Zytiga 250 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
ZYTIGA 250 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje abirateroni acetas 250 mg.
Pomocné látky se známým účinkem
Jedna tableta obsahuje 189 mg laktosy a 6,8 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Bílé až téměř bílé oválné tablety 15,9 x 9,5 mm označené na jedné straně AA250.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
ZYTIGA je indikována spolu s prednisonem nebo prednisolonem:
• k léčbě metastazujícího karcinomu prostaty rezistentního na kastraci u dospělých mužů, kteří jsou asymptomatičtí nebo mírně symptomatičtí po selhání androgenní deprivační léčby a
u nichž dosud nebyla chemoterapie klinicky indikována (viz bod 5.1);
• k léčbě metastazujícího karcinomu prostaty rezistentního na kastraci u dospělých mužů, jejichž onemocnění progredovalo při chemoterapeutickém režimu založeném na docetaxelu nebo po něm.
4.2 Dávkování a způsob podání
Tento přípravek je předepisován lékařem se specializací v příslušném oboru.
Dávkování
Doporučená dávka je 1 000 mg (čtyři 250mg tablety) jako jednorázová denní dávka, která se nesmí užít s jídlem (viz „Způsob podání“ níže). Užívání tablet s jídlem zvyšuje systémovou expozici abirateronu (viz body 4.5 a 5.2).
ZYTIGA se užívá s nízkou dávkou prednisonu nebo prednisolonu. Doporučená dávka prednisonu nebo prednisolonu je 10 mg denně.
U pacientů bez chirurgické kastrace musí léková kastrace pomocí analog hormonu uvolňujícího luteinizační hormon (LHRH) během léčby pokračovat.
Před zahájením léčby, každé dva týdny během prvních tří měsíců léčby a dále jednou měsíčně je nutno měřit hodnoty transamináz v séru. Jednou měsíčně je nutno kontrolovat krevní tlak, hladinu draslíku v séru a retenci tekutin. Pacienty se závažným rizikem městnavého srdečního selhání je však nutno během prvních třech měsíců léčby monitorovat každé 2 týdny a dále měsíčně (viz bod 4.4).
U pacientů s preexistující hypokalemií nebo u pacientů, u kterých se během léčby přípravkem ZYTIGA vyvine hypokalemie, je nutno zvážit udržování hladin draslíku pacienta > 4,0 mmol/l.
U pacientů, u kterých se vyvinou toxicity stupně > 3, včetně hypertenze, hypokalemie, otoku a jiných nemineralokortikoidních toxicit, je nutno léčbu ukončit a zahájit potřebná léčebná opatření. Léčbu
přípravkem ZYTIGA nelze obnovit, dokud se příznaky toxicity nezlepší na stupeň 1 nebo k počátečním hodnotám.
V případě vynechání dávky buď přípravku ZYTIGA nebo prednisonu či prednisolonu se v léčbě pokračuje další den obvyklou denní dávkou.
Hepatotoxicita
U pacientů, u nichž se během léčby vyvine hepatotoxicita [zvýšení alaninaminotransferázy (ALT) nebo aspartátaminotransferázy (AST) nad 5násobek horní hranice normálu (ULN)], je nutno okamžitě přerušit léčbu (viz bod 4.4). Obnovení léčby po návratu funkčních jaterních testů k výchozímu stavu u pacienta může být provedeno sníženou dávkou 500 mg (dvě tablety) jednou denně. U pacientů, u nichž byla léčba znovu zahájena, je nutno monitorovat transaminázy alespoň jednou za dva týdny po dobu tří měsíců a dále jednou za měsíc. Objeví-li se hepatotoxicita i u snížené dávky 500 mg denně, má být léčba ukončena.
Objeví-li se závažná hepatotoxicita (ALT nebo AST 20násobná oproti ULN) kdykoli během léčby, je nutno léčbu ukončit a u těchto pacientů nemá být léčba znovu zahájena.
Porucha funkce jater
U pacientů s již existující mírnou poruchou funkce jater, Child-Pugh třídy A, není nutná úprava dávky.
Ukázalo se, že středně závažná porucha funkce jater (Child-Pugh třídy B) zvyšuje systémovou expozici abirateronu po jednorázovém perorálním podání 1 000 mg abirateron-acetátu přibližně čtyřikrát (viz bod 5.2). Neexistují údaje o klinické bezpečnosti a účinnosti opakovaných dávek abirateron-acetátu po podání pacientům se středně závažnou nebo závažnou poruchou funkce jater (Child-Pugh třídy B nebo C). Nelze předpokládat žádnou úpravu dávky. U pacientů se středně závažnou poruchou funkce jater je nutno užití přípravku ZYTIGA důkladně posoudit, přínos by měl jasně převažovat možné riziko (viz body 4.2 a 5.2). Pacientům se závažnou poruchou funkce jater se přípravek ZYTIGA nesmí podávat (viz body 4.3, 4.4 a 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin není nutná úprava dávky (viz bod 5.2). U pacientů s karcinomem prostaty a závažnou poruchou funkce ledvin však nejsou žádné klinické zkušenosti. U těchto pacientů je nutná opatrnost (viz bod 4.4).
Pediatrická populace
Neexistuje žádné relevantní použití přípravku ZYTIGA u pediatrické populace.
Způsob podání
Přípravke ZYTIGA je určen k perorálnímu podání.
ZYTIGA se užívá alespoň dvě hodiny po jídle a po užití tablet se nesmí nic jíst po dobu alespoň jedné hodiny. Tablety se polykají celé a zapíjejí se vodou.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
- Těhotné ženy nebo ženy, které mohou otěhotnět (viz bod 4.6).
- Závažná porucha funkce jater [Child-Pugh třídy C (viz body 4.2, 4.4 a 5.2)].
4.4 Zvláštní upozornění a opatření pro použití
Hypertenze, hypokalemie, retence tekutin a srdeční selhání způsobené vzestupem mineralokortikoidů ZYTIGA může způsobit hypertenzi, hypokalemii a retenci tekutin (viz bod 4.8) jako důsledek zvýšených hladin mineralokortikoidů, které se objeví na základě inhibice CYP17 (viz bod 5.1). Při současném podání s kortikoidem dojde ke snížení vylučování adrenokortikotropního hormonu (ACTH), což má za následek snížení incidence a závažnosti těchto nežádoucích účinků. Opatrnost je nutná při léčbě pacientů, u nichž může zvýšení krevního tlaku, hypokalemie (např. u pacientů
užívajících srdeční glykosidy) nebo retence tekutin (např. u pacientů se srdečním selháním), závažná nebo nestabilní angina pectoris, recentní infarkt myokardu nebo komorová arytmie negativně ovlivnit jejich základní onemocnění, a u pacientů se závažnou poruchou funkce ledvin.
Přípravek ZYTIGA je nutno užívat s opatrností u pacientů s anamnézou kardiovaskulárního onemocnění. Hodnocení fáze 3 u přípravku ZYTIGA vyloučila pacienty s nekontrolovanou hypertenzí, klinicky významným onemocněním srdce, které se klinicky manifestovalo jako infarkt myokardu nebo arteriální trombotické příhody v posledních 6 měsících, pacienty se závažnou nebo nestabilní anginou nebo selháváním srdce třídy III nebo IV (hodnocení 301) podle New York Heart Association (NYHA) nebo srdečním selháváním třídy II až IV (hodnocení 302) nebo se snížením ejekční frakce pod 50 %. Z hodnocení 302 byli vyloučeni pacienti s fibrilací síní nebo dalšími srdečními arytmiemi vyžadujícími podávání léčiv. Bezpečnost přípravku ZYTIGA u pacientů s ejekční frakcí levé komory (LVEF) < 50 % nebo třídy III nebo IV NYHA srdečního selhání (v hodnocení 301) nebo třídy II až IV srdečního selhání (v hodnocení 302) nebyla stanovena (viz body 4.8 a 5.1).
Před zahájením léčby pacientů s významným rizikem městnavého srdečního selhání (např. s anamnézou srdečního selhání, nekontrolované hypertenze nebo srdečních příhod, jako například ischemická choroba srdeční) je nutno zvážit zhodnocení srdečních funkcí (např. echokardiogram).
Před zahájením léčby přípravkem ZYTIGA je nutno léčit srdeční selhání a optimalizovat funkci srdce. Je nutno upravit a kontrolovat hypertenzi, hypokalemii a retenci tekutin. Během léčby je nutno monitorovat krevní tlak, sérové hladiny draslíku, retenci tekutin (přírůstek tělesné hmotnosti, periferní otoky) a další známky a příznaky městnavého srdečního selhání každé 2 týdny během 3 měsíců a dále měsíčně, a abnormality korigovat. U pacientů s hypokalemií bylo pozorováno prodloužení QT intervalu v souvislosti s léčbou přípravkem ZYTIGA. Posouzení funkce srdce se provede, jak je klinicky indikováno, zahájí se vhodná léčba, a je-li přítomen klinicky významný pokles funkce srdce (viz bod 4.2), zváží se ukončení léčby tímto přípravkem.
Hepatotoxicita a porucha funkce jater
V kontrolovaných klinických hodnoceních se vyskytla významná zvýšení hodnot jaterních enzymů, což vedlo k ukončení léčby nebo změnám dávkování (viz bod 4.8). Před zahájením léčby, každé dva týdny během prvních tří měsíců léčby a dále jednou měsíčně je nutno měřit hodnoty transamináz v séru. Objeví-li se klinické příznaky, které ukazují na hepatotoxicitu, je nutno okamžitě stanovit transaminázy v séru. Pokud kdykoli dojde ke zvýšení ALT nebo AST nad 5násobek ULN, je nutno léčbu přerušit a pečlivě sledovat funkci jater. Obnovení léčby lze provést po navrácení jaterních testů pacienta k normálu a s podáváním nižší dávky (viz bod 4.2).
Vyvine-li se u pacientů kdykoli během léčby závažná hepatotoxicita (ALT nebo AST odpovídající 20násobku ULN), je nutno léčbu ukončit a u těchto pacientů se nemá léčba znovu zahajovat.
Pacienti s aktivní nebo symptomatickou virovou hepatitidou byli z klinických hodnocení vyloučeni; neexistují tedy údaje, které by použití přípravku ZYTIGA v této populaci podporovaly.
Nejsou dostupné údaje o klinické bezpečnosti a účinnosti opakovaných dávek abirateron-acetátu při podání pacientům se středně závažnou nebo závažnou poruchou funkce jater (Child-Pugh třídy B nebo C). U pacientů se středně závažnou poruchou funkce jater je nutno užití přípravku ZYTIGA důkladně posoudit, přínos by měl jasně převažovat možné riziko (viz body 4.2 a 5.2). Pacientům se závažnou poruchou funkce jater se přípravek ZYTIGA nesmí podávat (viz body 4.2, 4.3 a 5.2).
Během postmarketingového sledování byly vzácně hlášeny případy akutního selhání jater a fulminantní hepatitidy, některé s fatálními následky (viz bod 4.8).
Ukončení léčby kortikosteroidy a zvládání stresových situací
Je-li ukončeno podávání prednisonu nebo prednisolonu, je nutná opatrnost a pacienty je nutno monitorovat, zda se u nich nerozvíjí adrenokortikální nedostatečnost. Pokračuje-li se v léčbě přípravkem ZYTIGA po vysazení kortikosteroidů, je nutno pacienty sledovat, zda se u nich neobjeví příznaky zvýšené hladiny mineralokortikoidů (viz informace výše).
U pacientů, kterým je podáván prednison nebo prednisolon a kteří jsou vystaveni neobvyklému stresu, může být indikováno zvýšené dávkování kortikosteroidů před stresovou situací, během ní i po ní.
Kostní denzita
U mužů s metastazujícím pokročilým karcinomem prostaty (karcinomem prostaty rezistentním na kastraci) se může vyskytnout snížená kostní denzita. Podávání přípravku ZYTIGA v kombinaci s glukokortikoidy může tento účinek zvýšit.
Pacienti, kterým byl dříve podáván ketokonazol
U pacientů, kterým byl k léčbě karcinomu prostaty dříve podáván ketokonazol, lze očekávat snížený počet odpovědí.
Hyperglykemie
Užívání glukokortikoidů může zvyšovat hyperglykemii, proto je u pacientů s diabetem nutno často měřit glykemii.
Užívání s chemoterapií
Bezpečnost a účinnost současného užívání přípravku ZYTIGA s cytotoxickou chemoterapií nebyly stanoveny (viz bod 5.1).
Nesnášenlivost pomocných látek
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat. Tento léčivý přípravek obsahuje také více než 1 mmol (nebo 27,2 mg) sodíku na dávku čtyř tablet. To je nutno zohlednit u pacientů s dietou se sníženým přísunem sodíku.
Potenciální rizika
U mužů s metastazujícím karcinomem prostaty rezistentním na kastraci, včetně pacientů léčených přípravkem ZYTIGA, se mohou objevit anemie a sexuální dysfunkce.
Účinky na kosterní svalstvo
U pacientů léčených přípravkem ZYTIGA byly hlášeny případy myopatie. U některých pacientů se vyskytla rhabdomyolýza s renálním selháním. Většina případů se vyvinula během prvního měsíce léčby a ustoupila po ukončení podávání přípravku ZYTIGA. U pacientů léčených současně léčivými přípravky, o kterých je známo, že jsou spojeny s myopatií/rhabdomyolýzou, se doporučuje opatrnost.
Interakce s jinými léčivými přípravky
Z důvodu rizika snížené expozice abirateronem (viz bod 4.5) je nutné vyvarovat se během léčby podávání silných induktorů CYP3A4, ledaže by nebyla dispozici jiná alternativní terapeutická léčba.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vliv potravy na abirateron-acetát
Podání s jídlem významně zvyšuje absorpci abirateron-acetátu. Účinnost a bezpečnost při podávání s jídlem nebyla stanovena, proto se nesmí tento přípravek užívat s jídlem (viz body 4.2 a 5.2).
Interakce s jinými léčivými přípravky
Potenciál jiných léčivých přípravků ovlivňovat expozice abirateron-acetátu V klinické studii farmakokinetických interakcí na zdravých dobrovolnících, kteří byli předléčeni rifampicinem, silným induktorem CYP3A4, v dávce 600 mg denně po dobu 6 dní, po které následovala jednorázová dávka 1 000 mg abirateron-acetátu, se průměrná plazmatická AUC^ abirateronu snížila o 55 %.
Během léčby je nutno se vyvarovat podávání silných induktorů CYP3A4 (např. fenytoinu, karbamazepinu, rifampicinu, rifabutinu, rifapentinu, fenobarbitalu, třezalce tečkované [Hypericum perforatum]), ledaže by nebyla k dispozici jiná alternativní léčba.
V samostatné klinické studii farmakokinetických interakcí na zdravých dobrovolnících nemělo současné podávání ketokonazolu, silného inhibitoru CYP3A4, klinicky významný účinek na farmakokinetiku abirateronu.
Potenciál ovlivňovat expozice jiných léčivých přípravků
Abirateron je inhibitorem jaterních léky metabolizujících enzymů CYP2D6 a CYP2C8. V hodnocení, kde se stanovovaly účinky abirateron-acetátu (podaného spolu s prednisonem) na jednorázovou dávku dextromethorfanu, který je substrátem CYP2D6, byla systémová expozice (AUC) dextromethorfanu zvýšena přibližně 2,9násobně. AUC24 dextrorfanu, aktivního metabolitu dextromethorfanu, byla zvýšena přibližně o 33 %.
Opatrnost je nutná při současném podávání s léčivými přípravky aktivovanými nebo metabolizovanými CYP2D6, zejména s léčivými přípravky, které mají úzkou terapeutickou šíři. Je nutno zvážit snížení dávek léčivých přípravků metabolizovaných CYP2D6 s úzkou terapeutickou šíří. Příkladem léčivých přípravků metabolizovaných CYP2D6 jsou metoprolol, propranolol, desipramin, venlafaxin, haloperidol, risperidon, propafenon, flekainid, kodein, oxykodon a tramadol (poslední tři léčivé látky potřebují CYP2D6 k vytvoření svých účinných analgetických metabolitů).
V klinické studii lékových interakcí týkajících se CYP2C8 u zdravých jedinců byla systémová expozice AUC pioglitazonu zvýšena o 46 % a AUC aktivních metabolitů pioglitazonu MIII a MIV byla u každého snížena o 10 %, pokud byl pioglitazon podáván společně s jednorázovou dávkou 1000 mg abirateron-acetátu. Ačkoli tyto výsledky naznačují, že se neočekává žádné klinicky významné zvýšení expozice, pokud je přípravek ZYTIGA podáván v kombinaci s léčivými přípravky, které jsou převážně eliminovány pomocí CYP2C8, mají být pacienti sledováni pro známky toxicity související se substráty CYP2C8 s úzkým terapeutickým indexem, jsou-li užívány současně.
Údaje získané in vitro ukázaly, že hlavní metabolity abirateron sulfát a N-oxid abirateron sulfát inhibují jaterní transportér OATP1B1 a v důsledku toho může dojít ke zvýšení koncentrace léčivých látek vylučovaných OATP1B1. Nejsou k dispozici žádné klinické údaje, které by potvrdili interakce s tímto transportérem.
Užívání s přípravky, které prodlužují QT interval
Vzhledem k tomu, že androgen-deprivační léčba může prodlužovat QT interval je doporučena opatrnost, pokud je přípravek ZYTIGA užíván spolu s léčivými přípravky, které prodlužují interval QT nebo s léčivými přípravky, které mohou indukovat Torsade de pointes, jako jsou antiarytmika třídy IA (např.chinidin, disopyramid) nebo třídy III (např. amiodaron, sotalol, dofetilid, ibutilid), methadon, moxifloxacin, antipsychotika atd.
Užívání se spironolaktonem
Spironolakton se váže na androgenní receptory a může zvyšovat hladiny prostatického specifického antigenu (PSA). Užívání s přípravkem ZYTIGA se nedoporučuje (viz bod 5.1).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Neexistují údaje o podávání přípravku ZYTIGA v těhotenství a tento přípravek není určen k podávání ženám v plodném věku.
Antikoncepce u mužů a žen
Není známo, zda jsou abirateron nebo jeho metabolity přítomny v semeni. Při sexuální aktivitě s těhotnou ženou musí pacient použít kondom. Při sexuální aktivitě s ženou v plodném věku musí pacient použít kondom a zároveň další účinnou antikoncepční metodu. Studie u zvířat ukázaly reprodukční toxicitu (viz bod 5.3).
ZYTIGA není určena ženám a je kontraindikována u těhotných žen nebo u žen, které by mohly otěhotnět (viz body 4.3 a 5.3).
Kojení
ZYTIGA není určena k podávání ženám.
Fertilita
Abirateron ovlivňoval fertilitu u samců i samic potkanů, ale tyto účinky byly plně reverzibilní (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek ZYTIGA nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejčastěji pozorovanými nežádoucími účinky jsou periferní otok, hypokalemie, hypertenze a infekce močových cest.
Další závažné nežádoucí účinky zahrnují srdeční onemocnění, hepatotoxicitu, zlomeniny a alergickou alveolitidou.
ZYTIGA může způsobit hypertenzi, hypokalemii a retenci tekutin; jedná se o farmakodynamický následek jejího mechanismu účinku. V klinických hodnoceních byly předpokládané mineralokortikoidní nežádoucí účinky pozorovány častěji u pacientů léčených abirateron-acetátem než u pacientů léčených placebem: hypokalemie 21 % vs. 11 %, hypertenze 16 % vs. 11 % a retence tekutin (periferní otok) 26 % vs. 20 %. U pacientů léčených abirateron-acetátem se hypokalemie CTCAE (verze 3.0) stupňů 3 a 4 a hypertenze CTCAE (verze 3.0) stupňů 3 a 4 vyskytly u 4 %, resp.
2 % pacientů. Mineralokortikoidní účinky byly většinou lékařsky úspěšně zvládnutelné. Současné podávání kortikosteroidů incidenci a závažnost těchto nežádoucích účinků snižuje (viz bod 4.4).
Tabulkový seznam nežádoucích účinků
V hodnoceních u pacientů s pokročilým metastazujícím karcinomem prostaty, kterým byl podáván analog LHRH nebo pacientů dříve léčených orchiektomií, byla ZYTIGA podávána v dávce 1 000 mg denně v kombinaci s prednisonem nebo prednisolonem (10 mg denně).
Nežádoucí účinky pozorované v klinických hodnoceních a během postmarketingového sledování s přípravkem ZYTIGA jsou uvedeny dále dle kategorií četnosti. Kategorie četnosti jsou definovány jako: velmi časté (>1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000) a není známo (z dostupných údajů nelze určit).
V každé skupině četnosti jsou nežádoucí účinky uvedeny s klesající závažností.
Tabulka 1: Nežádoucí účinky zjištěné v klinických hodnoceních a postmarketingovém sledování
|
Třídy orgánových systémů |
Frekvence |
|
Infekce a infestace |
Velmi časté: infekce močových cest Časté: sepse |
|
Endokrinní poruchy |
Méně časté: adrenální insuficience |
|
Poruchy metabolismu a výživy |
Velmi časté: hypokalemie Časté: hypertriglyceridemie |
|
Srdeční poruchy |
Časté: srdeční selhání*, angina pectoris, arytmie, fibrilace síní, tachykardie Není známo: infarkt myokardu, prodloužení QT intervalu (viz body 4.4 a 4.5) |
|
Respirační, hrudní a mediastinální poruchy |
Vzácné: alergická alveolitida3 |
|
Cévní poruchy |
Velmi časté: hypertenze |
|
Gastrointestinální poruchy | |
|
Poruchy jater a žlučových cest |
Časté: zvýšení ALT, zvýšení AST Vzácné: fulminantní hepatitida, aktutní selhání jater |
|
Poruchy kůže a podkožní tkáně |
Časté: vyrážka |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Méně časté: myopatie, rhabdomyolýza |
|
Poruchy ledvin a močových cest |
Časté: hematurie |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté: periferní otok |
|
Poranění, otravy a procedurální komplikace |
Časté: zlomeniny** |
* Srdeční selhání zahrnuje také městnavé srdeční selhání, dysfunkci levé komory a snížení ejekční frakce ** Zlomeniny zahrnují všechny zlomeniny s výjimkou patologických zlomenin
spontánní hlášení z postmarketingového sledování
U pacientů léčených abirateron-acetátem se vyskytly následující nežádoucí účinky CTCAE (verze 3.0) stupně 3 závažnosti: hypokalemie 3 %, infekce močových cest, zvýšení alaninaminotrasferázy, hypertenze, zvýšení aspartátaminotransferázy, zlomeniny 2 %, periferní otok, srdeční selhání a fibrilace síní, vše s frekvencí 1 %. Hypertriglyceridemie a angina pectoris CTCAE (verze 3.0) stupně 3 se vyskytly u < 1 % pacientů. Periferní otok, hypokalemie, infekce močových cest, srdeční selhání a zlomeniny CTCAE (verze 3.0) stupně 4 se vyskytly u < 1 % pacientů.
Popis vybraných nežádoucích účinků
Kardiovaskulární účinky
Z obou hodnocení fáze 3 byli vyloučeni pacienti s nekontrolovanou hypertenzí, klinicky významným onemocněním srdce, které se klinicky manifestovalo jako infarkt myokardu nebo arteriální trombotické příhody v předchozích 6 měsících, pacienti se závažnou nebo nestabilní anginou pectoris nebo selháváním srdce NYHA třídy III nebo IV (hodnocení 301) nebo třídy II až IV (hodnocení 302) nebo s naměřenou ejekční frakcí < 50 %. Všichni zahrnutí pacienti (jak pacienti s aktivní medikací tak i pacienti na placebu) byli zároveň léčeni antiandrogenní léčbou zejména s použitím analogů LHRH, což bylo spojeno s diabetem, infarktem myokardu, cerebrovaskulární příhodou a náhlou zástavou srdce. Výskyt kardiovaskulárních nežádoucích účinků v hodnoceních fáze 3 u pacientů užívajících abirateron-acetát byl ve srovnání s placebem následující: hypertenze 14,5 % vs. 10,5 %, fibrilace síní 3,4 % vs. 3,4 %, tachykardie 2,8 % vs. 1,7 %, angina pectoris 1,9 % vs. 0,9 %, srdeční selhání 1,9 % vs. 0,6 % a arytmie 1,1 % vs. 0,4 %.
Hepatotoxicita
U pacientů léčených abirateron-acetátem byla hlášena hepatotoxicita se zvýšením ALT, AST a celkového bilirubinu. V rámci všech klinických hodnocení bylo zvýšení jaterních testů (zvýšení ALT nebo AST > 5násobek horní hranice normálu nebo zvýšení bilirubinu > 1,5násobek horní hranice normálu) hlášeno u 4 % pacientů léčených abirateron-acetátem, typicky během prvních 3 měsíců po zahájení léčby. V hodnocení 301 bylo zhoršení jaterních testů pravděpodobnější u pacientů, jejichž jaterní testy byly zvýšeny již na počátku, ve srovnání s pacienty, jejichž jaterní testy byly na počátku normální. Došlo-li ke zvýšení ALT nebo AST na > 5násobek horní hranice normálu nebo ke zvýšení bilirubinu na > 3násobek horní hranice normálu, bylo podávání abirateron-acetátu ukončeno. Ve dvou případech došlo k významnému zvýšení hodnot výsledků jaterních testů (viz bod 4.4). U těchto dvou pacientů s normální funkcí jater na počátku léčby došlo ke zvýšení ALT nebo AST na 15 až 40násobek horní hranice normálu a ke zvýšení bilirubinu na 2 až 6násobek horní hranice normálu. Po ukončení léčby došlo u obou pacientů k normalizaci jaterních testů a jeden z pacientů byl znovu léčen bez opakovaného zvýšení jejich hodnot. V hodnocení 302 byla zvýšení ALT nebo AST stupně 3 nebo 4 pozorována u 35 (6,5 %) pacientů léčených abirateron-acetátem. Zvýšené hladiny aminotransferáz se vrátily k normálu u všech kromě 3 pacientů (2 s novými mnohočetnými metastázami v játrech a 1 se zvýšením AST přibližně 3 týdny po poslední dávce abirateron-acetátu). Ukončení léčby kvůli zvýšením ALT nebo AST byla hlášena u 1,7 % a 1,3 % pacientů léčených abirateron-acetátem, resp. u 0,2 % a 0 % pacientů léčených placebem; kvůli hepatotoxicitě nebyla hlášena žádná úmrtí.
V klinických hodnoceních bylo riziko hepatotoxicity omezeno vyloučením pacientů s výchozí hepatitidou nebo významnými abnormalitami v jaterních testech. Z hodnocení 301 byli vyloučeni pacienti s výchozí hodnotou ALT a AST > 2,5násobek horní hranice normálu bez přítomnosti metastáz v játrech a pacientů s výchozí hodnotou ALT a AST > 5násobek horní hranice normálu, pokud v játrech byly přítomny metastázy. Pro hodnocení 302 nebyli pacienti s metastázami v játrech vhodní a pacienti s výchozí hodnotou ALT a AST > 2,5násobek horní hranice normálu byli z hodnocení vyloučeni. Objevující se zvýšení jaterních testů u pacientů v klinických hodnoceních bylo rázně řešeno požadavkem přerušení léčby a povolením jejího obnovení po návratu jaterních testů na výchozí hodnoty (viz bod 4.2). U pacientů s ALT nebo AST > 20násobek horní hranice normálu nebyla léčba znovu zahájena. Bezpečnost znovuzahájení léčby u takovýchto pacientů není známa. Mechanismus hepatotoxicity nebyl vysvětlen.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s předávkováním u člověka jsou omezené.
Specifické antidotum neexistuje. V případě předávkování je nutno ukončit podávání a zahájit obecná podpůrná opatření včetně monitorování arytmií, hypokalemie a známek a příznaků retence tekutin. Je také nutno vyšetřit funkci jater.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: endokrinní léčba, jiní antagonisté hormonů a příbuzné látky, ATC kód: L02BX03
Mechanismus účinku
Abirateron-acetát (ZYTIGA) se in vivo metabolizuje na abirateron, inhibitor biosyntézy androgenů. Abirateron selektivně inhibuje enzym 17a-hydroxylázu/C17, 20-lyázu (CYP17). Tento enzym je exprimován a je nutný pro biosyntézu androgenů ve tkáních varlat, nadledvin a v nádorovém tkanivu prostaty. CYP17 katalyzuje přeměnu pregnenolonu a progesteronu na prekurzory testosteronu, DHEA, resp. androstendionu 17a-hydroxylací a štěpením vazby C17, 20. Inhibice CYP17 vede též ke zvýšené produkci mineralokortikoidů nadledvinami (viz bod 4.4).
Androgen-senzitivní karcinom prostaty reaguje na léčbu, která snižuje hladiny androgenů. Terapie, které snižují hladinu androgenů, jako léčba analogy LHRH nebo orchiektomie, snižují produkci androgenů ve varlatech, ale neovlivňují produkci androgenů nadledvinami nebo v tumoru. Léčba přípravkem ZYTIGA, je-li podána s analogy LHRH (nebo s orchiektomií), snižuje hladinu testosteronu v séru na nedetekovatelné hodnoty (za použití komerčních metod stanovení).
Farmakodynamické účinky
ZYTIGA snižuje testosteron a jiné androgeny v séru na hladiny nižší, než které jsou dosaženy samotnými analogy LHRH nebo orchiektomií. To je způsobeno selektivní inhibicí enzymu CYP17 potřebného k biosyntéze androgenů. U pacientů s karcinomem prostaty slouží jako specifický biomarker PSA. Ve fázi 3 klinického hodnocení u pacientů, u kterých selhala předchozí léčba taxany, došlo k poklesu hladin PSA alespoň o 50 % oproti výchozímu stavu u 38 % pacientů léčených abirateron-acetátem ve srovnání s 10 % pacientů léčených placebem.
Klinická účinnost a bezpečnost
Účinnost přípravku ZYTIGA byla stanovena ve dvou randomizovaných placebem kontrolovaných multicentrických hodnoceních fáze 3 (hodnocení 301 a 302) u pacientů s metastazujícím karcinomem prostaty rezistentním na kastraci. V hodnocení 302 byli zahrnuti pacienti bez předchozí léčby docetaxelem; zatímco v hodnocení 301 byli zahrnuti pacienti, kteří dostávali dříve docetaxel. Pacienti užívali analog LHRH nebo podstoupili orchiektomii. V rameni s aktivní léčbou byla ZYTIGA podávána v dávce 1 000 mg denně v kombinaci s nízkou dávkou prednisonu nebo prednisolonu 5 mg dvakrát denně. Kontrolní skupinu tvořili pacienti, kteří dostávali placebo a nízkou dávkou prednisonu nebo prednisolonu 5 mg dvakrát denně.
Změny v sérových koncentracích PSA samy o sobě ne vždy předpovídají klinický přínos. V obou hodnoceních bylo tedy doporučeno, aby pacienti dostávali léčbu až do dosažení kritérií pro ukončení, jak jsou specifikována pro každé hodnocení dále.
V obou hodnoceních nebylo užívání se spironolaktonem povoleno, protože spironolakton se váže na androgenní receptor a může zvyšovat hladiny PSA.
Hodnocení 302 (pacienti bez předchozí chemoterapie)
Toto hodnocení zahrnovalo pacienty, kteří dříve nedostávali chemoterapii a kteří byli asymptomatičtí nebo mírně symptomatičtí a u nichž chemoterapie dosud nebyla klinicky indikována. Skóre 0-1 na Brief Pain Inventory-Short Form (BPI-SF) nejhorší bolesti během posledních 24 hodin bylo považováno za asymptomatické a skóre 2-3 za mírně symptomatické.
V hodnocení 302 (n = 1 088) byl u pacientů léčených přípravkem ZYTIGA a prednisonem nebo prednisolonem medián věku 71 let a u pacientů léčených placebem a prednisonem nebo prednisolonem byl medián věku 70 let. Počet pacientů léčených přípravkem ZYTIGA byl podle rasy 520 bělochů (95,4 %), 15 černochů (2,8 %), 4 asiaté (0,7 %) a 6 ostatních (1,1 %). Eastern Cooperative Oncoly Group (ECOG) skóre bylo 0 u 76 % pacientů a 1 u 24 % pacientů v obou ramenech. Padesát procent pacientů mělo pouze metastázy v kostech, dalších 31 % pacientů mělo metastázy v kostech a měkkých tkáních nebo lymfatických uzlinách a 19 % pacientů mělo metastázy pouze v měkkých tkáních nebo lymfatických uzlinách. Pacienti s viscerálními metastázami byli vyloučeni. Společnými primárními cíli bylo celkové přežití a přežití bez radiografické progrese (rPFS). Navíc k hodnocení společných primárních cílů byl přínos hodnocen také za použití doby do použití opioidu pro nádorovou bolest, doby do zahájení cytotoxické chemoterapie, doby do zhoršení ECOG skóre o > 1 stupeň a doby do progrese PSA založené na kritériích Prostate Cancer Working Group-2 (PCWG2). Podání léčby v hodnocení bylo ukončeno v době jednoznačné klinické progrese. Léčbu bylo také možno ukončit v době potvrzené radiografické progrese, podle uvážení zkoušejícího.
Přežití bez radiografické progrese (rPFS) bylo hodnoceno s použitím sekvenčního zobrazovacího sledování tak, jak jsou definována kritérii PCWG2 (pro kostní léze) a modifikovanými kritérii Response Evaluation Criteria In Solid Tumors (RECIST) (pro léze měkkých tkání). Analýza rPFS používala centrální vyhodnocování radiografické progrese.
V plánované rPFS analýze bylo 401 příhod, 150 (28 %) pacientů léčených přípravkem ZYTIGA a 251 (46 %) pacientů léčených placebem mělo radiografický průkaz progrese nebo zemřeli. Byl pozorován významný rozdíl mezi rPFS mezi skupinami léčby (viz Tabulka 2 a Obrázek 1).
Tabulka 2: Hodnocení 302: Přežití bez radiografické progrese u pacientů léčených buď
přípravkem ZYTIGA nebo placebem v kombinaci s prednisonem nebo prednisolonem a analogem LHRH nebo předchozí orchiektomií_
|
ZYTIGA (N = 546) |
Placebo (N = 542) | |
|
Přežití bez radiografické progrese (rPFS) | ||
|
Progrese nebo úmrtí |
150 (28 %) |
251 (46%) |
|
Medián rPFS v měsících |
Nedosaženo |
8,3 |
|
(95% CI) |
(11,66; NE) |
(8,12; 8,54) |
|
hodnota p* |
<0,0001 |
|
Poměr rizik** (95% CI) |
0,425 (0,347; 0,522) |
NE = nebylo stanoveno
* p-hodnota je odvozena z log-rank testu stratifikovaného podle výchozího ECOG skóre (0 nebo 1)
** Poměr rizik < 1je ve prospěch přípravku ZYTIGA
Obrázek 1: Kaplan Meierovy křivky přežití bez radiografické progrese u pacientů léčených buď přípravkem ZYTIGA nebo placebem v kombinaci s prednisonem nebo prednisolonem a analogem LHRH nebo předchozí orchiektomií
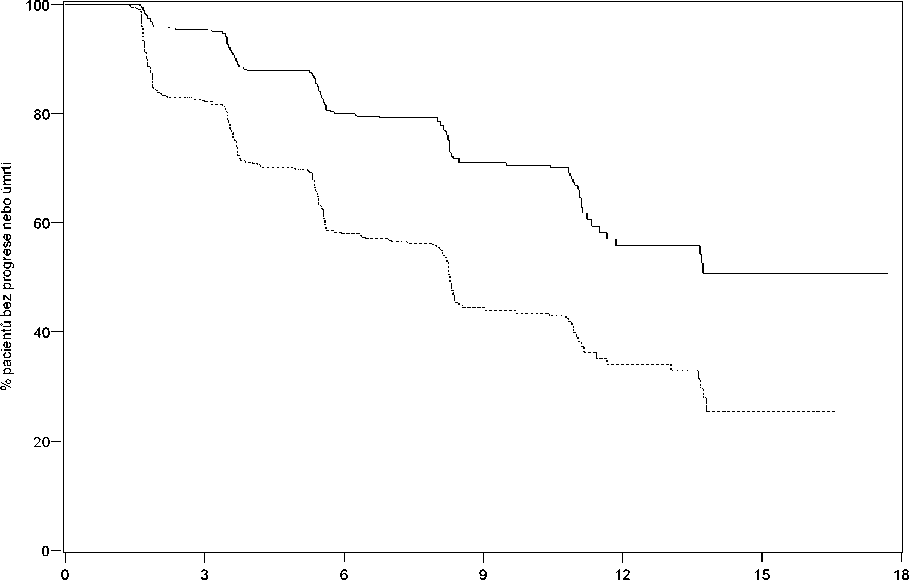
Měsíce od randomizace
AA 546 489 340 164 46 12 0
Placebo 542 400 204 90 30 3 0
“ Placebo AA
AA = ZYTIGA
Avšak údaje pacientů byly sbírány až do druhé průběžné analýzy celkového přežití (Overall survival = OS). Radiografické hodnocení rPFS provedené zkoušejícím následně po analýze senzitivity je uvedeno v Tabulce 3 a na Obrázku 2.
Šest set sedm (607) pacientů mělo radiografickou progresi nebo zemřelo: 271 (50 %) ve skupině s abirateron-acetátem a 336 (62 %) ve skupině s placebem. Léčba abirateron-acetátem snižovala riziko radiografické progrese nebo úmrtí o 47 % ve srovnání s placebem (poměr rizik = 0,530; 95% CI: [0,451 - 0,623]; p < 0,0001). Medián rPFS byl 16,5 měsíce ve skupině s abirateron-acetátem a 8,3 měsíce ve skupině s placebem.
Tabulka 3: Hodnocení 302: Přežití bez radiografické progrese u pacientů léčených buď
přípravkem ZYTIGA nebo placebem v kombinaci s prednisonem nebo prednisolonem a analogem LHRH nebo předchozí orchiektomií (při druhé průběžné analýze ^ hodnocení OS zkoušejícím) _
|
ZYTIGA |
Placebo | |
|
(N = 546) |
(N = 542) | |
|
Přežití bez radiografické | ||
|
progrese (rPFS) | ||
|
Progrese nebo úmrtí |
271 (50%) |
336 (62 %) |
|
Medián rPFS v měsících |
16,5 |
8,3 | |
|
(95% CI) |
(13,80; 16,79) |
(8,05; 9,43) | |
|
hodnota p* |
<0,0001 | ||
|
Poměr rizik** (95% CI) |
0,530 (0,451; 0,623) | ||
* p-hodnota je odvozena z log-rank testu stratifikovaného podle výchozího ECOG skóre (0 nebo 1) ** Poměr rizik < 1je ve prospěch přípravku ZYTIGA
Plánovaná předběžná analýza (interim analysis = IA) OS byla provedena po 333 pozorovaných úmrtích. Hodnocení bylo na základě pozorovaného významného klinického přínosu odslepeno a pacientům ve skupině s placebem byla nabídnuta léčba přípravkem ZYTIGA. Celkové přežití bylo delší u přípravku ZYTIGA než u placeba s 25% snížením rizika úmrtí (poměr rizik = 0,752; 95% CI: [0,606; 0,934], p=0,0097), ale data k celkovému přežití nebyla dostatečně zralá a průběžné výsledky nedosáhly předem specifikovanou hodnotu statistické významnosti (viz Tabulka 4). Přežití bylo po této předběžné analýze dále sledováno.
Po zaznamenání 741 úmrtí byla provedena plánovaná konečná analýza celkového přežití (medián sledování byl 49 měsíců). Zemřelo šedesát pět procent (354 z 546) pacientů léčených přípravkem ZYTIGA ve srovnání se 71% (387 z 542) pacientů léčených placebem. Byl prokázán statisticky významný přínos v celkovém přežití ve prospěch skupiny léčené přípravkem ZYTIGA se snížením rizika úmrtí o 19,4 % (HR=0,806; 95% CI: [0,697; 0,931], p=0,0033) a zlepšením mediánu celkového přežití o 4,4 měsíce (ZYTIGA 34,7 měsíce, placebo 30,3 měsíců) (viz Tabulka 4 a Obrázek 3). Toto zlepšení bylo prokázáno navzdory tomu, že 44 % pacientů v rameni s placebem užívalo přípravek ZYTIGA jako následnou léčbu.
Tabulka 4: Hodnocení 302: Celkové přežití pacientů léčených buď přípravkem ZYTIGA nebo placebem v kombinaci s prednisonem nebo prednisolonem a analogem LHRH
|
nebo předchozí orc |
hiektomií | |
|
ZYTIGA (N = 546) |
Placebo (N = 542) | |
|
Předběžná analýza celkového přežití | ||
|
Úmrtí (%) |
147 (27 %) |
186 (34 %) |
|
Medián přežití (měsíce) (95% CI) |
Nedosaženo (NE, NE) |
27,2 (25,95; NE) |
|
hodnota p* |
0,0097 | |
|
Poměr rizik** (95% CI) |
0,752 (0,606; 0,934) | |
|
Závěrečná analýza celkového přežití | ||
|
Úmrtí (%) |
354 (65 %) |
387 (71 %) |
|
Medián přežití (měsíce) (95% CI) |
34,7 (32,7; 36,8) |
30,2 (28,7; 33,3) |
|
hodnota p* |
0,0033 | |
|
Poměr rizik** (95% CI) |
0,806 (0,697; 0,931) | |
NE = nebylo stanoveno
* p-hodnota je odvozena z log-rank testu stratifikovaného podle výchozího ECOG skóre (0 nebo 1) ** Poměr rizik < 1je ve prospěch přípravku ZYTIGA
Obrázek 3: Kaplan Meierovy křivky přežití u pacientů léčených buď přípravkem ZYTIGA
nebo placebem v kombinaci s prednisonem nebo prednisolonem a analogem LHRH nebo předchozí orchiektomií, závěrečná analýza
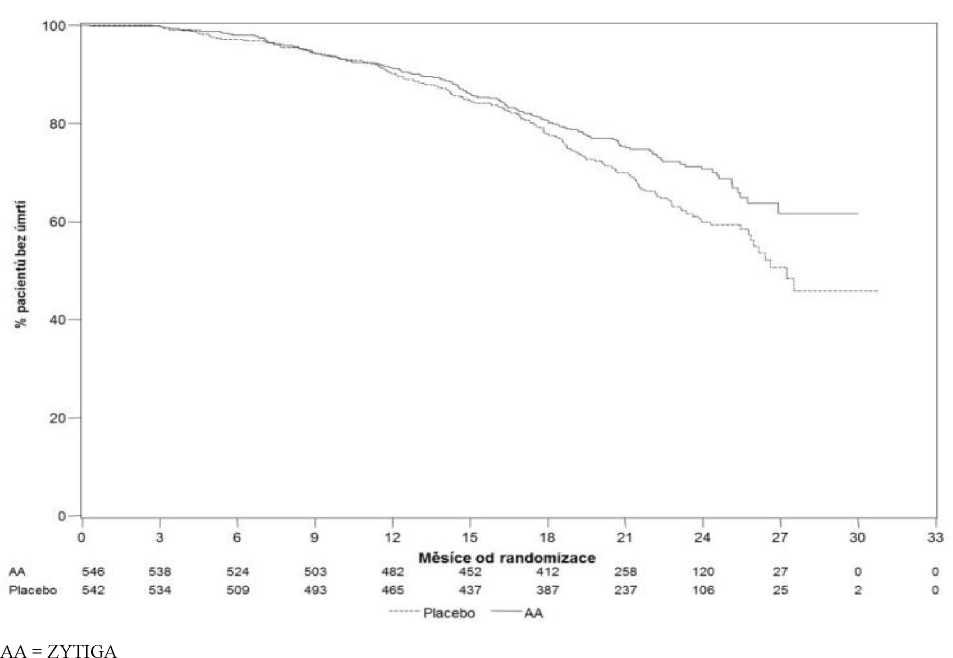
Navíc k pozorovaným zlepšením celkového přežití a rPFS byl u léčby přípravkem ZYTIGA oproti placebu prokázán přínos ve všech měřených sekundárních cílových parametrech, jak je uvedeno dále:
Doba do progrese založená na kritériích PCWG2: Medián doby do progrese PSA byl 11,1 měsíce u pacientů dostávajících přípravek ZYTIGA a 5,6 měsíce u pacientů dostávajících placebo [poměr rizik = 0,488; 95% CI: (0,420; 0,568), p < 0,0001]. Doba do PSA progrese byla při léčbě přípravkem ZYTIGA přibližně dvojnásobná (poměr rizik = 0,488). Podíl pacientů s potvrzenou PSA odpovědí byl vyšší ve skupině s přípravkem ZYTIGA než ve skupině s placebem (62 % vs. 24 %; p < 0,0001).
U pacientů s měřitelným onemocněním měkkých tkání byly při léčbě přípravkem ZYTIGA pozorovány významně vyšší počty kompletních nebo parciálních odpovědí nádoru.
Doba do použití opiodu kvůli nádorové bolesti: Medián doby do použití opioidu kvůli bolesti spojené s nádorem prostaty byl v době konečné analýzy 33,4 měsíce u pacientů dostávajících přípravek ZYTIGA a 23,4 měsíce u pacientů dostávajících placebo [poměr rizik = 0,721 95%CI: [0,614; 0,846],
p = 0,0001).
Doba do zahájení cytotoxické léčby: Medián doby do zahájení cytotoxické léčby byl u pacientů dostávajících přípravek ZYTIGA 25,2 měsíce a 16,8 měsíce u pacientů dostávajících placebo [poměr rizik = 0,580; 95%CI: (0,487; 0,691), p < 0,0001].
Doba do zhoršení skóre ECOG o >1 bod: Medián doby do zhoršení skóre ECOG o >1 bod byl u pacientů dostávajících přípravek ZYTIGA 12,3 měsíce a 10,9 měsíce u pacientů dostávajících placebo [poměr rizik = 0,821; 95%CI: (0,714; 0,943), p = 0,0053].
Následující výstupy hodnocení prokázaly statisticky významnou výhodu ve prospěch léčby přípravkem ZYTIGA:
Objektivní odpověď: Objektivní odpověď byla definována jako podíl pacientů s měřitelným onemocněním, kteří dosáhli kompletní nebo parciální odpovědi podle RECIST kritérií (pro hodnocení lymfatických uzlin jako cílových lézí byla vyžadována počáteční velikost > 2 cm). Podíl pacientů s měřitelným onemocněním na počátku, kteří měli objektivní odpověď, byl ve skupině s přípravkem ZYTIGA 36 % a ve skupině s placebem 16 % (p < 0,0001).
Bolest: Léčba přípravkem ZYTIGA významně snižovala riziko progrese průměrné intenzity bolesti o 18 % ve srovnání s placebem (p = 0,0490). Medián doby do progrese byl 26,7 měsíce ve skupině s přípravkem ZYTIGA a 18,4 měsíce ve skupině s placebem.
Doba do snížení FACT-P (celkové skóre): Léčba přípravkem ZYTIGA snižovala ve srovnání s placebem riziko snížení FACT-P (celkové skóre) o 22 % (p = 0,0028). Medián doby do snížení FACT-P (celkové skóre) byl 12,7 měsíce ve skupině s přípravkem ZYTIGA a 8,3 měsíce ve skupině s placebem.
Hodnocení 301 (pacienti, kteří dříve dostávali chemoterapii)
Hodnocení 301 zahrnovalo pacienty, kteří byli dříve léčeni docetaxelem. Nebylo požadováno, aby pacienti dosáhli progrese během léčby docetaxelem, protože toxicita této chemoterapie může vést k vysazení. Pacienti dostávali v hodnocení léčbu až do doby, než byla pozorována progrese PSA (potvrzený 25% vzestup nad pacientův výchozí stav/nadir) zároveň s protokolem definovanou radiologickou progresí a symptomatickou nebo klinickou progresí. Z tohoto hodnocení byli vyloučeni pacienti s předchozí léčbou karcinomu prostaty ketokonazolem. Primárním cílovým parametrem účinnosti bylo celkové přežití.
Medián věku pacientů zahrnutých do studie byl 69 let (rozpětí 39 - 95). Počet pacientů léčených přípravkem ZYTIGA podle rasy byl následující: 737 bělochů (93,2 %), 28 černochů (3,5 %), 11 asiatů (1,4 %) a 14 ostatních (1,8 %). Jedenáct procent zahrnutých pacientů mělo ECOG skóre 2; u 70 % existoval radiologický průkaz progrese onemocnění buď se zvýšením PSA, nebo bez něj; 70 % dostávalo v minulosti cytotoxickou chemoterapii a 30 % podstoupilo dvě chemoterapie. Metastázy v játrech byly přítomny u 11 % pacientů léčených přípravkem ZYTIGA.
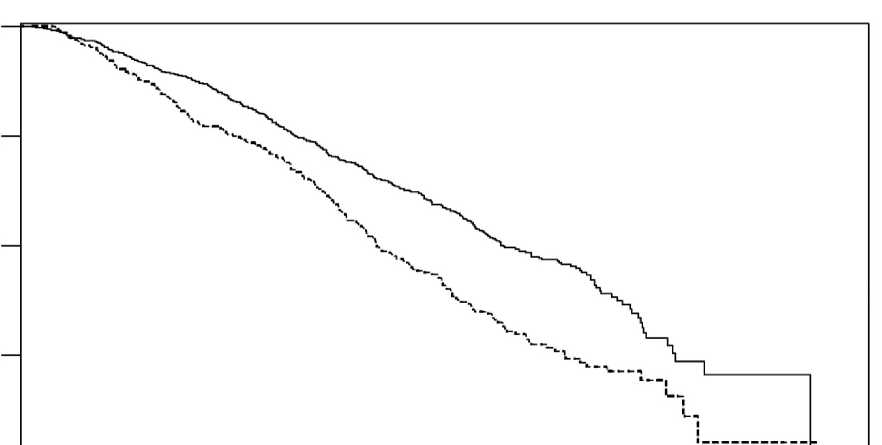
Podle plánované analýzy provedené po 552 pozorovaných úmrtích zemřelo ve skupině léčené přípravkem ZYTIGA 42 % (333 z 797) pacientů ve srovnání s placebem, kde zemřelo 55 % (219 z 398). U pacientů léčených přípravkem ZYTIGA bylo pozorováno statisticky významné zlepšení mediánu celkového přežití (viz Tabulka 5).
Tabulka 5: Celkové přežití pacientů léčených buď přípravkem ZYTIGA nebo placebem
v kombinaci s prednisonem nebo prednisolonem a zároveň analogem LHRH nebo předchozí orchiektomií __
|
ZYTIGA |
Placebo | |
|
(N = 797) |
(N = 398) | |
|
Primární analýza přežití | ||
|
Úmrtí (%) |
333 (42 %) |
219 (55 %) |
|
Medián přežití (měsíce) |
14,8(14,1; 15,4) |
10,9 (10,2; 12,0) |
|
(95% CI) | ||
|
p hodnota3 |
< 0,0001 | |
|
Poměr rizik (95% CI)b |
0,646 (0,543; 0,768) | |
|
Aktualizovaná analýza přežití | ||
|
Úmrtí (%) |
501 (63 %) |
274 (69 %) |
|
Medián přežití (měsíce) |
15,8 |
11,2 |
|
(95% CI) |
(14,8; 17,0) |
(10,4; 13,1) |
|
Poměr rizik (95% CI)b |
0,740 (0,638; 0,859) | |
a
p-hodnota je odvozena z log-rank testu stratifikovaného podle ECOG skóre účinnosti (0-1 vs. 2), skóre bolesti (chybějící vs. přítomna), počtu předchozích chemoterapeutických režimů (1 vs. 2), a typu progrese onemocnění (pouze PSA vs. radiografická).
Poměr rizik je odvozen ze stratifikovaného proporcionálního modelu rizika. Poměr rizik < 1 je ve prospěch přípravku
b
ZYTIGA.
Ve všech časových bodech vyhodnocení po několika úvodních měsících léčby přežívalo více pacientů léčených přípravkem ZYTIGA ve srovnání s pacienty léčenými placebem (viz Obrázek 4).
přípravkem ZYTIGA nebo placebem v kombinaci s prednisonem nebo prednisolonem a analogem LHRH nebo předchozí orchiektomií (při druhé průběžné analýze hodnocení OS zkoušejícím)
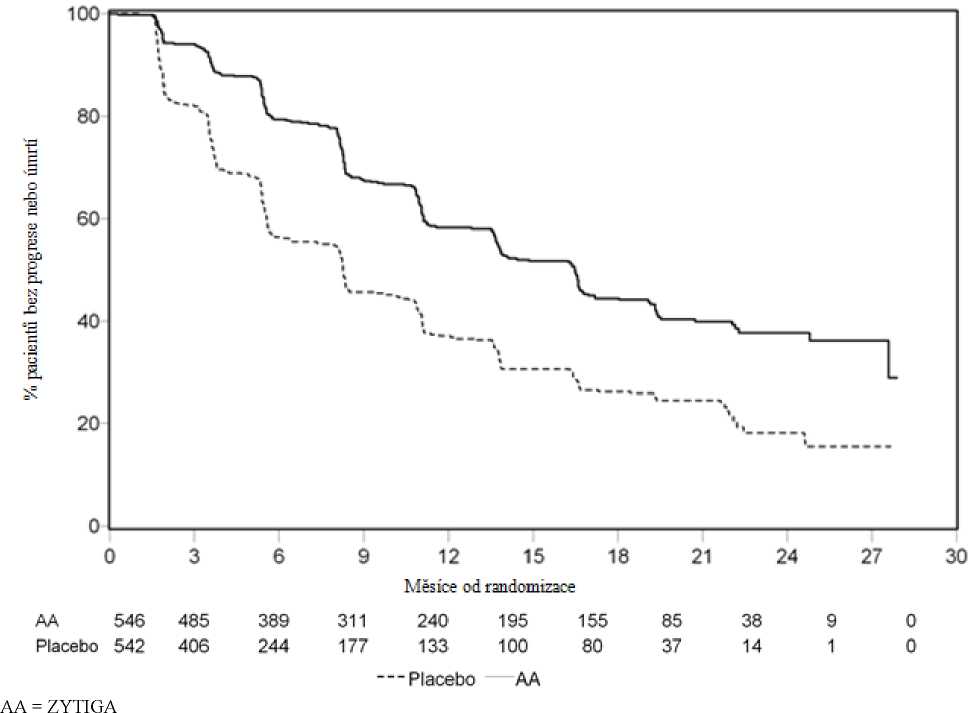
Obrázek 4: Kaplan Meierovy křivky přežití pro pacienty léčené buď přípravkem ZYTIGA nebo placebem v kombinaci s prednisonem nebo prednisolonem a zároveň analogem LHRH nebo předchozí orchiektomií
80
100
p
Ř
E 60
z
T
í
£
40 20—
0—!-
|
3 |
6 |
9 Doba do úmrtí. |
12 měsíce |
15 |
18 |
21 |
|
736 |
657 |
520 |
282 |
68 |
2 |
0 |
|
355 |
306 |
210 |
105 |
30 |
3 |
0 |
----Placebo -AA
0
AA 797 Placebo 398
AA = ZYTIGA
Analýza přežití u podskupin ukázala přínos léčby přípravkem ZYTIGA pro přežití (viz Obrázek 5). Obrázek 5: Celkové přežití podle podskupin: poměr rizik a 95% interval spolehlivosti
Medián (měsíce)
Proměnná Podskupina - ^ HR 95% C.l. N
|
Všechny subjekty |
Všichni |
14.8 |
10.9 |
i—*—i ! i |
G 66 |
(0.56, |
0 79) |
1195 |
|
Počáteční BCOG |
0-1 |
15.3. |
11.7 |
i—•—i | I |
0.64 |
(0.53, |
0.73) |
1060 |
|
2 |
7.3 |
7 |
i-*—!-1 l |
0.81 |
(0.53, |
1.24) |
127 | |
|
Počáteční BPI |
--A |
1 S.2 |
13 |
i W ! I |
0.64 |
(0.50, |
0.82) |
659 |
|
:==ř4 |
12.6 |
6.9 |
0.68 |
(0.53, |
0.85) |
536 | ||
|
Počet předchozích cherno- |
1 |
15.4 |
11.5 |
i-S—1 i |
0.63 |
(0.51, |
G.78) |
833- |
|
terapeutických režimů |
i | |||||||
|
2 |
14 |
10.3 |
i—*-1 i |
0.74 |
(0.55, |
0.99) |
362 | |
|
Typ progrese |
Pouze PSA |
NF |
12.3 |
1—*—1 i |
0.59 |
(0.42, |
0.82) |
:3S3- |
|
Radiografická |
14.2 |
104 |
i ♦ i |
0.69 |
(0.56, |
0.84) |
832 | |
|
Viscerální choroba na počátku |
ANO |
12.6 |
8.4 |
i—*-i! I |
0.70 |
(0.52, |
0 94). |
;3S3. |
|
NE |
15.4 |
11.2 |
1—•—i | i-1-1- |
0.62 _i |
(0.50, |
0.76} |
842 |
0.5 0.75 1 1.5
Lepší výsledek ^^ Lepší výsledek
s AA s placebem
AA = ZYTIGA; BPI = Brief Pain Inventory (dotazník bolesti); C.I. = interval spolehlivosti; ECOG = skóre účinnosti východní pracovní onkologické skupiny; HR = poměr rizik; NE = nelze vyhodnotit
Navíc k pozorovanému zlepšení celkového přežití byly všechny sekundární cílové parametry studie lepší pro přípravek ZYTIGA a zlepšení bylo po úpravě pro opakované testování statisticky signifikantní:
U pacientů dostávajících přípravek ZYTIGA se objevil významně vyšší celkový počet odpovědí týkající se PSA (definováno jako 50% snížení oproti výchozímu stavu), a to 38 % oproti placebu, kde byl podíl odpovědi 10 %, p < 0,0001.
Medián doby do progrese PSA byl 10,2 měsíce u pacientů léčených přípravkem ZYTIGA a 6,6 měsíce u pacientů léčených placebem [HR = 0,580; 95% CI: (0,462; 0,728), p < 0,0001].
Medián přežití bez radiologické progrese byl 5,6 měsíce u pacientů léčených přípravkem ZYTIGA a 3,6 měsíce u pacientů, kteří dostávali placebo [HR= 0,673; 95% CI: (0,585; 0,776), p < 0,0001].
Bolest
Podíl pacientů se zmírněním bolesti byl statisticky významně vyšší ve skupině s přípravkem ZYTIGA oproti skupině s placebem (44 % vs. 27 %, p = 0,0002). Pacient se zmírněním bolesti byl definován jako ten, u kterého došlo během 24 hodin alespoň ke 30% zmírnění od výchozího stavu dle BPI-SF skóre intenzity nejhorší bolesti, aniž by tomuto pacientovi byla podávána další analgetika, při čemž toto zlepšení bylo pozorováno ve dvou po sobě jdoucích vyhodnoceních s odstupem čtyř týdnů. Úleva od bolesti byla hodnocena pouze u pacientů s výchozím skóre bolesti >4 a nejméně jedním skóre bolesti po zahájení léčby (n = 512).
Ke zhoršení bolesti po 6 měsících (22 % vs. 28 %), po 12 měsících (30 % vs. 38 %) a po 18 měsících (35 % vs. 46 %) došlo u menšího podílu pacientů léčených přípravkem ZYTIGA oproti placebu. Zhoršení bolesti bylo definováno jako zvýšení BPI-SF skóre intenzity nejhorší bolesti oproti výchozímu stavu o > 30 % během předchozích 24 hodin, aniž došlo ke snížení podávání analgetik a které bylo pozorováno ve dvou po sobě jdoucích návštěvách. Doba do progrese bolesti u 25. percentilu byla 7,4 měsíce ve skupině s přípravkem ZYTIGA oproti 4,7 měsíce ve skupině s placebem.
Skeletální účinky
Nižší podíl skeletálních účinků se projevil u skupiny pacientů léčených přípravkem ZYTIGA oproti skupině s placebem po 6 měsících (18 % vs.28 %), po 12 měsících (30 % vs.40 %) a po 18 měsících (35 % vs. 40 %). Doba do prvního skeletálního účinku na 25. percentilu u skupiny s přípravkem ZYTIGA (9,9 měsíce) byla dvojnásobná oproti kontrolní skupině (4,9 měsíce). Skeletální účinek byl definován jako patologická fraktura, komprese páteře, paliativní radiace kostí, chirurgie kostí.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ZYTIGA u všech podskupin pediatrické populace u pokročilého karcinomu prostaty. Informace o pediatrickém použití viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Farmakokinetika abirateronu a abirateron-acetátu po perorálním podání abirateron-acetátu byla studována u zdravých subjektů, pacientů s metastazujícím pokročilým karcinomem prostaty a u subjektů bez zhoubného nádoru s poruchou funkce jater nebo ledvin. Abirateron-acetát je in vivo rychle metabolizován na abirateron, inhibitor biosyntézy androgenů (viz bod 5.1).
Absorpce
Po perorálním podání abirateron-acetátu nalačno je doba do dosažení maximální koncentrace abirateronu v plazmě přibližně 2 hodiny.
Podání abirateron-acetátu s potravou vede ve srovnání s podáním nalačno až k 10násobnému (AUC) a až 17násobnému (Cmax) vzestupu střední hodnoty systémové expozice abirateronu v závislosti na obsahu tuku v potravě. Při obvyklém složení potravy může podání přípravku ZYTIGA vést k velmi variabilním expozicím. Proto se ZYTIGA nesmí užívat spolu s jídlem. Je nutno ji užívat alespoň dvě hodiny po jídle a další jídlo se nesmí konzumovat alespoň jednu hodinu po užití přípravku ZYTIGA. Tablety se polykají celé spolu s vodou (viz bod 4.2).
Distribuce v organismu
Vazba 14C-abirateronu na plazmatické bílkoviny je 99,8 %. Zdánlivý distribuční objem je přibližně 5 630 l, což ukazuje, že abirateron je extenzivně distribuován do periferních tkání.
Biotransformace
Po perorálním podání 14C-abirateron-acetátu v tobolce se abirateron-acetát hydrolyzuje na abirateron; ten je potom dále metabolizován, včetně sulfatace, hydroxylace a oxidace, převážně v játrech. Většina cirkulující radioaktivity (přibližně 92 %) je nalezena ve formě metabolitů abirateronu.
Z 15 detekovatelných metabolitů jsou 2 metabolity hlavní, abirateron-sulfát a N-oxid abirateron-sulfátu, z nichž každý vykazuje přibližně 43 % celkové radioaktivity.
Eliminace z organismu
Průměrný plazmatický poločas abirateronu na základě stanovení u zdravých subjektů je přibližně 15 hodin. Po perorálním podání 1 000 mg 14C-abirateron-acetátu se přibližně 88 % radioaktivity objeví ve stolici a přibližně 5 % v moči. Nejčastější sloučeniny přítomné ve stolici jsou nezměněný abirateron-acetát a abirateron (přibližně 55 %, resp. 22 % podané dávky).
Porucha funkce jater
Farmakokinetika abirateron-acetátu byla zkoumána u subjektů s mírnou nebo středně závažnou poruchou funkce jater (Child-Pugh tříd A a B) a u zdravých kontrolních subjektů. Systémová expozice abirateronu po jednorázovém perorálním podání dávky 1 000 mg se u pacientů s mírnou poruchou funkce jater zvýšila přibližně o 11 % a u pacientů se středně závažnou poruchou funkce jater přibližně o 260 %. Průměrný poločas abirateronu je prodloužen přibližně na 18 hodin u pacientů s mírnou poruchou funkce jater a na 19 hodin u pacientů se středně závažnou poruchou funkce jater.
V další studii byla zkoumána farmakokinetika abirateronu u subjektů s již existující závažnou (n = 8) poruchou funkce jater (Child- Pugh třída C) a v kontrolní skupině u 8 zdravých subjektů s normální funkcí jater. AUC abirateronu se zvýšilo přibližně o 600 % a podíl volného léčiva se zvýšil o 80 % u subjektů se závažnou poruchou funkce jater ve srovnání s jedinci s normální funkcí jater.
U pacientů s mírnou poruchou funkce jater není nutná úprava dávkování.
U pacientů se středně závažnou poruchou funkce jater je nutno užití abirateron-acetátu důkladně posoudit, přínos by měl jasně převažovat možné riziko (viz body 4.2 a 4.4). Pacientům se závažnou poruchou funkce jater se abirateron-acetát nesmí podávat (viz body 4.2, 4.3 a 4.4).
U pacientů, u kterých vznikne hepatotoxicita během léčby, může být nutné léčbu přerušit nebo dávku upravit (viz body 4.2 a 4.4).
Porucha funkce ledvin
Farmakokinetika abirateron-acetátu byla srovnávána u pacientů s onemocněním ledvin v konečném stadiu stabilizovaných na hemodialýze proti kontrolním subjektům s normální funkcí ledvin. Systémová expozice abirateronu po jednorázovém perorálním podání dávky 1 000 mg se u pacientů s onemocněním ledvin v konečném stadiu na hemodialýze nezvýšila. Podání u pacientů s poruchou funkce ledvin, včetně závažné poruchy funkce ledvin, nevyžaduje snížení dávky (viz bod 4.2).
U pacientů s karcinomem prostaty a závažnou poruchou funkce ledvin však není klinická zkušenost.
U těchto pacientů se doporučuje opatrnost.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ve všech studiích toxicity na zvířatech byly hladiny cirkulujícího testosteronu výrazně sníženy.
V důsledku toho byly pozorovány snížení hmotnosti orgánů, morfologické a/nebo histopatologické změny reprodukčních orgánů, nadledvin, hypofýzy a mléčných žláz. Všechny změny byly úplně nebo částečně reverzibilní. Změny na reprodukčních orgánech a na orgánech citlivých na androgeny jsou konzistentní s farmakologií abirateronu. Všechny hormonální změny související s léčbou se znormalizovaly nebo se zdálo, že se normalizují po 4týdenním rekonvalescenčním období.
Ve studiích fertility jak u samců tak i samic potkanů snižoval abirateron-acetát fertilitu, což bylo kompletně reverzibilní mezi 4. až 16. týdnem po ukončení jeho podávání.
Ve studiích vývojové toxicity u potkanů ovlivňoval abirateron-acetát březost, včetně snížení hmotnosti plodu a přežití. Byly pozorovány účinky na externí pohlavní orgány, ačkoli abirateron-acetát nebyl teratogenní.
V těchto studiích fertility a vývojové toxicity provedených u potkanů byly všechny účinky spojené s farmakologickou aktivitou abirateronu.
Kromě změn na reprodukčních orgánech pozorovaných ve studiích toxicity na zvířatech neukazují předklinické údaje založené na konvenčních studiích bezpečnosti, toxicity po opakovaném podání, genotoxicity a karcinogenního poteciálu na zvláštní riziko pro člověka. Abirateron-acetát nebyl karcinogenní v 6-měsíční studii transgenních myší (Tg.rasH2). Ve 24-měsíční studii karcinogenity u potkanů zvýšil abirateron-acetát incidenci nádorů z intersticiálních buněk varlat. Tento nález je dáván do souvislosti s farmakologickým působením abirateronu a je považován za specifický pro potkany. Abirateron-acetát nebyl karcinogenní u samic potkanů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mikrokrystalická celulosa
Sodná sůl kroskarmelosy
Monohydrát laktosy
Magnesium-stearát
Povidon K 29-32
Koloidní bezvodý oxid křemičitý
Natrium-lauryl-sulfát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a obsah balení
Kulaté bílé HDPE lahvičky uzavřené polypropylenovým dětským bezpečnostním uzávěrem obsahující 120 tablet. Každé balení obsahuje jednu lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Vzhledem k mechanismu účinku může tento léčivý přípravek poškodit vyvíjející se plod; proto by těhotné ženy a ženy, které mohou otěhotnět, neměly zacházet s přípravkem bez ochrany, např. rukavic.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/714/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 05. září 2011 Datum posledního prodloužení:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Janssen-Cilag S.p.A
Via C. Janssen
IT-04100 Borgo San Michele
Latina
Itálie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovnaých zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměrů přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ZYTIGA 250 mg tablety Abirateroni acetas
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje abirateroni acetas 250 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu a sodík.
Další údaje viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
120 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Užijte přípravek ZYTIGA alespoň dvě hodiny po jídle a nic nejezte alespoň jednu hodinu po užití přípravku ZYTIGA.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Těhotné ženy a ženy, které mohou otěhotnět, nesmějí zacházet s přípravkem ZYTIGA bez rukavic.
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
Nepoužitý odpad zlikvidujte v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/714/001
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ZYTIGA
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ZYTIGA 250 mg tablety Abirateroni acetas
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje abirateroni acetas 250 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu a sodík.
Další údaje viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
120 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Užijte přípravek ZYTIGA alespoň dvě hodiny po jídle a nic nejezte alespoň jednu hodinu po užití přípravku ZYTIGA.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Těhotné ženy a ženy, které mohou otěhotnět, nesmějí zacházet s přípravkem ZYTIGA bez rukavic.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
Nepoužitý odpad zlikvidujte v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/714/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
ZYTIGA 250 mg tablety
abirateroni acetas
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je ZYTIGA a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek ZYTIGA užívat
3. Jak se ZYTIGA užívá
4. Možné nežádoucí účinky
5. Jak přípravek ZYTIGA uchovávat
6. Obsah balení a další informace
1. Co je ZYTIGA a k čemu se používá
ZYTIGA obsahuje léčivou látku zvanou abirateron-acetát. Užívá se k léčbě rakoviny prostaty u dospělých mužů, která se rozšířila do dalších částí těla. ZYTIGA brání Vašemu tělu produkovat testosteron; to může zpomalit růst zhoubného nádoru prostaty.
Užíváte-li tento léčivý přípravek, lékař Vám také předepíše další léčivý přípravek nazývaný prednison nebo prednisolon. To sníží riziko vysokého krevního tlaku, zadržování tekutin v těle (retence tekutin) nebo nízkých hladin chemické látky známé jako draslík v krvi.
2. Čemu musíte věnovat pozornost, než začnete přípravek ZYTIGA užívat Neužívejte přípravek ZYTIGA
- Jestliže jste alergický na abirateron-acetát nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodu 6).
- Pokud jste žena, a zejména pokud jste těhotná. ZYTIGA je určena pouze pro podání mužům..
- Máte-li závažnou poruchu funkce jater.
Neužívejte tento léčivý přípravek, pokud se Vás cokoli z výše uvedeného týká. Nejste-li si jistý, poraďte se před užíváním tohoto léčivého přípravku s lékařem nebo lékárníkem.
Upozornění a opatření
Před užitím přípravku Zytiga se poraďte se svým lékařem:
- pokud máte problémy s játry;
- pokud Vám bylo sděleno, že máte vysoký krevní tlak nebo srdeční selhání nebo nízkou hladinu draslíku v krvi (nízká hladina draslíku v krvi může zvýšit riziko poruchy srdečního rytmu);
- pokud máte jiné problémy se srdcem nebo cévami;
- pokud máte nepravidelný nebo rychlý srdeční tep;
- pokud jste dušný;
- pokud jste rychle přibral na tělesné hmotnosti;
- otékají-li Vám chodidla, kotníky nebo dolní končetiny;
- pokud jste dříve na rakovinu prostaty užíval léčivý přípravek zvaný ketokonazol;
- o potřebě užívat tento léčivý přípravek s prednisonem nebo prednisolonem;
- o možných účincích na kosti;
- pokud máte vysokou hladinu cukru v krvi.
Informujte svého lékaře, pokud Vám bylo sděleno, že máte jakékoli srdeční nebo cévní onemocnění, včetně potíží se srdečním rytmem (arytmie) nebo užíváte přípravky k léčbě těchto onemocnění.
Informujte svého lékaře, pokud máte zežloutnutí kůže nebo očí, ztmavnutí moči, silný pocit na zvracení nebo zvracení, protože tyto příznaky mohou být známkami nebo symptomy jaterních potíží. Vzácně se může objevit selhání jaterních funkcí (nazývané akutní selhání jater), což může vést k úmrtí.
Může se vyskytnout pokles počtu červených krvinek, snížení sexuální touhy, svalová slabost a/nebo bolest svalů.
Pokud si nejste jistý, zda se Vás něco z výše uvedeného týká, poraďte se před užíváním tohoto léčivého přípravku s lékařem nebo lékárníkem.
Kontroly krve
ZYTIGA může mít vliv na játra, ale nemusejí se objevit žádné příznaky. Užíváte-li tento léčivý přípravek, bude lékař pravidelně kontrolovat Vaši krev a sledovat účinky na Vaše játra.
Děti a dospívající
Tento přípravek není určen k používání u dětí a dospívajících. Pokud by nedopatřením došlo ke spolknutí přípravku ZYTIGA dítětem nebo dospívajícím, okamžitě vyhledejte nemocniční zařízení a vezměte s sebou příbalovou informaci, abyste ji ukázali na pohotovosti lékaři.
Další léčivé přípravky a ZYTIGA
Poraďte se s lékařem nebo lékárníkem před užíváním jakéhokoli léčivého přípravku.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste užíval v nedávné době, nebo které možná budete užívat. Je to důležité, protože ZYTIGA může zvyšovat účinek řady léčivých přípravků včetně léčivých přípravků na srdce, utišujících přípravků, rostlinných přípravků (např. třezalka tečkovaná) a dalších. Lékař může chtít změnit dávkování těchto léčivých přípravků. Některé léčivé přípravky mohou také zvýšit nebo snížit účinek přípravku ZYTIGA. To pak může vést k nežádoucím účinkům nebo ZYTIGA nebude účinkovat tak, jak by měla.
Další přípravky užívané s přípravkem ZYTIGA
Užívání androgen - deprivačních přípravků může zvyšovat riziko poruchy se srdečního rytmu Informujte svého lékaře, pokud užíváte přípravky
- užívané k léčbě poruch srdečního rytmu (např. chinidin, prokainamid, amiodaron, sotalol);
- o kterých je známo, že zvyšují riziko poruch srdečního rytmu [např. methadon (používaný k úlevě od bolesti a jako součást detoxikační léčby u drogových závislostí), moxifloxacin (antibiotikum), antipsychotika (užívaná k léčbě závažných duševních onemocnění)].
ZYTIGA s jídlem
- Tento léčivý přípravek se nesmí užívat s jídlem (viz bod 3 „Užívání léčivého přípravku“).
- Užívání přípravku ZYTIGA s jídlem může způsobovat nežádoucí účinky.
Těhotenství a kojení
ZYTIGA není určena k podání ženám.
- Tento léčivý přípravek může uškodit nenarozenému dítěti, pokud je užíván těhotnými ženami.
- Těhotné ženy nebo ženy, které by mohly být těhotné, musejí nosit rukavice, pokud potřebují zacházet s přípravkem ZYTIGA nebo se ho dotýkat.
- Máte-li pohlavní styk se ženou, která může otěhotnět, použijte kondom a jinou účinnou metodu kontroly početí. Máte-li pohlavní styk s těhotnou ženou, použijte kondom, abyste ochránil nenarozené dítě.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by tento léčivý přípravek ovlivňoval Vaši schopnost řídit a obsluhovat stroje.
ZYTIGA obsahuje laktosu a sodík
- ZYTIGA obsahuje laktosu (druh cukru). Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
- Tento léčivý přípravek obsahuje také přibližně 27 mg sodíku ve čtyřech tabletách denní dávky. To je nutno vzít v úvahu u pacientů, kteří mají dietu s omezeným přísunem sodíku.
3. Jak se ZYTIGA užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý, poraďte se se svým lékařem nebo lékárníkem.
Kolik se užívá
Doporučená dávka je 1 000 mg (čtyři tablety) jednou denně.
Užívání léčivého přípravku
- Přípravek užívejte perorálně.
- Neužívejte přípravek ZYTIGA s jídlem.
- Užijte přípravek ZYTIGA alespoň dvě hodiny po jídle a nic nejezte alespoň jednu hodinu po užití přípravku ZYTIGA (viz bod 2 „ZYTIGA s jídlem“).
- Tablety polkněte celé a zapijte vodou.
- Tablety nelamte.
- ZYTIGA se užívá spolu s léčivým přípravkem nazývaným prednison nebo prednisolon.
Užívejte prednison nebo prednisolon přesně dle pokynů svého lékaře.
- Během užívání přípravku ZYTIGA je nutno užívat prednison nebo prednisolon každý den.
- Je možné, že z naléhavých zdravotních důvodů bude třeba dávku prednisonu nebo prednisolonu změnit. Lékař Vám řekne, pokud bude potřeba změnit množství užívaného prednisonu nebo prednisolonu. Nepřestávejte užívat prednison nebo prednisolon, pokud Vám to lékař nedoporučil.
Během užívání přípravku ZYTIGA a prednisonu nebo prednisolonu Vám může lékař předepsat i jiné léčivé přípravky.
Jestliže jste užil více přípravku ZYTIGA, než jste měl
Jestliže jste užil více přípravku, než jste měl, poraďte se s lékařem nebo jděte okamžitě do nemocnice. Jestliže jste zapomněl užít přípravek ZYTIGA
- Jestliže jste zapomněl užít přípravek ZYTIGA nebo prednison nebo prednisolon, užijte obvyklou dávku následující den.
- Jestliže jste zapomněl užívat přípravek ZYTIGA nebo prednison nebo prednisolon po dobu více než jednoho dne, poraďte se okamžitě s lékařem.
Jestliže jste přestal užívat přípravek ZYTIGA
Nepřestávejte užívat přípravek ZYTIGA nebo prednison nebo prednisolon, pokud Vám to lékař nedoporučil.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte užívat přípravek ZYTIGA a okamžitě vyhledejte lékaře, pokud se objeví cokoli z dále uvedeného:
- Svalová slabost, svalové záškuby nebo bušení srdce (palpitace). Mohou to být příznaky nízké hladiny draslíku v krvi.
Další nežádoucí účinky jsou:
Velmi časté (mohou postihnout více než 1 z 10 osob):
Hromadění tekutiny v dolních končetinách, nízká hladina draslíku v krvi, vysoký krevní tlak, infekce močových cest, průjem.
Časté (mohou postihnout až 1 z 10 osob):
Vysoké hladiny tuků v krvi, zhoršení výsledků jaterních testů, bolest na hrudi, poruchy srdečního rytmu, srdeční selhání, rychlý srdeční tep, závažné infekce nazývané sepse, zlomeniny kostí, poruchy trávení, krev v moči, vyrážka.
Méně časté (mohou postihnout až 1 ze 100 osob):
Problémy s nadledvinami (spojeno s problémy se solí a vodou), svalová slabost a/nebo bolest svalů. Vzácné (mohou postihnout až 1 z 1 000 osob):
Podráždění plic (také nazýváno alergická alveolitida).
Selhání jaterních funkcí (také nazývané aktuní selhání jater).
Není známo (frekvenci z dostupných údajů nelze určit)
Infarkt, změny na EKG - elektrokardiogramu (prodloužení QT intervalu).
U mužů léčených kvůli nádoru prostaty se může vyskytnout úbytek kostní hmoty. ZYTIGA v kombinaci s prednisonem nebo prednisolonem může úbytek kostní hmoty zvýšit.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání informací o bezpečnosti tohoto přípravku.
5. Jak přípravek ZYTIGA uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za výrazem „Použitelné do“ a na štítku lahvičky za zkratkou „EXP“. Doba použitelnosti se vztahuje
k poslednímu dni uvedeného měsíce.
- Uchovávejte při teplotě do 30 °C.
- Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co ZYTIGA obsahuje
- Léčivou látkou je abirateroni acetas. Jedna tableta obsahuje abirateroni acetas 250 mg.
- Pomocnými látkami jsou mikrokrystalická celulosa, sodná sůl kroskarmelosy, monohydrát laktosy, magnesium-stearát, povidon K 29-32, koloidní bezvodý oxid křemičitý a natrium-lauryl-sulfát (viz bod 2 „ZYTIGA obsahuje laktosu a sodík“).
Jak ZYTIGA vypadá a co obsahuje toto balení
ZYTIGA jsou bílé až téměř bílé oválné tablety označené na jedné straně „AA250“.
- Tablety se dodávají v plastové lahvičce s dětským bezpečnostním uzávěrem. Každá lahvička obsahuje 120 tablet. Každá krabička obsahuje jednu lahvičku.
Držitel rozhodnutí o registraci
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
Výrobce
Janssen-Cilag SpA Via C. Janssen Borgo San Michele I-04100 Latina, Itálie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Tel/Tél: +32 14 64 94 11 |
Lietuva UAB "JOHNSON & JOHNSON' Geležinio Vilko g. 18A LT-08104 Vilnius Tel: +370 5 278 68 88 |
|
Eunrapnu „„fl^OHCbH & fl^OHCtH Etarapna” EOOfl ^.k. Mnagocr 4 En3Hec napK Co$na, crpaga 4 Co^na 1766 Ten.: +359 2 489 94 00 |
Luxembourg/Luxemburg Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Belgique/Belgien Tel/Tél: +32 14 64 94 11 |
|
Česká republika Janssen-Cilag s.r.o. Karla Engliše 3201/06 CZ-150 00 Praha 5 Tel. +420 227 012 227 |
Magyarország Janssen-Cilag Kft. Nagyenyed u. 8-14 H-Budapest, 1123 Tel.: +36 1 884 2858 |
|
Danmark Janssen-Cilag A/S Bregneredvej 133 DK-3460 Birkered Tlf: +45 45 94 82 82 |
Malta Am Mangion Ltd. Mangion Building, Triq Gdida fi Triq Valletta MT - Luqa LQA 6000 Tel: +356 2397 6000 |
|
Deutschland Janssen-Cilag GmbH Johnson & Johnson Platz 1 D-41470 Neuss Tel: +49 2137 955 955 |
Nederland Janssen-Cilag BV Dr. Paul Janssenweg 150 NL-5026 RH Tilburg Tel: +31 13 583 73 73 |
|
Eesti UAB "JOHNSON & JOHNSON" Eesti filiaal LSStsa 2 EE-11415 Tallinn Tel: +372 617 7410 |
Norge Janssen-Cilag AS Drammensveien 288 NO-0283 Oslo Tlf: +47 24 12 65 00 |
|
EXXáSa Janssen-Cilag Oap^aKsuxix^ A.E.B.E. Asm^ópog Eip^vn? 56 GR-151 21 nsÚKn, A0^va Tn^: +30 210 80 90 000 |
Osterreich Janssen-Cilag Pharma GmbH VorgartenstraBe 206B AT-1020 Wien Tel: +43 1 610 300 |
|
Espaňa Janssen-Cilag, S.A. Paseo de las Doce Estrellas, 5-7 Campo de las Naciones E-28042 Madrid Tel: +34 91 722 81 00 |
Polska Janssen-Cilag Polska Sp. z o.o. ul. Uzecka 24 PL-02-135 Warszawa Tel.+48 22 237 60 00 |
|
France Janssen-Cilag 1, rue Camille Desmoulins, TSA 91003 F-92787 Issy Les Moulineaux, Cedex 9 Tél: 0 800 25 50 75 / +33 1 55 00 40 03 |
Portugal Janssen-Cilag Farmaceutica, LDA. Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835 |
|
Hrvatska Johnson & Johnson S.E. d.o.o. Oreškoviceva 6h 10010 Zagreb Tel: +385 1 6610 700 |
Románia Johnson & Johnson Románia SRL Strada Tipografilor Nr. 11-15, Cládirea S-Park, Corp B3-B4, Etaj 3 013714 Bucure§ti. RO Tel: +40 21 207 18 00 |
|
Ireland Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG - UK Tel: +44 1494 567 444 |
Slovenija Johnson & Johnson d.o.o. Šmartinska 53 SI-1000 Ljubljana Tel: +386 1 401 18 30 |
|
Ísland Janssen-Cilag c/o Vistor hf. Horgatún 2 IS-210 Garóab^r Sími: +354 535 7000 |
Slovenská republika Johnson & Johnson, s.r.o. CBC III, Karadžičova 12 SK--821 08 Bratislava Tel: +421 232 408 400 |
|
Italia Janssen-Cilag SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI Tel: +39 02 2510 1 |
Suomi/Finland Janssen-Cilag Oy Vaisalantie/Vaisalavágen 2 FI-02130 Espoo/Esbo Puh/Tel: +358 207 531 300 |
|
Kúnpoq Aiavs^sxai anó: BapváPag Xax^nnavay^g AxS Asm^ópog riávvou KpaviSiróxn 226 CY-2234 AsuKmoía Tn^: ++357 22 207 700 |
Sverige Janssen-Cilag AB Box 4042 SE-16904 Solna Tel: +46 8 626 50 00 |
Latvija
United Kingdom
Janssen-Cilag Ltd.
50-100 Holmers Farm Way High Wycombe
Buckinghamshire HP12 4EG - UK Tel: +44 1494 567 444
UAB "JOHNSON & JOHNSON" filiale Latvija Mukusalas iela 101 Riga, LV-1004 Tel: +371 678 93561
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
36