem.
8. Injekční lahvičkou jemně míchejte, až se přípravek dokonale rozpustí.
9. Oddělte injekční stříkačku/filtr od nádobky/adaptéru. Do inekční stříkačky nasajte přibližně stejné množství vzduchu jako je objem roztoku. Injekční stříkačku/filtr znovu spojte s nádobkou/adaptérem a vytlačte vzduch.
10. Obraťte injekční lahvičku a roztok natáhněte do injekční stříkačky.
11. Připravte místo vpichu. Injekční stříkačku oddělte od filtru s adaptérem a přípravek aplikujte. Rychlost intravenózní aplikace nesmí překročit 0,08 ml/kg/min.
Rekonstituovaný roztok by se měl před aplikací vizuálně zkontrolovat, zda neobsahuje částice a zda nejsou patrné změny barvy. Roztok by měl být čirý, nebo nepatrně opalizující. Nepoužívejte roztoky, které jsou zakalené, nebo mají usazeniny.
Nespotřebovaný přípravek se nesmí uchovávat pro pozdější použití.
Aplikační soupravy opakovaně nepoužívejte.
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Instituto Grifols, S.A.
Can Guasch, 2 - Parets del Valles 08150 Barcelona- Španělsko
8. REGISTRAČNÍ ČÍSLO
75/221/90-C
9. DATUM PRVNÍ REGISTRACE/DATUM PRODLOUŽENÍ REGISTRACE
10.09.1990 / 9.7.2014
10. DATUM REVIZE TEXTU
9.7.2014
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Státního ústavu pro kontrolu léčiv (SUKL) - www.sukl.cz
-OCDC
cr>
M
IDO-