Zydelig 100 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Zydelig 100 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje idelalisibum 100 mg.
Pomocná látka/pomocné látky se známým účinkem: Jedna tableta obsahuje 0,1 mg oranžové žluti (E110) (viz bod 4.4).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Oranžová, oválná, potahovaná tableta o rozměrech 9,7 mm krát 6,0 mm, s vyraženým označením „GSI“ na jedné straně a „100“ na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Zydelig je indikován v kombinaci s rituximabem k léčbě dospělých pacientů s chronickou lymfatickou leukémií (CLL):
• kteří podstoupili alespoň jednu předchozí léčbu (viz bod 4.4) nebo
• jako pokračující léčba u pacientů s delecí 17p nebo mutací TP53, kteří nejsou vhodní pro chemo-imunoterapii a kteří již zahájili léčbu přípravkem Zydelig v rámci léčby první linie (viz bod 4.4).
Přípravek Zydelig je indikován v monoterapii k léčbě dospělých pacientů s folikulárním lymfomem (FL), který je refrakterní na dvě předchozí linie léčby (viz bod 4.4).
4.2 Dávkování a způsob podání
Léčbu přípravkem Zydelig má provádět lékař se zkušenostmi s protinádorovou léčbou.
Dávkování
Doporučená dávka přípravku Zydelig je 150 mg perorálně, dvakrát denně. V léčbě je třeba pokračovat až do progrese onemocnění nebo nepřijatelné toxicity.
Jestliže pacient vynechá dávku přípravku Zydelig a uplynulo méně než 6 hodin od doby, kdy je přípravek obvykle užíván, pacient má užít vynechanou dávku co nejdříve a vrátit se k obvyklému rozvrhu dávkování. Jestliže pacient vynechá dávku a uplynulo více než 6 hodin, pacient nemá užít vynechanou dávku, ale jednoduše se vrátit k obvyklému rozvrhu dávkování.
Úpravy dávkování
Zvýšení hladin jaterních transamináz
V případě, že se objeví zvýšení hladin aminotransferáz 3. nebo 4. stupně (alaninaminotransferáza [ALT]/aspartátaminotransferáza [AST] o > 5 x horní hranice normy [ULN, Upper Limit of Normal]), musí se léčba přípravkem Zydelig přerušit. Jakmile se hodnoty vrátí na 1. stupeň nebo níže (ALT/AST < 3 x ULN), může se léčba obnovit v dávce 100 mg dvakrát denně.
Pokud se příhoda nezopakuje, může ošetřující lékař podle svého uvážení znovu zvýšit dávku na 150 mg dvakrát denně.
Jestliže se příhoda zopakuje, musí se léčba přípravkem Zydelig přerušit až do návratu hodnot 1. stupně nebo níže, a poté může ošetřující lékař podle svého uvážení začít znovu podávat dávku 100 mg dvakrát denně (viz body 4.4 a 4.8).
Průjem/kolitida
V případě, že se objeví průjem/kolitida 3. nebo 4. stupně, musí se léčba přípravkem Zydelig přerušit. Jakmile se průjem/kolitida upraví na 1. stupeň nebo lepší, může se léčba obnovit v dávce 100 mg dvakrát denně. Pokud se průjem/kolitida nevrátí, může ošetřující lékař podle svého uvážení znovu zvýšit dávku na 150 mg dvakrát denně (viz bod 4.8).
Pneumonitida
V případě, že se předpokládá výskyt pneumonitidy, musí se léčba přípravkem Zydelig přerušit.
Jakmile pneumonitida ustoupí a pokud je vhodná opakovaná léčba, může se zvážit obnovení léčby v dávce 100 mg dvakrát denně (viz body 4.4 a 4.8).
V případě, že se objeví vyrážka 3. nebo 4. stupně, musí se léčba přípravkem Zydelig přerušit. Jakmile se vyrážka upraví na 1. stupeň nebo lepší, může se léčba obnovit v dávce 100 mg dvakrát denně.
Pokud se vyrážka nevrátí, může ošetřující lékař podle svého uvážení znovu zvýšit dávku na 150 mg dvakrát denně (viz bod 4.8).
Neutropenie
Léčba přípravkem Zydelig musí být přerušena u pacientů s absolutním počtem neutrofilů (ANC) nižším než 500/mm3. ANC musí být monitorován minimálně jednou týdně, dokud není ANC > 500/mm3, poté může být léčba znovu zahájena dávkou 100 mg dvakrát denně (viz bod 4.4).
|
ANC 1 000 až < 1 500/mm3 |
ANC 500 až < 1 000/mm3 |
ANC < 500/mm3 |
|
Udržujte dávkování přípravku Zydelig. |
Udržujte dávkování přípravku Zydelig. Monitorujte ANC minimálně jednou týdně. |
Přerušte podávání přípravku Zydelig. Monitorujte ANC minimálně jednou týdně dokud ANC není > 500/mm3 a poté můžete znovu zahájit podávání přípravku Zydelig v dávce 100 mg dvakrát denně. |
Zvláštní populace pacientů Starší pacienti
U starších pacientů (ve věku > 65 let) není nutná specifická úprava dávkování (viz bod 5.2).
Porucha funkce ledvin
U pacientů s mírnou, středně těžkou nebo těžkou poruchou funkce ledvin není nutná úprava dávkování (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou nebo středně těžkou poruchou funkce jater není při zahájení léčby přípravkem Zydelig nutná úprava dávkování, doporučuje se však intenzivnější sledování nežádoucích účinků (viz body 4.4 a 5.2).
Údaje pro doporučení dávky u pacientů s těžkou poruchou funkce jater nejsou dostatečné. Proto se doporučuje zvýšená opatrnost a intenzivnější sledování nežádoucích účinků při podávání přípravku Zydelig u této populac e (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Zydelig u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Zydelig je určen k perorálnímu podání. Pacienty je třeba poučit, aby tablety polykali celé. Potahované tablety se nesmí rozkousnout ani rozdrtit. Potahované tablety se mohou užívat spolu s jídlem nebo bez jídla (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Závažné infekce
Léčba přípravkem Zydelig se nesmí zahájit pokud má pacient známky jakékoliv systémové bakteriální, mykotické nebo virové infekc e.
Při podávání idelalisibu se vyskytly závažné a fatální infekce, včetně oportunních infekcí, jako je pneumonie vyvolaná Pneumocystisjirovecii (PJP) a cytomegalovirem (CMV). Proto během léčby idelalisibem musí být všem pacientům podávána profylaxe PJP.
U pacientů mají být v průběhu léčby monitorovány známky a symptomy respirační infekce. Pacienty je třeba poučit, aby ihned nahlásili nové respirační příznaky.
Má být prováděn pravidelný klinický a laboratorní screening na infekci vyvolanou CMV. Léčba přípravkem Zydelig má být ukončena u pacientů s průkazem infekce nebo virémie.
Neutropenie
U pacientů léčených idelalisibem docházelo ve spojení s léčbou k neutropenii stupně 3 nebo 4, včetně febrilní neutropenie. Po dobu prvních 6 měsíců léčby idelalisibem je třeba monitorovat krevní obraz u všech pacientů minimálně každé 2 týdny a u pacientů s ANC nižším než 1 000/mm3 minimálně jednou týdně (viz bod 4.2).
Zvýšení hladin transamináz
V klinických studiích s idelalisibem bylo pozorováno zvýšení hladin ALT a AST 3. a 4. stupně (> 5 x ULN). Tyto laboratorní nálezy byly obecně pozorovány během prvních 12 týdnů léčby, nálezy byly obecně asymptomatické a po přerušení podávání přípravku byly reverzibilní. U většiny pacientů byla léčba obnovena v nižší dávce bez recidivy (viz bod 4.2). Po dobu prvních 3 měsíců léčby je nutné u všech pacientů každé 2 týdny sledovat hladiny ALT, AST a celkového bilirubinu, poté se mají tyto hodnoty sledovat podle klinické indikace. Pokud se zjistí zvýšení hladin ALT a/nebo AST 2. stupně nebo více, je nutné pacienty sledovat každý týden až do poklesu hodnot na 1. stupeň nebo níže.
Průjem/kolitida
Případy těžké kolitidy související s lékem se vyskytovaly relativně pozdě (v řádu měsíců) po zahájení léčby, někdy s rychlým zhoršením, ustoupily však do několika týdnů po přerušení podávání léku a dodatečné symptomatické léčbě (např. protizánětlivá léčiva, jako je enterálně podávaný budesonid).
K dispozici je velmi málo zkušeností s léčbou pacientů s anamnézou zánětlivého střevního onemocnění.
Pneumonitida
V klinických studiích s idelalisibem byly hlášeny případy pneumonitidy. Pacienti, u nichž se objeví závažné plicní příhody, které nereagují na konvenční antibakteriální léčbu, se mají vyšetřit na léky indukovanou pneumonitidu. Pokud se předpokládá pneumonitida, je třeba léčbu idelalisibem přerušit a pacienta vhodně léčit. Při středně těžké nebo těžké symptomatické pneumonitidě se musí léčba ukončit.
Stevens-Johnsonův syndrom a toxická epidermální nekrolýza
Případy Stevens-Johnsonova syndromu (SJS) a toxické epidermální nekrolýzy (TEN) s fatálními následky byly hlášeny tehdy, když byl idelalisib podáván společně s jinými léčivými přípravky spojovanými s těmito syndromy. Pokud je podezření na SJS nebo TEN, je třeba léčbu idelalisibem okamžitě přerušit a pacienta vhodně léčit.
Induktory CYP3A
Při současném podávání s induktory CYP3A, jako jsou rifampicin, fenytoin, třezalka tečkovaná (Hypericumperforatum) nebo karbamazepin, se expozice idelalisibu může snížit. Jelikož snížení plazmatické koncentrace idelalisibu může vést ke snížené účinnosti přípravku, je třeba se vyhnout současnému podávání přípravku Zydelig se středně silnými nebo silnými induktory CYP3A (viz bod 4.5).
Substráty CYP3A
Primární metabolit idelalisibu, GS-563117, je silný inhibitor CYP3A4. Idelalisib má proto potenciál vzájemně působit s léčivými přípravky, které jsou metabolizovány CYP3A, což může vést ke zvýšeným sérovým koncentracím dalšího přípravku (viz bod 4.5). Při podávání idelalisibu současně s jinými léčivými přípravky je třeba postupovat podle doporučení ohledně jejich současného podávání s inhibitory CYP3A4 uvedených v souhrnu údajů o přípravku (SPC) těchto přípravků. Je třeba se vyhnout současné léčbě idelalisibem se substráty CYP3A způsobující závažné a/nebo život ohrožující nežádoucí účinky (např. alfuzosin, amiodaron, cisaprid, pimozid, chinidin, ergotamin, dihydroergotamin, kvetiapin, lovastatin, simvastatin, sildenafil, midazolam, triazolam), a pokud je to možné, je třeba použít alternativní léčivé přípravky, které jsou méně citlivé na inhibici CYP3A4.
Porucha funkce jater
U pacientů s poruchou funkce jater se doporučuje intenzivnější sledování nežádoucích účinků, protože se u této populace očekává zvýšená expozice, a to zvláště u pacientů se závažnou poruchou funkce jater. Do klinických studií s idelalisibem nebyli zařazeni žádní pacienti s těžkou poruchou funkce jater. Při podávání přípravku Zydelig u této populac e se doporučuje zvýšená opatrnost.
Chronická hepatitida
Idelalisib nebyl studován u pacientů s chronickou aktivní hepatitidou, včetně virové hepatitidy. Při podávání přípravku Zydelig pacientům s aktivní hepatitidou je třeba postupovat opatrně.
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby idelalisibem a ještě 1 měsíc po ukončení terapie používat vysoce účinnou antikoncepci (viz bod 4.6). Ženy užívající hormonální antikoncepci mají navíc používat bariérovou metodu jako druhou formu antikoncepce, protože v současné době není známo, jestli idelalisib snižuje účinnost hormonální antikoncepce.
Pomocné látky
Přípravek Zydelig obsahuje azobarvivo oranžovou žluť (E110), které může způsobovat alergické reakce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Idelalisib je primárně metabolizován aldehydoxidázou a v menší míře CYP3A a glukuronidací (UGT1A4). Jeho primárním metabolitem je farmakologicky neaktivní GS-563117. Idelalisib a GS-563117 jsou substráty P-gp a BCRP.
Vliv jiných léčivých přípravků na farmakokinetické vlastnosti idelalisibu
Induktory CYP3A
Klinická studie lékových interakcí zjistila, že současné podání jedné dávky 150 mg idelalisibu s rifampicinem (silný induktor CYP3A) vedlo k ~75% snížení hodnoty AUCinf idelalisibu. Je třeba se vyhnout současnému podávání přípravku Zydelig se středně silnými nebo silnými induktory CYP3A, jako jsou rifampicin, fenytoin, třezalka tečkovaná nebo karbamazepin, jelikož to může vést ke snížené účinnosti přípravku (viz bod 4.4).
Inhibitory CYP3A/P-gp
Klinická studie lékových interakcí zjistila, že současné podání jedné dávky 400 mg idelalisibu s ketokonazolem (silný inhibitor CYP3A, P-gp a BCRP) v dávce 400 mg jednou denně vedlo k 26% zvýšení hodnoty Cmax a 79% zvýšení hodnoty AUCinf idelalisibu. Při současném podávání idelalisibu s inhibitory CYP3A/P-gp se počáteční úprava dávkování idelalisibu nepovažuje za nutnou, doporučuje se však intenzivnějsí sledování nežádoucích účinků.
Vliv idelalisibu na farmakokinetické vlastnosti jiných léčivých přípravků
Substráty CYP3A
Primární metabolit idelalisibu, GS-563117, je silný inhibitor CYP3A Klinická studie lékových interakcí zjistila, že současné podávání idelalisibu s midazolamem (citlivý substrát CYP3A) vedlo k ~140% zvýšení hodnoty Cmax a ~440% zvýšení hodnoty AUCinf midazolamu v důsledku inhibice CYP3A způsobené GS-563117. Současné podávání idelalisibu se substráty CYP3A může zvyšovat jejich systémové expozice a zvyšovat nebo prodlužovat jejich terapeutickou aktivitu a nežádoucí účinky. In vitro byla inhibice CYP3A4 ireverzibilní a proto se návrat k normální enzymatické aktivitě očekává až za několik dnů po ukončení podávání idelalisibu.
Možné interakce mezi idelalisibem a současně podávanými léčivými přípravky, které jsou substráty CYP3A, jsou uvedeny v tabulce 1 (zvýšení je uvedeno jako „t”). Tento seznam není vyčerpávající a slouží pouze jako návod. Obecně je třeba postupovat podle doporučení ohledně jejich současného podávání s inhibitory CYP3A4 uvedených v SPC těchto přípravků (viz bod 4.4).
Tabulka 1: Interakce mezi idelalisibem a dalšími léčivými přípravky, které jsou substráty CYP3A
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
ANTAGONISTE ALFA-1 ADRENORECEPTORU | ||
|
Alfuzosin |
| koncentrace v séru |
Idelalisib se nemá podávat současně s alfuzosinem. |
|
ANALGETIKA | ||
|
Fentanyl, alfentanil, metadon, buprenorfin/naloxon |
| koncentrace v séru |
Doporučuje se pečlivé sledování nežádoucích účinků (např. respirační útlum, sedace). |
|
ANTIARYTMIKA | ||
|
Amiodaron, chinidin |
| koncentrace v séru |
Idelalisib se nemá podávat současně s amiodaronem ani chinidinem. |
|
Bepridil, disopyramid, lidokain |
| koncentrace v séru |
Doporučuje se klinické sledování. |
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
ANTINEOPLASTIKA | ||
|
Inhibitory tyrosinkinázy, jako je dasatinib a nilotinib, také vinkristin a vinblastin |
| koncentrace v séru |
Doporučuje se pečlivé sledování snášenlivosti těchto antineoplastik. |
|
ANTIKOAGULANCIA | ||
|
Warfarin |
| koncentrace v séru |
Při současném podávání a po ukončení léčby idelalisibem se doporučuje sledování hodnot mezinárodního normalizovaného poměru (INR) |
|
ANTIKONVULZIVA | ||
|
Karbamazepin |
| koncentrace v séru |
Je třeba sledovat hladiny antikonvulziv. |
|
ANTIDEPRESIVA | ||
|
Trazodon |
| koncentrace v séru |
Doporučuje se pečlivá titrace dávky antidepresiv a sledování antidepresivní odpovědi. |
|
LEKY PROTI DNE | ||
|
Kolchicin |
| koncentrace v séru |
Může být nutné snížení dávky kolchicinu. Idelalisib se nemá podávat současně s kolchicinem pacientům s poruchou funkce ledvin nebo jater. |
|
ANTIHYPERTENZIVA | ||
|
Amlodipin, diltiazem, felodipin, nifedipin, nikardipin |
| koncentrace v séru |
Doporučuje se klinické sledování terapeutického účinku a nežádoucích účinků. |
|
ANTIINFEKTIVA | ||
|
Antimykotika | ||
|
Ketokonazol, itrakonazol, posakonazol, vorikonazol |
| koncentrace v séru |
Doporučuje se klinické sledování. |
|
Antimykobakteriální látky | ||
|
Rifabutin |
| koncentrace v séru |
Doporučuje se zvýšené sledování nežádoucích účinků souvisejících s rifabutinem, včetně neutropenie a uveitidy. |
|
Inhibitory HCV proteázy | ||
|
Boceprevir, telaprevir |
| koncentrace v séru |
Doporučuje se klinické sledování. |
|
Makrolidová antibiotika | ||
|
Klarithromycin, telithromycin |
| koncentrace v séru |
U pacientů s normální funkcí ledvin nebo mírnou poruchou funkce ledvin (clearance kreatininu [CrCl] 60-90 ml/min) není nutná žádná úprava dávky klarithromycinu. U pacientů s CrCl < 90 ml/min se doporučuje klinické sledování. U pacientů s CrCl < 60 ml/min je třeba zvážit použití alternativního antibiotika. U telithromycinu se doporučuje klinické sledování. |
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
ANTIPSYCHOTIKA/NEUROLEPTIKA | ||
|
Kvetiapin, pimozid |
| koncentrace v séru |
Idelalisib se nemá podávat současně s kvetiapinem ani pimozidem. Mohou se zvážit alternativní léčivé přípravky, jako je olanzapin. |
|
Z 0 1 % < |
3VÝCH RECEPTORŮ | |
|
Bosentan |
| koncentrace v séru |
Je třeba postupovat opatrně a pacienty je třeba pečlivě sledovat s ohledem na toxicitu související s bosentanem. |
|
ERGOTAMINOVÉ ALKALOIDY | ||
|
Ergotamin, dihydroergotamin |
| koncentrace v séru |
Idelalisib se nemá podávat současně s ergotaminem ani dihydroergotaminem. |
|
PROKINETIKA | ||
|
Cisaprid |
| koncentrace v séru |
Idelalisib se nemá podávat současně s cisapridem. |
|
GLUKOKORTIKOIDY | ||
|
Inhalační/nazální kortikosteroidy: budesonid, flutikason Perorální budesonid |
| koncentrace v séru | koncentrace v séru |
Doporučuje se klinické sledování. Doporučuje se klinické sledování ohledně zvýšených známek/příznaků účinků kortikosteroidů. |
|
INHIBITORY HMG CO-A REDUKTÁZY | ||
|
Lovastatin, simvastatin Atorvastatin |
| koncentrace v séru | koncentrace v séru |
Idelalisib se nemá podávat současně s lovastatinem ani simvastatinem. Doporučuje se klinické sledování a může se zvážit nižší počáteční dávka atorvastatinu. Může být rovněž zvážen přechod na pravastatin, rosuvastatin nebo pitavastatin. |
|
IMUNOSUPRESIVA | ||
|
Cyklosporin, sirolimus, takrolimus |
| koncentrace v séru |
Doporučuje se terapeutické sledování. |
|
INHALAČNÍ BETA-AGONISTÉ | ||
|
Salmeterol |
| koncentrace v séru |
Současné podávání salmeterolu a idelalisibu se nedoporučuje. Kombinace může vést ke zvýšenému riziku kardiovaskulárních nežádoucích účinků souvisejících se salmeterolem, včetně prodloužení QT intervalu, palpitací a sinusové tachykardie. |
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
INHIBITORY FOSFODIESTERÁZY | ||
|
Plicní arteriální hypertenze: | ||
|
Sildenafil |
| koncentrace v séru |
Idelalisib se nemá podávat současně se sildenafilem. |
|
Tadalafil |
| koncentrace v séru |
Při současném podávání tadalafilu s idelalisibem je třeba postupovat opatrně, včetně zvážení snížení dávky. |
|
Erektilní dysfunkce: | ||
|
Sildenafil, tadalafil |
| koncentrace v séru |
Při předepisování sildenafilu nebo tadalafilu s idelalisibem je nutná zvýšená opatrnost a může se zvážit snížení dávky a zvýšené sledování nežádoucích účinků. |
|
SEDATIVA/HYPNOTIKA | ||
|
Midazolam (perorální), triazolam |
| koncentrace v séru |
Idelalisib se nemá podávat současně s midazolamem (perorální) ani triazolamem. |
|
Buspiron, klorazepát, diazepam, estazolam, flurazepam, zolpidem |
| koncentrace v séru |
Doporučuje se sledování koncentrace sedativ/hypnotik a může se zvážit snížení dávky. |
Substráty CYP2C8
In vitro idelalisib inhiboval a indukoval CYP2C8, není ale známo, zda to vede k účinku in vivo na substráty CYP2C8. Doporučuje se postupovat opatrně při podávání přípravku Zydelig současně s léky s úzkým terapeutickým indexem, které jsou substráty CYP2C8 (paklitaxel).
Substráty indukovatelných enzymů (např. CYP2C9, CYP2C19, CYP2B6 a UGT)
In vitro byl idelalisib induktorem několika enzymů a není možné vyloučit riziko snížené expozice a tím snížené účinnosti substrátů indukovatelných enzymů, jako je CYP2C9, CYP2C19, CYP2B6 a UGT. Doporučuje se postupovat opatrně při podávání přípravku Zydelig současně s léky s úzkým terapeutickým indexem, které jsou substráty těchto enzymů (warfarin, fenytoin, S-mefenytoin).
SubstrátyBCRP, OATP1B1, OATP1B3 aP-gp
Současné podávání vícečetných dávek idelalisibu 150 mg dvakrát denně zdravým jedincům vedlo ke srovnatelným expozicím u rosuvastatinu (AUC 90% IS: 87, 121) a digoxinu (AUC 90% IS: 98, 111), což nenaznačuje žádnou klinicky významnou inhibici BCRP, OATP1B1/1B3 nebo systémového P-gp idelalisibem. Není možné vyloučit riziko inhibice P-gp v gastrointestinálním traktu, která by mohla vést ke zvýšené expozici citlivých substrátů intestinálního P-gp, jako je dabigatran-etexilát.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Na základě zjištění u zvířat může idelalisib způsobit poškození plodu. Ženy by se měly během užívání přípravku Zydelig a do 1 měsíce po ukončení léčby vyhnout otěhotnění. Proto musí ženy ve fertilním věku během léčby přípravkem Zydelig a ještě 1 měsíc po ukončení terapie používat vysoce účinnou antikoncepci. V současné době není známo, jestli idelalisib snižuje účinnost hormonální antikoncepce, a proto ženy užívající hormonální antikoncepci mají navíc používat bariérovou metodu jako druhou formu antikoncepce.
Údaje o podávání idelalisibu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Podávání přípravku Zydelig se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se idelalisib a jeho metabolity vylučují do lidského mateřského mléka.
Riziko pro kojené novorozence/děti nelze vyloučit.
Kojení má být během léčby přípravkem Zydelig přerušeno.
Fertilita
Údaje o účinku idelalisibu na fertilitu u člověka nejsou k dispozici. Studie na zvířatech naznačují možné škodlivé účinky idelalisibu na fertilitu a vývoj plodu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Zydelig nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí profilu bezpečnosti
Hodnocení nežádoucích účinků je založeno na jedné studii fáze 3 a sedmi studiích fáze 1 a 2. Studie 312-0116 fáze 3 byla randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie, ve které bylo 220 pacientů s dříve léčenou CLL randomizováno v poměru 1:1 pro užívání idelalisibu a rituximabu nebo placeba a rituximabu. Studie fáze 1 a 2 hodnotily bezpečnost idelalisibu u 490 pacientů s hematologickými malignitami, včetně 354 pacientů užívajících idelalisib (v jakékoli dávce) jako samostatné léčivo a 136 pacientů užívajících idelalisib v kombinaci s monoklonálními protilátkami anti-CD20.
Nejčastěji hlášené nežádoucí účinky během léčby idelalisibem jsou uvedeny v tabulce 2.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky hlášené pro idelalisib užívaný samostatně nebo v kombinaci s monoklonálními protilátkami anti-CD20 uvádí tabulka 2. Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů a četnosti. Četnosti jsou definovány následujícím způsobem: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 2: Nežádoucí účinky hlášené v klinických studiích u pacientů s hematologickými malignitami užívajících idelalisib
|
Nežádoucí účinek |
Jakýkoli stupeň |
Stupeň > 3 |
|
Infekce a infestace | ||
|
Infekce |
Velmi časté |
Velmi časté |
|
Poruchy krve a lymfatického systému | ||
|
Neutropenie |
Velmi časté |
Velmi časté |
|
Respirační, hrudní a mediastinálníporuchy | ||
|
Pneumonitida |
Časté |
Časté |
|
Gastrointestinální poruchy | ||
|
Průjem/kolitida |
Velmi časté |
Velmi časté |
|
Poruchy jater a žlučových cest | ||
|
Zvýšené hladiny transamináz |
Velmi časté |
Velmi časté |
|
Poruchy kůže a podkožní tkáně | ||
|
Vyrážka* |
Velmi časté |
Časté |
|
Stevens - Johns onův |
Vzácné |
Vzácné |
|
syndrom/toxická epidermální | ||
|
nekrolýza | ||
|
Celkové poruchy a reakce v místě aplikace | ||
|
Pyrexie |
Velmi časté |
Časté |
|
Vyšetření | ||
|
Zvýšené hladiny triglyceridů |
Velmi časté |
Časté |
* Zahrnuje preferované termíny exfoliativní dermatitida, léková erupce, vyrážka, erytematózní vyrážka, generalizovaná vyrážka, makulární vyrážka, makulopapulámí vyrážka, papulární vyrážka, svědivá vyrážka a exfoliativní vyrážka.
Popis vybraných nežádoucích účinků
Vyrážka byla obecně mírná až středně těžká a měla za následek ukončení léčby přibližně u 2 % pacientů. Ve studii 312-0116 se vyrážka (hlášená jako exfoliativní dermatitida, léková erupce, vyrážka, erytematózní vyrážka, generalizovaná vyrážka, makulární vyrážka, makulopapulámí vyrážka, papulární vyrážka a svědivá vyrážka) objevila u 24,5 % pacientů, kterým byl podáván idelalisib a rituximab, a u 6,5 % pacientů, jimž bylo podáváno placebo a rituximab. Z toho mělo 3,6 % pacientů, jimž byl podáván idelalisib a rituximab, a 0,9 % pacientů, kterým bylo podáváno placebo a rituximab, vyrážku 3. stupně a žádný pacient neměl nežádoucí účinek 4. stupně. Vyrážka obvykle ustoupila po léčbě (např. místní a/nebo perorální steroidy, difenhydramin) a v těžkých případech po přerušení podávání přípravku (viz bod 5.3, fototoxicita).
Stevens-Johnsonův syndrom a toxická epidermální nekrolýza (viz bod 4.4)
Případy SJS a TEN se vzácně vyskytly tehdy, když byl idelalisib podáván souběžně s jinými léčivými přípravky spojovanými s těmito syndromy (bendamustin, rituximab, alopurinol a amoxicilin). SJS nebo TEN se objevily do jednoho měsíce od podání kombinace léků a měly fatální následky.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování musí být pacient sledován, zda nevykazuje známky toxicity (viz bod 4.8). Léčba předávkování přípravkem Zydelig zahrnuje obecná podpůrná opatření včetně sledování vitálních funkcí a pozorování klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorová léčiva, další protinádorová léčiva, ATC kód: L01XX47 Mechanismus účinku
Idelalisib inhibuje fosfatidylinositol 3-kinázu p1105 (PI3K5), která je hyperaktivní u malignit B-buněk a je centrem mnoha signálních drah, které řídí proliferaci, přežití, usídlení (,ftoming“) a retenci maligních buněk v lymfoidních tkáních a kostní dřeni. Idelalisib je selektivní inhibitor vazby adenosin-5’-trifosfátu (ATP) na katalytickou doménu PI3K5, vedoucí k inhibici fosforylace fosfatidylinositolu, který je klíčovým lipidovým druhým poslem („secondmessenger“), a k zabránění fosforylace Akt (proteinkinázy B).
Idelalisib indukuje apoptózu a inhibuje proliferaci v buněčných liniích odvozených od maligních B-buněk a v primárních nádorových buňkách. Inhibicí signalizace prostřednictvím chemokinových receptorů CXCR4 a CXCR5, indukované chemokiny CXCL12 a CXCL13, způsobuje idelalisib inhibici usídlení (,fioming“) a retence maligních B-buněk v nádorovém mikroprostředí, včetně lymfoidních tkáních a kostní dřeně.
Farmakodynamické účinky
Účinek idelalisibu (150 mg a 400 mg) na interval QT/QTc byl hodnocen v placebem a aktivně (moxifloxacin 400 mg) kontrolované zkřížené studii u 40 zdravých jedinců. Při dávce 2,7násobně vyšší než je maximální doporučená dávka nezpůsobil idelalisib prodloužení intervalu QT/QTc (tj.
< 10 ms).
Klinická účinnost u chronické lymfatické leukémie
Idelalisib v kombinaci s imunoterapií
Studie 312-0116 byla randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze 3 zahrnující 220 pacientů s dříve léčenou CLL, kteří potřebovali léčbu, ale cytotoxická chemoterapie pro ně nebyla považována za vhodnou. Pacienti byli randomizováni v poměru 1:1 pro podání 8 cyklů rituximabu (první cyklus v dávce 375 mg/m2 povrchu těla [body surface area, BSA], následující cykly v dávce 500 mg/m2 BSA) v kombinaci buď s perorálně podávaným placebem dvakrát denně nebo s idelalisibem 150 mg užívaným dvakrát denně, až do progrese onemocnění nebo nepřijatelné toxicity.
Medián věku byl 71 let (rozsah: 47 až 92), přičemž 78,2 % pacientů bylo starších než 65 let; 65,5 % byli muži a 90,0 % byli běloši; 64,1 % mělo klinické stadium III nebo IV podle Raie a 55,9 % mělo klinické stadium C podle Bineta. U většiny pacientů byly přítomny nepříznivé cytogenetické prognostické faktory: 43,2 % mělo chromozomovou deleci 17p a/nebo mutaci nádorového proteinu 53 (Tumour Protein 53, TP53) a 83,6 % mělo nemutované geny pro variabilní oblast těžkého imunoglobulinového řetězce (Immunoglobulin Heavy Chain Variable region, IGHV). Medián doby od diagnózy CLL do randomizace byl 8,5 roku. Pacienti měli medián hodnocení funkční zdatnosti (Cumulative Illness Rating Score, CIRS) rovný 8. Medián počtu předchozích terapií byl 3,0. Téměř všem (95,9 %) pacientům byly dříve podány monoklonální protilátky anti-CD20. Primárním cílovým parametrem bylo přežití bez progrese (Progression Free Survival, PFS). Výsledky účinnosti jsou shrnuty v tabulkách 3 a 4. Kaplan-Meierovu křivku pro PFS uvádí obrázek 1.
Ve srovnání s kombinací rituximabu a placeba vedla léčba kombinací idelalisibu a rituximabu ke statisticky významnému i klinicky zásadnímu zlepšení tělesného, sociálního a funkčního stavu, jakož i hodnot podstupnic specifických pro leukemii podle indexu funkčního hodnocení léčby rakoviny (Functional Assessment of Cancer Therapy: Leukaemia, FACT-LEU) a ke statisticky významnému a klinicky zásadnímu zlepšení aspektů úzkosti, deprese a obvyklých aktivit hodnocených podle indexu EuroQoL Five-Dimensions (EQ-5D).
Tabulka 3: Výsledky účinnosti ze studie 312-0116
|
Idelalisib + R N = 110 |
Placebo + R N = 110 | |
|
PFS Medián (měsíce) (95% IS) |
19,4 (12,3; NR) |
6,5 (4,0; 7,3) |
|
Poměr rizik (95% IS) |
0,15 (0,09; 0,25) | |
|
P-hodnota |
< 0,0001 | |
|
ORR* n (%) (95% IS) |
92 (83,6 %) (75,4; 90,0) |
17 (15,5 %) (9,3; 23,6) |
|
Poměr šancí (95% IS) |
27,76 (13,40; 57,49) | |
|
P-hodnota |
< 0,0001 | |
|
LNR** n/N (%) (95% IS) |
102/106 (96,2 %) (90,6; 99,0) |
7/104 (6,7 %) (2,7; 13,4) |
|
Poměr šancí (95% IS) |
225,83 (65,56; 777,94) | |
|
P-hodnota |
< 0,0001 | |
|
OSA Medián (měsíce) (95% IS) |
NR (NR, NR) |
20,8 (14,8; NR) |
|
Poměr rizik (95% IS) |
0,34 (0,19; 0,60) | |
|
P-hodnota |
0,0001 | |
IS: interval spolehlivosti; R rituximab; n: počet pacientů s odpovědí; N: počet pacientů ve skupině, NR: nedosaženo. Analýzy PFS, celkové četnosti odpovědi na léčbu (OverallResponseRate, ORR) a četnosti odpovědi týkající se lymfatických uzlin (Lymph Node Response Rate, LNR) vycházely z hodnocení nezávislou hodnotící komisí (IndependentReview Committee, IRC).
* Hodnota ORRje definovánajako podíl pacientů, kteří dosáhli kompletní odpovědi (Complete Response, CR) nebo částečné odpovědi (Partial Response, PR) na základě kritérií odpovědi stanovených Národní onkologickou sítí (National Comprehensive Cancer Network, NCCN) (2013) a podle Chesona (2012).
** Hodnota LNRje definována jako podíl pacientů, kteří dosáhli > 50 % zmenšení součtu součinů největších kolmých průměrů indexovaných lézí. Do analýzy byli zahrnuti pouze pacienti, u nichž bylo k dispozici výchozí hodnocení a > 1 platné hodnocení po zahájení léčby.
A Analýza celkového přežití (OverallSurvival, OS) zahrnuje data od pacientů, kteří dostali placebo + R ve studii 312-0116 a následně dostali idelalisib v prodloužené studii na základě analýzy podle původního léčebného záměru (intent-to-treat).
Tabulka 4: Souhrn hodnot PFS a četnosti odpovědí v předem stanovených podskupinách ze studie 312-0116
|
Idelalisib + R |
Placebo + R | |
|
Delece 17p/mutace TP53 |
N = 46 |
N = 49 |
|
Medián PFS (měsíce) (95% IS) |
NR (12,3; NR) |
4,0 (3,7; 5,7) |
|
Poměr rizik (95% IS) |
0,13 (0,07; 0,27) | |
|
ORR (95% IS) |
84,8 % (71,1; 93,7) |
12,2 % (4,6; 24,8) |
|
Nemutovaný IGHV |
N = 91 |
N = 93 |
|
Medián PFS (měsíce) (95% IS) |
19,4 (13,9, NR) |
5,6 (4,0; 7,2) |
|
Poměr rizik (95% IS) |
0,14 (0,08; 0,23) | |
|
ORR (95% IS) |
82,4 % (73,0; 89,6) |
15,1 % (8,5; 24,0) |
|
Věk > 65 let |
N = 89 |
N = 83 |
|
Medián PFS (měsíce) (95% IS) |
19,4 (12,3; NR) |
5,7 (4,0; 7,3) |
|
Poměr rizik (95% IS) |
0,14 (0,08; 0,25) | |
|
ORR (95% IS) |
84,3 % (75,0; 91,1) |
16,9 % (9,5; 26,7) |
IS: interval spolehlivosti; R: rituximab; N: počet pacientů ve skupině; NR: nedosaženo
Obrázek 1: Kaplan-Meierova křivka PFS ze studie 312-0116 (populace podle původního léčebného záměru (intent-to-treat))
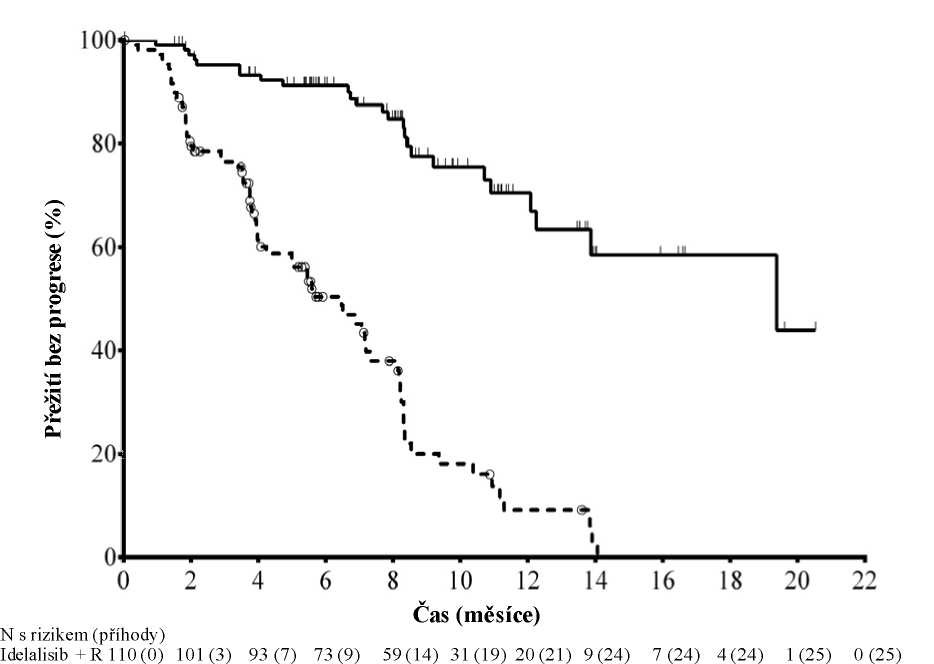
Placebo + R 110 (0) 84 (21) 48 (38) 29 (46) 20 (53) 9 (63) 4 (67) 1 (69) 0 (70) 0 (70) 0 (70) 0 (70)
Plná čára: idelalisib + R (N = 110), přerušovaná čára: placebo + R (N = 110)
R: rituximab; N: počet pacientů ve skupině
Analýza PFS vycházela z hodnocení nezávislou hodnotící komisí (Independent Review Committee, IRC). Pro pacienty ve skupině placeba + R obsahuje souhrn data až do podání první dávky idelalisibu v prodloužené studii. Medián PFS: idelalisib + R = 19,4 měsíce, placebo + R = 6,5 měsíce. Poměr rizik = 0,15; 95 % IS (0,09; 0,24); p < 0,0001.
Do studie 101-08/99 bylo zařazeno 64 pacientů s dosud neléčenou CLL, včetně 5 pacientů s lymfomem z malých lymfocytů (SLL). Pacienti dostávali idelalisib 150 mg dvakrát denně a rituximab 375 mg/m2 BSA jednou týdně. Hodnota ORR byla 96,9 %, přičemž 12 pacientů mělo CR (18,8 %) a 50 pacientů mělo PR (78,1 %), včetně 3 CR a 6 PR u pacientů s delecí 17p a/nebo mutací TP53 a 2 CR a 34 PR u pacientů s nemutovaným IGHV. Mediánu trvání odpovědi (Duration Of Response, DOR) nebylo dosaženo.
Klinická účinnost u folikulárního lymfomu
Bezpečnost a účinnost idelalisibu byly hodnoceny v jednoramenné multicentrické klinické studii (studie 101-09) prováděné u 125 pacientů s indolentním non-Hodgkinovým lymfomem z B-buněk (iNHL, včetně FL, n = 72; SLL, n = 28; lymfoplasmocytového lymfomu/Waldenstromovy makroglobulinémie [LPL/WM], n = 10; a lymfomu marginální zóny [MZL], n = 15). Všichni pacienti byli refrakterní na rituximab a 124 ze 125 pacientů bylo refrakterních nejméně na jednu alkylační látku. 112 (89,6 %) pacientů bylo refrakterních na poslední režim léčby před vstupem do studie.
Ze 125 zařazených pacientů 80 (64 %) byli muži, medián věku byl 64 let (rozsah: 33 až 87) a 110 (89 %) byli běloši. Pacientům byl podáván idelalisib 150 mg perorálně dvakrát denně až do progrese onemocnění nebo nepřijatelné toxicity.
Primárním cílovým parametrem byla hodnota ORR definovaná jako podíl pacientů, kteří dosáhli CR nebo PR (podle revidovaných kritérií odpovědi na léčbu maligního lymfomu [Cheson]), a u pacientů s Waldenstromovou makroglobulinémií nízkou odpověď (Minor Response, MR) (podle hodnocení odpovědi na léčbu u Waldenstromovy makroglobulinémie [Owen]). Hodnota DOR byla sekundárním cílovým parametrem a byla definována jako doba od první zdokumentované odpovědi (CR, PR nebo MR) do prvního zaznamenání progrese onemocnění nebo úmrtí pacienta z jakékoli příčiny. Výsledky účinnosti jsou shrnuty v tabulce 5.
Tabulka 5: Souhrn odpovědí u pacientů s FL léčených idelalisibem (podle hodnocení IRC)
|
Charakteristika |
Pacienti ve studii n (%) |
|
ORR (folikulární lymfom)* |
39 (54,2) |
|
95% IS |
42,0 - 66,0 |
|
ORR (všichni pacienti)* |
71 (56,8) |
|
95% IS |
47,6 - 65,6 |
|
Kategorie odpovědi (folikulární lymfom)* CR |
6 (8,3) |
|
PR |
33 (45,8) |
IS: interval spolehlivosti; n: počet pacientů s odpovědí
* Odpověď byla stanovena nezávislou hodnotící komisí (IRC); ORR = kompletní odpověď (CR) + částečná odpověď (PR).
Odhad mediánu DOR byl pro všechny pacienty 12,5 měsíce (12,5 měsíce pro pacienty s SLL, hodnoty nedosaženo u pacientů s FL, LPL/WM a MZL). Ze 122 pacientů s měřitelnými lymfatickými uzlinami ve výchozím stavu a po zahájení léčby bylo u 67 pacientů (54,9 %) dosaženo > 50% snížení součtu součinů průměrů indexovaných lézí. Z pacientů, kteří neodpověděli na léčbu, mělo 10 (8,0 %) progresivní onemocnění jako nejlepší odpověď a 2 (1,6 %) nebyli hodnotitelní. Medián OS, včetně dlouhodobého sledování u všech 125 pacientů, byl 20,3 měsíce.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s idelalisibem u jedné nebo více podskupin pediatrické populace v léčbě nádorů zralých B-buněk (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání jedné dávky idelalisibu byly maximální plazmatické koncentrace pozorovány za ~2 až 4 hodiny po podání v sytém stavu a za 0,5 až 1,5 hodiny po podání nalačno.
Po podávání 150 mg idelalisibu dvakrát denně byla průměrná hodnota (rozsah) a AUC v ustáleném stavu 1,953 (272; 3 905) ng/ml a 10 439 (2 349; 29 315) ng»h/ml pro idelalisib a 4 039 (669; 10 897) ng/ml a 39 744 (6 002; 119 770) ng»h/ml pro GS-563117. Plazmatické expozice idelalisibu (C^ a AUC) jsou v rozmezí dávek 50 mg až 100 mg přibližně úměrné dávce a při dávkách nad 100 mg jsou nižší, než by bylo úměrné dávce.
Vliv jídla
V porovnání s podáváním nalačno nevedlo podávání tobolek idelalisibu formulovaných v počáteční fázi vývoje s jídlem s vysokým obsahem tuku k žádné změně hodnoty a způsobilo 36 % zvýšení průměrné hodnoty AUCinf Proto lze idelalisib podávat bez ohledu na jídlo.
Distribuce
Při koncentracích, které jsou pozorovány v klinické praxi, je idelalisib z 93 % až 94 % navázán na proteiny lidské plazmy. Průměrný poměr koncentrací v krvi ke koncentracím v plazmě byl přibližně 0,5. Zdánlivý distribuční objem idelalisibu (průměrná hodnota) byl přibližně 96 l.
Biotransformace
Idelalisib je primárně metabolizován aldehydoxidázou a v menší míře CYP3A a UGT1A4. Primární a jediný cirkulující metabolit GS-563117 nevykazuje aktivitu proti PI3K5.
Eliminace
Terminální poločas eliminace idelalisibu po perorálním podání 150 mg idelalisibu dvakrát denně byl
8.2 (rozsah: 1,9; 37,2) hodiny a zdánlivá clearance idelalisibu byla 14,9 (rozsah: 5,1; 63,8) l/h. Po podání jedné perorální dávky 150 mg idelalisibu značeného [14C] se přibližně 78 % vyloučí stolicí
a 15 % močí. Idelalisib v nezměněné podobě tvoří 23 % celkové radioaktivity vyloučené močí během 48 hodin a 12 % celkové radioaktivity vyloučené stolicí během 144 hodin.
Údaje o interakcích in vitro
In vitro údaje naznačily, že idelalisib není inhibitorem metabolických enzymů CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A nebo UGT1A1 ani transportních proteinů OAT1, OAT3 nebo OCT2.
GS-563117 není inhibitorem metabolických enzymů CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 nebo UGT1A1 ani transportních proteinů P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3 nebo OCT2.
Zvláštní populac e
Pohlaví a etnikum
Populační farmakokinetické analýzy naznačily, že pohlaví a etnikum nemají klinicky významný vliv na expozici idelalisibu nebo GS-563117.
Starší pacienti
Populační farmakokinetické analýzy naznačily, že věk nemá klinicky významný vliv na expozici idelalisibu nebo GS-563117, a to ani při porovnání starších jedinců (ve věku 65 let a starší) s mladšími jedinci.
Porucha funkce ledvin
Studie farmakokinetiky a bezpečnosti idelalisibu byly provedeny u zdravých jedinců a u jedinců s těžkou poruchou funkce ledvin (odhadovaná CrCl 15 až 29 ml/min). Po podání jedné dávky 150 mg nebyly u jedinců s těžkou poruchou funkce ledvin pozorovány klinicky významné změny v expozicích idelalisibu nebo GS-563117 ve srovnání se zdravými jedinci.
Porucha funkce jater
Studie farmakokinetiky a bezpečnosti idelalisibu byly provedeny u zdravých jedinců a u jedinců se středně těžkou (stupeň B podle Child-Pughovy klasifikace) nebo těžkou (stupeň C podle Child-Pughovy klasifikace) poruchou funkce jater. Po podání jedné dávky 150 mg byly hodnoty AUC idelalisibu (celkové, tj. navázaný plus volný idelalisib) ve srovnání s odpovídajícími kontrolami o ~ 60 % vyšší u pacientů se středně těžkou a těžkou poruchou funkce jater. Hodnoty AUC idelalisibu (volného) po započtení rozdílů ve vazbě na proteiny byly o ~ 80 % (1,8násobně) vyšší u středně těžké a o ~ 152 % (2,5násobně) vyšší u těžké poruchy ve srovnání s odpovídajícími kontrolami.
Pediatrická populace
Farmakokinetika idelalisibu u pediatrické populace nebyla stanovena (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
T oxicita po opakovaném podání
Idelalisib vyvolával lymfoidní depleci sleziny, thymu, lymfatických uzlin a lymfatické tkáně střev. Obecně byly více postiženy oblasti závislé na B-lymfocytech než oblasti závislé na T-lymfocytech.
U potkanů má idelalisib potenciál inhibovat protilátkovou odpověď zprostředkovanou T-lymfocyty. Idelalisib však neinhiboval normální imunitní odpověď na Staphylococcus aureus a nezvyšoval myelosupresivní účinek cyklofosfamidu. Idelalisib není považován za látku s širokou imunosupresivní aktivitou.
Idelalisib vyvolával zánětlivé změny u potkanů a psů. Ve studiích na potkanech a psech trvajících až 4 týdny byla zaznamenána hepatická nekróza při expozici 7 až 5násobně vyšší na základě hodnot AUC, než je expozice u člověka. Zvýšení hladin sérových transamináz korelovalo s hepatickou nekrózou u psů, nebylo však pozorováno u potkanů. Ve studiích trvajících 13 týdnů a déle nebyly u potkanů ani psů pozorovány poruchy funkce jater ani chronické zvýšení hladin transamináz.
Genotoxicita
Idelalisib nezpůsoboval mutace v testu mikrobiální mutageneze (Amesův test), neměl klastogenní účinky v in vitro testu chromozomových aberací s použitím lymfocytů periferní lidské krve a nebyl genotoxický v in vivo mikronukleárním testu u potkanů.
Karcinogenita
Studie hodnotící karcinogenitu idelalisibu nebyly provedeny.
Reprodukční a vývojová toxicita
Ve studii embryofetálního vývoje u potkanů byly pozorovány zvýšené poimplantační ztráty embryí, malformace (absence ocasních obratlů a v některých případech také křížových obratlů), kosterní odchylky a nižší tělesná hmotnost plodu. Malformace byly pozorovány na základě hodnot AUC při expozicích od 12násobku expozice u člověka. Účinky na embryofetální vývoj nebyly studovány u jiného druhu.
V 2- až 13týdenních studiích s opakovaným podáváním u psů a potkanů byla pozorována degenerace semenotvorných kanálků ve varlatech, tento účinek však nebyl pozorován ve studiích trvajících 26 týdnů a déle. Ve studii fertility u samců potkanů bylo pozorováno snížení hmotnosti nadvarlat a varlat, avšak nebyly pozorovány nežádoucí účinky na ukazatele páření nebo fertility ani degenerace nebo úbytek spermatogeneze. Fertilita samic nebyla u potkanů ovlivněna.
Fototoxicita
Hodnocení možné fototoxicity v buněčné linii BALB/c 3T3 myších embryonálních fibroblastů bylo neprůkazné pro idelalisib v důsledku cytotoxicity v in vitro testu. Hlavní metabolit GS-563117 může zvyšovat fototoxicitu, pokud jsou buňky současně vystaveny UVA záření. Existuje potenciální riziko, že idelalisib může prostřednictvím svého hlavního metabolitu GS-563117 způsobit fotosenzitivitu u léčených pacientů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulóza Hyprolóza (E463)
Sodná sůl kroskarmelózy Sodná sůl karboxymethylškrobu Magnesium-stearát
Potah tablety
Polyvinylalkohol (E1203)
Makrogol 3350 (E1521)
Oxid titaničitý (E171)
Mastek (E553B)
Oranžová žluť (E110)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) s polypropylenovým dětským bezpečnostním uzávěrem obsahující 60 potahovaných tablet a polyesterovou vatu.
Každá krabička obsahuje 1 lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/938/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18. září 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Zydelig 150 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje idelalisibum 150 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Růžová, oválná, potahovaná tableta o rozměrech 10,0 mm krát 6,8 mm, s vyraženým označením „GSI“ na jedné straně a „150“ na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Zydelig je indikován v kombinaci s rituximabem k léčbě dospělých pacientů s chronickou lymfatickou leukémií (CLL):
• kteří podstoupili alespoň jednu předchozí léčbu (viz bod 4.4) nebo
• jako pokračující léčba u pacientů s delecí 17p nebo mutací TP53, kteří nejsou vhodní pro chemo-imunoterapii a kteří již zahájili léčbu přípravkem Zydelig v rámci léčby první linie (viz bod 4.4).
Přípravek Zydelig je indikován v monoterapii k léčbě dospělých pacientů s folikulárním lymfomem (FL), který je refrakterní na dvě předchozí linie léčby (viz bod 4.4).
4.2 Dávkování a způsob podání
Léčbu přípravkem Zydelig má provádět lékař se zkušenostmi s protinádorovou léčbou.
Dávkování
Doporučená dávka přípravku Zydelig je 150 mg perorálně, dvakrát denně. V léčbě je třeba pokračovat až do progrese onemocnění nebo nepřijatelné toxicity.
Jestliže pacient vynechá dávku přípravku Zydelig a uplynulo méně než 6 hodin od doby, kdy je přípravek obvykle užíván, pacient má užít vynechanou dávku co nejdříve a vrátit se k obvyklému rozvrhu dávkování. Jestliže pacient vynechá dávku a uplynulo více než 6 hodin, pacient nemá užít vynechanou dávku, ale jednoduše se vrátit k obvyklému rozvrhu dávkování.
Úpravy dávkování
Zvýšení hladin jaterních transamináz
V případě, že se objeví zvýšení hladin aminotransferáz 3. nebo 4. stupně (alaninaminotransferáza [ALT]/aspartátaminotransferáza [AST] o > 5 x horní hranice normy [ULN, Upper Limit ofNormal]), musí se léčba přípravkem Zydelig přerušit. Jakmile se hodnoty vrátí na 1. stupeň nebo níže (ALT/AST < 3 x ULN), může se léčba obnovit v dávce 100 mg dvakrát denně.
Pokud se příhoda nezopakuje, může ošetřující lékař podle svého uvážení znovu zvýšit dávku na 150 mg dvakrát denně.
Jestliže se příhoda zopakuje, musí se léčba přípravkem Zydelig přerušit až do návratu hodnot 1. stupně nebo níže, a poté může ošetřující lékař podle svého uvážení začít znovu podávat dávku 100 mg dvakrát denně (viz body 4.4 a 4.8).
Průjem/kolitida
V případě, že se objeví průjem/kolitida 3. nebo 4. stupně, musí se léčba přípravkem Zydelig přerušit. Jakmile se průjem/kolitida upraví na 1. stupeň nebo lepší, může se léčba obnovit v dávce 100 mg dvakrát denně. Pokud se průjem/kolitida nevrátí, může ošetřující lékař podle svého uvážení znovu zvýšit dávku na 150 mg dvakrát denně (viz bod 4.8).
Pneumonitida
V případě, že se předpokládá výskyt pneumonitidy, musí se léčba přípravkem Zydelig přerušit.
Jakmile pneumonitida ustoupí a pokud je vhodná opakovaná léčba, může se zvážit obnovení léčby v dávce 100 mg dvakrát denně (viz body 4.4 a 4.8).
V případě, že se objeví vyrážka 3. nebo 4. stupně, musí se léčba přípravkem Zydelig přerušit. Jakmile se vyrážka upraví na 1. stupeň nebo lepší, může se léčba obnovit v dávce 100 mg dvakrát denně.
Pokud se vyrážka nevrátí, může ošetřující lékař podle svého uvážení znovu zvýšit dávku na 150 mg dvakrát denně (viz bod 4.8).
Neutropenie
Léčba přípravkem Zydelig by musí být přerušena u pacientů s absolutním počtem neutrofilů (ANC) nižším než 500/mm3. ANC musí být monitorován minimálně jednou týdně, dokud není ANC > 500/mm3, poté může být léčba znovu zahájena dávkou 100 mg dvakrát denně (viz bod 4.4).
|
ANC 1 000 až < 1 500/mm3 |
ANC 500 až < 1 000/mm3 |
ANC < 500/mm3 |
|
Udržujte dávkování přípravku Zydelig. |
Udržujte dávkování přípravku Zydelig. Monitorujte ANC minimálně jednou týdně. |
Přerušte podávání přípravku Zydelig. Monitorujte ANC minimálně jednou týdně dokud ANC není > 500/mm3 a poté můžete znovu zahájit podávání přípravku Zydelig v dávce 100 mg dvakrát denně. |
Zvláštní populace pacientů Starší pacienti
U starších pacientů (ve věku > 65 let) není nutná specifická úprava dávkování (viz bod 5.2).
Porucha funkce ledvin
U pacientů s mírnou, středně těžkou nebo těžkou poruchou funkce ledvin není nutná úprava dávkování (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou nebo středně těžkou poruchou funkce jater není při zahájení léčby přípravkem Zydelig nutná úprava dávkování, doporučuje se však intenzivnější sledování nežádoucích účinků (viz body 4.4 a 5.2).
Údaje pro doporučení dávky u pacientů s těžkou poruchou funkce jater nejsou dostatečné. Proto se doporučuje zvýšená opatrnost a intenzivnější sledování nežádoucích účinků při podávání přípravku Zydelig u této populace (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Zydelig u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Zydelig je určen k perorálnímu podání. Pacienty je třeba poučit, aby tablety polykali celé. Potahované tablety se nesmí rozkousnout ani rozdrtit. Potahované tablety se mohou užívat spolu s jídlem nebo bez jídla (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Závažné infekce
Léčba přípravkem Zydelig se nesmí zahájit pokud má pacient známky jakékoliv systémové bakteriální, mykotické nebo virové infekce.
Při podávání idelalisibu se vyskytly závažné a fatální infekce, včetně oportunních infekcí, jako je pneumonie vyvolaná Pneumocystisjirovecii (PJP) a cytomegalovirem (CMV). Proto během léčby idelalisibem musí být všem pacientům podávána profylaxe PJP.
U pacientů mají být v průběhu léčby monitorovány známky a symptomy respirační infekce. Pacienty je třeba poučit, aby ihned nahlásili nové respirační příznaky.
Má být prováděn pravidelný klinický a laboratorní screening na infekci vyvolanou CMV. Léčba přípravkem Zydelig má být ukončena u pacientů s průkazem infekce nebo virémie.
Neutropenie
U pacientů léčených idelalisibem docházelo ve spojení s léčbou k neutropenii stupně 3 nebo 4, včetně febrilní neutropenie. Po dobu prvních 6 měsíců léčby idelalisibem je třeba monitorovat krevní obraz u všech pacientů minimálně každé 2 týdny a u pacientů s ANC nižším než 1 000/mm3 minimálně jednou týdně (viz bod 4.2).
Zvýšení hladin transamináz
V klinických studiích s idelalisibem bylo pozorováno zvýšení hladin ALT a AST 3. a 4. stupně (> 5 x ULN). Tyto laboratorní nálezy byly obecně pozorovány během prvních 12 týdnů léčby, nálezy byly obecně asymptomatické a po přerušení podávání přípravku byly reverzibilní. U většiny pacientů byla léčba obnovena v nižší dávce bez recidivy (viz bod 4.2). Po dobu prvních 3 měsíců léčby je nutné u všech pacientů každé 2 týdny sledovat hladiny ALT, AST a celkového bilirubinu, poté se mají tyto hodnoty sledovat podle klinické indikace. Pokud se zjistí zvýšení hladin ALT a/nebo AST 2. stupně nebo více, je nutné pacienty sledovat každý týden až do poklesu hodnot na 1. stupeň nebo níže.
Průjem/kolitida
Případy těžké kolitidy související s lékem se vyskytovaly relativně pozdě (v řádu měsíců) po zahájení léčby, někdy s rychlým zhoršením, ustoupily však do několika týdnů po přerušení podávání léku a dodatečné symptomatické léčbě (např. protizánětlivá léčiva, jako je enterálně podávaný budesonid).
K dispozici je velmi málo zkušeností s léčbou pacientů s anamnézou zánětlivého střevního onemocnění.
Pneumonitida
V klinických studiích s idelalisibem byly hlášeny případy pneumonitidy. Pacienti, u nichž se objeví závažné plicní příhody, které nereagují na konvenční antibakteriální léčbu, se mají vyšetřit na léky indukovanou pneumonitidu. Pokud se předpokládá pneumonitida, je třeba léčbu idelalisibem přerušit a pacienta vhodně léčit. Při středně těžké nebo těžké symptomatické pneumonitidě se musí léčba ukončit.
Stevens-Johnsonův syndrom a toxická epidermální nekrolýza
Případy Stevens-Johnsonova syndromu (SJS) a toxické epidermální nekrolýzy (TEN) s fatálními následky byly hlášeny tehdy, když byl idelalisib podáván společně s jinými léčivými přípravky spojovanými s těmito syndromy. Pokud je podezření na SJS nebo TEN, je třeba léčbu idelalisibem okamžitě přerušit a pacienta vhodně léčit.
Induktory CYP3A
Při současném podávání s induktory CYP3A, jako jsou rifampicin, fenytoin, třezalka tečkovaná (Hypericumperforatum) nebo karbamazepin, se expozice idelalisibu může snížit. Jelikož snížení plazmatické koncentrace idelalisibu může vést ke snížené účinnosti přípravku, je třeba se vyhnout současnému podávání přípravku Zydelig se středně silnými nebo silnými induktory CYP3A (viz bod 4.5).
Substráty CYP3A
Primární metabolit idelalisibu, GS-563117, je silný inhibitor CYP3A4. Idelalisib má proto potenciál vzájemně působit s léčivými přípravky, které jsou metabolizovány CYP3A, což může vést ke zvýšeným sérovým koncentracím dalšího přípravku (viz bod 4.5). Při podávání idelalisibu současně s jinými léčivými přípravky je třeba postupovat podle doporučení ohledně jejich současného podávání s inhibitory CYP3A4 uvedených v souhrnu údajů o přípravku (SPC) těchto přípravků. Je třeba se vyhnout současné léčbě idelalisibem se substráty CYP3A způsobující závažné a/nebo život ohrožující nežádoucí účinky (např. alfuzosin, amiodaron, cisaprid, pimozid, chinidin, ergotamin, dihydroergotamin, kvetiapin, lovastatin, simvastatin, sildenafil, midazolam, triazolam), a pokud je to možné, je třeba použít alternativní léčivé přípravky, které jsou méně citlivé na inhibici CYP3A4.
Porucha funkce jater
U pacientů s poruchou funkce jater se doporučuje intenzivnější sledování nežádoucích účinků, protože se u této populace očekává zvýšená expozice, a to zvláště u pacientů se závažnou poruchou funkce jater. Do klinických studií s idelalisibem nebyli zařazeni žádní pacienti s těžkou poruchou funkce jater. Při podávání přípravku Zydelig u této populace se doporučuje zvýšená opatrnost.
Chronická hepatitida
Idelalisib nebyl studován u pacientů s chronickou aktivní hepatitidou, včetně virové hepatitidy. Při podávání přípravku Zydelig pacientům s aktivní hepatitidou je třeba postupovat opatrně.
Ženy ve fertilním věku
Zeny ve fertilním věku musí během léčby idelalisibem a ještě 1 měsíc po ukončení terapie používat vysoce účinnou antikoncepci (viz bod 4.6). Ženy užívající hormonální antikoncepci mají navíc používat bariérovou metodu jako druhou formu antikoncepce, protože v současné době není známo, jestli idelalisib snižuje účinnost hormonální antikoncepce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Idelalisib je primárně metabolizován aldehydoxidázou a v menší míře CYP3A a glukuronidací (UGT1A4). Jeho primárním metabolitem je farmakologicky neaktivní GS-563117. Idelalisib a GS-563117 jsou substráty P-gp a BCRP.
Vliv jiných léčivých přípravků na farmakokinetické vlastnosti idelalisibu
Induktory CYP3A
Klinická studie lékových interakcí zjistila, že současné podání jedné dávky 150 mg idelalisibu s rifampicinem (silný induktor CYP3A) vedlo k ~75% snížení hodnoty AUCinf idelalisibu. Je třeba se vyhnout současnému podávání přípravku Zydelig se středně silnými nebo silnými induktory CYP3A, jako jsou rifampicin, fenytoin, třezalka tečkovaná nebo karbamazepin, jelikož to může vést ke snížené účinnosti přípravku (viz bod 4.4).
Inhibitory CYP3A/P-gp
Klinická studie lékových interakcí zjistila, že současné podání jedné dávky 400 mg idelalisibu s ketokonazolem (silný inhibitor CYP3A, P-gp a BCRP) v dávce 400 mg jednou denně vedlo k 26% zvýšení hodnoty Cmax a 79% zvýšení hodnoty AUCinf idelalisibu. Při současném podávání idelalisibu s inhibitory CYP3A/P-gp se počáteční úprava dávkování idelalisibu nepovažuje za nutnou, doporučuje se však intenzivnějsí sledování nežádoucích účinků.
Vliv idelalisibu na farmakokinetické vlastnosti jiných léčivých přípravků
Substráty CYP3A
Primární metabolit idelalisibu, GS-563117, je silný inhibitor CYP3A Klinická studie lékových interakcí zjistila, že současné podávání idelalisibu s midazolamem (citlivý substrát CYP3A) vedlo k ~140% zvýšení hodnoty Cmax a ~440% zvýšení hodnoty AUCinf midazolamu v důsledku inhibice CYP3A způsobené GS-563117. Současné podávání idelalisibu se substráty CYP3A může zvyšovat jejich systémové expozice a zvyšovat nebo prodlužovat jejich terapeutickou aktivitu a nežádoucí účinky. In vitro byla inhibice CYP3A4 ireverzibilní a proto se návrat k normální enzymatické aktivitě očekává až za několik dnů po ukončení podávání idelalisibu.
Možné interakce mezi idelalisibem a současně podávanými léčivými přípravky, které jsou substráty CYP3A, jsou uvedeny v tabulce 1 (zvýšení je uvedeno jako „t”). Tento seznam není vyčerpávající a slouží pouze jako návod. Obecně je třeba postupovat podle doporučení ohledně jejich současného podávání s inhibitory CYP3A4 uvedených v SPC těchto přípravků (viz bod 4.4).
Tabulka 1: Interakce mezi idelalisibem a dalšími léčivými přípravky, které jsou substráty CYP3A
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
ANTAGONISTE ALFA-1 ADRENORECEPTORU | ||
|
Alfuzosin |
| koncentrace v séru |
Idelalisib se nemá podávat současně s alfuzosinem. |
|
ANALGETIKA | ||
|
Fentanyl, alfentanil, metadon, buprenorfin/naloxon |
| koncentrace v séru |
Doporučuje se pečlivé sledování nežádoucích účinků (např. respirační útlum, sedace). |
|
ANTIARYTMIKA | ||
|
Amiodaron, chinidin |
| koncentrace v séru |
Idelalisib se nemá podávat současně s amiodaronem ani chinidinem. |
|
Bepridil, disopyramid, lidokain |
| koncentrace v séru |
Doporučuje se klinické sledování. |
|
ANTINEOPLASTIKA | ||
|
Inhibitory tyrosinkinázy, jako je dasatinib a nilotinib, také vinkristin a vinblastin |
| koncentrace v séru |
Doporučuje se pečlivé sledování snášenlivosti těchto antineoplastik. |
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
ANTIKOAGULANCIA | ||
|
Warfarin |
| koncentrace v séru |
Při současném podávání a po ukončení léčby idelalisibem se doporučuje sledování hodnot mezinárodního normalizovaného poměru (INR) |
|
ANTIKONVULZIVA | ||
|
Karbamazepin |
| koncentrace v séru |
Je třeba sledovat hladiny antikonvulziv. |
|
ANTIDEPRESIVA | ||
|
Trazodon |
| koncentrace v séru |
Doporučuje se pečlivá titrace dávky antidepresiv a sledování antidepresivní odpovědi. |
|
LÉKY PROTI DNĚ | ||
|
Kolchicin |
| koncentrace v séru |
Může být nutné snížení dávky kolchicinu. Idelalisib se nemá podávat současně s kolchicinem pacientům s poruchou funkce ledvin nebo jater. |
|
ANTIHYPERTENZIVA | ||
|
Amlodipin, diltiazem, felodipin, nifedipin, nikardipin |
| koncentrace v séru |
Doporučuje se klinické sledování terapeutického účinku a nežádoucích účinků. |
|
ANTIINFEKTIVA | ||
|
Antimykotika | ||
|
Ketokonazol, itrakonazol, posakonazol, vorikonazol |
| koncentrace v séru |
Doporučuje se klinické sledování. |
|
Antimykobakteriální látky | ||
|
Rifabutin |
| koncentrace v séru |
Doporučuje se zvýšené sledování nežádoucích účinků souvisejících s rifabutinem, včetně neutropenie a uveitidy. |
|
Inhibitory HCV proteázy | ||
|
Boceprevir, telaprevir |
| koncentrace v séru |
Doporučuje se klinické sledování. |
|
Makrolidová antibiotika | ||
|
Klarithromycin, telithromycin |
| koncentrace v séru |
U pacientů s normální funkcí ledvin nebo mírnou poruchou funkce ledvin (clearance kreatininu [CrCl] 60-90 ml/min) není nutná žádná úprava dávky klarithromycinu. U pacientů s CrCl < 90 ml/min se doporučuje klinické sledování. U pacientů s CrCl < 60 ml/min je třeba zvážit použití alternativního antibiotika. U telithromycinu se doporučuje klinické sledování. |
|
ANTIPSYCHOTIKA/NEUROLEPTIKA | ||
|
Kvetiapin, pimozid |
| koncentrace v séru |
Idelalisib se nemá podávat současně s kvetiapinem ani pimozidem. Mohou se zvážit alternativní léčivé přípravky, jako je olanzapin. |
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
Z 0 1 % < |
3VÝCH RECEPTORŮ | |
|
Bosentan |
| koncentrace v séru |
Je třeba postupovat opatrně a pacienty je třeba pečlivě sledovat s ohledem na toxicitu související s bosentanem. |
|
ERGOTAMINOVÉ ALKALOIDY | ||
|
Ergotamin, dihydroergotamin |
| koncentrace v séru |
Idelalisib se nemá podávat současně s ergotaminem ani dihydroergotaminem. |
|
PROKUNEHKA | ||
|
Cisaprid |
| koncentrace v séru |
Idelalisib se nemá podávat současně s cisapridem. |
|
GLUKOKORTIKOIDY | ||
|
Inhalační/nazální kortikosteroidy: budesonid, flutikason Perorální budesonid |
| koncentrace v séru | koncentrace v séru |
Doporučuje se klinické sledování. Doporučuje se klinické sledování ohledně zvýšených známek/příznaků účinků kortikosteroidů. |
|
INHIBITORY HMG CO-A REDUKTÁZY | ||
|
Lovastatin, simvastatin Atorvastatin |
| koncentrace v séru | koncentrace v séru |
Idelalisib se nemá podávat současně s lovastatinem ani simvastatinem. Doporučuje se klinické sledování a může se zvážit nižší počáteční dávka atorvastatinu. Může být rovněž zvážen přechod na pravastatin, rosuvastatin nebo pitavastatin. |
|
IMUNOSUPRESIVA | ||
|
Cyklosporin, sirolimus, takrolimus |
| koncentrace v séru |
Doporučuje se terapeutické sledování. |
|
INHALAČNÍ BETA-AGONISTÉ | ||
|
Salmeterol |
| koncentrace v séru |
Současné podávání salmeterolu a idelalisibu se nedoporučuje. Kombinace může vést ke zvýšenému riziku kardiovaskulárních nežádoucích účinků souvisejících se salmeterolem, včetně prodloužení QT intervalu, palpitací a sinusové tachykardie. |
|
Léčivý přípravek |
Očekávaný účinek idelalisibu na hladiny léčivého přípravku |
Klinické doporučení týkající se současného podávání s idelalisibem |
|
INHIBITORY FOSFODIESTERÁZY | ||
|
Plicní arteriální hypertenze: | ||
|
Sildenafil |
| koncentrace v séru |
Idelalisib se nemá podávat současně se sildenafilem. |
|
Tadalafil |
| koncentrace v séru |
Při současném podávání tadalafilu s idelalisibem je třeba postupovat opatrně, včetně zvážení snížení dávky. |
|
Erektilní dysfunkce: | ||
|
Sildenafil, tadalafil |
| koncentrace v séru |
Při předepisování sildenafilu nebo tadalafilu s idelalisibem je nutná zvýšená opatrnost a může se zvážit snížení dávky a zvýšené sledování nežádoucích účinků. |
|
SEDATIVA/HYPNOTIKA | ||
|
Midazolam (perorální), triazolam |
| koncentrace v séru |
Idelalisib se nemá podávat současně s midazolamem (perorální) ani triazolamem. |
|
Buspiron, klorazepát, diazepam, estazolam, flurazepam, zolpidem |
| koncentrace v séru |
Doporučuje se sledování koncentrace sedativ/hypnotik a může se zvážit snížení dávky. |
Substráty CYP2C8
In vitro idelalisib inhiboval a indukoval CYP2C8, není ale známo, zda to vede k účinku in vivo na substráty CYP2C8. Doporučuje se postupovat opatrně při podávání přípravku Zydelig současně s léky s úzkým terapeutickým indexem, které jsou substráty CYP2C8 (paklitaxel).
Substráty indukovatelných enzymů (např. CYP2C9, CYP2C19, CYP2B6 a UGT)
In vitro byl idelalisib induktorem několika enzymů a není možné vyloučit riziko snížené expozice a tím snížené účinnosti substrátů indukovatelných enzymů, jako je CYP2C9, CYP2C19, CYP2B6 a UGT. Doporučuje se postupovat opatrně při podávání přípravku Zydelig současně s léky s úzkým terapeutickým indexem, které jsou substráty těchto enzymů (warfarin, fenytoin, S-mefenytoin).
SubstrátyBCRP, OATP1B1, OATP1B3 aP-gp
Současné podávání vícečetných dávek idelalisibu 150 mg dvakrát denně zdravým jedincům vedlo ke srovnatelným expozicím u rosuvastatinu (AUC 90% IS: 87, 121) a digoxinu (AUC 90% IS: 98, 111), což nenaznačuje žádnou klinicky významnou inhibici BCRP, OATP1B1/1B3 nebo systémového P-gp idelalisibem. Není možné vyloučit riziko inhibice P-gp v gastrointestinálním traktu, která by mohla vést ke zvýšené expozici citlivých substrátů intestinálního P-gp, jako je dabigatran-etexilát.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Na základě zjištění u zvířat může idelalisib způsobit poškození plodu. Ženy by se měly během užívání přípravku Zydelig a do 1 měsíce po ukončení léčby vyhnout otěhotnění. Proto musí ženy ve fertilním věku během léčby přípravkem Zydelig a ještě 1 měsíc po ukončení terapie používat vysoce účinnou antikoncepci. V současné době není známo, jestli idelalisib snižuje účinnost hormonální antikoncepce, a proto ženy užívající hormonální antikoncepci mají navíc používat bariérovou metodu jako druhou formu antikoncepce.
Údaje o podávání idelalisibu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Podávání přípravku Zydelig se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se idelalisib a jeho metabolity vylučují do lidského mateřského mléka.
Riziko pro kojené novorozence/děti nelze vyloučit.
Kojení má být během léčby přípravkem Zydelig přerušeno.
Fertilita
Údaje o účinku idelalisibu na fertilitu u člověka nejsou k dispozici. Studie na zvířatech naznačují možné škodlivé účinky idelalisibu na fertilitu a vývoj plodu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Zydelig nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí profilu bezpečnosti
Hodnocení nežádoucích účinků je založeno na jedné studii fáze 3 a sedmi studiích fáze 1 a 2. Studie 312-0116 fáze 3 byla randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie, ve které bylo 220 pacientů s dříve léčenou CLL randomizováno v poměru 1:1 pro užívání idelalisibu a rituximabu nebo placeba a rituximabu. Studie fáze 1 a 2 hodnotily bezpečnost idelalisibu u 490 pacientů s hematologickými malignitami, včetně 354 pacientů užívajících idelalisib (v jakékoli dávce) jako samostatné léčivo a 136 pacientů užívajících idelalisib v kombinaci s monoklonálními protilátkami anti-CD20.
Nejčastěji hlášené nežádoucí účinky během léčby idelalisibem jsou uvedeny v tabulce 2.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky hlášené pro idelalisib užívaný samostatně nebo v kombinaci s monoklonálními protilátkami anti-CD20 uvádí tabulka 2. Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů a četnosti. Četnosti jsou definovány následujícím způsobem: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 2: Nežádoucí účinky hlášené v klinických studiích u pacientů s hematologickými malignitami užívajících idelalisib
|
Nežádoucí účinek |
Jakýkoli stupeň |
Stupeň > 3 |
|
Infekce a infestace | ||
|
Infekce |
Velmi časté |
Velmi časté |
|
Poruchy krve a lymfatického systému | ||
|
Neutropenie |
Velmi časté |
Velmi časté |
|
Respirační, hrudní a mediastinálníporuchy | ||
|
Pneumonitida |
Časté |
Časté |
|
Gastrointestinální poruchy | ||
|
Průjem/kolitida |
Velmi časté |
Velmi časté |
|
Poruchy jater a žlučových cest | ||
|
Zvýšené hladiny transamináz |
Velmi časté |
Velmi časté |
|
Poruchy kůže a podkožní tkáně | ||
|
Vyrážka* |
Velmi časté |
Časté |
|
Stevens - Johns onův |
Vzácné |
Vzácné |
|
syndrom/toxická epidermální | ||
|
nekrolýza | ||
|
Celkové poruchy a reakce v místě aplikace | ||
|
Pyrexie |
Velmi časté |
Časté |
|
Vyšetření | ||
|
Zvýšené hladiny triglyceridů |
Velmi časté |
Časté |
* Zahrnuje preferované termíny exfoliativní dermatitida, léková erupce, vyrážka, erytematózní vyrážka, generalizovaná vyrážka, makulární vyrážka, makulopapulámí vyrážka, papulární vyrážka, svědivá vyrážka a exfoliativní vyrážka.
Popis vybraných nežádoucích účinků
Vyrážka byla obecně mírná až středně těžká a měla za následek ukončení léčby přibližně u 2 % pacientů. Ve studii 312-0116 se vyrážka (hlášená jako exfoliativní dermatitida, léková erupce, vyrážka, erytematózní vyrážka, generalizovaná vyrážka, makulární vyrážka, makulopapulámí vyrážka, papulární vyrážka a svědivá vyrážka) objevila u 24,5 % pacientů, kterým byl podáván idelalisib a rituximab, a u 6,5 % pacientů, jimž bylo podáváno placebo a rituximab. Z toho mělo 3,6 % pacientů, jimž byl podáván idelalisib a rituximab, a 0,9 % pacientů, kterým bylo podáváno placebo a rituximab, vyrážku 3. stupně a žádný pacient neměl nežádoucí účinek 4. stupně. Vyrážka obvykle ustoupila po léčbě (např. místní a/nebo perorální steroidy, difenhydramin) a v těžkých případech po přerušení podávání přípravku (viz bod 5.3, fototoxicita).
Stevens-Johnsonův syndrom a toxická epidermální nekrolýza (viz bod 4.4)
Případy SJS a TEN se vzácně vyskytly tehdy, když byl idelalisib podáván souběžně s jinými léčivými přípravky spojovanými s těmito syndromy (bendamustin, rituximab, alopurinol a amoxicilin). SJS nebo TEN se objevily do jednoho měsíce od podání kombinace léků a měly fatální následky.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování musí být pacient sledován, zda nevykazuje známky toxicity (viz bod 4.8). Léčba předávkování přípravkem Zydelig zahrnuje obecná podpůrná opatření včetně sledování vitálních funkcí a pozorování klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: protinádorová léčiva, další protinádorová léčiva, ATC kód: L01XX47 Mechanismus účinku
Idelalisib inhibuje fosfatidylinositol 3-kinázu p1105 (PI3K5), která je hyperaktivní u malignit B-buněk a je centrem mnoha signálních drah, které řídí proliferaci, přežití, usídlení („homing“) a retenci maligních buněk v lymfoidních tkáních a kostní dřeni. Idelalisib je selektivní inhibitor vazby adenosin-5’ -trifosfátu (ATP) na katalytickou doménu PI3K5, vedoucí k inhibici fosforylace fosfatidylinositolu, který je klíčovým lipidovým druhým poslem („secondmessenger“), a k zabránění fosforylace Akt (proteinkinázy B).
Idelalisib indukuje apoptózu a inhibuje proliferaci v buněčných liniích odvozených od maligních B-buněk a v primárních nádorových buňkách. Inhibicí signalizace prostřednictvím chemokinových receptorů CXCR4 a CXCR5, indukované chemokiny CXCL12 a CXCL13, způsobuje idelalisib inhibici usídlení („homing“) a retence maligních B-buněk v nádorovém mikroprostředí, včetně lymfoidních tkáních a kostní dřeně.
Farmakodynamické účinky
Účinek idelalisibu (150 mg a 400 mg) na interval QT/QTc byl hodnocen v placebem a aktivně (moxifloxacin 400 mg) kontrolované zkřížené studii u 40 zdravých jedinců. Při dávce 2,7násobně vyšší než je maximální doporučená dávka nezpůsobil idelalisib prodloužení intervalu QT/QTc (tj.
< 10 ms).
Klinická účinnost u chronické lymfatické leukémie
Idelalisib v kombinaci s imunoterapií
Studie 312-0116 byla randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie fáze 3 zahrnující 220 pacientů s dříve léčenou CLL, kteří potřebovali léčbu, ale cytotoxická chemoterapie pro ně nebyla považována za vhodnou. Pacienti byli randomizováni v poměru 1:1 pro podání 8 cyklů rituximabu (první cyklus v dávce 375 mg/m2 povrchu těla [body surface area, BSA], následující cykly v dávce 500 mg/m2 BSA) v kombinaci buď s perorálně podávaným placebem dvakrát denně nebo s idelalisibem 150 mg užívaným dvakrát denně, až do progrese onemocnění nebo nepřijatelné toxicity.
Medián věku byl 71 let (rozsah: 47 až 92), přičemž 78,2 % pacientů bylo starších než 65 let; 65,5 % byli muži a 90,0 % byli běloši; 64,1 % mělo klinické stadium III nebo IV podle Raie a 55,9 % mělo klinické stadium C podle Bineta. U většiny pacientů byly přítomny nepříznivé cytogenetické prognostické faktory: 43,2 % mělo chromozomovou deleci 17p a/nebo mutaci nádorového proteinu 53 (Tumour Protein 53, TP53) a 83,6 % mělo nemutované geny pro variabilní oblast těžkého imunoglobulinového řetězce (Immunoglobulin Heavy Chain Variable region, IGHV). Medián doby od diagnózy CLL do randomizace byl 8,5 roku. Pacienti měli medián hodnocení funkční zdatnosti (Cumulative Illness Rating Score, CIRS) rovný 8. Medián počtu předchozích terapií byl 3,0. Téměř všem (95,9 %) pacientům byly dříve podány monoklonální protilátky anti-CD20. Primárním cílovým parametrem bylo přežití bez progrese (Progression Free Survival, PFS). Výsledky účinnosti jsou shrnuty v tabulkách 3 a 4. Kaplan-Meierovu křivku pro PFS uvádí obrázek 1.
Ve srovnání s kombinací rituximabu a placeba vedla léčba kombinací idelalisibu a rituximabu ke statisticky významnému i klinicky zásadnímu zlepšení tělesného, sociálního a funkčního stavu, jakož i hodnot podstupnic specifických pro leukemii podle indexu funkčního hodnocení léčby rakoviny (Functional Assessment of Cancer Therapy: Leukaemia, FACT-LEU) a ke statisticky významnému a klinicky zásadnímu zlepšení aspektů úzkosti, deprese a obvyklých aktivit hodnocených podle indexu EuroQoL Five-Dimensions (EQ-5D).
Tabulka 3: Výsledky účinnosti ze studie 312-0116
|
Idelalisib + R N = 110 |
Placebo + R N = 110 | |
|
PFS Medián (měsíce) (95% IS) |
19,4 (12,3; NR) |
6,5 (4,0; 7,3) |
|
Poměr rizik (95% IS) |
0,15 (0,09; 0,25) | |
|
P-hodnota |
< 0,0001 | |
|
ORR* n (%) (95% IS) |
92 (83,6 %) (75,4; 90,0) |
17 (15,5 %) (9,3; 23,6) |
|
Poměr šancí (95% IS) |
27,76 (13,40; 57,49) | |
|
P-hodnota |
< 0,0001 | |
|
LNR** n/N (%) (95% IS) |
102/106 (96,2 %) (90,6; 99,0) |
7/104 (6,7 %) (2,7; 13,4) |
|
Poměr šancí (95% IS) |
225,83 (65,56; 777,94) | |
|
P-hodnota |
< 0,0001 | |
|
OSA Medián (měsíce) (95% IS) |
NR (NR, NR) |
20,8 (14,8; NR) |
|
Poměr rizik (95% IS) |
0,34 (0,19; 0,60) | |
|
P-hodnota |
0,0001 | |
IS: interval spolehlivosti; R rituximab; n: počet pacientů s odpovědí; N: počet pacientů ve skupině, NR: nedosaženo. Analýzy PFS, celkové četnosti odpovědi na léčbu (OverallResponseRate, ORR) a četnosti odpovědi týkající se lymfatických uzlin (Lymph Node Response Rate, LNR) vycházely z hodnocení nezávislou hodnotící komisí (IndependentReview Committee, IRC).
* Hodnota ORRje definovánajako podíl pacientů, kteří dosáhli kompletní odpovědi (Complete Response, CR) nebo částečné odpovědi (Partial Response, PR) na základě kritérií odpovědi stanovených Národní onkologickou sítí (National Comprehensive Cancer Network, NCCN) (2013) a podle Chesona (2012).
** Hodnota LNRje definována jako podíl pacientů, kteří dosáhli > 50 % zmenšení součtu součinů největších kolmých průměrů indexovaných lézí. Do analýzy byli zahrnuti pouze pacienti, u nichž bylo k dispozici výchozí hodnocení a > 1 platné hodnocení po zahájení léčby.
A Analýza celkového přežití (OverallSurvival, OS) zahrnuje data od pacientů, kteří dostali placebo + R ve studii 312-0116 a následně dostali idelalisib v prodloužené studii na základě analýzy podle původního léčebného záměru (intent-to-treat).
Tabulka 4: Souhrn hodnot PFS a četnosti odpovědí v předem stanovených podskupinách ze studie 312-0116
|
Idelalisib + R |
Placebo + R | |
|
Delece 17p/mutace TP53 |
N = 46 |
N = 49 |
|
Medián PFS (měsíce) (95% IS) |
NR (12,3; NR) |
4,0 (3,7; 5,7) |
|
Poměr rizik (95% IS) |
0,13 (0,07; 0,27) | |
|
ORR (95% IS) |
84,4 % (71,1; 93,7) |
12,2 % (4,6; 24,8) |
|
Nemutovaný IGHV |
N = 91 |
N = 93 |
|
Medián PFS (měsíce) (95% IS) |
19,4 (13,9, NR) |
5,6 (4,0; 7,2) |
|
Poměr rizik (95% IS) |
0,14 (0,08; 0,23) | |
|
ORR (95% IS) |
82,4 % (73,0; 89,6) |
15,1 % (8,5; 24,0) |
|
Věk > 65 let |
N = 89 |
N = 83 |
|
Medián PFS (měsíce) (95% IS) |
19,4 (12,3; NR) |
5,7 (4,0; 7,3) |
|
Poměr rizik (95% IS) |
0,14 (0,08; 0,25) | |
|
ORR (95% IS) |
84,3 % (75,0; 91,1) |
16,9 % (9,5; 26,7) |
IS: interval spolehlivosti; R rituxiniab; N: počet pacientů ve skupině; NR: nedosaženo
Obrázek 1: Kaplan-Meierova křivka PFS ze studie 312-0116 (populace podle původního léčebného záměru (intent-to-treat))
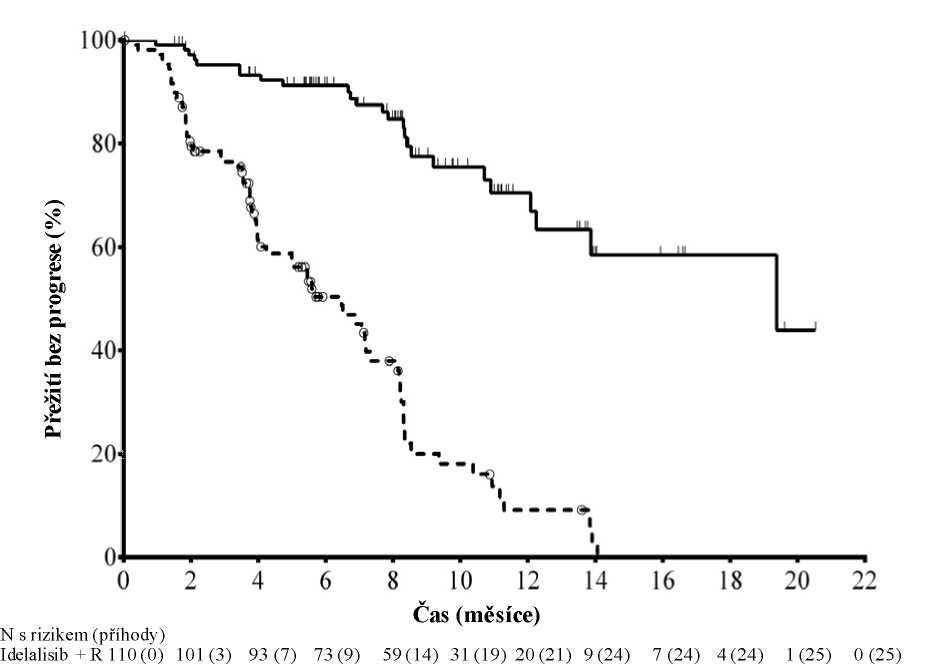
Placebo + R 110 (0) 84 (21) 48 (38) 29 (46) 20 (53) 9 (63) 4 (67) 1 (69) 0 (70) 0 (70) 0 (70) 0 (70)
Plná čára: idelalisib + R (N = 110), přerušovaná čára: placebo + R (N = 110)
R: rituximab; N: počet pacientů ve skupině
Analýza PFS vycházela z hodnocení nezávislou hodnotící komisí (Independent Review Committee, IRC). Pro pacienty ve skupině placeba + R obsahuje souhrn data až do podání první dávky idelalisibu v prodloužené studii. Medián PFS: idelalisib + R = 19,4 měsíce, placebo + R = 6,5 měsíce. Poměr rizik = 0,15; 95 % IS (0,09; 0,24); p < 0,0001.
Do studie 101-08/99 bylo zařazeno 64 pacientů s dosud neléčenou CLL, včetně 5 pacientů s lymfomem z malých lymfocytů (SLL). Pacienti dostávali idelalisib 150 mg dvakrát denně a rituximab 375 mg/m2 BSA jednou týdně. Hodnota ORR byla 96,9 %, přičemž 12 pacientů mělo CR (18,8 %) a 50 pacientů mělo PR (78,1 %), včetně 3 CR a 6 PR u pacientů s delecí 17p a/nebo mutací TP53 a 2 CR a 34 PR u pacientů s nemutovaným IGHV. Mediánu trvání odpovědi (Duration Of Response, DOR) nebylo dosaženo.
Klinická účinnost u folikulárního lymfomu
Bezpečnost a účinnost idelalisibu byly hodnoceny v jednoramenné multicentrické klinické studii (studie 101-09) prováděné u 125 pacientů s indolentním non-Hodgkinovým lymfomem z B-buněk (iNHL, včetně FL, n = 72; SLL, n = 28; lymfoplasmocytového lymfomu/Waldenstromovy makroglobulinémie [LPL/WM], n = 10; a lymfomu marginální zóny [MZL], n = 15). Všichni pacienti byli refrakterní na rituximab a 124 ze 125 pacientů bylo refrakterních nejméně na jednu alkylační látku. 112 (89,6 %) pacientů bylo refrakterních na poslední režim léčby před vstupem do studie.
Ze 125 zařazených pacientů 80 (64 %) byli muži, medián věku byl 64 let (rozsah: 33 až 87) a 110 (89 %) byli běloši. Pacientům byl podáván idelalisib 150 mg perorálně dvakrát denně až do progrese onemocnění nebo nepřijatelné toxicity.
Primárním cílovým parametrem byla hodnota ORR definovaná jako podíl pacientů, kteří dosáhli CR nebo PR (podle revidovaných kritérií odpovědi na léčbu maligního lymfomu [Cheson]), a u pacientů s Waldenstromovou makroglobulinémií nízkou odpověď (Minor Response, MR) (podle hodnocení odpovědi na léčbu u Waldenstromovy makroglobulinémie [Owen]). Hodnota DOR byla sekundárním cílovým parametrem a byla definována jako doba od první zdokumentované odpovědi (CR, PR nebo MR) do prvního zaznamenání progrese onemocnění nebo úmrtí pacienta z jakékoli příčiny. Výsledky účinnosti jsou shrnuty v tabulce 5.
Tabulka 5: Souhrn odpovědí u pacientů s FL léčených idelalisibem (podle hodnocení IRC)
|
Charakteristika |
Pacienti ve studii n (%) |
|
ORR (folikulární lymfom)* |
39 (54,2) |
|
95% IS |
42,0 - 66,0 |
|
ORR (všichni pacienti)* |
71 (56,8) |
|
95% IS |
47,6 - 65,6 |
|
Kategorie odpovědi (folikulární lymfom)* CR |
6 (8,3) |
|
PR |
33 (45,8) |
IS: interval spolehlivosti; n: počet pacientů s odpovědí
* Odpověď byla stanovena nezávislou hodnotící komisí (IRC); ORR = kompletní odpověď (CR) + částečná odpověď (PR).
Odhad mediánu DOR byl pro všechny pacienty 12,5 měsíce (12,5 měsíce pro pacienty s SLL, hodnoty nedosaženo u pacientů s FL, LPL/WM a MZL). Ze 122 pacientů s měřitelnými lymfatickými uzlinami ve výchozím stavu a po zahájení léčby bylo u 67 pacientů (54,9 %) dosaženo > 50% snížení součtu součinů průměrů indexovaných lézí. Z pacientů, kteří neodpověděli na léčbu, mělo 10 (8,0 %) progresivní onemocnění jako nejlepší odpověď a 2 (1,6 %) nebyli hodnotitelní. Medián OS, včetně dlouhodobého sledování u všech 125 pacientů, byl 20,3 měsíce.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s idelalisibem u jedné nebo více podskupin pediatrické populace v léčbě nádorů zralých B-buněk (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání jedné dávky idelalisibu byly maximální plazmatické koncentrace pozorovány za ~2 až 4 hodiny po podání v sytém stavu a za 0,5 až 1,5 hodiny po podání nalačno.
Po podávání 150 mg idelalisibu dvakrát denně byla průměrná hodnota (rozsah) C^ a AUC v ustáleném stavu 1,953 (272; 3 905) ng/ml a 10 439 (2 349; 29 315) ng»h/ml pro idelalisib a 4 039 (669; 10 897) ng/ml a 39 744 (6 002; 119 770) ng»h/ml pro GS-563117. Plazmatické expozice idelalisibu (C^ a AUC) jsou v rozmezí dávek 50 mg až 100 mg přibližně úměrné dávce a při dávkách nad 100 mg jsou nižší, než by bylo úměrné dávce.
Vliv jídla
V porovnání s podáváním nalačno nevedlo podávání tobolek idelalisibu formulovaných v počáteční fázi vývoje s jídlem s vysokým obsahem tuku k žádné změně hodnoty 0^ a způsobilo 36 % zvýšení průměrné hodnoty AUCinf Proto lze idelalisib podávat bez ohledu na jídlo.
Distribuce
Při koncentracích, které jsou pozorovány v klinické praxi, je idelalisib z 93 % až 94 % navázán na proteiny lidské plazmy. Průměrný poměr koncentrací v krvi ke koncentracím v plazmě byl přibližně 0,5. Zdánlivý distribuční objem idelalisibu (průměrná hodnota) byl přibližně 96 l.
Biotransformace
Idelalisib je primárně metabolizován aldehydoxidázou a v menší míře CYP3A a UGT1A4. Primární a jediný cirkulující metabolit GS-563117 nevykazuje aktivitu proti PI3K5.
Eliminace
Terminální poločas eliminace idelalisibu po perorálním podání 150 mg idelalisibu dvakrát denně byl
8.2 (rozsah: 1,9; 37,2) hodiny a zdánlivá clearance idelalisibu byla 14,9 (rozsah: 5,1; 63,8) l/h. Po podání jedné perorální dávky 150 mg idelalisibu značeného [14C] se přibližně 78 % vyloučí stolicí
a 15 % močí. Idelalisib v nezměněné podobě tvoří 23 % celkové radioaktivity vyloučené močí během 48 hodin a 12 % celkové radioaktivity vyloučené stolicí během 144 hodin.
Údaje o interakcích in vitro
In vitro údaje naznačily, že idelalisib není inhibitorem metabolických enzymů CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A nebo UGT1A1 ani transportních proteinů OAT1, OAT3 nebo OCT2.
GS-563117 není inhibitorem metabolických enzymů CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 nebo UGT1A1 ani transportních proteinů P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3 nebo OCT2.
Zvláštní populace
Pohlaví a etnikum
Populační farmakokinetické analýzy naznačily, že pohlaví a etnikum nemají klinicky významný vliv na expozici idelalisibu nebo GS-563117.
Starší pacienti
Populační farmakokinetické analýzy naznačily, že věk nemá klinicky významný vliv na expozici idelalisibu nebo GS-563117, a to ani při porovnání starších jedinců (ve věku 65 let a starší) s mladšími jedinci.
Porucha funkce ledvin
Studie farmakokinetiky a bezpečnosti idelalisibu byly provedeny u zdravých jedinců a u jedinců s těžkou poruchou funkce ledvin (odhadovaná CrCl 15 až 29 ml/min). Po podání jedné dávky 150 mg nebyly u jedinců s těžkou poruchou funkce ledvin pozorovány klinicky významné změny v expozicích idelalisibu nebo GS-563117 ve srovnání se zdravými jedinci.
Porucha funkce jater
Studie farmakokinetiky a bezpečnosti idelalisibu byly provedeny u zdravých jedinců a u jedinců se středně těžkou (stupeň B podle Child-Pughovy klasifikace) nebo těžkou (stupeň C podle Child-Pughovy klasifikace) poruchou funkce jater. Po podání jedné dávky 150 mg byly hodnoty AUC idelalisibu (celkové, tj. navázaný plus volný idelalisib) ve srovnání s odpovídajícími kontrolami o ~ 60 % vyšší u pacientů se středně těžkou a těžkou poruchou funkce jater. Hodnoty AUC idelalisibu (volného) po započtení rozdílů ve vazbě na proteiny byly o ~ 80 % (1,8násobně) vyšší u středně těžké a o ~ 152 % (2,5násobně) vyšší u těžké poruchy ve srovnání s odpovídajícími kontrolami.
Pediatrická populace
Farmakokinetika idelalisibu u pediatrické populace nebyla stanovena (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
T oxicita po opakovaném podání
Idelalisib vyvolával lymfoidní depleci sleziny, thymu, lymfatických uzlin a lymfatické tkáně střev. Obecně byly více postiženy oblasti závislé na B-lymfocytech než oblasti závislé na T-lymfocytech.
U potkanů má idelalisib potenciál inhibovat protilátkovou odpověď zprostředkovanou T-lymfocyty. Idelalisib však neinhiboval normální imunitní odpověď na Staphylococcus aureus a nezvyšoval myelosupresivní účinek cyklofosfamidu. Idelalisib není považován za látku s širokou imunosupresivní aktivitou.
Idelalisib vyvolával zánětlivé změny u potkanů a psů. Ve studiích na potkanech a psech trvajících až 4 týdny byla zaznamenána hepatická nekróza při expozici 7 až 5násobně vyšší na základě hodnot AUC, než je expozice u člověka. Zvýšení hladin sérových transamináz korelovalo s hepatickou nekrózou u psů, nebylo však pozorováno u potkanů. Ve studiích trvajících 13 týdnů a déle nebyly u potkanů ani psů pozorovány poruchy funkce jater ani chronické zvýšení hladin transamináz.
Genotoxicita
Idelalisib nezpůsoboval mutace v testu mikrobiální mutageneze (Amesův test), neměl klastogenní účinky v in vitro testu chromozomových aberací s použitím lymfocytů periferní lidské krve a nebyl genotoxický v in vivo mikronukleárním testu u potkanů.
Karcinogenita
Studie hodnotící karcinogenitu idelalisibu nebyly provedeny.
Reprodukční a vývojová toxicita
Ve studii embryofetálního vývoje u potkanů byly pozorovány zvýšené poimplantační ztráty embryí, malformace (absence ocasních obratlů a v některých případech také křížových obratlů), kosterní odchylky a nižší tělesná hmotnost plodu. Malformace byly pozorovány na základě hodnot AUC při expozicích od 12násobku expozice u člověka. Účinky na embryofetální vývoj nebyly studovány u jiného druhu.
V 2- až 13týdenních studiích s opakovaným podáváním u psů a potkanů byla pozorována degenerace semenotvorných kanálků ve varlatech, tento účinek však nebyl pozorován ve studiích trvajících 26 týdnů a déle. Ve studii fertility u samců potkanů bylo pozorováno snížení hmotnosti nadvarlat a varlat, avšak nebyly pozorovány nežádoucí účinky na ukazatele páření nebo fertility ani degenerace nebo úbytek spermatogeneze. Fertilita samic nebyla u potkanů ovlivněna.
Fototoxicita
Hodnocení možné fototoxicity v buněčné linii BALB/c 3T3 myších embryonálních fibroblastů bylo neprůkazné pro idelalisib v důsledku cytotoxicity v in vitro testu. Hlavní metabolit GS-563117 může zvyšovat fototoxicitu, pokud jsou buňky současně vystaveny UVA záření. Existuje potenciální riziko, že idelalisib může prostřednictvím svého hlavního metabolitu GS-563117 způsobit fotosenzitivitu u léčených pacientů.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulóza Hyprolóza (E463)
Sodná sůl kroskarmelózy Sodná sůl karboxymethylškrobu Magnesium-stearát
Potah tablety
Polyvinylalkohol (E1203)
Makrogol 3350 (E1521)
Oxid titaničitý (El71)
Mastek (E553B)
Červený oxid železitý (E172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu o vysoké hustotě (HDPE) s polypropylenovým dětským bezpečnostním uzávěrem obsahující 60 potahovaných tablet a polyesterovou vatu.
Každá krabička obsahuje 1 lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/938/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18. září 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Gilead Sciences Ireland UC
IDA Business & Technology Park
Carrigtohill
County Cork
Irsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 8 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Žadatel má předložit závěrečnou studijní zprávu pro prodlouženou studii fáze 3, GS-US-312-0117, hodnotící účinnost a bezpečnost idelalisibu (GS-1101) v kombinaci s rituximabem u dříve léčené CLL. Je třeba předložit aktualizované údaje týkající se PFS, OS a trvání odpovědi u pacientů s delecí 17p/mutací TP53 nebo bez delece 17p/mutace TP53 a u celkové populace. |
31. prosinec 2017 |
|
Žadatel má předložit závěrečnou studijní zprávu pro studii fáze 2, 101-09, hodnotící účinnost a bezpečnost idelalisibu u pacientů s indolentním NHL z B-buněk refrakterním na rituximab a alkylační látky. Je třeba předložit aktualizované výsledky týkající se bezpečnosti a účinnosti, včetně celkového přežití, a aktualizované údaje z analýz pacientů s výchozí lymfopenií. |
31. prosinec 2016 |
|
Žadatel má předložit závěrečnou studijní zprávu pro prodlouženou studii 101-99. |
30. září 2017 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zydelig 100 mg potahované tablety Idelalisibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje idelalisibum 100 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje oranžovou žluť (E110), pro další informace viz příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Potahované tablety 60 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
EU/1/14/938/001
Lot
Výdej léčivého přípravku vázán na lékařský předpis.

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zydelig 100 mg potahované tablety Idelalisibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje idelalisibum 100 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje oranžovou žluť (E110), pro další informace viz příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Potahované tablety 60 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
EU/1/14/938/001
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zydelig 150 mg potahované tablety Idelalisibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje idelalisibum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Potahované tablety 60 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
EU/1/14/938/002
Lot
Výdej léčivého přípravku vázán na lékařský předpis.

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zydelig 150 mg potahované tablety Idelalisibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje idelalisibum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Potahované tablety 60 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
EU/1/14/938/002
Lot
Výdej léčivého přípravku vázán na lékařský předpis.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Zydelig 100 mg potahované tablety
Idelalisibum
^^Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- T ento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Zydelig a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zydelig užívat
3. Jak se přípravek Zydelig užívá
4. Možné nežádoucí účinky
5. Jak přípravek Zydelig uchovávat
6. Obsah balení a další informace
1. Co je přípravek Zydelig a k čemu se používá
Zydelig je protinádorový léčivý přípravek, který obsahuje léčivou látku idelalisib. Účinkuje tak, že blokuje účinky enzymu, který se podílí na množení a přežívání určitých bílých krvinek nazývaných lymfocyty. Protože je aktivita tohoto enzymu v určitých rakovinných bílých krvinkách zvýšena, jeho blokováním přípravek Zydelig usmrcuje rakovinné buňky a snižuje jejich počet.
Přípravek Zydelig je možné používat k léčbě dvou různých druhů rakoviny:
Chronické lymfatické leukémie
Chronická lymfatická leukémie (CLL) je rakovina typu bílých krvinek nazývaných B-lymfocty.
U tohoto onemocnění se lymfocyty příliš rychle množí a žijí příliš dlouho, takže v krvi jich koluje příliš mnoho.
V případě CLL může léčba přípravkem Zydelig pokračovat v kombinaci s jiným léčivým přípravkem nazývaným rituximab u pacientů, kteří mají určité vysoce rizikové faktory a kteří již přípravek Zydelig užívají. Může být také používán v kombinaci s rituximabem u CLL pacientů, u kterých se rakovina vrátila po minimálně jedné předchozí léčbě.
Folikulárního lymfomu
Folikulární lymfom (FL) je rakovina typu bílých krvinek nazývaných B-lymfocyty. U folikulárního lymfomu se B-lymfocyty příliš rychle množí a žijí příliš dlouho, takže v lymfatických uzlinách je jich příliš mnoho. V případě FL se přípravek Zydelig používá samostatně u pacientů, jejichž rakovina nereagovala na léčbu dvěma předchozími typy léčby rakoviny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zydelig užívat Neužívejte přípravek Zydelig
• jestliže jste alergický(á) na idelalisib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
^ Pokud se Vás to týká, poraďte se se svým lékařem.
Upozornění a opatření
Před užitím přípravku Zydelig se poraďte se svým lékařem. Sdělte svému lékaři,
• jestliže máte problémy s játry,
• jestliže máte jakékoli jiné zdravotní potíže nebo onemocnění (zejména infekci nebo horečku).
U pacientů užívajících přípravek Zydelig se vyskytly závažné a smrtelné infekce. Během léčby přípravkem Zydelig užívejte také další přípravek, který Vám předepíše Váš lékař, aby se zabránilo vzniku infekc e. Váš lékař bude sledovat, zda se u Vás neprojevují známky infekce. Informujte ihned svého lékaře, pokud během užívání přípravku Zydelig onemocníte (zejména pokud budete mít horečku, kašel nebo obtíže s dýcháním).
Před léčbou přípravkem Zydelig a během této léčby budete potřebovat pravidelná vyšetření krve.
T a slouží k zjištění toho, že nemáte infekci, že Vaše játra správně fungují a že Váš krevní obraz je normální. Pokud to bude nutné, Váš lékař může rozhodnout, že léčbu na nějakou dobu přeruší, než opět zahájí léčbu stejnou nebo nižší dávkou. Váš lékař může rozhodnout, že léčbu přípravkem Zydelig trvale ukončí.
Přípravek Zydelig může způsobit těžký průjem. Při prvních známkách průjmu o tom okamžitě informujte svého lékaře.
Přípravek Zydelig může způsobit zánět plic. Okamžitě sdělte svému lékaři,
• jestliže se u Vás objeví kašel nebo se kašel zhorší,
• jestliže se u Vás objeví dušnost nebo obtíže s dýcháním.
Těžké případy tvorby puchýřů na pokožce byly hlášeny u některých osob, kterým byl přípravek Zydelig podáván současně s jinými přípravky, o nichž je známo, že mohou vyvolat potenciálně život ohrožující stavy. Puchýře se mohou vytvářet také na sliznici úst, pohlavních orgánů a/nebo očí. Odlupování kůže může vést k závažné infekci. Okamžitě sdělte svému lékaři:
• jestliže máte zarudlou kůži a tvoří se na ní puchýře,
• jestliže máte otok a puchýře na sliznici úst, pohlavních orgánů a/nebo očí.
Děti a dospívající
Nepodávejte tento přípravek dětem a dospívajícím ve věku do 18 let, protože nebyl u této věkové skupiny studován.
Další léčivé přípravky a přípravek Zydelig
Přípravek Zydelig se nemá užívat spolu s jinými přípravky, pokud Vám lékař nesdělil, že je to bezpečné.
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Je to nesmírně důležité, protože užívání více než jednoho léku současně může zesílit nebo zeslabit účinky těchto léků.
Užívání přípravku Zydelig současně s některými léky může způsobit, že tyto léky přestanou správně účinkovat nebo že se zhorší jejich nežádoucí účinky. Zejména informujte svého lékaře, pokud užíváte kterýkoli z následujících přípravků:
• alfuzosin, lék používaný k léčbě zvětšené prostaty
• dabigatran, warfarin, léky používané k ředění krve
• amiodaron, bepridil, disopyramid, lidokain, chinidin, léky používané k léčbě problémů se srdcem
• dihydroergotamin, ergotamin, léky používané k léčbě migrény
• cisaprid, lék používaný ke zmírnění určitých žaludečních problémů
• pimozid, lék používaný k léčbě neobvyklých myšlenek nebo pocitů
• midazolam, triazolam, užívané ústy ke zlepšení spánku a/nebo zmírnění úzkosti
• kvetiapin, lék používaný k léčbě schizofrenie, bipolární poruchy a závažné depresivní poruchy
• amlodipin, diltiazem, felodipin, nikardipin, nifedipin, léky používané k léčbě vysokého krevního tlaku a problémů se srdcem
• bosentan, lék používaný k léčbě plicní arteriální hypertenze
• sildenafil, tadalafil, léky používané k léčbě impotence a plicní hypertenze, onemocnění plic, které způsobuje potíže s dýcháním
• budesonid, flutikason, léky používané k léčbě senné rýmy a astmatu a salmeterol používaný k léčbě astmatu
• rifabutin, lék používaný k léčbě bakteriálních infekcí, včetně tuberkulózy
• itrakonazol, ketokonazol, posakonazol, vorikonazol, léky používané k léčbě plísňových infekcí
• boceprevir, telaprevir, léky používané k léčbě hepatitidy C
• karbamazepin, S-mefenytoin, fenytoin, léky používané k prevenci záchvatů
• rifampicin, lék používaný k prevenci a léčbě tuberkulózy a jiných infekcí
• třezalku tečkovanou (Hypericum perforatum), rostlinný přípravek používaný k léčbě deprese a úzkosti
• alfentanil, fentanyl, metadon, buprenorfin/naloxon, léky používané k úlevě od bolesti
• cyklosporin, sirolimus, takrolimus, léky používané k potlačení imunitní reakc e těla po transplantaci
• kolchicin, lék používaný k léčbě dny
• trazodon, lék používaný k léčbě deprese
• buspiron, klorazepát, diazepam, estazolam, flurazepam, zolpidem, léky používané k léčbě poruch nervového systému
• dasatinib, nilotinib, paklitaxel, vinblastin, vinkristin, léky používané k léčbě rakoviny
• ústy užívanou nebo implantovanou hormonální antikoncepci, používanou k zabránění otěhotnění
• klarithromycin, telithromycin, léky používané k léčbě bakteriálních infekcí
• atorvastatin, lovastatin, simvastatin, léky používané ke snížení hladiny cholesterolu
K léčbě CLL může být Zydelig předepsán v kombinaci s jinými přípravky. Je nesmírně důležité, abyste si přečetl(a) také příbalové informac e, které jsou součástí balení těchto přípravků.
Poraďte se se svým lékařem, máte-li jakékoli otázky týkající se kteréhokoli z Vašich léků.
Těhotenství a kojení
• Přípravek Zydelig se nesmí užívat v těhotenství. Údaje o bezpečnosti tohoto přípravku u těhotných žen nejsou k dispozici.
• Během léčby přípravkem Zydelig a 1 měsíc po posledním podání přípravku používejte spolehlivou metodu antikoncepce, abyste zabránila otěhotnění.
• Přípravek Zydelig může snížit účinnost antikoncepční „pilulky“ a implantované hormonální antikoncepce. Během užívání přípravku Zydelig a 1 měsíc po posledním podání přípravku musíte používat také bariérovou metodu antikoncepce, jako jsou kondomy nebo „spirála“.
• Pokud otěhotníte, okamžitě to sdělte svému lékaři.
Během užívání přípravku Zydelig nesmíte kojit. Jestliže v současné době kojíte, poraďte se před zahájením léčby se svým lékařem. Není známo, zda se léčivá látka obsažená v přípravku Zydelig vylučuje do lidského mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že by přípravek Zydelig ovlivňoval Vaši schopnost řídit nebo obsluhovat stroje. Přípravek Zydelig obsahuje oranžovou žluť (E110)
Jestliže máte alergii na oranžovou žluť (E110), sdělte to svému lékaři. Přípravek Zydelig obsahuje oranžovou žluť, která může způsobovat alergické reakce.
3. Jak se přípravek Zydelig užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Doporučená dávka je 150 mg ústy, dvakrát denně. Jestliže se však u Vás vyskytnou určité nežádoucí účinky, může Váš lékař tuto dávku snížit na 100 mg dvakrát denně.
Přípravek Zydelig lze užívat s jídlem nebo bez jídla.
Tabletu spolkněte celou. Tablety nekousejte ani nedrťte. Pokud máte problémy s polykáním tablet, s dělte to s vému lékaři.
Jestliže jste užil(a) více přípravku Zydelig, než jste měl(a)
Jestliže jste omylem užil(a) více přípravku Zydelig, než je doporučená dávka, může u Vás být zvýšené riziko výskytu nežádoucích účinků tohoto přípravku (viz bod 4, Možné nežádoucí účinky).
Okamžitě kontaktujte svého lékaře nebo nejbližší lékařskou pohotovost. Vezměte si s sebou lahvičku a tuto příbalovou informaci, abyste mohl(a) snadno popsat, jaké léky jste užil(a).
Jestliže jste zapomněl(a) užít přípravek Zydelig
Snažte se nevynechat žádnou dávku přípravku Zydelig. Jestliže vynecháte dávku a uplynulo méně než 6 hodin od doby, kdy jste ji měl(a) užít, vynechanou dávku ihned užijte. Poté užijte následující dávku v obvyklou dobu. Jestliže vynecháte dávku a uplynulo více než 6 hodin od doby, kdy jste ji měl(a) užít, počkejte a užijte následující dávku v obvyklou dobu.
Nepřestávejte užívat přípravek Zydelig
Nepřestávejte užívat tento léčivý přípravek, pokud Vám to neřekne Váš lékař.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Některé nežádoucí účinky mohou být závažné.
PŘESTAŇTE přípravek Zydelig užívat a okamžitě vyhledejte lékařskou pomoc, pokud se u Vás objeví některé z následujících příznaků:
• zarudnutí pokožky a tvorba puchýřů na pokožc e,
• otok a puchýře na sliznici úst, pohlavních orgánů a/nebo očí.
Další nežádoucí účinky
Velmi časté nežádoucí účinky
(mohou se vyskytnout u více než 1 z 10 osob)
• průjem/zánět tlustého střeva
• vyrážka
• snížení počtu bílých krvinek
• infekce
• horečka
Časté nežádoucí účinky
(mohou se vyskytnout až u 1 z 10 osob)
• zánět plic
Krevní testy mohou také ukázat:
• zvýšení hladin jaterních enzymů nebo tuku v krvi
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zydelig uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Zydelig obsahuje
• Léčivou látkou je idelalisib. Jedna potahovaná tableta obsahuje idelalisibum 100 mg.
• Dalšími složkami jsou:
Jádro tablety:
Mikrokrystalická celulóza, hyprolóza (E463), sodná sůl kroskarmelózy, sodná sůl karboxymethylškrobu, magnesium-stearát.
Potah tablety:
Polyvinylalkohol (E1203), makrogol 3350 (E1521), oxid titaničitý (E171), mastek (E553B), oranžová žluť (E110).
Jak přípravek Zydelig vypadá a co obsahuje toto balení
Potahované tablety jsou oranžové, oválné tablety s vyraženým označením „GSI“ na jedné straně a „100“ na druhé straně.
Dostupná je následující velikost balení: krabička obsahující 1 plastovou lahvičku s 60 potahovanými tabletami.
Držitel rozhodnutí o registraci
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
Výrobce
Gilead Sciences Ireland UC
IDA Business & Technology Park
Carrigtohill
County Cork
Irsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Gilead Sciences Belgium SPRL-BVBA Gilead Sciences Sweden AB
Tél/Tel: + 32 (0) 24 01 35 50 Tel: + 46 (0) 8 5057 1849
Et.irapHH
Gilead Sciences International Ltd Ten.: + 44 (0) 20 7136 8820
Česká republika
Gilead Sciences s.r.o.
Tel: + 420 910 871 986
Luxembourg/Luxemburg
Gilead Sciences Belgium SPRL-BVBA Tél/Tel: + 32 (0) 24 01 35 50
Magyarország
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Danmark
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
Deutschland
Gilead Sciences GmbH Tel: + 49 (0) 89 899890-0
Eesti
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
EM,á8a
Gilead Sciences EILá^ M. blik. Tr|L: + 30 210 8930 100
Espaňa
Gilead Sciences, S.L.
Tel: + 34 91 378 98 30
France
Gilead Sciences
Tél: + 33 (0) 1 46 09 41 00
Hrvatska
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Malta
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Nederland
Gilead Sciences Netherlands B.V. Tel: + 31 (0) 20 718 36 98
Norge
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
Osterreich
Gilead Sciences GesmbH Tel: + 43 1 260 830
Polska
Gilead Sciences Poland Sp. z o.o. Tel: + 48 22 262 8702
Portugal
Gilead Sciences, Lda.
Tel: + 351 21 7928790
Románia
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Ireland
Gilead Sciences Ltd Tel: + 44 (0) 1223 897555
island
Gilead Sciences Sweden AB Sími: + 46 (0) 8 5057 1849
Italia
Gilead Sciences S.r.l.
Tel: + 39 02 439201
Kúnpo^
Gilead Sciences M.ETIE.
Tr|X: + 30 210 8930 100
Latvija
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
Slovenija
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Slovenská republika
Gilead Sciences Slovakia s.r.o. Tel: + 421 232 121 210
Suomi/Finland
Gilead Sciences Sweden AB Puh/Tel: + 46 (0) 8 5057 1849
Sverige
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
United Kingdom
Gilead Sciences Ltd Tel: + 44 (0) 1223 897555
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Příbalová informace: informace pro pacienta
Zydelig 150 mg potahované tablety
Idelalisibum
^^Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- T ento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Zydelig a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zydelig užívat
3. Jak se přípravek Zydelig užívá
4. Možné nežádoucí účinky
5. Jak přípravek Zydelig uchovávat
6. Obsah balení a další informace
1. Co je přípravek Zydelig a k čemu se používá
Zydelig je protinádorový léčivý přípravek, který obsahuje léčivou látku idelalisib. Účinkuje tak, že blokuje účinky enzymu, který se podílí na množení a přežívání určitých bílých krvinek nazývaných lymfocyty. Protože je aktivita tohoto enzymu v určitých rakovinných bílých krvinkách zvýšena, jeho blokováním přípravek Zydelig usmrcuje rakovinné buňky a snižuje jejich počet.
Přípravek Zydelig je možné používat k léčbě dvou různých druhů rakoviny:
Chronické lymfatické leukémie
Chronická lymfatická leukémie (CLL) je rakovina typu bílých krvinek nazývaných B-lymfocty.
U tohoto onemocnění se lymfocyty příliš rychle množí a žijí příliš dlouho, takže v krvi jich koluje příliš mnoho.
V případě CLL může léčba přípravkem Zydelig pokračovat v kombinaci s jiným léčivým přípravkem nazývaným rituximab u pacientů, kteří mají určité vysoce rizikové faktory a kteří již přípravek Zydelig užívají. Může být také používán v kombinaci s rituximabem u CLL pacientů, u kterých se rakovina vrátila po minimálně jedné předchozí léčbě.
Folikulárního lymfomu
Folikulární lymfom (FL) je rakovina typu bílých krvinek nazývaných B-lymfocyty. U folikulárního lymfomu se B-lymfocyty příliš rychle množí a žijí příliš dlouho, takže v lymfatických uzlinách je jich příliš mnoho. V případě FL se přípravek Zydelig používá samostatně u pacientů, jejichž rakovina nereagovala na léčbu dvěma předchozími typy léčby rakoviny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zydelig užívat Neužívejte přípravek Zydelig
• jestliže jste alergický(á) na idelalisib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
^ Pokud se Vás to týká, poraďte se se svým lékařem.
Upozornění a opatření
Před užitím přípravku Zydelig se poraďte se svým lékařem. Sdělte svému lékaři,
• jestliže máte problémy s játry,
• jestliže máte jakékoli jiné zdravotní potíže nebo onemocnění (zejména infekci nebo horečku).
U pacientů užívajících přípravek Zydelig se vyskytly závažné a smrtelné infekce. Během léčby přípravkem Zydelig užívejte také další přípravek, který Vám předepíše Váš lékař, aby se zabránilo vzniku infekce. Váš lékař bude sledovat, zda se u Vás neprojevují známky infekce. Informujte ihned svého lékaře, pokud během užívání přípravku Zydelig onemocníte (zejména pokud budete mít horečku, kašel nebo obtíže s dýcháním).
Před léčbou přípravkem Zydelig a během této léčby budete potřebovat pravidelná vyšetření krve. Ta slouží k zjištění toho, že nemáte infekci, že Vaše játra správně fungují a že Váš krevní obraz je normální. Pokud to bude nutné, Váš lékař může rozhodnout, že léčbu na nějakou dobu přeruší, než opět zahájí léčbu stejnou nebo nižší dávkou. Váš lékař může rozhodnout, že léčbu přípravkem Zydelig trvale ukončí.
Přípravek Zydelig může způsobit těžký průjem. Při prvních známkách průjmu o tom okamžitě informujte svého lékaře.
Přípravek Zydelig může způsobit zánět plic. Okamžitě sdělte svému lékaři,
• jestliže se u Vás objeví kašel nebo se kašel zhorší,
• jestliže se u Vás objeví dušnost nebo obtíže s dýcháním.
Těžké případy tvorby puchýřů na pokožce byly hlášeny u některých os ob, kterým byl přípravek Zydelig podáván současně s jinými přípravky, o nichž je známo, že mohou vyvolat potenciálně život ohrožující stavy. Puchýře se mohou vytvářet také na sliznici úst, pohlavních orgánů a/nebo očí. Odlupování kůže může vést k závažné infekci. Okamžitě sdělte svému lékaři:
• jestliže máte zarudlou kůži a tvoří se na ní puchýře,
• jestliže máte otok a puchýře na sliznici úst, pohlavních orgánů a/nebo očí.
Děti a dospívající
Nepodávejte tento přípravek dětem a dospívajícím ve věku do 18 let, protože nebyl u této věkové skupiny studován.
Další léčivé přípravky a přípravek Zydelig
Přípravek Zydelig se nemá užívat spolu s jinými přípravky, pokud Vám lékař nesdělil, že je to bezpečné.
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Je to nesmírně důležité, protože užívání více než jednoho léku současně může zesílit nebo zeslabit účinky těchto léků.
Užívání přípravku Zydelig současně s některými léky může způsobit, že tyto léky přestanou správně účinkovat nebo že se zhorší jejich nežádoucí účinky. Zejména informujte svého lékaře, pokud užíváte kterýkoli z následujících přípravků:
• alfuzosin, lék používaný k léčbě zvětšené prostaty
• dabigatran, warfarin, léky používané k ředění krve
• amiodaron, bepridil, disopyramid, lidokain, chinidin, léky používané k léčbě problémů se srdcem
• dihydroergotamin, ergotamin, léky používané k léčbě migrény
• cisaprid, lék používaný ke zmírnění určitých žaludečních problémů
• pimozid, lék používaný k léčbě neobvyklých myšlenek nebo pocitů
• midazolam, triazolam, užívané ústy ke zlepšení spánku a/nebo zmírnění úzkosti
• kvetiapin, lék používaný k léčbě schizofrenie, bipolární poruchy a závažné depresivní poruchy
• amlodipin, diltiazem, felodipin, nikardipin, nifedipin, léky používané k léčbě vysokého krevního tlaku a problémů se srdcem
• bosentan, lék používaný k léčbě plicní arteriální hypertenze
• sildenafil, tadalafil, léky používané k léčbě impotence a plicní hypertenze, onemocnění plic, které způsobuje potíže s dýcháním
• budesonid, flutikason, léky používané k léčbě senné rýmy a astmatu a salmeterol, používaný k léčbě astmatu
• rifabutin, lék používaný k léčbě bakteriálních infekcí, včetně tuberkulózy
• itrakonazol, ketokonazol, posakonazol, vorikonazol, léky používané k léčbě plísňových infekcí
• boceprevir, telaprevir, léky používané k léčbě hepatitidy C
• karbamazepin, S-mefenytoin, fenytoin, léky používané k prevenci záchvatů
• rifampicin, lék používaný k prevenci a léčbě tuberkulózy a jiných infekcí
• třezalku tečkovanou (Hypericumperforatum), rostlinný přípravek používaný k léčbě deprese a úzkosti
• alfentanil, fentanyl, metadon, buprenorfin/naloxon, léky používané k úlevě od bolesti
• cyklosporin, sirolimus, takrolimus, léky používané k potlačení imunitní reakce těla po transplantaci
• kolchicin, lék používaný k léčbě dny
• trazodon, lék používaný k léčbě deprese
• buspiron, klorazepát, diazepam, estazolam, flurazepam, zolpidem, léky používané k léčbě poruch nervového systému
• dasatinib, nilotinib, paklitaxel, vinblastin, vinkristin, léky používané k léčbě rakoviny
• ústy užívanou nebo implantovanou hormonální antikoncepci, používanou k zabránění otěhotnění
• klarithromycin, telithromycin, léky používané k léčbě bakteriálních infekcí
• atorvastatin, lovastatin, simvastatin, léky používané ke snížení hladiny cholesterolu
K léčbě CLL může být Zydelig předepsán v kombinaci s jinými přípravky. Je nesmírně důležité, abyste si přečetl(a) také příbalové informace, které jsou součástí balení těchto přípravků.
Poraďte se se svým lékařem, máte-li jakékoli otázky týkající se kteréhokoli z Vašich léků.
Těhotenství a kojení
• Přípravek Zydelig se nesmí užívat v těhotenství. Údaje o bezpečnosti tohoto přípravku u těhotných žen nejsou k dispozici.
• Během léčby přípravkem Zydelig a 1 měsíc po posledním podání přípravku používejte spolehlivou metodu antikoncepce, abyste zabránila otěhotnění.
• Přípravek Zydelig může snížit účinnost antikoncepční „pilulky“ a implantované hormonální antikoncepce. Během užívání přípravku Zydelig a 1 měsíc po posledním podání přípravku musíte používat také bariérovou metodu antikoncepce, jako jsou kondomy nebo „spirála“.
• Pokud otěhotníte, okamžitě to sdělte svému lékaři.
Během užívání přípravku Zydelig nesmíte kojit. Jestliže v současné době kojíte, poraďte se před zahájením léčby se svým lékařem. Není známo, zda se léčivá látka obsažená v přípravku Zydelig vylučuje do lidského mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že by přípravek Zydelig ovlivňoval Vaši schopnost řídit nebo obsluhovat stroje.
3. Jak se přípravek Zydelig užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Doporučená dávka je 150 mg ústy, dvakrát denně. Jestliže se však u Vás vyskytnou určité nežádoucí účinky, může Váš lékař tuto dávku snížit na 100 mg dvakrát denně.
Přípravek Zydelig lze užívat s jídlem nebo bez jídla.
Tabletu spolkněte celou. Tablety nekousejte ani nedrťte. Pokud máte problémy s polykáním tablet, sdělte to svému lékaři.
Jestliže jste užil(a) více přípravku Zydelig, než jste měl(a)
Jestliže jste omylem užil(a) více přípravku Zydelig, než je doporučená dávka, může u Vás být zvýšené riziko výskytu nežádoucích účinků tohoto přípravku (viz bod 4, Možné nežádoucí účinky).
Okamžitě kontaktujte svého lékaře nebo nejbližší lékařskou pohotovost. Vezměte si s sebou lahvičku a tuto příbalovou informaci, abyste mohl(a) snadno popsat, jaké léky jste užil(a).
Jestliže jste zapomněl(a) užít přípravek Zydelig
Snažte se nevynechat žádnou dávku přípravku Zydelig. Jestliže vynecháte dávku a uplynulo méně než 6 hodin od doby, kdy jste ji měl(a) užít, vynechanou dávku ihned užijte. Poté užijte následující dávku v obvyklou dobu. Jestliže vynecháte dávku a uplynulo více než 6 hodin od doby, kdy jste ji měl(a) užít, počkejte a užijte následující dávku v obvyklou dobu.
Nepřestávejte užívat přípravek Zydelig
Nepřestávejte užívat tento léčivý přípravek, pokud Vám to neřekne Váš lékař.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Některé nežádoucí účinky mohou být závažné.
PŘESTAŇTE přípravek Zydelig užívat a okamžitě vyhledejte lékařskou pomoc, pokud se u Vás objeví některé z následujících příznaků:
• zarudnutí pokožky a tvorba puchýřů na pokožc e,
• otok a puchýře na sliznici úst, pohlavních orgánů a/nebo očí.
Další nežádoucí účinky
Velmi časté nežádoucí účinky
(mohou se vyskytnout u více než 1 z 10 osob)
• průjem/zánět tlustého střeva
• vyrážka
• snížení počtu bílých krvinek
• infekce
• horečka
Časté nežádoucí účinky
(mohou se vyskytnout až u 1 z 10 osob)
• zánět plic
Krevní testy mohou také ukázat:
• zvýšení hladin jaterních enzymů nebo tuku v krvi
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zydelig uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Zydelig obsahuje
• Léčivou látkou je idelalisib. Jedna potahovaná tableta obsahuje idelalisibum 150 mg.
• Dalšími složkami jsou:
Jádro tablety:
Mikrokrystalická celulóza, hyprolóza (E463), sodná sůl kroskarmelózy, sodná sůl karboxymethylškrobu, magnesium-stearát.
Potah tablety:
Polyvinylalkohol (E1203), makrogol 3350 (E1521), oxid titaničitý (E171), mastek (E553B), červený oxid železitý (E172).
Jak přípravek Zydelig vypadá a co obsahuje toto balení
Potahované tablety jsou růžové, oválné tablety s vyraženým označením „GSI“ na jedné straně a „150“ na druhé straně.
Dostupná je následující velikost balení: krabička obsahující 1 plastovou lahvičku s 60 potahovanými tabletami.
Držitel rozhodnutí o registraci
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Velká Británie
Výrobce
Gilead Sciences Ireland UC
IDA Business & Technology Park
Carrigtohill
County Cork
Irsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Gilead Sciences Belgium SPRL-BVBA Gilead Sciences Sweden AB
Tél/Tel: + 32 (0) 24 01 35 50 Tel: + 46 (0) 8 5057 1849
Et.irapHH
Gilead Sciences International Ltd Ten.: + 44 (0) 20 7136 8820
Česká republika
Gilead Sciences s.r.o.
Tel: + 420 910 871 986
Danmark
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
Deutschland
Gilead Sciences GmbH Tel: + 49 (0) 89 899890-0
Eesti
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
EM,á8a
Gilead Sciences EILá^ M. blik. Tr|L: + 30 210 8930 100
Es pana
Gilead Sciences, S.L.
Tel: + 34 91 378 98 30
France
Gilead Sciences
Tél: + 33 (0) 1 46 09 41 00
Hrvatska
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Luxembourg/Luxemburg
Gilead Sciences Belgium SPRL-BVBA Tél/Tel: + 32 (0) 24 01 35 50
Magyarország
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Malta
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Nederland
Gilead Sciences Netherlands B.V.
Tel: + 31 (0) 20 718 36 98
Norge
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
Osterreich
Gilead Sciences GesmbH Tel: + 43 1 260 830
Polska
Gilead Sciences Poland Sp. z o.o.
Tel: + 48 22 262 8702
Portugal
Gilead Sciences, Lda.
Tel: + 351 21 7928790
Románia
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Ireland
Gilead Sciences Ltd Tel: + 44 (0) 1223 897555
island
Gilead Sciences Sweden AB Sími: + 46 (0) 8 5057 1849
Italia
Gilead Sciences S.r.l.
Tel: + 39 02 439201
Kúnpo^
Gilead Sciences M.ETIE.
Tr|X: + 30 210 8930 100
Latvija
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
Slovenija
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Slovenská republika
Gilead Sciences Slovakia s.r.o. Tel: + 421 232 121 210
Suomi/Finland
Gilead Sciences Sweden AB Puh/Tel: + 46 (0) 8 5057 1849
Sverige
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
United Kingdom
Gilead Sciences Ltd Tel: + 44 (0) 1223 897555
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
63