Zoledronic Acid Hospira 4 Mg/100 Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Zoledronic Acid Hospira 4 mg/5 ml, koncentrát pro přípravu infuzního roztoku
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s 5 ml koncentrátu obsahuje acidum zoledronicum 4 mg (ve formě acidum zoledronicum monohydricum).
Jeden ml koncentrátu obsahuje acidum zoledronicum 0,8 mg (ve formě acidum zoledronicum monohydricum).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro přípravu infuzního roztoku Čirý a bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
- Prevence kostních příhod (patologických zlomenin, kompresivních zlomenin obratlů, radiační nebo chirurgická léčba kostí, nebo hyperkalcemie vyvolané nádorovým onemocněním) u dospělých pacientů s pokročilým maligním onemocněním postihujícím kosti.
- Léčba dospělých pacientů s hyperkalcemií vyvolanou nádorovým onemocněním (TIH).
4.2 Dávkování a způsob podání
Kyselinu zoledronovou musí předepisovat a podávat pacientům pouze zdravotničtí pracovníci se zkušenostmi s intravenózním podáváním bisfosfonátů. Pacientům léčeným kyselinou zoledronovou má být k dispozici příbalová informace a pacientská informační karta.
Dávkování
Prevence kostních _příhod u _pacientů s _pokročilým maligním onemocněním _postihujícím kosti Dospělí a starší pacienti
Doporučená dávka pro prevenci kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti je 4 mg kyseliny zoledronové každé 3 až 4 týdny.
Pacientům je též nutné denně doplňkově perorálně podávat 500 mg vápníku a 400 mj. vitamínu D.
Při rozhodování o léčbě pacientů s kostními metastázami za účelem prevence kostních příhod je nutné vzít v úvahu, že účinky léčby se projeví za 2-3 měsíce.
Léčba TIH
Dospělí a starší pacienti
Doporučená dávka při léčbě hyperkalcemie (albuminem korigovaný vápník > 12,0 mg/dl nebo 3,0 mmol/l) je 4 mg kyseliny zoledronové v jedné dávce.
Porucha funkce ledvin
TIH:
U pacientů s TIH, kteří zároveň trpí závažnou poruchou funkce ledvin, lze o léčbě kyselinou zoledronovou uvažovat až po zhodnocení rizika a přínosu léčby. Pacienti s hladinou kreatininu v séru > 400 pmol/l nebo > 4,5 mg/dl byli z klinických studií vyloučeni. U pacientů s TIH s hladinou kreatininu v séru < 400 pmol/l nebo < 4,5 mg/dl není nutná úprava dávkování (viz bod 4.4).
Prevence kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti:
U pacientů s mnohočetným myelomem nebo metastatickými lézemi solidních nádorů do kostí je nutné při zahájení léčby kyselinou zoledronovou stanovit sérový kreatinin a clearanci kreatininu (CLcr).
CLcr se vypočítá ze sérového kreatininu pomocí Cockcroft-Gaultova vzorce. Nedoporučuje se podávat kyselinu zoledronovou pacientům, kteří již před zahájením léčby trpí závažnou poruchou funkce ledvin, která je u této populace definována jako CLcr < 30 ml/min. Pacienti s hladinou kreatininu v séru > 265 pmol/l nebo > 3,0 mg/dl byli z klinických studií s kyselinou zoledronovou vyloučeni.
U pacientů s kostními metastázami, kteří před zahájením léčby trpěli mírnou nebo středně závažnou poruchou funkce ledvin, jež je pro tuto populaci definována jako CLcr 30-60 ml/min, se doporučuje následující dávkování kyseliny zoledronové (viz též bod 4.4):
|
Výchozí hodnoty clearance kreatininu (ml/min) |
Doporučená dávka kyseliny zoledronové* |
|
> 60 |
4,0 mg kyseliny zoledronové |
|
50-60 |
3,5 mg* kyseliny zoledronové |
|
40-49 |
3,3 mg* kyseliny zoledronové |
|
30-39 |
3,0 mg* kyseliny zoledronové |
*Dávky byly vypočteny z předpokládané cílové AUC 0,66 (mg^hod/l) (CLcr = 75 ml/min). Při podávání snížených dávek pacientům s poruchou funkce ledvin lze předpokládat dosažení stejné AUC, jako byla pozorována u pacientů s clearancí kreatininu 75 ml/min.
Po zahájení léčby je nutné měřit sérový kreatinin před podáním každé dávky kyseliny zoledronové a při zhoršení renální funkce musí být léčba přerušena. V klinických studiích bylo zhoršení renální funkce definováno následujícím způsobem:
- u pacientů s normální výchozí hodnotou sérového kreatininu (< 1,4 mg/dl nebo < 124 pmol/l) zvýšení o 0,5 mg/dl nebo 44 pmol/l,
- u pacientů s abnormální výchozí hodnotou sérového kreatininu (> 1,4 mg/dl nebo > 124 pmol/l) zvýšení o 1,0 mg/dl nebo 88 pmol/l.
V klinických studiích byla léčba kyselinou zoledronovou znovu zahájena teprve tehdy, když se hladina kreatininu vrátila do rozmezí, které se nelišilo o více než 10 % od výchozí hodnoty (viz bod 4.4).
Léčbu kyselinou zoledronovou je nutné obnovit ve stejné dávce, jako byla dávka podávaná před přerušením.
Pediatrická populace
Bezpečnost a účinnost kyseliny zoledronové u dětí ve věku od 1 do 17 let nebyla stanovena.
V současnosti dostupné údaje jsou popsány v bodě 5.1, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání
Intravenózní podání.
Přípravek Zoledronic Acid Hospira, dále naředěný ve 100 ml (viz bod 6.6), se má podávat jako jednorázová intravenózní infuze po dobu nejméně 15 minut.
U pacientů s mírnou až středně těžkou poruchou funkce ledvin se doporučuje snížení dávek kyseliny zoledronové (viz bod „Dávkování“ výše a bod .4.4).
Pokyny pro přípravu nižších dávek přípravku Zoledronic Acid Hospira
Odeberte potřebný odpovídající objem koncentrátu podle následujících pokynů:
- 4,4 ml pro dávku 3,5 mg,
- 4,1 ml pro dávku 3,3 mg,
- 3,8 ml pro dávku 3,0 mg.
Odebrané množství koncentrátu musí být dále naředěno ve 100 ml sterilního injekčního roztoku chloridu sodného 9 mg/ml (0,9%) nebo v 5% (w/v) roztoku glukózy. Dávka musí být podána v jedné intravenózní infuzi po dobu nejméně 15 minut.
Přípravek Zoledronic Acid Hospira nesmí být mísen s infuzními roztoky obsahujícími vápník nebo jiné dvoumocné kationty, jako je laktátový Ringerův roztok, a musí se podávat jako samostatný intravenózní roztok oddělenou infuzní linkou.
Pacienti musejí být před podáním přípravku Zoledronic Acid Hospira a poté dobře hydratováni.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku, jiné bisfosfonáty nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Obecná
Před podáním kyseliny zoledronové a poté musí být posouzen stav pacientů, aby se zajistila jejich odpovídající hydratace.
U pacientů s rizikem srdečního selhání je nutné zabránit nadměrné hydrataci.
Po zahájení léčby kyselinou zoledronovou se musí pečlivě sledovat standardní metabolické parametry související s hyperkalcemií, jako jsou sérové hladiny vápníku, fosfátu a hořčíku. Pokud se objeví hypokalcemie, hypofosfatemie nebo hypomagnesemie, může být nezbytná krátkodobá suplementační terapie. Pacienti s neléčenou hyperkalcemií mají obvykle do jisté míry poškozenou funkci ledvin, proto je nutné zvážit pečlivé sledování ledvinných funkcí.
K léčbě osteoporózy a Pagetovy choroby kostí jsou k dispozici i jiné přípravky obsahující jako léčivou látku kyselinu zoledronovou. Pacienti léčení přípravkem Zoledronic Acid Hospira nemají být současně léčeni těmito přípravky ani jiným bisfosfonátem, neboť kombinované účinky těchto látek nejsou známy.
Renální insuficience
U pacientů s TIH a zjištěným zhoršováním funkce ledvin je nutné odpovídajícím způsobem posoudit jejich stav a je nutné zvážit, zda potenciální přínos léčby kyselinou zoledronovou převáží možné riziko.
Při rozhodování o léčbě pacientů s kostními metastázami za účelem prevence kostních příhod je nutné vzít v úvahu, že účinky léčby se projeví za 2-3 měsíce.
U kyseliny zoledronové byla hlášena souvislost s dysfunkcí ledvin. Mezi faktory, které mohou zvyšovat možnost zhoršení funkce ledvin, patří dehydratace, již existující poruchy funkce ledvin, opakované cykly podávání kyseliny zoledronové a jiných bisfosfonátů, stejně jako užívání jiných nefrotoxických léčivých přípravků. I když je toto riziko sníženo při podání dávky kyseliny zoledronové po dobu 15 minut, může přesto dojít ke zhoršení funkce ledvin. Bylo hlášeno zhoršení funkce ledvin a progrese až k selhání ledvin a nutnosti dialýzy u pacientů po úvodní nebo jednorázové dávce 4 mg kyseliny zoledronové. Může též dojít ke zvýšení kreatininu v séru u některých pacientů po opakovaném podání kyseliny zoledronové v dávkách doporučených k prevenci kostních příhod, i když méně často.
Před aplikací každé dávky kyseliny zoledronové je nutné u pacientů stanovit hladinu sérového kreatininu. U pacientů s kostními metastázami a s mírnou až středně závažnou poruchou funkce ledvin se doporučuje zahájit léčbu nižšími dávkami kyseliny zoledronové. U pacientů, u kterých bylo v průběhu léčby prokázáno zhoršení funkce ledvin, musí být léčba kyselinou zoledronovou přerušena. Léčba kyselinou zoledronovou se má obnovit pouze tehdy, až se hladina sérového kreatininu vrátí do rozmezí, které se nebude lišit o více než 10 % od výchozí hodnoty. Léčbu kyselinou zoledronovou je nutné obnovit ve stejné dávce, jako byla dávka podávaná před přerušením.
Vzhledem k možnému vlivu kyseliny zoledronové na funkci ledvin, nedostatku klinických údajů o bezpečnosti podávání u pacientů s těžkou poruchou funkce ledvin již před zahájením léčby (v klinických studiích definované jako hladina kreatininu v séru > 400 ^mol/l nebo > 4,5 mg/dl u pacientů s TIH a > 265 ^mol/l nebo > 3,0 mg/dl u pacientů s karcinomem a kostními metastázami) a pouze omezenému množství farmakokinetických údajů těchto pacientů (clearance kreatininu < 30 ml/min) se podávání kyseliny zoledronové pacientům se závažnou poruchou funkce ledvin nedoporučuje.
Hepatální insuficience
U pacientů s těžkou jaterní insuficiencí je k dispozici jen omezené množství klinických údajů, a proto nelze pro tuto skupinu pacientů stanovit konkrétní doporučení.
Osteonekróza čelisti
U pacientů používajících kyselinu zoledronovou byla v klinických studiích a po uvedení přípravku na trh méně často hlášena osteonekróza čelisti.
U pacientů s nehojícími se lézemi měkkých tkání v ústech by mělo být s výjimkou akutních medicínských stavů zahájení léčby nebo nového cyklu léčby odloženo. Před zahájením léčby bisfosfonáty je u pacientů s konkomitantními rizikovými faktory doporučené příslušné vyšetření s preventivním ošetřením a individuálním vyhodnocením poměru prospěchu-rizika.
Při posuzování rizika rozvoje osteonekrózy čelisti u jednotlivých pacientů je třeba zvážit následující rizikové faktory:
- potenciál bisfosfonátů (vyšší riziko u vysoce účinných sloučenin), cesta podání (vyšší riziko při parenterálním podání) a kumulativní dávka bisfosfonátů,
- maligní nádorové onemocnění, komorbidity (např. anémie, koagulopatie, infekce), kouření.
- konkomitantní terapie:chemoterapie, inhibitory angiogeneze (viz bod 4.5), radioterapie hlavy a krku, kortikosteroidy.
- stomatologická onemocnění v anamnéze, špatná ústní hygiena, periodontální onemocnění, invazivní stomatologické zákroky (např. extrakce zubů)a špatně naléhající zubní protéza.
Všichni pacienti mají být vyzváni, aby během léčby kyselinou zoledronovou udržovali dobrou ústní hygienu, absolvovali rutinní vyšetření chrupu a okamžitě hlásili jakékoli příznaky v ústech, jako je kývání zubů, bolest nebo otoky nebo nehojící se vředy nebo výtok.
Během léčby mají být invazivní stomatologické procedury prováděny pouze po pečlivém vyhodnocení a při nastávajícím podávání kyseliny zoledronové mají být vyloučeny. U pacientů, u nichž se během bisfosfonátové léčby rozvine osteonekróza čelisti, může stomatochirurgický zákrok zhoršit stav. Neexistují údaje, které by dokládaly, zda vysazení léčby bisfosfonáty snižuje u pacientů vyžadujících stomatologický výkon riziko osteonekrózy čelisti. Plán léčby pacientů s OČ by měl být navržen v úzké spolupráci mezi ošetřujícím lékařem a dentistou nebo zubním chirurgem s odbornou znalostí OČ. Dokud se stav nezlepší a pokud možno nevymizí příspívající rizikové faktory, mělo by být zváženo dočasné přerušení léčby kyselinou zoledronovou.
Osteonekróza zevního zvukovodu
V souvislosti s léčbou bisfosfonáty byla hlášena osteonekróza zevního zvukovodu, zejména při dlouhodobém podávání. Mezi možné rizikové faktory osteonekrózy zevního zvukovodu patří používání steroidů a chemoterapie a/nebo lokální rizikové faktory, jako například infekce nebo trauma. Možnost vzniku osteonekrózy zevního zvukovodu je třeba zvážit u pacientů léčených bisfosfonáty, kteří mají ušní symptomy včetně chronických infekcí ucha.
Bolesti pohybového systému
Zkušenosti po uvedení přípravku na trh dokládají, že u pacientů, kteří užívali kyselinu zoledronovou, byly hlášeny silné bolesti kostí, kloubů a/nebo svalů, jež byly občas zneschopňující. Taková hlášení jsou však řídká. Doba do vzniku příznaků byla různá, a to od jednoho dne až do několika měsíců po zahájení léčby. Po ukončení léčby se většině pacientů od příznaků ulevilo. U podskupiny pacientů byl zaznamenán opětovný návrat příznaků poté, co byli znovu léčeni kyselinou zoledronovou nebo jiným bisfosfonátem.
Atypické zlomeniny femuru
V souvislosti s léčbou bisfosfonáty, a to zejména u pacientů dlouhodobě léčených pro osteoporózu, byly hlášeny atypické subtrochanterické a diafyzární fraktury femuru. Tyto příčné nebo krátké šikmé fraktury mohou vzniknout kdekoli na femuru od místa těsně pod malým trochanterem až do místa těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s ním a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“), týdny až měsíce před manifestací kompletní zlomeniny femuru. Fraktury jsou často oboustranné, a proto je u pacientů léčených bisfosfonáty, u nichž došlo ke zlomenině diafýzy femuru, nutné vyšetřit kontralaterální femur. Bylo též zaznamenáno špatné hojení těchto fraktur. U pacientů, u nichž existuje podezření na atypickou frakturu femuru, je nutné při hodnocení jejich stavu zvážit přerušení léčby bisfosfonáty, a to na základě individuálního posouzení rizik a přínosů této léčby.
Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, kyčle nebo třísla, a všechny pacienty, u nichž se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou neúplnou frakturu femuru.
Hypokalcemie
Hypokalcemie byla hlášena u pacientů léčených kyselinou zoledronovou. V důsledku těžké hypokalcemie byly hlášeny srdeční arytmie a neurologické nežádoucí účinky (včetně křečí, hypoestezie a tetanie). Byly hlášeny případy těžké hypokalcemie vyžadující hospitalizaci. V některých případech může být hypokalcemie život ohrožující (viz bod 4.8). Při podávání Zoledronic Acid Hospira spolu s léčivými přípravky, které mohou způsobit hypokalcemii, je nutné dbát opatrnosti, protože mohou mít synergní účinek vyúsťující v závažnou hypokalcemii (viz bod 4.5). Před zahájením léčby kyselinou zoledronovoumá být změřena hladina vápníku v séru a upravena hypokalcemie. Pacientům má být dodáváno přiměřené množství vápníku a vitamin D.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. je prakticky „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
V klinických studiích byla kyselina zoledronová podávána současně s běžně používanými protinádorovými léky, diuretiky, antibiotiky a analgetiky, aniž by byl pozorován výskyt klinicky zřejmých interakcí. Kyselina zoledronová nevykazuje žádnou vazbu na plazmatické bílkoviny a nebyla u ní zjištěna inhibice lidských enzymů P450 in vitro (viz bod 5.2), nebyly však provedeny žádné formální klinické studie interakcí.
Při aplikaci bisfosfonátů s aminoglykosidy, kalcitoninem nebo kličkovými (loop) diuretiky se doporučuje zvláštní opatrnost, neboť obě látky mohou mít aditivní účinek, jenž může vést k následnému snížení hladiny vápníku v séru na delší dobu, než je žádoucí (viz bod 4.4).
Pokud je kyselina zoledronová podávána spolu s dalšími potenciálně nefrotoxickými léčivými přípravky, je třeba postupovat opatrně. Je též nutné věnovat pozornost možnému vzniku hypomagnesemie během léčby.
U pacientů s mnohočetným myelomem se může zvýšit riziko renální dysfunkce, používá-li se kyselina zoledronová v kombinaci s thalidomidem.
Opatrnost je nutná, pokud je přípravek Zoledronic Acid Hospira podáván s antiangiogenními léčivými přípravky, protože u pacientů léčených současně těmito léčivými přípravky byl hlášen zvýšený výskyt osteonekrózy čelisti.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání kyseliny zoledronové těhotným ženám nejsou k dispozici. Reprodukční studie na zvířatech provedené s kyselinou zoledronovou prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známo.
Ženám ve fertilním věku má být doporučeno vyhnout se otěhotnění.
Kojení
Není známo, zda je kyselina zoledronová vylučována do lidského mateřského mléka. Podávání kyseliny zoledronové kojícím ženám je kontraindikováno (viz bod 4.3).
Fertilita
Potenciální nežádoucí účinky kyseliny zoledronové na fertilitu v parentální a F1 generaci byly hodnoceny u potkanů. Došlo k výrazným farmakologickým účinkům, o nichž se má za to, že souvisejí s inhibicí metabolizace vápníku v kostech touto sloučeninou, což se projevilo peripartální hypokalcemií (skupinovým účinkem bisfosfonátů), dystokií a časným ukončením studie. Tyto výsledky proto zabránily stanovení konečného účinku kyseliny zoledronové na lidskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nežádoucí účinky, jako jsou závratě a ospalost, mohou mít vliv na schopnost řídit nebo obsluhovat stroje, proto je třeba dbát opatrnosti při řízení a obsluze strojů během používání kyseliny zoledronové.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Během tří dnů po podání kyseliny zoledronové byly často hlášeny reakce akutní fáze s příznaky zahrnujícími bolest kostí, horečku, únavu, artralgii, myalgii, třes a artritida s následnými otoky kloubů. Tyto příznaky obvykle ustupují během několika dní (viz popis vybraných nežádoucích účinků).
Dále jsou uvedena významná identifikovaná rizika kyseliny zoledronové ve schválených indikacích: porucha funkce ledvin, osteonekróza čelisti, osteonekróza zevního zvukovodu, reakce akutní fáze, hypokalcemie, fibrilace síní, anafylaxe, intersticiální onemocnění plic . Frekvence každého z těchto identifikovaných rizik jsou obsaženy v tabulce 1.
Seznam nežádoucích účinků v tabulce
Následující nežádoucí účinky, uvedené v tabulce 1, byly získány z klinických studií a hlášení po uvedení přípravku na trh, a to převážně po dlouhodobé léčbě 4 mg kyseliny zoledronové:
Tabulka 1
Nežádoucí účinky jsou uvedeny podle frekvence výskytu, nejčastější jako první, podle následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Anemie | |
|
Méně časté: |
Trombocytopenie, leukopenie | |
|
Vzácné: |
Pancytopenie | |
|
Poruchy imunitního systému | ||
|
Méně časté: |
Hypersenzitivní reakce | |
|
Vzácné: |
Angioneurotický edém | |
|
Psychiatrické poruchy | ||
|
Méně časté: |
Úzkost, poruchy spánku | |
|
Vzácné: | ||
|
Poruchy nervového systému | ||
|
Časté: | ||
|
Méně časté: |
Závratě, parestezie, dysgeuzie, hypestezie, hyperestezie, tremor, ospalost | |
|
Velmi vzácné: |
Křeče, hypoestezie a tetanie (v důsledku hypokalcemie) | |
|
Poruchy oka | ||
|
Časté: |
Konjunktivitida | |
|
Méně časté: |
Neostré vidění, skleritida a zánět očnice | |
|
Vzácné: |
Uveitida | |
|
Velmi vzácné: |
Episkleritida | |
|
Poruchy ucha a labyrintu | ||
|
Velmi vzácné |
Osteonekróza zevního zvukovodu (skupinový nežádoucí účinek bisfosfonátů). | |
|
Srdeční poruchy | ||
|
Méně časté: |
Hypertenze, hypotenze, fibrilace síní, hypotenze vedoucí k synkopě nebo oběhovému kolapsu | |
|
Vzácné: |
Bradykardie, srdeční arytmie (v důsledku hypokalcemie) | |
|
Respirační, hrudní a mediastinálníporuchy | ||
|
Méně časté: | ||
|
Vzácné: |
Intersticiální plicní nemoc | |
|
Gastrointestinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Průjem, zácpa, bolesti břicha, dyspepsie, stomatitida, sucho v ústech | |
|
Poruchy kůže a podkožní tkáně | ||
|
Méně časté: |
Svědění, vyrážka (včetně erytematózní a makulární vyrážky), zvýšené pocení | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Bolest kostí, myalgie, artralgie, generalizovaná bolest | |
|
Méně časté: |
Svalové křeče, osteonekróza čelisti | |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Porucha funkce ledvin | |
|
Méně časté: |
Akutní selhání ledvin, hematurie, proteinurie | |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Časté: |
Horečka, chřipkové příznaky (včetně únavy, třesavky, malátnosti a návalů horka) | |
|
Méně časté: |
Astenie, periferní edém, reakce v místě vpichu (včetně bolesti, podráždění, otoku, indurace), bolest na hrudi, zvýšení tělesné hmotnosti, anafylaktická reakce/šok, kopřivka | |
|
Vzácné: |
Artritida a otoky kloubů jako symptom reakce akutní fáze | |
|
Vyšetření | ||
|
Velmi časté: |
Hypofosfatemie | |
|
Časté: |
Zvýšení kreatininu a močoviny v krvi, hypokalcemie | |
|
Méně časté: |
Hypomagnesemie, hypokalemie | |
|
Vzácné: |
Hyperkalemie, hypernatremie | |
Popis vybraných nežádoucích účinků
U kyseliny zoledronové byla hlášena souvislost s dysfunkcí ledvin. Ve sloučené analýze údajů o bezpečnosti získaných ze studií hodnotících kyselinu zoledronovou podávanou pro předcházení kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti, byla četnost nežádoucích účinků spojených s poruchou funkce ledvin, u níž existuje podezření na souvislost s podáváním kyseliny zoledronové, (nežádoucí reakce) následující: mnohočetný myelom (3,2 %), karcinom prostaty (3,1 %), karcinom prsu (4,3 %), karcinom plic a další solidní nádory (3,2 %). Mezi faktory, které mohou zvýšit možnost zhoršení funkce ledvin, patří dehydratace, již existující poruchy funkce ledvin, opakované cykly podávání kyseliny zoledronové nebo jiných bisfosfonátů, stejně jako současné užívání nefrotoxických léčivých přípravků nebo kratší doba aplikace infuze, než se doporučuje. Bylo hlášeno zhoršení funkce ledvin a progrese do selhání ledvin a nutnost dialýzy u pacientů po úvodní nebo jednorázové dávce 4 mg kyseliny zoledronové (viz bod 4.4).
Osteonekróza čelisti
Byly hlášeny případy osteonekrózy čelistí, a to především u pacientů s karcinomem, kteří byli léčeni léčivými přípravky inhibujícími resorpci kostí, jako je kyselina zoledronová (viz bod 4.4). Mnozí z těchto pacientů také dostávali chemoterapii a kortikosteroidy a měli známky lokální infekce, včetně osteomyelitidy. Většina hlášení se týkala pacientů s karcinomem po extrakci zubu nebo jiném stomatochirurgickém výkonu.
Fibrilace síní
V jedné tříleté, randomizované, dvojitě zaslepené kontrolované studii, která hodnotila účinnost a bezpečnost kyseliny zoledronové 5 mg podávané jednou ročně ve srovnání s placebem při léčbě postmenopauzální osteoporózy (PMO), byl celkový výskyt fibrilace síní 2,5 % (96 z 3862) u pacientek, jimž byla podávána kyselina zoledronová 5 mg, a 1,9 % (75 z 3852) u pacientek s placebem. Výskyt vážných nežádoucích účinků ve formě fibrilace síní byl 1,3 % (51 z 3862) u pacientek, jimž byla podávána kyselina zoledronová 5 mg, a 0,6 % (22 z 3852) u pacientek s placebem. Nerovnováha zjištěná v této studii nebyla zaznamenána v jiných studiích s kyselinou zoledronovou, včetně studií s kyselinou zoledronovou 4 mg podávanou každé 3-4 týdny onkologickým pacientům. Mechanismus původu zvýšeného výskytu fibrilace síní v této jediné klinické studii není známý.
Reakce akutní fáze
Tento nežádoucí účinek se skládá ze souboru příznaků, které zahrnují horečku, myalgii, bolest hlavy, bolest končetin, nauzeu, zvracení, průjem, artralgii a artritidu s následnými otoky kloubů. Nastupuje < 3 dny po infuzi kyseliny zoledronové a reakce je také známá pod názvy „chřipkové příznaky“ nebo „příznaky po podání dávky“.
Atypické zlomeniny _femuru
Během sledování po uvedení na trh byly hlášeny následující nežádoucí účinky (frekvence vzácná): atypické subtrochanterické a diafýzární zlomeniny femuru (skupinový nežádoucí účinek bisfosfonátů).
Nežádoucí účinky spojené s hypokalcemií
Hypokalcemie je důležitým známým rizikem podání kyseliny zoledronové ve schválených indikacích. Na základě hodnocení případů z klinických studií a případů po uvedení na trh existují dostatečné důkazy pro podporu souvislosti mezi léčbou kyselinou zoledronovou, hlášenými případy hypokalcemie a následným výskytem srdeční arytmie. Dále existují důkazy o souvislosti mezi hypokalcemií a následnými neurologickými nežádoucími účinky hlášenými v těchto případech, které zahrnují křeče, hypoestezii a tetanii (viz bod 4.4).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické zkušenosti s akutním předávkováním kyselinou zoledronovou jsou omezené. Bylo hlášeno chybné podávání kyseliny zoledronové v dávkách až do 48 mg. Pacienti, jimž byly aplikovány vyšší dávky, než je dávka doporučená (viz bod 4.2), musí být pečlivě sledováni, protože byla pozorována porucha funkce ledvin (včetně selhání ledvin) a odchylky v hladinách sérových koncentrací elektrolytů (včetně vápníku, fosforu a hořčíku). V případě hypokalcemie lze dle klinické indikace podat infuze kalcium-glukonátu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii nemocí kostí, bisfosfonáty, ATC kód: M05BA08
Kyselina zoledronová patří do skupiny bisfosfonátů a působí primárně na kosti. Je to inhibitor osteoklastické resorpce kostí.
Selektivní působení bisfosfonátů na kosti spočívá v jejich vysoké afinitě k mineralizované kosti, ale přesný molekulární mechanismus vedoucí k inhibici osteoklastické aktivity zůstává stále neobjasněn. V dlouhodobých studiích na zvířatech inhibovala kyselina zoledronová kostní resorpci, aniž by nežádoucím způsobem ovlivňovala tvorbu, mineralizaci nebo mechanické vlastnosti kostí.
Kromě silné inhibice kostní resorpce má kyselina zoledronová navíc některé protinádorové vlastnosti, které by mohly přispívat k její celkové účinnosti při léčbě kostních metastáz. V preklinických studiích byly doloženy následující vlastnosti:
- In vivo: inhibice osteoklastické kostní resorpce, která ovlivňuje vnitřní mikroprostředí kostní dřeně a zhoršuje tak podmínky pro růst nádorových buněk; antiangiogenní účinek a analgetický účinek.
- In vitro: Inhibice osteoblastické proliferace, přímý cytostatický a proapoptotický účinek na nádorové buňky, synergický cytostatický účinek spolu s dalšími protinádorovými léky, antiadhezivní/antiinvazivní působení.
Výsledky klinických studií prevence kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti
První randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie srovnávala kyselinu zoledronovou 4 mg s placebem v prevenci kostních příhod (SRE = skeletal related events) u pacientů s karcinomem prostaty. Kyselina zoledronová 4 mg významně snížila podíl pacientů, u nichž došlo k výskytu alespoň jedné kostní příhody (SRE), prodloužila medián času do první SRE o > 5 měsíců a snížila roční výskyt příhod na pacienta - kostní morbiditu. Analýza mnohočetných příhod ukázala ve srovnání s placebem 36% snížení rizika rozvoje SRE ve skupině, jíž byla podávána kyselina zoledronová 4 mg. Pacienti, jimž byla aplikována kyselina zoledronová 4 mg, uváděli nižší nárůst bolesti než ti, kteří dostávali placebo, a rozdíl dosáhl významnosti v měsících 3, 9, 21 a 24. U pacientů dostávajících kyselinu zoledronovou 4 mg byl nižší výskyt patologických zlomenin. U pacientů s blastickými lézemi byl léčebný efekt méně zřejmý. Výsledky účinnosti jsou uvedeny v tabulce 2.
Ve druhé studii, v níž byly zahrnuty jiné solidní nádory než karcinom prsu a prostaty, kyselina zoledronová 4 mg významně snížila podíl pacientů se SRE, prodloužila medián času do první kostní příhody o > 2 měsíce a snížila kostní morbiditu. Analýza mnohočetných příhod ukázala ve srovnání s
placebem 30,7% snížení rizika rozvoje SRE ve skupině, jíž byla podávána kyselina zoledronová 4 mg. Výsledky účinnosti jsou uvedeny v tabulce 3.
|
Tabulka 2: Výsledky účinnosti (pacienti s karcinomem prostaty užívající hormonální léčbu) | ||||||
|
Jakákoli SRE (+TIH) |
Fraktury* |
Radioterapie kostí | ||||
|
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo | |
|
n |
214 |
208 |
214 |
208 |
214 |
208 |
|
Podíl pacientů se SRE (%) |
38 |
49 |
17 |
25 |
26 |
33 |
|
Hodnota p |
0,028 |
0,052 |
0,119 | |||
|
Medián času do SRE (dny) |
488 |
321 |
NR |
NR |
NR |
640 |
|
Hodnota p |
0,009 |
0,020 |
0,055 | |||
|
Kostní morbidita |
0,77 |
1,47 |
0,20 |
0,45 |
0,42 |
0,89 |
|
Hodnota p |
0,005 |
0,023 |
0,060 | |||
|
Snížení rizika mnohočetných příhod** (%) |
36 |
- |
NA |
NA |
NA |
NA |
|
Hodnota p |
0,002 |
NA |
NA | |||
* Zahrnuje vertebrální i nevertebrální fraktury.
** Zahrnuje všechny kostní příhody, celkový počet i dobu do každé příhody během studie. NR Nedosaženo NA Neuplatňuje se
|
Tabulka 3: Výsledky účinnosti (solidní nádory - jiné než karcinom prsu a prostaty) | ||||||
|
Jakákoli SRE (+TIH) |
Fraktury* |
Radioterapie kostí | ||||
|
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo | |
|
n |
257 |
250 |
257 |
250 |
257 |
250 |
|
Podíl pacientů se SRE (%) |
39 |
48 |
16 |
22 |
29 |
34 |
|
Hodnota p |
0,039 |
0,064 |
0,173 | |||
|
Medián času do SRE (dny) |
236 |
155 |
NR |
NR |
424 |
307 |
|
Hodnota p |
0,009 |
0,020 |
0,079 | |||
|
Kostní |
1,74 |
2,71 |
0,39 |
0,63 |
1,24 |
1,89 |
|
morbidita | ||||||
|
Hodnota p |
0,012 |
0,066 |
0,099 | |||
|
Snížení rizika mnohočetných příhod1 (%) |
30,7 |
- |
NA |
NA |
NA |
NA |
|
Hodnota p |
0,003 |
NA |
NA | |||
* Zahrnuje vertebrální i nevertebrální fraktury.
** Zahrnuje všechny kostní příhody, celkový počet i dobu do každé příhody během studie.
NR Nedosaženo NA Neuplatňuje se
V randomizované dvojitě zaslepené studii fáze III byla srovnávána kyselina zoledronová 4 mg s 90 mg pamidronátu, které byly podávány jednou za 3 až 4 týdny pacientům s mnohočetným myelomem nebo karcinomem prsu s nejméně jednou kostní lézí. Výsledky ukázaly, že kyselina zoledronová 4 mg měla v prevenci SRE srovnatelnou účinnost jako 90 mg pamidronátu. Analýza mnohočetných příhod odhalila významné snížení rizika u pacientů léčených kyselinou zoledronovou 4 mg o 16 % ve srovnání s pacienty, kterým byl podáván pamidronát. Výsledky účinnosti jsou uvedeny v tabulce 4.
|
Tabulka 4: Výsledky účinnosti (pacienti s karcinomem prsu a mnohočetným myelomem) | ||||||
|
Jakákoli SRE (+TIH) |
Fraktury* |
Radioterapie kostí | ||||
|
kyselina zoledronová 4 mg |
Pam 90 mg |
kyselina zoledronová 4 mg |
Pam 90 mg |
kyselina zoledronová 4 mg |
Pam 90 mg | |
|
n |
561 |
555 |
561 |
555 |
561 |
555 |
|
Podíl pacientů se SRE (%) |
48 |
52 |
37 |
39 |
19 |
24 |
|
Hodnota p |
0,198 |
0,653 |
0,037 | |||
|
Medián času do SRE (dny) |
376 |
356 |
NR |
714 |
NR |
NR |
|
Hodnota p |
0,151 |
0,672 |
0,026 | |||
|
Kostní morbidita |
1,04 |
1,39 |
0,53 |
0,60 |
0,47 |
0,71 |
|
Hodnota p |
0,084 |
0,614 |
0,015 | |||
|
Snížení rizika mnohočetných příhod1 (%) |
16 |
- |
NA |
NA |
NA |
NA |
|
Hodnota p |
0,030 |
NA |
NA | |||
* Zahrnuje vertebrální i nevertebrální fraktury.
NA Neuplatňuje se
Kyselina zoledronová 4 mg byla též hodnocena ve dvojitě zaslepené, randomizované, placebem kontrolované studii u 228 pacientů s dokumentovanými kostními metastázami v důsledku karcinomu prsu. Hodnocen byl účinek kyseliny zoledronové 4 mg na procento výskytu kostních příhod (SRE), který byl vypočten jako podíl celkového počtu kostních příhod (s výjimkou hyperkalcemie a po zohlednění předchozích zlomenin) vůči celkovému období ohrožení. Pacientům byly po dobu jednoho roku jednou za čtyři týdny podávány buď 4 mg kyseliny zoledronové, nebo placebo. Pacienti byli rovnoměrně rozděleni do skupin léčených kyselinou zoledronovou nebo placebem.
Podíl SRE (kostní příhody/osoba/rok) činil 0,628 u kyseliny zoledronové a 1,096 u placeba. Podíl pacientů s nejméně jednou příhodou SRE (kromě hyperkalcemie) byl 29,8 % ve skupině léčené kyselinou zoledronovou, oproti 49,6 % ve skupině s placebem (p=0,003). Medián času do vzniku první kostní příhody nebyl v rameni léčby kyselinou zoledronovou na konci studie dosažen, a ve srovnání s placebem byl významně prodloužen (p=0,007). Kyselina zoledronová 4 mg snížila ve srovnání s placebem riziko SRE podle analýzy mnohočetných příhod o 41 % (podíl rizika=0,59, p=0,019).
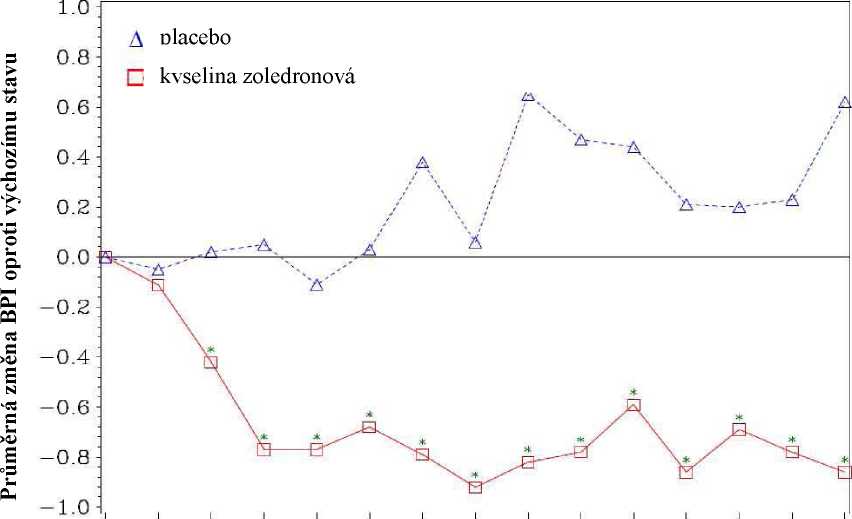
V průběhu studie bylo ve skupině léčené kyselinou zoledronovou po 4 týdnech a v každém následném časovém bodě zjištěno statisticky významné zlepšení skóre bolesti (pomocí stručných dotazníků o bolesti, [Brief Pain Inventory, BPI]) ve srovnání s placebem (obr. 1). Skóre bolesti při léčbě kyselinou zoledronovou bylo soustavně pod úrovní vstupního stavu, a snížení bolesti bylo doprovázeno trendem sníženého analgetického skóre.
Obrázek 1: Průměrná změna oproti výchozímu stavu u skóre BPI. Statisticky významné rozdíly jsou vyznačeny (*p<0,05) pro účely srovnání mezi typy léčby (4 mg kyseliny zoledronové vs. placebo)
0 a 4 S 12 16 20 24 28 32 36 40 44 48 52
Doba ve studii (týdny)
Výsledky klinického hodnocení léčby TIH
V klinických studiích hyperkalcemie vyvolané nádorovým onemocněním (TIH) bylo dokázáno, že účinek kyseliny zoledronové je charakterizován poklesem hladiny vápníku v séru a vylučování vápníku močí. Ve studiích fáze I pro stanovení dávky u pacientů s mírnou až středně závažnou
hyperkalcemií vyvolanou nádorovým onemocněním (TIH) se testované účinné dávky pohybovaly v rozmezí cca 1,2-2,5 mg.
Pro účely hodnocení účinků 4 mg kyseliny zoledronové ve srovnání s 90 mg pamidronátu byly v předem plánované analýze zkombinovány výsledky dvou stěžejních multicentrických studií u pacientů s TIH. Po dávce kyseliny zoledronové 8 mg byla zjištěna rychlejší normalizace korigovaných hodnot vápníku v séru 4. den a po dávce kyseliny zoledronové 4 mg a 8 mg 7. den. Byla pozorována následující míra odpovědi:
|
Tabulka 5: Podíl kompletních odpovědí v kombinovaných studiích TIH podle dne | |||
|
4. den |
7. den |
10. den | |
|
Kyselina zoledronová 4 mg (n=86) |
45,3 % (p=0,104) |
82,6 % (p=0,005)* |
88,4 % (p=0,002)* |
|
Kyselina zoledronová 8 mg (n=90) |
55,6 % (p=0,021)* |
83,3 % (p=0,010)* |
86,7 % (p=0,015)* |
|
Pamidronát 90 mg (n=99) |
33,3 % |
63,6 % |
69,7 % |
|
*Hodnoty p ve srovnání s pamidronátem. | |||
Medián doby k dosažení normální koncentrace vápníku byl 4 dny. Medián doby do relapsu (opakované zvýšení hladiny vápníku v séru korigované na albumin > 2,9 mmol/l) činil u pacientů léčených kyselinou zoledronovou 30 až 40 dnů oproti 17 dnům u pacientů léčených pamidronátem 90 mg (hodnoty p: 0,001 pro kyselinu zoledronovou 4 mg a 0,007 pro kyselinu zoledronovou 8 mg). Mezi těmito dvěma dávkami kyseliny zoledronové nebyl nalezen statisticky významný rozdíl.
V klinických studiích bylo 69 pacientů s relapsem nebo refrakterních na počáteční léčbu (kyselina zoledronová 4 mg, 8 mg nebo pamidronát 90 mg) přeléčeno kyselinou zoledronovou 8 mg. Odpověď u těchto pacientů činila přibližně 52 %. Vzhledem k tomu, že tito pacienti byli přeléčeni pouze dávkou
8 mg, nejsou k dispozici údaje umožňující srovnání s dávkou 4 mg kyseliny zoledronové.
V klinických studiích provedených u pacientů s hyperkalcemií vyvolanou nádorovým onemocněním (TIH) byl celkový bezpečnostní profil u všech 3 léčebných skupin (kyselina zoledronová 4 a 8 mg a pamidronát 90 mg) podobný v typech i závažnosti.
Pediatrická populace
Výsledky klinických studií v léčbě těžké _formy osteogenesis imperfecta u _pediatrických _pacientů ve věku 1 až 17 let
Účinky intravenózně podávané kyseliny zoledronové při léčbě pediatrických pacientů (ve věku 1 až 17 let) s těžkou formou osteogenesis imperfecta (typ I, III a IV) byly srovnány s intravenózně podávaným pamidronátem v jedné mezinárodní, multicentrické, randomizované otevřené studii, která hodnotila 74 pacientů ve skupině s kyselinou zoledronovou a 76 pacientů ve skupině s pamidronátem. Doba léčby ve studii byla 12 měsíců a léčbě předcházelo 4-9týdenní screeningové období, během něhož byl doplňkově podáván po dobu nejméně 2 týdnů vitamín D a vápník v elementární formě.
V klinickém programu byla pacientům ve věku 1 až < 3 roky podána jednou za 3 měsíce dávka 0,025 mg/kg kyseliny zoledronové (až do maximální jednotlivé dávky 0,35 mg) a pacientům ve věku 3-17 let bylo jednou za 3 měsíce podáno 0,05 mg/kg kyseliny zoledronové (až do maximální jednotlivé dávky 0,83 mg). Byla provedena pokračovací studie, jejímž cílem bylo hodnocení dlouhodobé celkové a renální bezpečnosti kyseliny zoledronové podávané jednou nebo dvakrát ročně v průběhu 12měsíční pokračovací léčby u dětí, které dokončily 1 rok léčby kyselinou zoledronovou nebo pamidronátem v základní studii.
Primárním cílovým parametrem této studie bylo zjištění procentuální změny oproti výchozímu stavu kostní denzity bederní páteře (BMD) po 12 měsících léčby. Očekávané účinky léčby na BMD byly podobné, ale koncepce studie nebyla dostatečně robustní, aby prokázala obdobnou účinnost (non-inferioritu) kyseliny zoledronové. Především nedošlo k jasnému prokázání účinnosti na výskyt fraktur nebo bolesti. Nežádoucí účinky ve formě fraktur dlouhých kostí dolních končetin byly hlášeny cca u 24 % (femur) a 14 % (tibia) pacientů s těžkou formou osteogenesis imperfecta léčených kyselinou zoledronovou oproti 12 % a 5 % pacientů s těžkou formou osteogenesis imperfecta léčených pamidronátem, a to bez ohledu na typ onemocnění a kauzalitu. Souhrnný výskyt fraktur u pacientů léčených kyselinou zoledronovou a pamidronátem byl však srovnatelný: 43 % (32/74) vs. 41 % (31/76). Interpretace rizika fraktur je ztížena skutečností, že fraktury jsou častým jevem u pacientů s těžkou formou osteogenesis imperfecta a jsou součástí průběhu tohoto onemocnění.
Typ nežádoucích účinků zjištěných u této populace byl podobný účinkům dříve zjištěným u dospělých pacientů s pokročilými malignitami zasahujícími kosti (viz bod 4.8). Nežádoucí účinky řazené podle četností jsou uvedené v tabulce 6. Je použita následující obvyklá klasifikace: velmi časté (> 1/10), časté (> 1/100, < 1/10), méně časté (> 1/1000, < 1/100), vzácné (> 1/10 000, <1 /1000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Tabulka 6: Nežádoucí účinky zjištěné u pediatrických pacientů s těžkou formou osteogenesis | ||
|
imperfecta1 | ||
|
Poruchy nervového systému | ||
|
Časté: | ||
|
Srdeční poruchy | ||
|
Časté: | ||
|
Respirační, hrudní a mediastinálníporuchy | ||
|
Časté: |
Nazofaryngitida | |
|
Gastrointestinální poruchy | ||
|
Velmi časté: | ||
|
Časté: | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Bolest končetin, artralgie, muskuloskeletální bolest | |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Pyrexie, únava | |
|
Časté: |
Reakce akutní fáze, bolest | |
|
Vyšetření | ||
|
Velmi časté: |
Hypokalcemie | |
|
Časté: |
Hypofosfatemie | |
1 Nežádoucí účinky s četností < 5 % byly zhodnoceny z medicínského hlediska a bylo prokázáno, že tyto případy odpovídají dobře známému bezpečnostnímu profilu kyseliny zoledronové (viz bod 4.8).
U pediatrických pacientů s těžkou formou osteogenesis imperfecta se zdá být použití kyseliny zoledronové ve srovnání s pamidronátem spojené s výraznějším rizikem reakce akutní fáze, hypokalcemie a neobjasněné tachykardie, ale tento rozdíl se snižuje po následných infuzích.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s referenčním léčivým přípravkem obsahujícím kyselinou zoledronovou u všech podskupin pediatrické
populace při léčbě hyperkalcemie vyvolané nádorovým onemocněním a prevenci kostních příhod u pacientů s pokročilou formou maligního onemocnění postihujícího kosti (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Následující farmakokinetické údaje, které byly získány po jednorázové a opakované 5 a 15minutové infuzi 2, 4, 8 a 16 mg kyseliny zoledronové u 64 pacientů s kostními metastázami, neprokázaly závislost na dávce.
Po zahájení infuze kyseliny zoledronové rychle stouply koncentrace kyseliny zoledronové v plazmě a dosáhly vrcholu na konci infuze; poté následoval rychlý pokles na < 10 % vrcholové koncentrace za 4 hodiny a < 1 % vrcholové koncentrace za 24 hodin. Pak následovalo dlouhé období velmi nízké koncentrace, která nepřesáhla 0,1 % vrcholové koncentrace před druhou infuzí kyseliny zoledronové 28. den.
Nitrožilně podaná kyselina zoledronová je vylučována třífázovým procesem: rychlé dvoufázové vymizení ze systémového krevního oběhu s poločasy t./2a 0,24 a t/2p 1,87 hodin, po němž následuje dlouhá fáze vylučování s konečným poločasem vylučování t/Y 146 hodin. Po opakovaném podávání každých 28 dnů nebyla zjištěna akumulace kyseliny zoledronové v plazmě. Kyselina zoledronová není metabolizována a je vylučována nezměněná ledvinami. Během prvních 24 hodin se močí vyloučí 39 ± 16 % podané dávky, zatímco zbytek je vázán převážně v kostní tkáni. Z kostní tkáně je kyselina zoledronová velmi pomalu uvolňována zpět do systémového krevního oběhu a vylučována ledvinami. Celková tělesná clearance je 5,04 ± 2,5 l/h a není závislá na dávce ani ovlivněna pohlavím, věkem, rasou a tělesnou hmotností. Prodloužení doby infuze z 5 na 15 minut způsobilo na konci infuze 30% pokles koncentrace kyseliny zoledronové, ale nemělo žádný vliv na plochu pod křivkou plazmatické koncentrace vs. čas.
Variabilita farmakokinetických parametrů kyseliny zoledronové mezi jednotlivými pacienty byla vysoká, stejně jako je tomu u ostatních bisfosfonátů.
U pacientů s hyperkalcemií nebo s jaterní insuficiencí nejsou pro kyselinu zoledronovou dostupné farmakokinetické údaje. Kyselina zoledronová neinhibuje lidské enzymy P450 in vitro, nevykazuje biotransformaci a ve studiích na zvířatech bylo < 3 % z aplikované dávky vyloučeno ve stolici, což svědčí o tom, že játra nehrají ve farmakokinetice kyseliny zoledronové významnou úlohu.
Ledvinná clearance kyseliny zoledronové korelovala s clearancí kreatininu, přičemž ledvinná clearance reprezentovala 75 ± 33 % clearance kreatininu. Střední hodnota u 64 hodnocených pacientů s nádorovým onemocněním činila 84 ± 29 ml/min (rozmezí 22 až 143 ml/min). Populační analýza ukázala, že u pacientů s clearancí kreatininu 20 ml/min (těžká porucha funkce ledvin) nebo 50 ml/min (středně těžká porucha) by odpovídající predikovaná clearance kyseliny zoledronové činila 37 %, resp. 72 % hodnoty pacientů s clearancí kreatininu 84 ml/min. U pacientů s těžkou ledvinnou insuficiencí (clearance kreatininu < 30 ml/min) je dostupné pouze omezené množství údajů.
V in vitro studii vykazovala kyselina zoledronová nízkou afinitu k buňkám lidské krve s průměrem poměru koncentrace v krvi ke koncentaci v plazmě 0,59 v rozmezí koncentrací 30 ng/ml až 5000 ng/ml. Vazba k plazmatickým proteinům je nízká, s nenavázanou frakcí v rozmezí od 60 % při 2 ng/ml do 77 % při 2000 ng/ml kyseliny zoledronové.
Zvláštní populace
Pediatričtí _ pacienti
Omezené farmakokinetické údaje u dětí s těžkou formou osteogenesis imperfecta naznačují, že farmakokinetika kyseliny zoledronové u dětí ve věku 3 až 17 let je při obdobném dávkování v mg/kg podobná farmakokinetice u dospělých pacientů. Zdá se, že věk, tělesná hmotnost, pohlaví a clearance kreatininu nemají na systémovou expozici kyseliny zoledronové žádný vliv.
5.3 Předklinické údaje vztahující se k bezpečnosti
Akutní toxicita
Nejvyšší jednorázová intravenózní dávka, která nezpůsobila žádné úmrtí zvířat, byla u myší 10 mg/kg a u potkanů 0,6 mg/kg tělesné hmotnosti.
Subchronická a chronická toxicita
Kyselina zoledronová byla dobře snášena, pokud byla aplikována subkutánně potkanům a intravenózně psům v dávce až do 0,02 mg/kg denně po dobu 4 týdnů. Dávka 0,001 mg/kg/den aplikovaná subkutánně potkanům a intravenózní dávka 0,005 mg/kg aplikovaná jednou za 2-3 dny psům až po dobu 52 týdnů byla také dobře snášena.
Nejčastějším nálezem ve studiích s opakovanými dávkami podávanými zvířatům v době růstu byl téměř při všech dávkách nárůst spongiózní tkáně v metafýzách dlouhých kostí, což je odrazem farmakologické antiresorpční aktivity této sloučeniny.
Hranice bezpečnosti z hlediska účinku na ledviny byly při dlouhodobém opakovaném parenterálním podávání ve studiích na zvířatech velmi úzké. Kumulativní hladiny, při nichž ještě nebyl pozorován nežádoucí účinek (NOAEL) po podání jednorázové dávky (1,6 mg/kg) a po opakovaném podávání až po dobu 1 měsíce (0,06 až 0,6 mg/kg/den), však nenaznačily účinek na ledviny při dávkách ekvivalentních nebo přesahujících nejvyšší terapeutické humánní dávky. Dlouhodobé opakované podávání kyseliny zoledronové v dávkách v rozmezí nejvyšší předpokládané terapeutické humánní dávky mělo toxické účinky na jiné orgány, včetně zažívacího traktu, jater, sleziny a plic a míst intravenózního vpichu.
Reprodukční toxicita
Kyselina zoledronová byla při subkutánním podání potkanům v dávce > 0,2 mg/kg teratogenní. Ačkoli teratogenita nebo fetotoxicita nebyla u králíků pozorována, byla zjištěna maternální toxicita. U potkanů byla pozorována při nejnižší testované dávce (0,01 mg/kg tělesné hmotnosti) dystokie.
Mutagenita a karcinogenní potenciál
Kyselina zoledronová nevykazovala v provedených testech mutagenity mutagenní účinek.
V provedených testech karcinogenity nebyly zjištěny žádné důkazy karcinogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mannitol
Dihydrát natrium-citrátu Voda na injekci
6.2 Inkompatibility
Aby se zamezilo potenciálním inkompatibilitám, musí se přípravek Zoledronic Acid Hospira naředit injekčním roztokem chloridu sodného 9 mg/ml (0,9%) nebo 5% (w/v) roztokem glukózy.
Tento léčivý přípravek nesmí být mísen s infuzními roztoky obsahujícími vápník nebo jiné dvoumocné kationty, jako je laktátový Ringerův roztok, a musí se podávat jako samostatný intravenózní roztok oddělenou infuzní linkou.
6.3 Doba použitelnosti
3 roky
Po naředění: Z mikrobiologického hlediska je nutné naředěný infuzní roztok použít okamžitě.
Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při teplotě 2 °C - 8 °C.
Chlazený roztok musí být před podáním vytemperován na pokojovou teplotu.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
6ml skleněná injekční lahvička typu I nebo 5ml plastová injekční lahvička, s halobutylovou zátkou potaženou fluoropolymerem s hliníkovým uzávěrem a flip-off diskem.
Přípravek Zoledronic Acid Hospira se dodává v balení, které obsahuje 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před podáním musí být 5,0 ml koncentrátu z jedné injekční lahvičky nebo objem koncentrátu odebraný dle potřeby dále naředěno 100 ml roztoku, který neobsahuje vápník (injekční roztok chloridu sodného 9 mg/ml (0,9%) nebo 5% (w/v) roztok glukózy).
Další informace o zacházení s přípravkem Zoledronic Acid Hospira, včetně pokynů pro přípravu nižších dávek jsou uvedeny v bodě 4.2.
Při přípravě infuze musí být dodržena aseptická technika. Určeno pouze k jednorázovému použití.
Smí se použít pouze čirý roztok bez obsahu částic a zabarvení.
Zdravotnickým pracovníkům se doporučuje, aby nepoužitý přípravek Zoledronic Acid Hospira nevyhazovali do domácího systému odpadních vod.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Hospira UK Limited
Horizon
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/800/001
EU/1/12/800/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
19. listopadu 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden vak o objemu 100 ml obsahuje acidum zoledronicum 4 mg (ve formě acidum zoledronicum monohydricum).
Jeden ml roztoku obsahuje acidum zoledronicum 0,04 mg (ve formě acidum zoledronicum monohydricum).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Infuzní roztok
Čirý a bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
- Prevence kostních příhod (patologických zlomenin, kompresivních zlomenin obratlů, radiační nebo chirurgická léčba kostí, nebo hyperkalcemie vyvolané nádorovým onemocněním) u dospělých pacientů s pokročilým maligním onemocněním postihujícím kosti.
- Léčba dospělých pacientů s hyperkalcemií vyvolanou nádorovým onemocněním (TIH).
4.2 Dávkování a způsob podání
Kyselinu zoledronovou musí předepisovat a podávat pacientům pouze zdravotničtí pracovníci se zkušenostmi s intravenózním podáváním bisfosfonátů. Pacientům léčeným kyselinou zoledronovou má být k dispozici příbalová informace a pacientská informační karta Dávkování
Prevence kostních _příhod u _pacientů s _pokročilým maligním onemocněním _postihujícím kosti Dospělí a starší pacienti
Doporučená dávka pro prevenci kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti je 4 mg kyseliny zoledronové každé 3 až 4 týdny.
Pacientům je též nutné denně doplňkově perorálně podávat 500 mg vápníku a 400 mj. vitamínu D.
Při rozhodování o léčbě pacientů s kostními metastázami za účelem prevence kostních příhod je nutné vzít v úvahu, že účinky léčby se projeví za 2-3 měsíce.
Léčba TIH
Dospělí a starší pacienti
Doporučená dávka při léčbě hyperkalcemie (albuminem korigovaný vápník > 12,0 mg/dl nebo 3,0 mmol/l) je 4 mg kyseliny zoledronové v jedné dávce.
Porucha funkce ledvin
TIH:
U pacientů s TIH, kteří zároveň trpí závažnou poruchou funkce ledvin, lze o léčbě kyselinou zoledronovou uvažovat až po zhodnocení rizika a přínosu léčby. Pacienti s hladinou kreatininu v séru > 400 ^mol/l nebo > 4,5 mg/dl byli z klinických studií vyloučeni. U pacientů s TIH s hladinou kreatininu v séru < 400 ^mol/l nebo < 4,5 mg/dl není nutná úprava dávkování (viz bod 4.4).
Prevence kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti:
U pacientů s mnohočetným myelomem nebo metastatickými lézemi solidních nádorů do kostí je nutné při zahájení léčby kyselinou zoledronovou stanovit sérový kreatinin a clearanci kreatininu (CLcr).
CLcr se vypočítá ze sérového kreatininu pomocí Cockcroft-Gaultova vzorce. Nedoporučuje se podávat kyselinu zoledronovou pacientům, kteří již před zahájením léčby trpí závažnou poruchou funkce ledvin, která je u této populace definována jako CLcr < 30 ml/min. Pacienti s hladinou kreatininu v séru > 265 ^mol/l nebo > 3,0 mg/dl byli z klinických studií s kyselinou zoledronovou vyloučeni.
Pacientům s normální funkcí ledvin (definovanou jako CLcr > 60 ml/min) lze infuzní roztok kyseliny zoledronové 4 mg/100 ml podávat přímo bez jakékoli další přípravy. U pacientů s kostními metastázami, kteří před zahájením léčby trpěli mírnou nebo středně závažnou poruchou funkce ledvin, jež je pro tuto populaci definována jako CLcr 30-60 ml/min, se doporučují snížené dávky přípravku Zoledronic Acid Hospira (viz též bod 4.4).
|
Výchozí hodnoty clearance kreatininu (ml/min) |
Doporučená dávka přípravku Zoledronic Acid Hospira* |
|
> 60 |
4,0 mg kyseliny zoledronové |
|
50-60 |
3,5 mg* kyseliny zoledronové |
|
40-49 |
3,3 mg* kyseliny zoledronové |
|
30-39 |
3,0 mg* kyseliny zoledronové |
*Dávky byly vypočteny z předpokládané cílové AUC 0,66 (mg^hod/l) (CLcr = 75 ml/min). Při podávání snížených dávek pacientům s poruchou funkce ledvin lze předpokládat dosažení stejné AUC, jako byla pozorována u pacientů s clearancí kreatininu 75 ml/min.
Po zahájení léčby je nutné měřit sérový kreatinin před podáním každé dávky přípravku Zoledronic Acid Hospira a při zhoršení renální funkce musí být léčba přerušena. V klinických studiích bylo zhoršení renální funkce definováno následujícím způsobem:
- u pacientů s normální výchozí hodnotou sérového kreatininu (< 1,4 mg/dl nebo < 124 ^mol/l) zvýšení o 0,5 mg/dl nebo 44 ^mol/l,
- u pacientů s abnormální výchozí hodnotou sérového kreatininu (> 1,4 mg/dl nebo > 124 ^mol/l) zvýšení o 1,0 mg/dl nebo 88 ^mol/l.
V klinických studiích byla léčba kyselinou zoledronovou znovu zahájena teprve tehdy, když se hladina kreatininu vrátila do rozmezí, které se nelišilo o více než 10 % od výchozí hodnoty (viz bod 4.4).
Léčbu přípravkem Zoledronic Acid Hospira je nutné obnovit ve stejné dávce, jako byla dávka podávaná před přerušením.
Pediatrická populace
Bezpečnost a účinnost kyseliny zoledronové u dětí ve věku od 1 do 17 let nebyla stanovena. V současnosti dostupné údaje jsou popsány v bodě 5.1, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání Intravenózní podání.
Přípravek Zoledronic Acid Hospira se má podávat jako jednorázová intravenózní infuze po dobu nejméně 15 minut.
U pacientů s normální funkcí ledvin, definovanou jako CLcr > 60 ml/min, se nesmí infuzní roztok kyseliny zoledronové 4 mg/100 ml dále ředit.
U pacientů s mírnou až středně těžkou poruchou funkce ledvin se doporučuje snížení dávek přípravku Zoledronic Acid Hospira (viz bod „Dávkování“ výše a bod 4.4).
Snížené dávky pro pacienty s výchozí hodnotou CLcr < 60 ml/min připravte podle tabulky 1 níže.
Před podáním odstraňte z vaku uvedený objem přípravku Zoledronic Acid Hospira.
Tabulka 1: Příprava snížených dávek přípravku Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok.
|
Výchozí hodnoty clearance kreatininu (ml/min) |
Odstraňte následující objem (ml) přípravku Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok |
Upravená dávka (mg kyseliny zoledronové) |
|
50-60 |
12,0 |
3,5 |
|
40-49 |
18,0 |
3,3 |
|
30-39 |
25,0 |
3,0 |
Přípravek Zoledronic Acid Hospira nesmí být mísen s jinými infuzními roztoky a musí se podávat jako samostatný intravenózní roztok oddělenou infuzní linkou.
Pacienti musejí být před podáním přípravku Zoledronic Acid Hospira a poté dobře hydratováni.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku, jiné bisfosfonáty nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Obecná
Před podáním kyseliny zoledronové a poté musí být posouzen stav pacientů, aby se zajistila jejich odpovídající hydratace.
U pacientů s rizikem srdečního selhání je nutné zabránit nadměrné hydrataci.
Po zahájení léčby kyselinou zoledronovou se musí pečlivě sledovat standardní metabolické parametry související s hyperkalcemií, jako jsou sérové hladiny vápníku, fosfátu a hořčíku. Pokud se objeví hypokalcemie, hypofosfatemie nebo hypomagnesemie, může být nezbytná krátkodobá suplementační terapie. Pacienti s neléčenou hyperkalcemií mají obvykle do jisté míry poškozenou funkci ledvin, proto je nutné zvážit pečlivé sledování ledvinných funkcí.
K léčbě osteoporózy a Pagetovy choroby kostí jsou k dispozici i jiné přípravky obsahující jako léčivou látku kyselinu zoledronovou. Pacienti léčení přípravkem Zoledronic Acid Hospira nemají být současně léčeni těmito přípravky ani jiným bisfosfonátem, neboť kombinované účinky těchto látek nejsou známé.
Renální insuficience
U pacientů s TIH a zjištěným zhoršováním funkce ledvin je nutné odpovídajícím způsobem posoudit jejich stav a je nutné zvážit, zda potenciální přínos léčby kyselinou zoledronovou převáží možné riziko.
Při rozhodování o léčbě pacientů s kostními metastázami za účelem prevence kostních příhod je nutné vzít v úvahu, že účinky léčby se projeví za 2-3 měsíce.
U kyseliny zoledronové byla hlášena souvislost s dysfunkcí ledvin. Mezi faktory, které mohou zvyšovat možnost zhoršení funkce ledvin, patří dehydratace, již existující poruchy funkce ledvin, opakované cykly podávání kyseliny zoledronové a jiných bisfosfonátů, stejně jako užívání jiných nefrotoxických léčivých přípravků. I když je toto riziko sníženo při podání dávky kyseliny zoledronové po dobu 15 minut, může přesto dojít ke zhoršení funkce ledvin. Bylo hlášeno zhoršení funkce ledvin a progrese až k selhání ledvin a nutnosti dialýzy u pacientů po úvodní nebo jednorázové dávce 4 mg kyseliny zoledronové. Může též dojít ke zvýšení kreatininu v séru u některých pacientů po opakovaném podání kyseliny zoledronové v dávkách doporučených k prevenci kostních příhod, i když méně často.
Před aplikací každé dávky kyseliny zoledronové je nutné u pacientů stanovit hladinu sérového kreatininu. U pacientů s kostními metastázami a s mírnou až středně závažnou poruchou funkce ledvin se doporučuje zahájit léčbu nižšími dávkami kyseliny zoledronové. U pacientů, u kterých bylo v průběhu léčby prokázáno zhoršení funkce ledvin, musí být léčba kyselinou zoledronovou přerušena. Léčba kyselinou zoledronovou se má obnovit pouze tehdy, až se hladina sérového kreatininu vrátí do rozmezí, které se nebude lišit o více než 10 % od výchozí hodnoty. Léčbu kyselinou zoledronovou je nutné obnovit ve stejné dávce, jako byla dávka podávaná před přerušením.
Vzhledem k možnému vlivu kyseliny zoledronové na funkci ledvin, nedostatku klinických údajů o bezpečnosti podávání u pacientů s těžkou poruchou funkce ledvin již před zahájením léčby (v klinických studiích definované jako hladina kreatininu v séru > 400 ^mol/l nebo > 4,5 mg/dl u pacientů s TIH a > 265 ^mol/l nebo > 3,0 mg/dl u pacientů s karcinomem a kostními metastázami) a pouze omezenému množství farmakokinetických údajů těchto pacientů (clearance kreatininu < 30 ml/min) se podávání kyseliny zoledronové pacientům se závažnou poruchou funkce ledvin nedoporučuje.
Hepatální insuficience
U pacientů s těžkou jaterní insuficiencí je k dispozici jen omezené množství klinických údajů, a proto nelze pro tuto skupinu pacientů stanovit konkrétní doporučení.
Osteonekróza čelisti
U pacientů používajících kyselinu zoledronovou byly v klinických studiích a po uvedení přípravku na trh méně často hlášeny případy osteonekrózy čelisti (OČ).
U pacientů s nehojícími se lézemi měkkých tkání v ústech by mělo být s výjimkou akutních medicínských stavů zahájení léčby nebo nového cyklu léčby odloženo. Před zahájením léčby bisfosfonáty je u pacientů s konkomitantními rizikovými faktory doporučené příslušné vyšetření s preventivním ošetřením a individuálním vyhodnocením poměru prospěchu-rizika.
Při posuzování rizika rozvoje osteonekrózy čelisti u jednotlivých pacientů je třeba zvážit následující rizikové faktory:
- potenciál bisfosfonátů (vyšší riziko u vysoce účinných sloučenin), cesta podání (vyšší riziko při parenterálním podání) a kumulativní dávka bisfosfonátů,
- maligní nádorové onemocnění, komorbidity (např. anémie, koagulopatie, infekce), kouření.
- Konkomitantní terapie :chemoterapie, inhibitory angiogeneze (viz bod 4.5), radioterapie krku a hlavy, kortikosteroidy.
- stomatologická onemocnění v anamnéze, špatná ústní hygiena, periodontální onemocnění, invazivní stomatologické zákroky (např. extrakce zubů) a špatně naléhající zubní protéza.
Všichni pacienti mají být vyzváni, aby během léčby kyselinou zoledronovou udržovali dobrou ústní hygienu, absolvovali rutinní vyšetření chrupu a okamžitě hlásili jakékoli příznaky v ústech, jako je kývání zubů, bolest nebo otoky nebo nehojící se vředy nebo výtok.
Během léčby mají být invazivní stomatologické procedury prováděny pouze po pečlivém vyhodnocení a při nastávajícím podávání kyseliny zoledronové mají být vyloučena.
U pacientů, u nichž se během bisfosfonátové léčby rozvine osteonekróza čelisti, může stomatochirurgický zákrok zhoršit stav. Neexistují údaje, které by dokládaly, zda vysazení léčby bisfosfonáty snižuje u pacientů vyžadujících stomatologický výkon riziko osteonekrózy čelisti.
Plán léčby pacientů s OČ by měl být navržen v úzké spolupráci mezi ošetřujícím lékařem a dentistou nebo zubním chirurgem s odbornou znalostí OČ. Dokud se stav nezlepší a pokud možno nevymizí příspívající rizikové faktory, mělo by být zváženo dočasné přerušení léčby kyselinou zoledronovou.
Osteonekróza zevního zvukovodu
V souvislosti s léčbou bisfosfonáty byla hlášena osteonekróza zevního zvukovodu, zejména při dlouhodobém podávání. Mezi možné rizikové faktory osteonekrózy zevního zvukovodu patří používání steroidů a chemoterapie a/nebo lokální rizikové faktory, jako například infekce nebo trauma. Možnost vzniku osteonekrózy zevního zvukovodu je třeba zvážit u pacientů léčených bisfosfonáty, kteří mají ušní symptomy včetně chronických infekcí ucha.
Bolesti pohybového systému
Zkušenosti po uvedení přípravku na trh dokládají, že u pacientů, kteří užívali kyselinu zoledronovou, byly hlášeny silné bolesti kostí, kloubů a/nebo svalů, jež byly občas zneschopňující. Taková hlášení jsou však řídká. Doba do vzniku příznaků byla různá, a to od jednoho dne až do několika měsíců po zahájení léčby. Po ukončení léčby se většině pacientů od příznaků ulevilo. U podskupiny pacientů byl zaznamenán opětovný návrat příznaků poté, co byli znovu léčeni kyselinou zoledronovou nebo jiným bisfosfonátem.
Atypické zlomeniny femuru
V souvislosti s léčbou bisfosfonáty, a to zejména u pacientů dlouhodobě léčených pro osteoporózu, byly hlášeny atypické subtrochanterické a diafyzární fraktury femuru. Tyto příčné nebo krátké šikmé fraktury mohou vzniknout kdekoli na femuru od místa těsně pod malým trochanterem až do místa těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s ním a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“), týdny až měsíce před manifestací kompletní zlomeniny femuru. Fraktury jsou často oboustranné, a proto je u pacientů léčených bisfosfonáty, u nichž došlo ke zlomenině diafýzy femuru, nutné vyšetřit kontralaterální femur. Bylo též zaznamenáno špatné hojení těchto fraktur. U pacientů, u nichž existuje podezření na atypickou frakturu femuru, je nutné při hodnocení jejich stavu zvážit přerušení léčby bisfosfonáty, a to na základě individuálního posouzení rizik a přínosů této léčby.
Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, kyčle nebo třísla, a všechny pacienty, u nichž se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou neúplnou frakturu femuru.
Hypokalcemie
Hypokalcemie byla hlášena u pacientů léčených kyselinou zoledronovou.V důsledku těžké hypokalcemie byly hlášeny srdeční arytmie a neurologické nežádoucí účinky (včetně křečí, hypoestezii a tetanie) byly hlášeny jako sekundární v případech těžké hypokalcemie. Byly hlášeny případy těžké hypokalcemie vyžadující hospitalizaci. V některých případech může být hypokalcemie život ohrožující (viz bod 4.8). Při podávání Zoledronic Acid Hospira spolu s léčivými přípravky, které mohou způsobit hypokalcemii, je nutné dbát opatrnosti, protože mohou mít synergní účinek vyúsťující v závažnou hypokalcemii (viz bod 4.5). Před zahájením léčby přípravkem Zoledronic Acid Hospira má být změřena hladina vápníku v séru a upravena hypokalcemie. Pacientům má být dodáváno přiměřené množství vápníku a vitamínu D.
Tento léčivý přípravek obsahuje 16 mmol (neboli 360 mg) sodíku v jedné dávce. To je třeba vzít v úvahu u pacientů na dietě s nízkým příjmem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
V klinických studiích byla kyselina zoledronová podávána současně s běžně používanými protinádorovými léky, diuretiky, antibiotiky a analgetiky, aniž by byl pozorován výskyt klinicky zřejmých interakcí. Kyselina zoledronová nevykazuje žádnou vazbu na plazmatické bílkoviny a nebyla u ní zjištěna inhibice lidských enzymů P450 in vitro (viz bod 5.2), nebyly však provedeny žádné formální klinické studie interakcí.
Při aplikaci bisfosfonátů s aminoglykosidy, kalcitoninem nebo kličkovými (loop) diuretiky se doporučuje zvláštní opatrnost, neboť obě látky mohou mít aditivní účinek, jenž může vést k následnému snížení hladiny vápníku v séru na delší dobu, než je žádoucí (viz bod 4.4).
Pokud je kyselina zoledronová podávána spolu s dalšími potenciálně nefrotoxickými léčivými přípravky, je třeba postupovat opatrně. Je též nutné věnovat pozornost možnému vzniku hypomagnesemie během léčby.
U pacientů s mnohočetným myelomem se může zvýšit riziko renální dysfunkce, používá-li se kyselina zoledronová v kombinaci s thalidomidem.
Opatrnost je nutná, pokud je přípravek Zoledronic Acid Hospira podáván s antiangiogenními léčivými přípravky, protože u pacientů léčených současně těmito léčivými přípravky byl hlášen zvýšený výskyt osteonekrózy čelisti.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání kyseliny zoledronové těhotným ženám nejsou k dispozici. Reprodukční studie na zvířatech provedené s kyselinou zoledronovou prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známo.
Ženám ve fertilním věku má být doporučeno vyhnout se otěhotnění.
Kojení
Není známo, zda je kyselina zoledronová vylučována do lidského mateřského mléka. Podávání kyseliny zoledronové kojícím ženám je kontraindikováno (viz bod 4.3).
Fertilita
Potenciální nežádoucí účinky kyseliny zoledronové na fertilitu v parentální a F1 generaci byly hodnoceny u potkanů. Došlo k výrazným farmakologickým účinkům, o nichž se má za to, že souvisejí s inhibicí metabolizace vápníku v kostech touto sloučeninou, což se projevilo peripartální hypokalcemií (skupinovým účinkem bisfosfonátů), dystokií a časným ukončením studie. Tyto výsledky proto zabránily stanovení konečného účinku kyseliny zoledronové na lidskou fertilitu.
Nežádoucí účinky, jako j sou závratě a ospalost, mohou mít vliv na schopnost řídit nebo obsluhovat stroje, proto je třeba dbát opatrnosti při řízení a obsluze strojů během používání kyseliny zoledronové.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Během tří dnů po podání kyseliny zoledronové byly často hlášeny reakce akutní fáze s příznaky zahrnujícími bolest kostí, horečku, únavu, artralgii, myalgii a třes a artritida s následnými otoky kloubů. Tyto příznaky obvykle ustupují během několika dní (viz popis vybraných nežádoucích účinků).
Dále jsou uvedena významná identifikovaná rizika kyseliny zoledronové ve schválených indikacích: porucha funkce ledvin, osteonekróza čelisti, osteonekróza zevního zvukovodu, reakce akutní fáze, hypokalcemie, fibrilace síní, anafylaxe, intersticiální onemocnění plic. Frekvence každého z těchto identifikovaných rizik jsou obsaženy v tabulce 2.
Seznam nežádoucích účinků v tabulce
Následující nežádoucí účinky, uvedené v tabulce 2, byly získány z klinických studií a hlášení po uvedení přípravku na trh, a to převážně po dlouhodobé léčbě 4 mg kyseliny zoledronové:
Tabulka 2
Nežádoucí účinky jsou uvedeny podle frekvence výskytu, nejčastější jako první, podle následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
|
Poruchy krve a lymfatického systému | ||
|
Časté: |
Anemie | |
|
Méně časté: |
Trombocytopenie, leukopenie | |
|
Vzácné: |
Pancytopenie | |
|
Poruchy imunitního systému | ||
|
Méně časté: |
Hypersenzitivní reakce | |
|
Vzácné: |
Angioneurotický edém | |
|
Psychiatrické poruchy | ||
|
Méně časté: |
Úzkost, poruchy spánku | |
|
Vzácné: | ||
|
Poruchy nervového systému | ||
|
Časté: | ||
|
Méně časté: |
Závratě, parestezie, dysgeuzie, hypestezie, hyperestezie, tremor, ospalost | |
|
Velmi vzácné: |
Křeče, hypoestezie a tetanie (v důsledku hypokalcemie) | |
|
Poruchy oka | ||
|
Časté: |
Konjunktivitida | |
|
Méně časté: |
Neostré vidění, skleritida a zánět očnice | |
|
Vzácné: |
Uveitida | |
|
Velmi vzácné: |
Episkleritida | |
|
Poruchy ucha a labyrintu | ||
|
Velmi vzácné |
Osteonekróza zevního zvukovodu (skupinový nežádoucí účinek bisfosfonátů). | |
|
Srdeční poruchy | ||
|
Méně časté: |
Hypertenze, hypotenze, fibrilace síní, hypotenze vedoucí k synkopě nebo oběhovému kolapsu | |
|
Vzácné: |
Bradykardie, srdeční arytmie (v důsledku hypokalcemie) | |
|
Respirační, hrudní a mediastinálníporuchy | ||
|
Méně časté: | ||
|
Vzácné |
Intersticiální plicní nemoc | |
|
Gastrointestinální poruchy | ||
|
Časté: | ||
|
Méně časté: |
Průjem, zácpa, bolesti břicha, dyspepsie, stomatitida, sucho v ústech | |
|
Poruchy kůže a podkožní tkáně | ||
|
Méně časté: |
Svědění, vyrážka (včetně erytematózní a makulární vyrážky), zvýšené pocení | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Bolest kostí, myalgie, artralgie, generalizovaná bolest | |
|
Méně časté: |
Svalové křeče, osteonekróza čelisti | |
|
Poruchy ledvin a močových cest | ||
|
Časté: |
Porucha funkce ledvin | |
|
Méně časté: |
Akutní selhání ledvin, hematurie, proteinurie | |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Časté: |
Horečka, chřipkové příznaky (včetně únavy, třesavky, malátnosti a návalů horka) | |
|
Méně časté: |
Astenie, periferní edém, reakce v místě vpichu (včetně bolesti, podráždění, otoku, indurace), bolest na hrudi, zvýšení tělesné hmotnosti, anafylaktická reakce/šok, kopřivka | |
|
Vzácné |
Artritida a otoky kloubů jako symptom reakce akutní fáze | |
|
Vyšetření | ||
|
Velmi časté: |
Hypofosfatemie | |
|
Časté: |
Zvýšení kreatininu a močoviny v krvi, hypokalcemie | |
|
Méně časté: |
Hypomagnesemie, hypokalemie | |
|
Vzácné: |
Hyperkalemie, hypernatremie | |
Popis vybraných nežádoucích účinků
Porucha _ funkce ledvin
U kyseliny zoledronové byla hlášena souvislost s dysfunkcí ledvin. Ve sloučené analýze údajů o bezpečnosti získaných ze studií hodnotících kyselinu zoledronovou podávanou pro předcházení kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti, byla četnost nežádoucích účinků spojených s poruchou funkce ledvin, u níž existuje podezření na souvislost s podáváním kyseliny zoledronové, (nežádoucí reakce) následující: mnohočetný myelom (3,2 %), karcinom prostaty (3,1 %), karcinom prsu (4,3 %), karcinom plic a další solidní nádory (3,2 %). Mezi faktory, které mohou zvýšit možnost zhoršení funkce ledvin, patří dehydratace, již existující poruchy funkce ledvin, opakované cykly podávání kyseliny zoledronové nebo jiných bisfosfonátů, stejně jako současné užívání nefrotoxických léčivých přípravků nebo kratší doba aplikace infuze, než se doporučuje. Bylo hlášeno zhoršení funkce ledvin a progrese do selhání ledvin a nutnost dialýzy u pacientů po úvodní nebo jednorázové dávce 4 mg kyseliny zoledronové (viz bod 4.4).
Osteonekróza čelisti
Byly hlášeny případy osteonekrózy čelistí, a to především u pacientů s karcinomem, kteří byli léčeni léčivými přípravky inhibujícími resorpci kostí, jako je kyselina zoledronová (viz bod 4.4). Mnozí z těchto pacientů také dostávali chemoterapii a kortikosteroidy a měli známky lokální infekce, včetně osteomyelitidy. Většina hlášení se týkala pacientů s karcinomem po extrakci zubu nebo jiném stomatochirurgickém výkonu.
Fibrilace síní
V jedné tříleté, randomizované, dvojitě zaslepené kontrolované studii, která hodnotila účinnost a bezpečnost kyseliny zoledronové 5 mg podávané jednou ročně ve srovnání s placebem při léčbě postmenopauzální osteoporózy (PMO), byl celkový výskyt fibrilace síní 2,5 % (96 z 3862) u pacientek, jimž byla podávána kyselina zoledronová 5 mg, a 1,9 % (75 z 3852) u pacientek s placebem. Výskyt vážných nežádoucích účinků ve formě fibrilace síní byl 1,3 % (51 z 3862) u pacientek, jimž byla podávána kyselina zoledronová 5 mg, a 0,6 % (22 z 3852) u pacientek s placebem. Nerovnováha zjištěná v této studii nebyla zaznamenána v jiných studiích s kyselinou zoledronovou, včetně studií s kyselinou zoledronovou 4 mg podávanou každé 3-4 týdny onkologickým pacientům. Mechanismus původu zvýšeného výskytu fibrilace síní v této jediné klinické studii není známý.
Reakce akutní _fáze
Tento nežádoucí účinek se skládá ze souboru příznaků, které zahrnují horečku, myalgii, bolest hlavy, bolest končetin, nauzeu, zvracení, průjem, artralgii a artritidu s následnými otoky kloubů. Nastupuje < 3 dny po infuzi kyseliny zoledronové a reakce je také známá pod názvy „chřipkové příznaky“ nebo „příznaky po podání dávky“.
Atypické zlomeniny _femuru
Během sledování po uvedení na trh byly hlášeny následující nežádoucí účinky (frekvence vzácná): atypické subtrochanterické a diafýzární zlomeniny femuru (skupinový nežádoucí účinek bisfosfonátů).
Nežádoucí účinky spojené s hypokalcemií
Hypokalcemie je důležitým známým rizikem podání kyseliny zoledronové ve schválených indikacích. Na základě hodnocení případů z klinických studií a případů po uvedení na trh existují dostatečné důkazy pro podporu souvislosti mezi léčbou kyselinou zoledronovou, hlášenými případy hypokalcemie a následným výskytem srdeční arytmie. Dále existují důkazy o souvislosti mezi hypokalcemií a následnými neurologickými nežádoucími účinky hlášenými v těchto případech, které zahrnují křeče, hypoestezii a tetami (viz bod 4.4).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické zkušenosti s akutním předávkováním kyselinou zoledronovou jsou omezené. Bylo hlášeno chybné podávání kyseliny zoledronové v dávkách až do 48 mg. Pacienti, jimž byly aplikovány vyšší dávky, než je dávka doporučená (viz bod 4.2), musí být pečlivě sledováni, protože byla pozorována porucha funkce ledvin (včetně selhání ledvin) a odchylky v hladinách sérových koncentrací elektrolytů (včetně vápníku, fosforu a hořčíku). V případě hypokalcemie lze dle klinické indikace podat infuze kalcium-glukonátu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii nemocí kostí, bisfosfonáty, ATC kód: M05BA08
Kyselina zoledronová patří do skupiny bisfosfonátů a působí primárně na kosti. Je to inhibitor osteoklastické resorpce kostí.
Selektivní působení bisfosfonátů na kosti spočívá v jejich vysoké afinitě k mineralizované kosti, ale přesný molekulární mechanismus vedoucí k inhibici osteoklastické aktivity zůstává stále neobjasněn. V dlouhodobých studiích na zvířatech inhibovala kyselina zoledronová kostní resorpci, aniž by nežádoucím způsobem ovlivňovala tvorbu, mineralizaci nebo mechanické vlastnosti kostí.
Kromě silné inhibice kostní resorpce má kyselina zoledronová navíc některé protinádorové vlastnosti, které by mohly přispívat k její celkové účinnosti při léčbě kostních metastáz. V preklinických studiích byly doloženy následující vlastnosti:
- In vivo: inhibice osteoklastické kostní resorpce, která ovlivňuje vnitřní mikroprostředí kostní dřeně a zhoršuje tak podmínky pro růst nádorových buněk; antiangiogenní účinek a analgetický účinek.
- In vitro: Inhibice osteoblastické proliferace, přímý cytostatický a proapoptotický účinek na nádorové buňky, synergický cytostatický účinek spolu s dalšími protinádorovými léky, antiadhezivní/antiinvazivní působení.
Výsledky klinických studií prevence kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti
První randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie srovnávala kyselinu zoledronovou 4 mg s placebem v prevenci kostních příhod (SRE = skeletal related events) u pacientů s karcinomem prostaty. Kyselina zoledronová 4 mg významně snížila podíl pacientů, u nichž došlo k výskytu alespoň jedné kostní příhody (SRE), prodloužila medián času do první SRE o > 5 měsíců a snížila roční výskyt příhod na pacienta - kostní morbiditu. Analýza mnohočetných příhod ukázala ve srovnání s placebem 36% snížení rizika rozvoje SRE ve skupině, jíž byla podávána kyselina zoledronová 4 mg. Pacienti, jimž byla aplikována kyselina zoledronová 4 mg, uváděli nižší nárůst bolesti než ti, kteří dostávali placebo a rozdíl dosáhl významnosti v měsících 3, 9, 21 a 24. U pacientů dostávajících kyselinu zoledronovou 4 mg byl nižší výskyt patologických zlomenin. U pacientů s blastickými lézemi byl léčebný efekt méně zřejmý. Výsledky účinnosti jsou uvedeny v tabulce 3.
Ve druhé studii, v níž byly zahrnuty jiné solidní nádory než karcinom prsu a prostaty, kyselina zoledronová 4 mg významně snížila podíl pacientů se SRE, prodloužila medián času do první kostní příhody o > 2 měsíce a snížila kostní morbiditu. Analýza mnohočetných příhod ukázala ve srovnání s placebem 30,7% snížení rizika rozvoje SRE ve skupině, jíž byla podávána kyselina zoledronová 4 mg. Výsledky účinnosti jsou uvedeny v tabulce 4.
|
Tabulka 3: Výsledky účinnosti (pacienti s karcinomem prostaty užívající hormonální léčbu) | ||||||
|
Jakákoli SRE (+TIH) |
Fraktury* |
Radioterapie kostí | ||||
|
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo | |
|
n |
214 |
208 |
214 |
208 |
214 |
208 |
|
Podíl pacientů se SRE (%) |
38 |
49 |
17 |
25 |
26 |
33 |
|
Hodnota p |
0,028 |
0,052 |
0,119 | |||
|
Medián času do SRE (dny) |
488 |
321 |
NR |
NR |
NR |
640 |
|
Hodnota p |
0,009 |
0,020 |
0,055 | |||
|
Kostní morbidita |
0,77 |
1,47 |
0,20 |
0,45 |
0,42 |
0,89 |
|
Hodnota p |
0,005 |
0,023 |
0,060 | |||
|
Snížení rizika mnohočetných příhod** (%) |
36 |
NA |
NA |
NA |
NA | |
|
Hodnota p |
0,002 |
NA |
NA | |||
* Zahrnuje vertebrální i nevertebrální fraktury.
** Zahrnuje všechny kostní příhody, celkový počet i dobu do každé příhody během studie. NR Nedosaženo NA Neuplatňuje se
|
Tabulka 4: Výsledky účinnosti (solidní nádory - jiné než karcinom prsu a prostaty) | ||||||
|
Jakákoli SRE (+TIH) |
Fraktury* |
Radioterapie kostí | ||||
|
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo |
kyselina zoledronová 4 mg |
placebo | |
|
n |
257 |
250 |
257 |
250 |
257 |
250 |
|
Podíl pacientů se SRE (%) |
39 |
48 |
16 |
22 |
29 |
34 |
|
Hodnota p |
0,039 |
0,064 |
0,173 | |||
|
Medián času do SRE (dny) |
236 |
155 |
NR |
NR |
424 |
307 |
|
Hodnota p |
0,009 |
0,020 |
0,079 | |||
|
Kostní morbidita |
1,74 |
2,71 |
0,39 |
0,63 |
1,24 |
1,89 |
|
Hodnota p |
0,012 |
0,066 |
0,099 | |||
|
Snížení rizika mnohočetných příhod** (%) |
30,7 |
- |
NA |
NA |
NA |
NA |
|
Hodnota p |
0,003 |
NA |
NA | |||
* Zahrnuje vertebrální i nevertebrální fraktury.
** Zahrnuje všechny kostní příhody, celkový počet i dobu do každé příhody během studie.
NR Nedosaženo NA Neuplatňuje se
V randomizované dvojitě zaslepené studii fáze III byla srovnávána kyselina zoledronová 4 mg s 90 mg pamidronátu, které byly podávány jednou za 3 až 4 týdny pacientům s mnohočetným myelomem nebo karcinomem prsu s nejméně jednou kostní lézí. Výsledky ukázaly, že kyselina zoledronová 4 mg měla v prevenci SRE srovnatelnou účinnost jako 90 mg pamidronátu. Analýza mnohočetných příhod odhalila významné snížení rizika u pacientů léčených kyselinou zoledronovou 4 mg o 16 % ve srovnání s pacienty, kterým byl podáván pamidronát. Výsledky účinnosti j sou uvedeny v tabulce 5.
|
Tabulka 5: Výsledky účinnosti (pacienti s karcinomem prsu a mnohočetným myelomem) | ||||||
|
Jakákoli SRE (+TIH) |
Fraktury* |
Radioterapie kostí | ||||
|
kyselina zoledronov á 4 mg |
Pam 90 mg |
kyselina zoledronov á 4 mg |
Pam 90 mg |
kyselina zoledronov á 4 mg |
Pam 90 mg | |
|
n |
561 |
555 |
561 |
555 |
561 |
555 |
|
Podíl pacientů se SRE (%) |
48 |
52 |
37 |
39 |
19 |
24 |
|
Hodnota p |
0,198 |
0,653 |
0,037 | |||
|
Medián času do SRE (dny) |
376 |
356 |
NR |
714 |
NR |
NR |
|
Hodnota p |
0,151 |
0,672 |
0,026 | |||
|
Kostní morbidita |
1,04 |
1,39 |
0,5 3 |
0,6 0 |
0,4 7 |
0,7 1 |
|
Hodnota p |
0,084 |
0,614 |
0,015 | |||
|
Snížení rizika mnohočetnýc h příhod** (%) |
16 |
NA |
NA |
NA |
NA | |
Hodnota p 0,030
* Zahrnuje vertebrální i nevertebrální fraktury.
NA
NA
** Zahrnuje všechny kostní příhody, celkový počet i dobu do každé příhody během studie.
NR Nedosaženo NA Neuplatňuje se
Kyselina zoledronová 4 mg byla též hodnocena ve dvojitě zaslepené, randomizované, placebem kontrolované studii u 228 pacientů s dokumentovanými kostními metastázami v důsledku karcinomu prsu. Hodnocen byl účinek kyseliny zoledronové 4 mg na procento výskytu kostních příhod (SRE), který byl vypočten jako podíl celkového počtu kostních příhod (s výjimkou hyperkalcemie a po zohlednění předchozích zlomenin) vůči celkovému období ohrožení. Pacientům byly po dobu jednoho roku jednou za čtyři týdny podávány buď 4 mg kyseliny zoledronové, nebo placebo. Pacienti byli rovnoměrně rozděleni do skupin léčených kyselinou zoledronovou nebo placebem.
Podíl SRE (kostní příhody/osoba/rok) činil 0,628 u kyseliny zoledronové a 1,096 u placeba. Podíl pacientů s nejméně jednou příhodou SRE (kromě hyperkalcemie) byl 29,8 % ve skupině léčené kyselinou zoledronovou, oproti 49,6 % ve skupině s placebem (p=0,003). Medián času do vzniku první kostní příhody nebyl v rameni léčby kyselinou zoledronovou na konci studie dosažen, a ve srovnání s placebem byl významně prodloužen (p=0,007). Kyselina zoledronová 4 mg snížila ve srovnání s placebem riziko SRE podle analýzy mnohočetných příhod o 41 % (podíl rizika=0,59, p=0,019).
V průběhu studie bylo ve skupině léčené kyselinou zoledronovou po 4 týdnech a v každém následném časovém bodě zjištěno statisticky významné zlepšení skóre bolesti (pomocí stručných dotazníků o bolesti, [Brief Pain Inventory, BPI]) ve srovnání s placebem (obr. 1). Skóre bolesti při léčbě kyselinou zoledronovou bylo soustavně pod úrovní vstupního stavu, a snížení bolesti bylo doprovázeno trendem sníženého analgetického skóre.
Obrázek 1: Průměrná změna oproti výchozímu stavu u skóre BPI. Statisticky významné rozdíly jsou vyznačeny (*p<0,05) pro účely srovnání mezi typy léčby (4 mg kyseliny zoledronové vs. placebo)
-1.0
|
A |
placebo | ||
|
□ |
kyselina zoledronová |
A |
é |
o.a 0.6 0.4 0.2 0.0 -0.2 -0.4 -0.6 -0.8
0 2 4 8 12 16 20 24 28 32 36 40 44 48 52
Doba ve studii (týdny)
Výsledky klinického hodnocení léčby TIH
V klinických studiích hyperkalcemie vyvolané nádorovým onemocněním (TIH) bylo dokázáno, že účinek kyseliny zoledronové je charakterizován poklesem hladiny vápníku v séru a vylučování vápníku močí. Ve studiích fáze I pro stanovení dávky u pacientů s mírnou až středně závažnou hyperkalcemií vyvolanou nádorovým onemocněním (TIH) se testované účinné dávky pohybovaly v rozmezí cca 1,2-2,5 mg.
Pro účely hodnocení účinků 4 mg kyseliny zoledronové ve srovnání s 90 mg pamidronátu byly v předem plánované analýze zkombinovány výsledky dvou stěžejních multicentrických studií u pacientů s TIH. Po dávce kyseliny zoledronové 8 mg byla zjištěna rychlejší normalizace korigovaných hodnot vápníku v séru 4. den a po dávce kyseliny zoledronové 4 mg a 8 mg 7. den. Byla pozorována následující míra odpovědi:
|
Tabulka 6: Podíl kompletních odpovědí v kombinovaných studiích TIH podle dne | |||
|
4. den |
7. den |
10. den | |
|
Kyselina zoledronová 4 mg (n=86) |
45,3 % (p=0,104) |
82,6 % (p=0,005)* |
88,4 % (p=0,002)* |
|
Kyselina zoledronová 8 mg (n=90) |
55,6 % (p=0,021)* |
83,3 % (p=0,010)* |
86,7 % (p=0,015)* |
|
Pamidronát 90 mg (n=99) |
33,3 % |
63,6 % |
69,7 % |
|
*Hodnoty p ve srovnání s pamidronátem. | |||
Medián doby k dosažení normální koncentrace vápníku byl 4 dny. Medián doby do relapsu (opakované zvýšení hladiny vápníku v séru korigované na albumin > 2,9 mmol/l) činil u pacientů léčených kyselinou zoledronovou 30 až 40 dnů oproti 17 dnům u pacientů léčených pamidronátem 90 mg (hodnoty p: 0,001 pro kyselinu zoledronovou 4 mg a 0,007 pro kyselinu zoledronovou 8 mg). Mezi těmito dvěma dávkami kyseliny zoledronové nebyl nalezen statisticky významný rozdíl.
V klinických studiích bylo 69 pacientů s relapsem nebo refraktemích na počáteční léčbu (kyselina zoledronová 4 mg, 8 mg nebo pamidronát 90 mg) přeléčeno kyselinou zoledronovou 8 mg. Odpověď u těchto pacientů činila přibližně 52 %. Vzhledem k tomu, že tito pacienti byli přeléčeni pouze dávkou
8 mg, nejsou k dispozici údaje umožňující srovnání s dávkou 4 mg kyseliny zoledronové.
V klinických studiích provedených u pacientů s hyperkalcemií vyvolanou nádorovým onemocněním (TIH) byl celkový bezpečnostní profil u všech 3 léčebných skupin (kyselina zoledronová 4 a 8 mg a pamidronát 90 mg) podobný v typech i závažnosti.
Pediatrická populace
Výsledky klinických studií v léčbě těžké _ formy osteogenesis imperfecta u pediatrických pacientů ve věku 1 až 17 let
Účinky intravenózně podávané kyseliny zoledronové při léčbě pediatrických pacientů (ve věku 1 až 17 let) s těžkou formou osteogenesis imperfecta (typ I, III a IV) byly srovnány s intravenózně podávaným pamidronátem v jedné mezinárodní, multicentrické, randomizované otevřené studii, která hodnotila 74 pacientů ve skupině s kyselinou zoledronovou a 76 pacientů ve skupině s pamidronátem. Doba léčby ve studii byla 12 měsíců a léčbě předcházelo 4-9týdenní screeningové období, během něhož byl doplňkově podáván po dobu nejméně 2 týdnů vitamín D a vápník v elementární formě.
V klinickém programu byla pacientům ve věku 1 až < 3 roky podána jednou za 3 měsíce dávka 0,025 mg/kg kyseliny zoledronové (až do maximální jednotlivé dávky 0,35 mg) a pacientům ve věku 3-17 let bylo jednou za 3 měsíce podáno 0,05 mg/kg kyseliny zoledronové (až do maximální jednotlivé dávky 0,83 mg). Byla provedena pokračovací studie, jejímž cílem bylo hodnocení dlouhodobé celkové a renální bezpečnosti kyseliny zoledronové podávané jednou nebo dvakrát ročně v průběhu 12měsíční pokračovací léčby u dětí, které dokončily 1 rok léčby kyselinou zoledronovou nebo pamidronátem v základní studii.
Primárním cílovým parametrem této studie bylo zjištění procentuální změny oproti výchozímu stavu kostní denzity bederní páteře (BMD) po 12 měsících léčby. Očekávané účinky léčby na BMD byly podobné, ale koncepce studie nebyla dostatečně robustní, aby prokázala obdobnou účinnost (non-inferioritu) kyseliny zoledronové. Především nedošlo k jasnému prokázání účinnosti na výskyt fraktur nebo bolesti. Nežádoucí účinky ve formě fraktur dlouhých kostí dolních končetin byly hlášeny cca u 24 % (femur) a 14 % (tibia) pacientů s těžkou formou osteogenesis imperfecta léčených kyselinou zoledronovou oproti 12 % a 5 % pacientů s těžkou formou osteogenesis imperfecta léčených pamidronátem, a to bez ohledu na typ onemocnění a kauzalitu. Souhrnný výskyt fraktur u pacientů léčených kyselinou zoledronovou a pamidronátem byl však srovnatelný: 43 % (32/74) vs. 41 % (31/76). Interpretace rizika fraktur je ztížena skutečností, že fraktury jsou častým jevem u pacientů s těžkou formou osteogenesis imperfecta a jsou součástí průběhu tohoto onemocnění.
Typ nežádoucích účinků zjištěných u této populace byl podobný účinkům dříve zjištěným u dospělých pacientů s pokročilými malignitami zasahujícími kosti (viz bod 4.8). Nežádoucí účinky řazené podle četností jsou uvedené v tabulce 7. Je použita následující obvyklá klasifikace: velmi časté (> 1/10), časté (> 1/100, < 1/10), méně časté (> 1/1000, < 1/100), vzácné (> 1/10 000, <1 /1000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Tabulka 7: Nežádoucí účinky zjištěné u pediatrických pacientů s těžkou formou osteogenesis imperfecta1 | |
|
Poruchy nervového systému | |
|
Časté: | |
|
Srdeční poruchy | |
|
Časté: | |
|
Respirační, hrudní a mediastinálníporuchy | ||
|
Časté: |
Nazofaryngitida | |
|
Gastrointestinální poruchy | ||
|
Velmi časté: | ||
|
Časté: | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Časté: |
Bolest končetin, artralgie, muskuloskeletální bolest | |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté: |
Pyrexie, únava | |
|
Časté: |
Reakce akutní fáze, bolest | |
|
Vyšetření | ||
|
Velmi časté: |
Hypokalcemie | |
|
Časté: |
Hypofosfatemie | |
1 Nežádoucí účinky s četností < 5 % byly zhodnoceny z medicínského hlediska a bylo prokázáno, že tyto případy odpovídají dobře známému bezpečnostnímu profilu kyseliny zoledronové (viz bod 4.8).
U pediatrických pacientů s těžkou formou osteogenesis imperfecta se zdá být použití kyseliny zoledronové ve srovnání s pamidronátem spojené s výraznějším rizikem reakce akutní fáze, hypokalcemie a neobjasněné tachykardie, ale tento rozdíl se snižuje po následných infuzích.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s referenčním léčivým přípravkem obsahujícím kyselinou zoledronovou u všech podskupin pediatrické populace při léčbě hyperkalcemie vyvolané nádorovým onemocněním a prevenci kostních příhod u pacientů s pokročilou formou maligního onemocnění postihujícího kosti (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Následující farmakokinetické údaje, které byly získány po jednorázové a opakované 5 a 15minutové infuzi 2, 4, 8 a 16 mg kyseliny zoledronové u 64 pacientů s kostními metastázami, neprokázaly závislost na dávce.
Po zahájení infuze kyseliny zoledronové rychle stouply koncentrace kyseliny zoledronové v plazmě a dosáhly vrcholu na konci infuze; poté následoval rychlý pokles na < 10 % vrcholové koncentrace za 4 hodiny a < 1 % vrcholové koncentrace za 24 hodin. Pak následovalo dlouhé období velmi nízké koncentrace, která nepřesáhla 0,1 % vrcholové koncentrace před druhou infuzí kyseliny zoledronové 28. den.
Nitrožilně podaná kyselina zoledronová je vylučována třífázovým procesem: rychlé dvoufázové vymizení ze systémového krevního oběhu s poločasy t./2a 0,24 a t/2p 1,87 hodin, po němž následuje dlouhá fáze vylučování s konečným poločasem vylučování t/Y 146 hodin. Po opakovaném podávání každých 28 dnů nebyla zjištěna akumulace kyseliny zoledronové v plazmě. Kyselina zoledronová není metabolizována a je vylučována nezměněná ledvinami. Během prvních 24 hodin se močí vyloučí 39 ± 16 % podané dávky, zatímco zbytek je vázán převážně v kostní tkáni. Z kostní tkáně je kyselina zoledronová velmi pomalu uvolňována zpět do systémového krevního oběhu a vylučována ledvinami. Celková tělesná clearance je 5,04 ± 2,5 l/h a není závislá na dávce ani ovlivněna pohlavím, věkem, rasou a tělesnou hmotností. Prodloužení doby infuze z 5 na 15 minut způsobilo na konci infuze 30% pokles koncentrace kyseliny zoledronové, ale nemělo žádný vliv na plochu pod křivkou plazmatické koncentrace vs. čas.
Variabilita farmakokinetických parametrů kyseliny zoledronové mezi jednotlivými pacienty byla vysoká, stejně jako je tomu u ostatních bisfosfonátů.
U pacientů s hyperkalcemií nebo s jaterní insuficiencí nejsou pro kyselinu zoledronovou dostupné farmakokinetické údaje. Kyselina zoledronová neinhibuje lidské enzymy P450 in vitro, nevykazuje biotransformaci a ve studiích na zvířatech bylo < 3 % z aplikované dávky vyloučeno ve stolici, což svědčí o tom, že játra nehrají ve farmakokinetice kyseliny zoledronové významnou úlohu.
Ledvinná clearance kyseliny zoledronové korelovala s clearance kreatininu, přičemž ledvinná clearance reprezentovala 75 ± 33 % clearance kreatininu. Střední hodnota u 64 hodnocených pacientů s nádorovým onemocněním činila 84 ± 29 ml/min (rozmezí 22 až 143 ml/min). Populační analýza ukázala, že u pacientů s clearancí kreatininu 20 ml/min (těžká porucha funkce ledvin) nebo 50 ml/min (středně těžká porucha) by odpovídající predikovaná clearance kyseliny zoledronové činila 37 %, resp. 72 % hodnoty pacientů s clearancí kreatininu 84 ml/min. U pacientů s těžkou ledvinnou insuficiencí (clearance kreatininu < 30 ml/min) je dostupné pouze omezené množství údajů.
V in vitro studii vykazovala kyselina zoledronová nízkou afinitu k buňkám lidské krve s průměrem poměru koncentrace v krvi ke koncentaci v plazmě 0,59 v rozmezí koncentrací 30 ng/ml až 5000 ng/ml. Vazba k plazmatickým proteinům je nízká, s nenavázanou frakcí v rozmezí od 60 % při 2 ng/ml do 77 % při 2000 ng/ml kyseliny zoledronové.
Zvláštní populace:
Pediatričtí pacienti
Omezené farmakokinetické údaje u dětí s těžkou formou osteogenesis imperfecta naznačují, že farmakokinetika kyseliny zoledronové u dětí ve věku 3 až 17 let je při obdobném dávkování v mg/kg podobná farmakokinetice u dospělých pacientů. Zdá se, že věk, tělesná hmotnost, pohlaví a clearance kreatininu nemají na systémovou expozici kyseliny zoledronové žádný vliv.
5.3 Předklinické údaje vztahující se k bezpečnosti
Akutní toxicita.
Nejvyšší jednorázová intravenózní dávka, která nezpůsobila žádné úmrtí zvířat, byla u myší 10 mg/kg a u potkanů 0,6 mg/kg tělesné hmotnosti.
Subchronická a chronická toxicita
Kyselina zoledronová byla dobře snášena, pokud byla aplikována subkutánně potkanům a intravenózně psům v dávce až do 0,02 mg/kg denně po dobu 4 týdnů. Dávka 0,001 mg/kg/den aplikovaná subkutánně potkanům a intravenózní dávka 0,005 mg/kg aplikovaná jednou za 2-3 dny psům až po dobu 52 týdnů byla také dobře snášena.
Nejčastějším nálezem ve studiích s opakovanými dávkami podávanými zvířatům v době růstu byl téměř při všech dávkách nárůst spongiózní tkáně v metafýzách dlouhých kostí, což je odrazem farmakologické antiresorpční aktivity této sloučeniny.
Hranice bezpečnosti z hlediska účinku na ledviny byly při dlouhodobém opakovaném parenterálním podávání ve studiích na zvířatech velmi úzké. Kumulativní hladiny, při nichž ještě nebyl pozorován nežádoucí účinek (NOAEL) po podání jednorázové dávky (1,6 mg/kg) a po opakovaném podávání až po dobu 1 měsíce (0,06 až 0,6 mg/kg/den), však nenaznačily účinek na ledviny při dávkách ekvivalentních nebo přesahujících nejvyšší terapeutické humánní dávky. Dlouhodobé opakované podávání kyseliny zoledronové v dávkách v rozmezí nejvyšší předpokládané terapeutické humánní dávky mělo toxické účinky na jiné orgány, včetně zažívacího traktu, jater, sleziny a plic a míst intravenózního vpichu.
Reprodukční toxicita
Kyselina zoledronová byla při subkutánním podání potkanům v dávce > 0,2 mg/kg teratogenní. Ačkoli teratogenita nebo fetotoxicita nebyla u králíků pozorována, byla zjištěna maternální toxicita. U potkanů byla pozorována při nejnižší testované dávce (0,01 mg/kg tělesné hmotnosti) dystokie.
Mutagenita a karcinogenní potenciál
Kyselina zoledronová nevykazovala v provedených testech mutagenity mutagenní účinek.
V provedených testech karcinogenity nebyly zjištěny žádné důkazy karcinogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mannitol
Dihydrát natrium-citrátu Chlorid sodný Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí přijít do styku s roztoky obsahujícími vápník, a nesmí být mísen ani intravenózně podáván s jiným léčivým přípravkem stejnou infuzní linkou.
6.3 Doba použitelnosti
Neotevřený vak: 2 roky.
Po prvním otevření: Z mikrobiologického hlediska je nutné přípravek použít okamžitě. Pokud není použit okamžitě, zodpovídá za délku a podmínky jeho uchovávání po otevření před použitím uživatel; tato doba by neměla překročit 24 hodin při teplotě 2 °C - 8 °C. Chlazený roztok musí být před podáním vytemperován na pokojovou teplotu.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
100ml polypropylenové vaky s polypropylenovým šroubovacím portem opatřeným uzávěrem, s polyester/polypropylenovým přebalem.
Velikost balení
Přípravek Zoledronic Acid Hospira se dodává v balení, které obsahuje 1 vak.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Při přípravě infuze musí být dodržena aseptická technika. Určeno pouze k jednorázovému použití. Smí se použít pouze čirý roztok bez obsahu částic a zabarvení.
Zdravotnickým pracovníkům se doporučuje, aby nepoužitý přípravek Zoledronic Acid Hospira nevyhazovali do domácího systému odpadních vod.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Hospira UK Limited
Horizon
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/800/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
19. listopadu 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
1. NÁZEV PŘÍPRAVKU
Zoledronic Acid Hospira 5 mg/100 ml infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden vak o objemu 100 ml obsahuje acidum zoledronicum 5 mg (ve formě acidum zoledronicum monohydricum).
Jeden ml roztoku obsahuje bezvodé acidum zoledronicum 0,05 mg (ve formě acidum zoledronicum monohydricum).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Infuzní roztok
Čirý a bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba Pagetovy choroby kosti u dospělých.
4.2 Dávkování a způsob podání
Dávkování
Před podáním přípravku Zoledronic Acid Hospira musí být pacient dostatečně hydratován. Je to zvláště důležité u starších pacientů a u pacientů, kteří užívají diuretika.
Při podávání přípravku Zoledronic Acid Hospira se doporučuje podávat adekvátní dávku kalcia a vitaminu D.
K léčbě Pagetovy choroby by měli kyselinu zoledronovou předepisovat pouze lékaři se zkušenostmi s léčbou Pagetovy choroby kostí. Doporučená dávka je jednorázová intravenózní infuze 5 mg kyseliny zoledronové. Navíc se důrazně doporučuje, aby pacienti s Pagetovou kostní chorobou dostávali po dobu nejméně 10 dnů po aplikaci přípravku Zoledronic Acid Hospira adekvátní doplňkovou dávku kalcia odpovídající alespoň 500 mg kalcia 2x denně (viz bod 4.4).
Opakovaná léčba Pagetovy choroby: po úvodní léčbě Pagetovy choroby kyselinou zoledronovou lze pozorovat prodloužené období remise u pacientů odpovídajících na léčbu. Opakovaná léčba se skládá z další intravenózní infuze 5 mg kyseliny zoledronové po uplynutí jednoho roku nebo více od úvodní léčby u pacientů, u nichž došlo k relapsu. O opakované léčbě Pagetovy choroby jsou k dispozici omezené údaje (viz bod 5.1).
Zvláštní _ populace
Pacienti s poruchou funkce ledvin
Kyselina zoledronová je kontraindikována u pacientů s clearancí kreatininu < 35 ml/min (viz body 4.3 a 4.4).
U pacientů s clearancí kreatininu > 35 ml/min není třeba žádná úprava dávkování.
Pacienti s poruchou funkce jater
Není třeba žádná úprava dávkování (viz bod 5.2).
Starší lidé (> 65 let)
Není třeba žádná úprava dávkování, neboť biologická dostupnost, distribuce a eliminace byly u starších pacientů i mladších subjektů podobné.
Pediatrická populace
Bezpečnost a účinnost kyseliny zoledronové u dětí a dospívajících do 18 let nebyla stanovena.
Nejsou dostupné žádné údaje.
Způsob podání
Intravenózní podání.
Přípravek Zoledronic Acid Hospira (5 mg ve 100 ml infuzního roztoku k okamžitému použití) se podává infuzní linkou při stálé rychlosti infuze. Doba infuze musí být nejméně 15 minut. Informace o infuzi přípravku Zoledronic Acid Hospira naleznete v bodě 6.6.
Pacientům léčeným Zoledronic Acid Hospira má být k dispozici příbalová informace a pacientská informační karta.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku, na jakékoli bisfosfonáty nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Pacienti s hypokalcemií (viz bod 4.4).
- Závažná porucha funkce ledvin s hodnotou clearance kreatininu < 35 ml/min (viz bod 4.4).
- Těhotenství a kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití Funkce ledvin
Použití přípravku Zoledronic Acid Hospira u pacientů se závažnou poruchou funkce ledvin (clearance kreatininu < 35 ml/min) je kontraindikováno vzhledem ke zvýšenému riziku selhání ledvin v této populaci.
Byla pozorována porucha funkce ledvin po podání kyseliny zoledronové (viz bod 4.8), zejména u pacientů s již dříve existující dysfunkcí ledvin nebo jinými riziky zahrnujícími pokročilý věk, souběžné užívání nefrotoxických léčivých přípravků, souběžnou diuretickou léčbu (viz bod 4.5) nebo dehydrataci vyskytující se po podání kyseliny zoledronové. Zhoršení funkce ledvin bylo u pacientů zaznamenáno po jediném podání. Zřídka se u pacientů se základní poruchou funkce ledvin nebo s některým z výše popsaných rizikových faktorů vyskytlo selhání ledvin vyžadující dialýzu nebo vedoucí k úmrtí.
Aby se minimalizovalo riziko nežádoucích renálních účinků, je nutné vzít v úvahu následující bezpečnostní opatření:
• Před podáním každé dávky přípravku Zoledronic Acid Hospira je nutné spočítat clearanci
kreatininu na základě aktuální tělesné hmotnosti s použitím Cockcroft-Gaultova vzorce.
• Přechodné zvýšení hladiny sérového kreatininu může být větší u pacientů se základní poruchou
funkce ledvin.
• U rizikových pacientů je nutné zvážit sledování hladiny kreatininu v séru.
• Kyselina zoledronová se má podávat s obezřetností, pokud se užívá současně s jinými léčivými
přípravky, které by mohly ovlivnit funkci ledvin (viz bod 4.5).
• Pacienti, zejména starší pacienti a ti, kteří užívají diuretika, by měli být před podáním kyseliny
zoledronové přiměřeně hydratováni.
• Jednotlivá dávka kyseliny zoledronové nesmí přesáhnout 5 mg a délka infuze musí být alespoň
15 minut (viz bod 4.2).
Hypokalcemie
Již existující hypokalcemie se musí léčit dostatečným přísunem vápníku a vitamínu D před zahájením léčby kyselinou zoledronovou (viz bod 4.3). Také jiné poruchy metabolismu minerálů se musí účinně léčit (např. nedostatečnost příštítných tělísek, malabsorpce vápníku ve střevě). Lékaři by u těchto pacientů měli zvážit klinické sledování.
Zvýšená přestavba kostní tkáně je charakteristickým příznakem Pagetovy choroby kostí. Díky rychlému nástupu účinku kyseliny zoledronové na přestavbu kostní tkáně se může rozvinout přechodná hypokalcemie, někdy symptomatická, která obvykle dosahuje maxima během prvních 10 dnů po infuzi kyseliny zoledronové (viz bod 4.8).
V souvislosti s podáváním kyseliny zoledronové se doporučuje dostatečný přísun vápníku a vitamínu D. Dále se u pacientů s Pagetovou chorobou důrazně doporučuje zajistit dostatečné doplnění vápníku odpovídající alespoň 500 mg elementárního vápníku dvakrát denně nejméně po dobu 10 dní po podání kyseliny zoledronové (viz bod 4.2). Pacienty je nutné informovat o příznacích hypokalcemie a dostatečně klinicky sledovat během rizikového období. U pacientů s Pagetovou chorobou se doporučuje před infuzí kyseliny zoledronové změřit hladinu vápníku v séru.
U pacientů, kteří užívali bisfosfonáty, včetně kyseliny zoledronové, byly vzácně hlášeny silné bolesti kostí, kloubů a/nebo svalů, jež byly občas zneschopňující (viz bod 4.8).
Osteonekróza čelisti (OČ)
Osteonekróza čelisti byla hlášena v postmarketingovém období u pacientů užívajících kyselinu zoledronovou k léčbě osteoporózy (viz bod 4.8).
U pacientů s nehojícími se lézemi měkkých tkání v ústech by mělo být s výjimkou akutních medicínských stavů zahájení léčby nebo nového cyklu léčby odloženo. Před zahájením léčby Zoledronic Acid Hospira je u pacientů s konkomitantními rizikovými faktory doporučené příslušné vyšetření s preventivním ošetřením a individuálním vyhodnocením poměru prospěchu-rizika.
Je nutno vzít v úvahu při hodnocení rizika vzniku OČ u pacienta následující:
- Účinek léčivého přípravku, který inhibuje resorpci kostí (vyšší riziko u vysoce účinných látek), cestu podání (vyšší riziko u parenterálního podání) a kumulativní dávku při terapii kostní resorpce.
- Maligní nádorové onemocnění, komorbidity (např. anémie, koagulopatie, infekce), kouření.
- Konkomitantní terapie: kortikosteroidy, chemoterapie, inhibitory angiogeneze, radioterapie krku a hlavy.
- Špatná ústní hygiena, periodontální onemocnění, špatně naléhající zubní protézy,
stomatologická onemocnění v anamnéze, invazivní stomatologické zákroky (např. extrakce zubů).
Všichni pacienti mají být vyzváni, aby během léčby kyselinou zoledronovou udržovali dobrou ústní hygienu, absolvovali rutinní vyšetření chrupu a okamžitě hlásili jakékoli příznaky v ústech, jako je kývání zubů, bolest nebo otoky nebo nehojící se vředy nebo výtok. Během léčby mají být invazivní stomatologické procedury prováděny pouze po pečlivém vyhodnocení a při nastávajícím podávání kyseliny zoledronové mají být vyloučena.
Plán léčby pacientů s OČ by měl být navržen v úzké spolupráci mezi ošetřujícím lékařem a dentistou nebo zubním chirurgem s odbornou znalostí OČ. Dokud se stav nezlepší a pokud možno nevymizí příspívající rizikové faktory, mělo by být zváženo dočasné přerušení léčby kyselinou zoledronovou.
Osteonekróza vnějšího zvukovodu
V souvislosti s léčbou bisfosfonáty byla hlášena osteonekróza zevního zvukovodu, zejména při dlouhodobém podávání. Mezi možné rizikové faktory osteonekrózy zevního zvukovodu patří používání steroidů a chemoterapie a/nebo lokální rizikové faktory, jako například infekce nebo trauma. Možnost vzniku osteonekrózy zevního zvukovodu je třeba zvážit u pacientů léčených bisfosfonáty, kteří mají ušní symptomy včetně chronických infekcí ucha.
Atypické zlomeniny femuru
V souvislosti s léčbou bisfosfonáty, a to zejména u pacientů dlouhodobě léčených pro osteoporózu, byly hlášeny atypické subtrochanterické a diafyzární fraktury femuru. Tyto příčné nebo krátké šikmé fraktury mohou vzniknout kdekoli na femuru od místa těsně pod malým trochanterem až do místa těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s ním a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“), týdny až měsíce před manifestací kompletní zlomeniny femuru. Fraktury jsou často oboustranné, a proto je u pacientů léčených bisfosfonáty, u nichž došlo ke zlomenině diafýzy femuru, nutné vyšetřit kontralaterální femur. Bylo též zaznamenáno špatné hojení těchto fraktur. U pacientů, u nichž existuje podezření na atypickou frakturu femuru, je nutné při hodnocení jejich stavu zvážit přerušení léčby bisfosfonáty, a to na základě individuálního posouzení rizik a přínosů této léčby.
Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, kyčle nebo třísla, a všechny pacienty, u nichž se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou neúplnou frakturu femuru.
Obecně
Výskyt příznaků po podání dávky, které se objevují během prvních tří dnů po podání přípravku Zoledronic Acid Hospira, lze snížit podáním paracetamolu nebo ibuprofenu bezprostředně po podání přípravku Zoledronic Acid Hospira.
Pro onkologické indikace jsou k dispozici jiné přípravky obsahující kyselinu zoledronovou. Pacienti, kteří jsou léčeni přípravkem Zoledronic Acid Hospira, by neměli být souběžně léčeni takovými přípravky ani jakýmkoli jiným bisfosfonátem, neboť kombinované účinky těchto látek nejsou známy.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí s jinými léčivými přípravky. Kyselina zoledronová není systémově metabolizována a in vitro nepůsobí na enzymy lidského cytochromu P450 (viz bod 5.2). Kyselina zoledronová se neváže vysokou měrou na plazmatické proteiny (váže se přibližně ze 4355 %) a interakce způsobené vytěsněním léků vysoce vázaných na proteiny jsou proto nepravděpodobné.
Kyselina zoledronová je eliminována prostřednictvím exkrece ledvinami. Pokud se kyselina zoledronová podává spolu s léčivými přípravky, které mohou významně ovlivnit funkci ledvin (např. aminoglykosidy nebo diuretika, které mohou způsobit dehydrataci), je třeba postupovat obezřetně (viz bod 4.4).
U pacientů s poruchou funkce ledvin se může zvýšit systémová expozice souběžným léčivým přípravkům, které jsou primárně vylučovány ledvinami.
4.6 Fertilita, těhotenství a kojení
Přípravek Zoledronic Acid Hospira je kontraindikován během těhotenství (viz bod 4.3). Adekvátní údaje o podávání kyseliny zoledronové těhotným ženám nejsou k dispozici. Studie s kyselinou zoledronovou na zvířatech prokázaly reprodukční toxikologické účinky, včetně malformací (viz bod 5.3). Potenciální riziko pro člověka není známo.
Kojení
Podávání přípravku Zoledronic Acid Hospira je během kojení kontraindikováno (viz bod 4.3).
Není známo, zda se kyselina zoledronová vylučuje do lidského mateřského mléka.
Ženy ve fertilním věku.
Podávání kyseliny zoledronové ženám ve fertilním věku se nedoporučuje.
Fertilita
Potenciální nežádoucí účinky kyseliny zoledronové na fertilitu v parentální a F1 generaci byly hodnoceny u potkanů. Došlo k výrazným farmakologickým účinkům, o nichž se má za to, že souvisejí s inhibicí mobilizace vápníku v kostech touto sloučeninou, což se projevilo peripartální hypokalcemií (skupinovým účinkem bisfosfonátů), dystokií a časným ukončením studie. Tyto výsledky proto zabránily stanovení konečného účinku kyseliny zoledronové na lidskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Zoledronic Acid Hospira nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Nežádoucí účinky, jako je závrať, mohou ovlivnit schopnost řídit nebo obsluhovat stroje, přestože studie tohoto účinku kyseliny zoledronové nebyly provedeny.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Celkové procento pacientů, u nichž se vyskytly nežádoucí účinky, činilo 44,7 % po první infuzi,
16,7 % po druhé a 10,2 % po třetí infuzi. Výskyt jednotlivých nežádoucích účinků po první infuzi činil: horečka (17,1 %), myalgie (7,8 %), chřipkovité příznaky (6,7 %), artralgie (4,8 %) a bolest hlavy (5,1 %). Výskyt těchto účinků výrazně klesl při následujících každoročních dávkách kyseliny zoledronové. Většina těchto účinků se vyskytla během prvních tří dnů po podání kyseliny zoledronové. Většina těchto účinků byla mírná až středně závažná a vymizela během tří dnů od nástupu příhody. Procento pacientů, u nichž se vyskytly nežádoucí účinky, bylo nižší v menší studii (19,5 % po první, 10,4 % po druhé, 10,7 % po třetí infuzi), v níž se použila profylaxe proti nežádoucím účinkům.
Ve studii HORIZON - Hlavní studii fraktur [PFT] (viz bod 5.1) činil celkový výskyt fibrilace síní
2,5 % (96 z 3862) u pacientů užívajících kyselinu zoledronovou a 1,9 % (75 z 3852) u pacientů užívajících placebo. Výskyt vážných nežádoucích příhod ve formě fibrilace síní byl vyšší (1,3 %) (51 z 3862) u pacientek, jimž byla podána kyselina zoledronová v porovnání s pacientkami užívajícími placebo (0,6 %) (22 z 3852). Mechanismus původu zvýšeného výskytu fibrilace síní není známý. Ve studiích osteoporózy (PFT, HORIZON - Studie opakovaných fraktur [RFT]) byl souhrnný výskyt fibrilace síní mezi kyselinou zoledronovou (2,6 %) a placebem (2,1 %) srovnatelný. Souhrnný výskyt závažných nežádoucích příhod v podobě fibrilace síní činil 1,3 % u kyseliny zoledronové a 0,8 % u placeba.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky v tabulce 1 jsou seřazeny podle třídy orgánových systémů MedDRA a kategorie frekvence. Kategorie frekvencí jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100, < 1/10), méně časté (> 1/1000, < 1/100), vzácné (> 1/10 000, <1 /1000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1
|
Infekce a infestace |
Méně časté |
Chřipka, nazofaryngitida |
|
Poruchy krve a lymfatického systému |
Méně časté |
Anemie |
|
Poruchy imunitního systému |
Není známo** |
Hypersenzitivní reakce včetně vzácných případů bronchokonstrikce, kopřivky a angioedému a velmi vzácných případů anafylaktické reakce/šoku |
|
Poruchy metabolismu a výživy |
Časté |
Hypokalcemie* |
|
Méně časté |
Anorexie, snížená chuť k jídlu | |
|
Psychiatrické poruchy |
Méně časté | |
|
Poruchy nervového systému |
Časté |
Bolest hlavy, závrať |
|
Méně časté |
Letargie, parestezie, ospalost, třes, synkopa, porucha chuti k jídlu | |
|
Poruchy oka |
Časté |
Hyperémie oka |
|
Méně časté |
Konjunktivitida, bolest oka | |
|
Vzácné |
Uveitida, episkleritida, iritida | |
|
Není známo** |
Skleritida a zánět očnice | |
|
Poruchy ucha a labyrintu |
Méně časté | |
|
Velmi vzácné |
Osteonekróza zevního zvukovodu (skupinový nežádoucí účinek bisfosfonátů). | |
|
Srdeční poruchy |
Časté |
Fibrilace síní |
|
Méně časté |
Palpitace | |
|
Cévní poruchy |
Méně časté |
Hypertenze, návaly horka |
|
Není známo** |
Hypotenze (někteří pacienti měli základní rizikové faktory) | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Časté | |
|
Méně časté |
Dyspepsie, bolest v nadbřišku, bolest břicha, gastroezofageální reflux, zácpa, sucho v ústech, ezofagitida, bolest zubů, gastritida# | |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Vyrážka, hyperhidróza, pruritus, erytém |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Časté |
Myalgie, artralgie, bolest kostí, bolest zad, bolest končetin |
|
Méně časté |
Bolest krku, myoskeletální ztuhlost, otoky kloubů, svalové křeče, bolest ramene, myoskeletální bolest na hrudi, myoskeletální bolest, ztuhlost kloubů, artritida, svalová slabost | |
|
Vzácné |
Atypické subtrochanterické a diafyzární fraktury femuruf (skupinový nežádoucí účinek bisfosfonátů) | |
|
Není známo** |
Osteonekróza čelisti (viz body 4.4 a 4.8 Účinky dle tříd) | |
|
Poruchy ledvin a močových cest |
Méně časté |
Zvýšená hladina kreatininu v krvi, polakisurie, proteinurie |
|
Není známo** |
Porucha funkce ledvin. Byly hlášeny vzácné případy selhání ledvin vyžadující dialýzu a vzácné případy vedoucí k úmrtí u pacientů s již existující dysfunkcí ledvin nebo jinými rizikovými faktory, jako je pokročilý věk, souběžně podávané nefrotoxické léčivé přípravky, souběžná léčba diuretiky nebo dehydratace v období po infuzi (viz body 4.4 a 4.8 Účinky dle tříd) | |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté | |
|
Časté |
Chřipkovité příznaky, zimnice, únava, astenie, bolest, malátnost, reakce v místě podání infuze | |
|
Méně časté |
Periferní edém, žízeň, reakce akutní fáze, bolest na hrudi jiného než srdečního původu | |
|
Není známo** |
Dehydratace v důsledku příznaků po podání dávky, jako je horečka, zvracení a průjem | |
|
Vyšetření |
Časté |
Zvýšená hladina C-reaktivního proteinu |
|
Méně časté |
Snížená hladina vápníku v krvi |
#Pozorováno u pacientů užívajících souběžně glukokortikoidy.
* Běžné pouze u Pagetovy choroby.
** Na základě zpráv po uvedení přípravku na trh. Četnost nelze z dostupných údajů určit. f Zjištěno po uvedení přípravku na trh.
Popis vybraných nežádoucích účinků Účinky dle tříd:
Porucha funkce ledvin
Kyselina zoledronová byla spojena s poruchou funkce ledvin projevující se jako zhoršení funkce ledvin (tj. zvýšená hladina kreatininu v séru) a ve vzácných případech s akutním selháním ledvin. Byla pozorována porucha funkce ledvin po podání kyseliny zoledronové, zejména u pacientů s již existující dysfunkcí ledvin nebo dalšími rizikovými faktory (např. pokročilý věk, onkologičtí pacienti na chemoterapii, souběžné užívání nefrotoxických léčivých přípravků, souběžná léčba diuretiky, závažná dehydratace), z nichž většině byla podávána dávka 4 mg každé 3-4 týdny, avšak byla pozorována i u pacientů po jednorázovém podání.
V klinických studiích u osteoporózy byla změna clearance kreatininu (měřená jednou ročně před podáním dávky) a výskyt selhání a poruchy funkce ledvin srovnatelná u léčebné skupiny s kyselinou zoledronovou a u skupiny s placebem během období tří let. V průběhu 10 dnů došlo k přechodnému zvýšení hladiny kreatininu v séru u 1,8 % pacientů léčených kyselinou zoledronovou oproti
0,8 pacientům léčených placebem.
Hypokalcemie
V klinických studiích u osteoporózy došlo po podání kyseliny zoledronové u přibližně 0,2 % pacientů k značnému poklesu hladiny vápníku v séru (méně než 1,87 mmol/l). Nebyly pozorovány žádné symptomatické případy hypokalcemie.
Ve studiích Pagetovy choroby byla pozorována symptomatická hypokalcemie u přibližně 1 % pacientů a u všech těchto pacientů odezněla.
Dle laboratorního zhodnocení se vyskytly přechodné asymptomatické hladiny vápníku pod normálním referenčním rozmezím (méně než 2,10 mmol/l) u 2,3 % pacientů léčených kyselinou zoledronovou ve velké klinické studii v porovnání s 21 % pacientů léčených kyselinou zoledronovou ve studiích Pagetovy choroby. Četnost výskytu hypokalcemie byla značně nižší po následujících infuzích.
Všem pacientům ve studii postmenopauzální osteoporózy, studii prevence klinických fraktur po fraktuře kyčle a ve studiích Pagetovy choroby byly podány dostatečné doplňující dávky vitamínu D a vápníku (viz také bod 4.2). Ve studii prevence klinických fraktur po nedávné fraktuře kyčle se hladiny vitamínu D rutinně neměřily, avšak většina pacientů obdržela před podáním kyseliny zoledronové nárazovou dávku vitamínu D (viz bod 4.2).
Místní reakce
Ve velké klinické studii byly hlášeny místní reakce v místě aplikace infuze (0,7 %), jako je zčervenání, otok a/nebo bolest, po podání kyseliny zoledronové.
Osteonekróza čelisti
Byly hlášeny případy osteonekrózy čelisti, především u pacientů s karcinomem léčených přípravky, které inhibují resorpci kostí, včetně kyseliny zoledronové (viz bod 4.4). Ve velké klinické studii se 7736 pacienty byla hlášena osteonekróza čelisti u jednoho pacienta léčeného kyselinou zoledronovou a u jednoho pacienta léčeného placebem. Případy OČ byly hlášeny v postmarketingovém období u pacientů užívajících kyselinu zoledronovou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické zkušenosti s akutním předávkováním jsou omezené. Pacienty, kterým byly podány vyšší dávky než doporučené, je nutné pečlivě sledovat. V případě předávkování vedoucího ke klinicky významné hypokalcemii je možné dosáhnout obratu perorálním podáním doplňků vápníku a/nebo intravenózní infuzí glukonátu vápenatého.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii nemocí kostí, bisfosfonáty, ATC kód: M05BA08 Mechanismus účinku
Kyselina zoledronová patří do třídy bisfosfonátů obsahujících dusík a působí primárně na kosti. Je to inhibitor osteoklastické resorpce kostí.
Farmakodynamické účinky
Selektivní účinek bisfosfonátů na kost je založen na jejich vysoké afinitě k minerální kosti.
Hlavním molekulovým cílem kyseliny zoledronové v osteoklastu je enzym farnesyl-pyrofosfát-syntáza. Dlouhodobé působení kyseliny zoledronové lze přičíst její vysoké vazebné afinitě k aktivnímu místu farnesyl-pyrofosfát (FPP) syntázy a její silné vazebné afinitě ke kostním minerálům.
Klinická účinnost při léčbě Pagetovy choroby kostí
Kyselina zoledronová byla studována u mužských a ženských pacientů starších 30 let s primárně mírnou až středně závažnou Pagetovou chorobou kostí (průměrná hladina sérové alkalické fosfatázy v době zařazení do studie činila 2,6-3,0násobek horního limitu normálního referenčního rozmezí specifického pro daný věk) potvrzenou radiograficky.
Účinnost jedné infuze 5 mg kyseliny zoledronové oproti denní dávce 30 mg risedronátu po dobu 2 měsíců byla prokázána ve dvou 6měsíčních srovnávacích studiích. Po 6 měsících došlo u pacientů léčených kyselinou zoledronovou k odpovědi u 96 % (169/176) a k normalizaci sérové alkalické fosfatázy (SAP) u 89 % (156/176) v porovnání s risedronátem, kde bylo odpovědi dosaženo u 74 % (127/171) a normalizace SAP u 58 % (99/171) (všechny p<0,001).
Ve sloučených výsledcích bylo u kyseliny zoledronové a risedronátu pozorováno podobné snížení skóre intenzity bolesti a interference bolesti oproti výchozí hodnotě během 6 měsíců.
Pacienti, kteří byli klasifikování jako odpovídající na léčbu na konci 6měsíční hlavní studie, byli způsobilí k zařazení do prodlouženého období následného sledování. Ze 153 pacientů léčených kyselinou zoledronovou a 115 pacientů léčených risedronátem, kteří byli zařazeni do prodloužené observační studie, byl podíl pacientů, jež ukončili období prodlouženého sledování v důsledku nutnosti opakované léčby (na základě klinického posudku), po mediánu trvání sledování 3,8 let
od okamžiku podání dávky, vyšší u risedronátu (48 pacientů, neboli 41,7 %) v porovnání s kyselinou zoledronovou (11 pacientů, neboli 7,2 %). Průměrná doba ukončení období prodlouženého sledování v důsledku nutnosti opakované léčby Pagetovy choroby od podání úvodní dávky byla delší u kyseliny zoledronové (7,7 let) než u risedronátu (5,1 let).
Šest pacientů, u kterých bylo dosaženo odpovědi na léčbu 6 měsíců po léčbě kyselinou zoledronovou a později u nich došlo k relapsu onemocnění během období prodlouženého sledování, bylo znovu přeléčeno kyselinou zoledronovou po průměrné době 6,5 let od původní léčby. Pět z těchto 6 pacientů mělo v 6. měsíci hodnotu SAP v normálním rozmezí (pozdní provedené sledování, LOCF).
U 7 pacientů s Pagetovou chorobou bylo provedeno histologické vyšetření kostní tkáně 6 měsíců po léčbě 5 mg kyseliny zoledronové. Výsledky kostní biopsie prokázaly kostní tkáň normální kvality bez známek poškozené remodelace kostní tkáně a bez známek poruch mineralizace. Tyto výsledky se shodovaly s biochemickými markery průkazu normalizace přestavby kostní tkáně.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s referenčním léčivým přípravkem obsahujícím kyselinu zoledronovou u všech podskupin pediatrické populace s Pagetovou chorobou kostí (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Následující farmakokinetické údaje, které byly získány po jednorázové a opakované 5 a 15minutové infuzi 2, 4, 8 a 16 mg kyseliny zoledronové u 64 pacientů vedly k následujícím farmakokinetickým údajům, které byly shledány nezávislými na dávce.
Distribuce
Po zahájení infuze kyseliny zoledronové rychle stouply koncentrace léčivé látky v plazmě a dosáhly vrcholu na konci infuze; poté následoval rychlý pokles na < 10 % vrcholové koncentrace za 4 hodiny a < 1 % vrcholové koncentrace za 24 hodin. Pak následovalo dlouhé období velmi nízké koncentrace, která nepřesáhla 0,1 % vrcholové koncentrace.
Eliminace
Nitrožilně podaná kyselina zoledronová je vylučována třífázovým procesem: rychlé dvoufázové vymizení ze systémového krevního oběhu s poločasy t./2a 0,24 a t/2p 1,87 hodin, po němž následuje dlouhá fáze vylučování s konečným poločasem vylučování t/Y 146 hodin. Po opakovaném podávání dávek jednou za 28 dnů nebyla zjištěna akumulace léčivé látky v plazmě. Časné fáze dispozice ( a a P,
s výše uvedenými hodnotami t/2) patrně vyjadřují rychlou absorpci kostní tkání a exkreci ledvinami.
Kyselina zoledronová není metabolizována a je vylučována nezměněná ledvinami. Během prvních 24 hodin se močí vyloučí 39 ± 16 % podané dávky, zatímco zbytek je vázán převážně v kostní tkáni. Tato absorpce kostní tkání je běžná pro všechny bisfosfonáty a dochází k ní patrně v důsledku strukturní podobnosti s pyrofosfátem. Stejně jako u ostatních bisfosfonátů je doba retence kyseliny zoledronové v kostní tkáni velmi dlouhá. Z kostní tkáně je kyselina zoledronová velmi pomalu uvolňována zpět do systémového krevního oběhu a vylučována ledvinami. Celková tělesná clearance je 5,04 ± 2,5 l/h a není závislá na dávce ani ovlivněna pohlavím, věkem, rasou a tělesnou hmotností. Bylo zjištěno, že interindividuální variabilita plazmatické clearance kyseliny byla 36 % a intraindividuální variabilita 34 %. Prodloužení doby infuze z 5 na 15 minut způsobilo na konci infuze 30% pokles koncentrace kyseliny zoledronové, ale nemělo žádný vliv na plochu pod křivkou plazmatické koncentrace vs. čas.
F armakoki n eti cké/farmakodynamické vztahy
Nebyly provedeny žádné studie interakcí kyseliny zoledronové s jinými léčivými přípravky. Protože kyselina zoledronová není v lidském organismu metabolizována a zjistilo se, že tato látka má velmi malou nebo žádnou schopnost působit jako přímý inhibitor enzymů P450 a/nebo inhibitor enzymů P450 ireverzibilně závislý na metabolismu, je nepravděpodobné, že by kyselina zoledronová snižovala metabolickou clearanci látek, které jsou metabolizovány prostřednictvím systémů enzymů P450. Kyselina zoledronová se neváže vysokou měrou na plazmatické proteiny (váže se přibližně ze 43-55 %) a proces vázání nezávisí na koncentraci. Proto jsou interakce způsobené vytěsněním léků vysoce vázaných na proteiny nepravděpodobné.
Zvláštní populace (viz bod 4.2)
Porucha funkce ledvin
Ledvinná clearance kyseliny zoledronové korelovala s clearancí kreatininu - ledvinná clearance reprezentovala 75 ± 33 % clearance kreatininu. Střední hodnota u 64 hodnocených pacientů činila 84 ± 29 ml/min (rozmezí 22 až 143 ml/min). Malá pozorovaná zvýšení AUC(0-24hod) o přibližně 30 % až 40 % u mírné až středně závažné poruchy funkce ledvin v porovnání s pacienty s normální funkcí ledvin a absence akumulace léku po opakovaném podání bez ohledu na funkci ledvin naznačují, že úprava dávkování kyseliny zoledronové u mírné (Clcr = 50-80 ml/min) a středně závažné poruchy funkce ledvin až po hodnotu clearance kreatininu 35 ml/min není nutná. Použití kyseliny zoledronové u pacientů se závažnou poruchou funkce ledvin (clearance kreatininu < 35 ml/min) je kontraindikováno vzhledem ke zvýšenému riziku selhání ledvin v této populaci.
5.3 Předklinické údaje vztahující se k bezpečnosti
Akutní toxicita
Nejvyšší jednorázová intravenózní dávka, která nezpůsobila žádné úmrtí zvířat, byla u myší 10 mg/kg a u potkanů 0,6 mg/kg tělesné hmotnosti. Ve studiích jednorázového podání infuze psům, byla dávka 1,0 mg/kg (6násobek doporučené terapeutické expozice u lidí na základě AUC) podaná po dobu 15 minut dobře snášena a neměla žádné účinky na ledviny.
Subchronická a chronická toxicita
Ve studiích intravenózní infuze byla prokázána renální snášenlivost kyseliny zoledronové u potkanů při dávce 0,6 mg/kg podané celkem šestkrát jako 15minutová infuze v 3denních intervalech (kumulativní dávka, která odpovídala přibližně 6násobku hladiny AUC terapeutické expozice lidí) a psi dobře snášeli pět 15minutových infuzí o dávce 0,25 mg/kg podávaných ve 2-3týdenních intervalech (kumulativní dávka, která odpovídala 7násobku terapeutické expozice u lidí). Ve studiích
s intravenózním podáním bolusu klesaly dobře snášené dávky s narůstající délkou trvání studie: denní dávka 0,2 mg/kg u potkanů a 0,02 mg/kg u psů byla dobře snášena po dobu 4 týdnů, avšak pokud byla kyselina zoledronová podávána po dobu 52 týdnů, činila dobře snášená dávka pouze 0,01 mg/kg
u potkanů a 0,005 mg/kg u psů.
Dlouhodobé opakované podávání v kumulativních dávkách dostatečně přesahujících maximální zamýšlenou expozici člověka vyvolalo toxikologické účinky v jiných orgánech, včetně gastrointestinálního traktu a jater, a v místě podání intravenózní infuze. Klinický význam těchto zjištění není znám. Nejčastějším nálezem po opakovaném podání studovaných dávek zvířatům v době růstu byl téměř při všech dávkách nárůst spongiózní tkáně v metafýzách dlouhých kostí, což je odrazem farmakologické antiresorpční aktivity této sloučeniny.
Reprodukční toxicita
Byly provedeny teratologické studie u dvou druhů, obě při subkutánním podání. Teratogenita byla pozorována u potkanů při dávkách > 0,2 mg/kg a projevovala se externími, viscerálními a skeletálními malformacemi. U potkanů byla pozorována při nejnižší testované dávce (0,01 mg/kg tělesné hmotnosti) dystokie. U králíků nebyly pozorovány žádné teratologické účinky ani účinky na embryo/fetus, přestože byla při dávce 0,1 mg/kg zaznamenána toxicita pro matku v důsledku snížení hladiny vápníku v séru.
Mutagenita a karcinogenní potenciál
Kyselina zoledronová nevykazovala v provedených testech mutagenity mutagenní účinek.
V provedených testech karcinogenity nebyly zjištěny žádné důkazy karcinogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Mannitol
Dihydrát natrium-citrátu Voda na injekci
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné 100ml dávce přípravku Zoledronic Acid Hospira, tj. je prakticky „bez sodíku“.
6.2 Inkompatibility
Tento léčivý přípravek nesmí přijít do styku s roztoky obsahujícími vápník. Přípravek Zoledronic Acid Hospira nesmí být mísen ani podáván intravenózně s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřený vak: 2 roky Po otevření: 24 hodin při 2 °C - 8 °C
Z mikrobiologického hlediska je nutné přípravek použít okamžitě. Pokud není použit okamžitě, zodpovídá za délku a podmínky jeho uchovávání po otevření před použitím uživatel; tato doba by neměla obvykle překročit 24 hodin při teplotě 2 °C - 8 °C.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání. Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6. 3.
6.5 Druh obalu a obsah balení
100ml polypropylenové vaky s polypropylenovým šroubovacím portem opatřeným uzávěrem, s polyester/polypropylenovým přebalem
Přípravek Zoledronic Acid Hospira se dodává v balení, které obsahuje 1 vak.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Určeno pouze k jednorázovému použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Smí se použít pouze čirý roztok bez obsahu částic a zabarvení.
Pokud je přípravek uchováván v chladničce, nechte chlazený roztok před podáním vytemperovat na pokojovou teplotu. Při přípravě infuze musí být dodržena aseptická technika.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Hospira UK Limited
Horizon
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/800/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
19. listopadu 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Hospira Enterprises B.V.
Randstad 22-11 NL-1316 BN Almere Nizozemsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci musí zajistit implemetaci pacientské informační karty pojednávající o osteonekróze čelisti.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
KRABIČKA NA 1 INJEKČNÍ LAHVIČKU JAKO KUSOVÉ BALENÍ (VČETNĚ “BLUE BOX”)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zoledronic Acid Hospira 4 mg/5 ml, koncentrát pro přípravu infuzního roztoku. Acidum zoledronicum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje acidum zoledronicum 4 mg (ve formě acidum zoledronicum monohydricum).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje rovněž mannitol, dihydrát natrium-citrátu a vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro přípravu infuzního roztoku 4 mg/5 ml 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze intravenózní použití.
Před použitím nařeďte.
Pouze pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP.:
Po naředění stabilní po 24 hodin při 2 °C - 8 °C.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Hospira UK Limited Hurley SL6 6RJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/800/001
EU/1/12/800/002
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Zoledronic Acid Hospira 4 mg/5 ml, sterilní koncentrát i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP.:
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Před použitím nařeďte.
KRABICE NA 1 VAK JAKO KUSOVÉ BALENÍ (VČETNĚ “BLUE BOX”)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok. Acidum zoledronicum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden vak obsahuje acidum zoledronicum 4 mg (ve formě acidum zoledronicum monohydricum).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje rovněž mannitol, dihydrát natrium-citrátu, vodu na injekci a roztok chloridu sodného.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Infuzní roztok
4 mg/100 ml (uvést na kroužku)
1 intravenózní vak
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze intravenózní použití.
Pouze pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP.:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Hospira UK Limited Hurley SL6 6RJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/800/003
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Zoledronic Acid Hospira 4 mg/100 ml (uvést na kroužku) infuzní roztok Acidum zoledronicum Pouze intravenózní použití
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP.:
4. ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
(zahrnuto v textu v bodě 1)
6. JINÉ
KRABICE NA 1 VAK JAKO KUSOVÉ BALENÍ (VČETNĚ “BLUE BOX”)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zoledronic Acid Hospira 5 mg/100 ml infuzní roztok Acidum zoledronicum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden vak o objemu 100 ml obsahuje acidum zoledronicum 5 mg (ve formě acidum zoledronicum monohydricum).
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol, dihydrát natrium-citrátu a voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Infuzní roztok 1 intravenózní vak
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze intravenózní použití.
Pouze pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP.:
Po otevření: 24 hodin při 2 °C - 8 °C.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Hospira UK Limited Hurley SL6 6RJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/800/004
13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
ŠTÍTEK VAKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Zoledronic Acid Hospira 5 mg/100 ml (uvést na kroužku) infuzní roztok Acidum zoledronicum Pouze intravenózní použití.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP.:
4. ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
(zahrnuto v textu v bodě 1)
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Zoledronic Acid Hospira 4 mg/5 ml, koncentrát pro přípravu infuzního roztoku
Acidum zoledronicum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Zoledronic Acid Hospira a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zoledronic Acid Hospira používat
3. Jak se přípravek Zoledronic Acid Hospira používá
4. Možné nežádoucí účinky
5. Jak přípravek Zoledronic Acid Hospira uchovávat
6. Obsah balení a další informace
1. Co je Zoledronic Acid Hospira a k čemu se používá
Léčivou látkou v přípravku Zoledronic Acid Hospira je kyselina zoledronová, která patří do skupiny látek nazývaných bisfosfonáty. Kyselina zoledronová působí tak, že se sama naváže na kosti a zpomaluje rychlost kostního obratu. Používá se k:
• prevenci kostních komplikací, např. zlomenin u dospělých pacientů (rozsev karcinomu z původního místa do kosti),
• snížení množství vápníku v krvi u dospělých pacientů s příliš vysokou hladinou vápníku způsobenou nádorovým onemocněním. Nádory mohou zrychlit normální kostní obrat tak, že dojde ke zvýšenému uvolňování vápníku z kostí. Tento stav je známý jako hyperkalcemie vyvolaná nádorovým onemocněním (TIH).
2. Čemu musíte věnovat pozornost, než začnete přípravek Zoledronic Acid Hospira používat
Pečlivě dodržujte všechny pokyny, které Vám dal Váš lékař.
Lékař Vám před zahájením léčby přípravkem Zoledronic Acid Hospira provede vyšetření krve a bude v pravidelných intervalech kontrolovat Vaši odpověď na léčbu.
Nepoužívejte přípravek Zoledronic Acid Hospira:
- jestliže kojíte,
- jestliže jste alergický(á) na kyselinu zoledronovou, jiný bisfosfonát (skupina
látek, do které patří kyselina zoledronová) nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Zoledronic Acid Hospira se poraďte se svým lékařem:
- jestliže trpíte nebo jste trpěl(a) onemocněním ledvin,
- jestliže trpíte nebo jste trpěl(a) bolestí, otokem nebo znecitlivěním čelisti a pocitem ztěžknutí čelisti nebo vikláním zubu. Před zahájením léčby přípravkem Zoledronic Acid Hospira Vám může lékař doporučit, abyste absolvoval(a) stomatologické vyšetření.
- jestliže proděláváte ošetření zubů nebo máte podstoupit zubní chirurgický výkon, sdělte svému zubnímu lékaři, že jste léčen(a) přípravkem Zoledronic Acid Hospira a informujte Vašeho lékaře o léčbě vašeho chrupu.
Během léčby přípravkem Zoledronic Acid Hospira byste měl(a) dodržovat pečlivou ústní hygienu (včetně pravidelného čištění zubů) a podstoupit pravidelné zubní vyšetření.
Kontaktujte okamžitě svého lékaře a stomatologa, pokud se u Vás objeví jakékoli obtíže v ústní dutině nebo zubní potíže, jako je padání zubů, bolest nebo otoky, nebo nehojící se vřídky nebo výtok, protože může jít o příznaky stavu zvaného osteonekróza čelistí.
Pacienti, kteří podstupují chemoterapii a/nebo radioterapii, pacienti užívající kortikosteroidy, pacienti, kteří absolvovali zubní zákrok, kteří nepodstupují pravidelnou zubní péči, kteří mají potíže s dásněmi, kuřáci nebo pacienti, kteří byli dříve léčeni bisfosfonáty (užívanými k léčbě nebo prevenci kostních onemocnění) mohou mít vyšší riziko vzniku osteonekrózy čelisti.
U pacientů léčených zoledronovou kyselinou byly hlášeny snížené hladiny vápníku v krvi (hypokalcemie), které někdy vedou ke svalovým křečím, suché kůži a pocitům pálení. Jako sekundární (druhotné) příznaky při závažné hypokalcemii byly hlášeny nepravidelný srdeční tep (srdeční arytmie), záchvaty, křeče a svalové záškuby (tetanie). V některých případech může být hypokalcemie život ohrožující. Pokud se některý z výše uvedených příznaků u Vás objeví, okamžitě informujte svého lékaře. Pokud jste před zahájením léčby trpěl(a) hypokalcémií, musí být hypokalcémie před podáním první dávky Zoledronic Acid Hospira upravena. Bude vám podáváno přiměřené množství vápníku a vitamínu D.
Další léčivé přípravky a přípravek Zoledronic Acid Hospira
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Především je důležité, abyste svému lékaři sdělil(a), zda též užíváte:
- aminoglykosidy (léčivé přípravky používané k léčbě závažných infekcí), kalcitonin (druh léku používaného k léčbě postmenopauzálníl osteoporózy a hyperkalcemie), kličková (loop) diuretika (druh léku určeného k léčbě vysokého krevního tlaku nebo edému) nebo jiné léky snižující hladinu vápníku, neboť kombinace těchto přípravků s bisfosfonáty může způsobit příliš velké snížení hladiny vápníku v krvi,
- thalidomid (léčivý přípravek užívaný k léčbě určitých typů rakoviny krve postihující kosti) nebo jiné léčivé přípravky, které mohou poškodit ledviny,
- jakékoli jiné léčivé přípravky, které obsahují kyselinu zoledronovou a používají se k léčbě osteoporózy a jiných onemocnění kostí nenádorového původu nebo jiný bisfosfonát, protože kombinovaný účinek těchto léků užívaných současně s přípravkem Zoledronic Acid Hospira není známý,
- antiangiogenní léčivé přípravky (používané k léčbě rakoviny), neboť jejich kombinace s kyselinou zoledronovou je spojována se zvýšeným rizikem výskytu osteonekrózy čelisti (OČ).
Pacienti ve věku 65 let a starší
Přípravek Zoledronic Acid Hospira může být podáván lidem ve věku 65 a více let. Neexistují žádné důkazy, které by svědčily o tom, že jsou nutná jakákoli zvláštní opatření.
Děti a dospívající
Přípravek Zoledronic Acid Hospira se nedoporučuje podávat dospívajícím a dětem mladším 18 let. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, nesmíte používat přípravek Zoledronic Acid Hospira.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Ve velmi vzácných případech byla při použití přípravku Zoledronic Acid Hospira zaznamenána mátožnost a ospalost. Při řízení dopravních prostředků, obsluze strojů nebo vykonávání jiné činnosti, která vyžaduje plnou pozornost, byste proto měl(a) být opatrný/opatrná.
Přípravek Zoledronic Acid Hospira obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. je prakticky „bez sodíku“.
3. Jak se přípravek Zoledronic Acid Hospira používá
- Přípravek Zoledronic Acid Hospira smí podávat pouze zdravotničtí pracovníci zaškolení v intravenózním podávání bisfosfonátů, tj. do žíly (zvané rovněž „i.v.“ podání).
- Lékař Vám doporučí, abyste před každou léčbou vypil(a) dostatečné množství vody, aby se zabránilo dehydrataci.
- Pečlivě dodržujte všechny další pokyny, které Vám dal Váš lékař, zdravotní sestra nebo lékárník.
Jaké množství přípravku Zoledronic Acid Hospira se podává
- Obvyklá jednorázová dávka je 4 mg.
- Jestliže trpíte onemocněním ledvin, lékař Vám podle závažnosti Vašeho onemocnění podá nižší dávku.
Jak často se přípravek Zoledronic Acid Hospira podává
- Pokud j ste léčen(a) za účelem prevence kostních komplikací způsobených metastázami v kostech, bude Vám podávána jedna infuze přípravku Zoledronic Acid Hospira jednou za tři až čtyři týdny.
- Pokud jste léčen(a) za účelem snížení množství vápníku v krvi, bude Vám zpravidla podána pouze jedna infuze přípravku Zoledronic Acid Hospira.
Jak se přípravek Zoledronic Acid Hospira podává
- Přípravek Zoledronic Acid Hospira se podává formou kapačky (infuze) do žíly, která musí trvat nejméně 15 minut a musí být podávána jako samostatný nitrožilní roztok oddělenou infuzní linkou.
Pacientům, jejichž hladina vápníku v krvi není příliš vysoká, budou též předepsány doplňkové denní dávky vápníku a vitamínu D.
Jestliže jste užil(a) více přípravku Zoledronic Acid Hospira, než jste měl(a)
Pokud Vám byly podány vyšší než doporučené dávky, musíte být pečlivě sledován(a) lékařem. Je tomu tak proto, že může dojít k abnormálním změnám v hladině elektrolytů v séru (např. abnormální hladina vápníku, fosforu a hořčíku) a/nebo ke změnám funkce ledvin, včetně těžkého poškození ledvin. Jestliže se Vaše hladina vápníku příliš sníží, může Vám být vápník doplněn infuzí.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Nejčastější nežádoucí účinky jsou obvykle mírné a po krátké době pravděpodobně vymizí.
Pokud se u Vás projeví následující závažné nežádoucí účinky, okamžitě informujte svého lékaře:
Časté (postihují 1 až 10 uživatelů ze 100):
- Těžké poškození ledvin (obvykle je zjistí Váš lékař určitými speciálními krevními testy).
- Nízká hladina vápníku v krvi.
Méně časté (postihují 1 až 10 uživatelů z 1 000):
- Bolest v ústech, bolest zubů a/nebo čelisti, otok nebo nehojící se vředy v ústech nebo na dásních, necitlivost nebo pocit ztěžknutí čelisti nebo viklání zubu. Mohlo by jít o známky kostního poškození čelisti (osteonekróza). Pokud se u Vás projeví tyto příznaky během léčby přípravkem Zoledronic Acid Hospira nebo po ukončení léčby, okamžitě to sdělte svému lékaři a zubnímu lékaři.
- U pacientek, které užívaly kyselinu zoledronovou k léčbě postmenopauzální osteoporózy, byl pozorován nepravidelný srdeční rytmus (fibrilace síní). V současné době není jasné, zda kyselina zoledronová tento nepravidelný srdeční rytmus způsobuje, ale pokud se u Vás tyto příznaky po podání kyseliny zoledronové projeví, informujte svého lékaře.
- Závažné alergické reakce: dušnost, otoky, zejména otok obličeje a krku.
Vzácné (mohou postihnout až 1 z 1000 lidí):
-Jako následek nízkých hodnot vápníku: nepravidelný srdeční tep (srdeční arytmie; sekundárně po hypokalcémii).
Velmi vzácné (mohou postihnout až 1 z 10 000 lidí):
- Jako následek nízkých hodnot vápníku: křeče, pocit necitlivosti a tetanie (sekundárně po hypokalcemii).
- Poraďte se se svým lékařem, pokud máte bolest ucha, výtok z ucha a/nebo infekci ucha. Mohlo by se jednat o známky poškození kosti v uchu.
Pokud se u Vás vyskytne kterýkoli z následujících nežádoucích účinků, co nejdříve to sdělte svému lékaři:
Velmi časté (postihují více než 1 uživatele z 10):
- Nízká hladina fosfátů v krvi.
Časté (postihují 1 až 10 uživatelů ze 100):
- Bolest hlavy a chřipkové příznaky, jako je horečka, únava, slabost, mátožnost, zimnice, bolesti kostí, kloubů a/nebo svalů. Ve většině případů není třeba žádná specifická léčba a příznaky po krátké době vymizí (po několika hodinách nebo dnech).
- Zažívací potíže, jako je např. nevolnost, zvracení a ztráta chuti k jídlu.
- Zánět spojivek.
- Nízký počet červených krvinek (anemie).
Méně časté (postihují 1 až 10 uživatelů z 1 000):
- Hypersenzitivní reakce.
- Nízký krevní tlak.
- Bolest na hrudi.
- Kožní reakce (zarudnutí a otok) v místě zavedení infuze, vyrážka, svědění.
- Vysoký krevní tlak, dušnost, závrať, úzkost, poruchy spánku, třes, brnění nebo necitlivost rukou nebo nohou, průjem, zácpa, bolest břicha, sucho v ústech.
- Nízký počet bílých krvinek a krevních destiček.
- Nízká hladina hořčíku a draslíku v krvi. Lékař bude tyto hladiny sledovat a učiní nezbytná opatření.
- Ospalost.
- Zvýšení tělesné hmotnosti.
- Zvýšené pocení.
- Rozmazané vidění, slzení očí, citlivost očí na světlo.
- Náhlý pocit chladu a mdloby, ochablost nebo kolaps.
- Dýchací obtíže se sípáním nebo kašlem.
- Kopřivka.
Vzácné (postihují 1 až 10 uživatelů z 10 000):
- Pomalý rytmus srdce.
- Zmatenost.
- Vzácně může vzniknout neobvyklá zlomenina stehenní kosti zvláště u pacientů dlouhodobě léčených pro osteoporózu. Jestliže se u Vás projeví bolest, slabost nebo máte nepříjemný pocit v oblasti stehna, kyčle nebo třísla, obraťte se na svého lékaře, neboť se může jednat o časný příznak možné zlomeniny stehenní kosti.
- Intersticiální plicní choroba (zánět tkáně v okolí plicních sklípků v plicích).
- Příznaky podobné chřipce, včetně artritidy a otoky kloubů.
- Bolestivé zarudnutí a/nebo otok očí.
Velmi vzácné (postihují méně než 1 uživatele z 10 000):
- Mdloba způsobená nízkým krevním tlakem.
- Silná bolest kostí, svalů a/nebo kloubů, občas zneschopňující.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zoledronic Acid Hospira uchovávat
Váš lékař, zdravotní sestra nebo lékárník vědí, jak se přípravek Zoledronic Acid Hospira správně uchovává (viz bod 6).
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu za EXP.
6. Obsah balení a další informace
Co přípravek Zoledronic Acid Hospira obsahuje
- Léčivou látkou je acidum zoledronicum.
Jedna injekční lahvička obsahuje 4 mg kyseliny zoledronové, což odpovídá 4,264 mg acidum zoledronicum monohydricum.
- Dalšími složkami jsou: mannitol, dihydrát natrium-citrátu a voda na injekci.
Jak přípravek Zoledronic Acid Hospira vypadá a co obsahuje toto balení
Přípravek Zoledronic Acid Hospira se dodává jako tekutý koncentrát (zvaný „koncentrát pro přípravu infuzního roztoku“ nebo „sterilní koncentrát“) v injekční lahvičce. Jedna injekční lahvička obsahuje acidum zoledronicum 4 mg.
Každé balení obsahuje jednu injekční lahvičku koncentrátu.
Držitel rozhodnutí o registraci
Hospira UK Limited
Horizon
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Velká Británie
Výrobce
Hospira Enterprises B.V.
Randstad 22-11 1316 BN Almere Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
AT
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1521 15-0
BG/CZ/EE/EL/HU/LT/LV/ MT / PL / RO / SI / SK / UK
Hospira UK Limited Tel: + 44 (0) 1628 515500
DE
Pfizer Pharma PFE GmbH Tel: + 49 (0)800 8535555
ES
Pfizer GEP, S.L.
Tel: +34 91 490 99 00
HR
Alvogen d.o.o.
Tél/Tel: + 385 1 6641 830
BE/LU/ NL Pfizer S.A. / N.V.
Tél/Tel: +32 2 554 62 11
CY
Name: N.Karoullas Pharmaceutical Trading Co Ltd 33, Artemidos avenue, 6025 Larnaca Tel: 24656165/ Mob.: 99403969 Email: n.karoullas@ptc-ltd.com
DK / FI / IS / NO / SE Hospira Nordic AB Tel: + 46 (0) 8 672 85 00
FR
Hospira France
Tél: + 33 (0) 1 40 83 82 00
IE
Hospira Ireland Sales Limited Tel: 1800 633 363 (toll free)
+44 (0) 1304 616161
PT
Hospira Portugal Lda (+351) 21 423 55 00
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Jak připravit a podávat přípravek Zoledronic Acid Hospira
- Chcete-li připravit infuzní roztok obsahující 4 mg kyseliny zoledronové, dále nařeďte koncentrát přípravku Zoledronic Acid Hospira (5,0 ml) 100 ml infuzního roztoku bez obsahu vápníku nebo jiných dvojmocných kationtů. Jestliže je nutná nižší dávka přípravku Zoledronic Acid Hospira, nejdříve odeberte odpovídající objem, jak je uvedeno níže, a poté dále nařeďte 100 ml infuzního roztoku. Aby se zabránilo možné inkompatibilitě, musí být infuzním roztokem použitým pro ředění buď injekční roztok chloridu sodného 9 mg/ml (0,9%), nebo 5% (w/v) roztok glukózy.
Nemíchejte koncentrát Zoledronic Acid Hospira s roztoky obsahujícími vápník nebo jiné dvojmocné kationty, jako je laktátový Ringerův roztok.
Pokyny pro přípravu nižších dávek přípravku Zoledronic Acid Hospira:
Odeberte odpovídající objem tekutého koncentrátu podle následujících pokynů:
- 4,4 ml pro dávku 3,5 mg,
- 4,1 ml pro dávku 3,3 mg, - 3,8 ml pro dávku 3,0 mg.
- Určeno pro jednorázové použití. Veškerý nepoužitý roztok zlikvidujte. Smí se použít pouze čirý roztok bez obsahu částic a zabarvení. Při přípravě infuze musí být dodržena aseptická technika.
- Z mikrobiologického hlediska je nutné naředěný infuzní roztok použít okamžitě. Pokud není použit okamžitě, zodpovídá za délku a podmínky jeho uchovávání po otevření před použitím uživatel. Tato doba by neměla překročit 24 hodin při teplotě 2 °C - 8 °C.
Chlazený roztok musí být před podáním vytemperován na pokojovou teplotu.
- Roztok obsahující kyselinu zoledronovou se podává jako jednorázová 15minutová nitrožilní infuze v samostatné infuzní lince. Před podáním kyseliny zoledronové a poté musí být posouzena hydratace pacienta, aby bylo zajištěno jeho odpovídající zavodnění.
- Studie s několika druhy infuzních linek vyrobených z polyvinylchloridu, polyetylenu a polypropylenu neprokázaly inkompatibilitu s kyselinou zoledronovou.
- Vzhledem k tomu, že nejsou k dispozici žádné údaje o kompatibilitě kyseliny zoledronové s dalšími nitrožilně podávanými látkami, nesmí se přípravek Zoledronic Acid Hospira mísit s jinými léčivými přípravky/látkami a vždy je nutné jej podávat samostatnou infuzní linkou.
Jak přípravek Zoledronic Acid Hospira uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah a dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu za EXP.
- Neotevřená injekční lahvička nevyžaduje žádné zvláštní podmínky uchovávání.
- Naředěný infuzní roztok přípravku Zoledronic Acid Hospira je nutné použít okamžitě, aby se zabránilo mikrobiální kontaminaci.
Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok
Acidum zoledronicum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Zoledronic Acid Hospira a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zoledronic Acid Hospira používat
3. Jak se přípravek Zoledronic Acid Hospira používá
4. Možné nežádoucí účinky
5. Jak přípravek Zoledronic Acid Hospira uchovávat
6. Obsah balení a další informace
1. Co je Zoledronic Acid Hospira a k čemu se používá
Léčivou látkou v přípravku Zoledronic Acid Hospira je kyselina zoledronová, která patří do skupiny látek nazývaných bisfosfonáty. Kyselina zoledronová působí tak, že se sama naváže na kosti a zpomaluje rychlost kostního obratu. Používá se k:
• prevenci kostních komplikací, např. zlomenin u dospělých pacientů (rozsev karcinomu z původního místa do kosti),
• snížení množství vápníku v krvi u dospělých pacientů s příliš vysokou hladinou vápníku způsobenou nádorovým onemocněním. Nádory mohou zrychlit normální kostní obrat tak, že dojde ke zvýšenému uvolňování vápníku z kostí. Tento stav je známý jako hyperkalcemie vyvolaná nádorovým onemocněním (TIH).
2. Čemu musíte věnovat pozornost, než začnete přípravek Zoledronic Acid Hospira používat
Pečlivě dodržujte všechny pokyny, které Vám dal Váš lékař.
Lékař Vám před zahájením léčby přípravkem Zoledronic Acid Hospira provede vyšetření krve a bude v pravidelných intervalech kontrolovat Vaši odpověď na léčbu.
Nepoužívejte přípravek Zoledronic Acid Hospira:
- jestliže kojíte,
- jestliže jste alergický(á) na kyselinu zoledronovou, jiný bisfosfonát (skupina látek, do které patří kyselina zoledronová) nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Zoledronic Acid Hospira se poraďte se svým lékařem:
- jestliže trpíte nebo jste trpěl(a) onemocněním ledvin,
- jestliže trpíte nebo jste trpěl(a) bolestí, otokem nebo znecitlivěním čelisti a pocitem ztěžknutí čelisti nebo vikláním zubu. Před zahájením léčby přípravkem Zoledronic Acid Hospira Vám může lékař doporučit, abyste absolvoval(a) stomatologické vyšetření.
- jestliže proděláváte ošetření zubů nebo máte podstoupit zubní chirurgický výkon, sdělte svému zubnímu lékaři, že jste léčen(a) přípravkem Zoledronic Acid Hospira a informujte Vašeho lékaře o léčbě vašeho chrupu.
Během léčby přípravkem Zoledronic Acid Hospira byste měl(a) dodržovat pečlivou ústní hygienu (včetně pravidelného čištění zubů) a podstoupit pravidelné zubní vyšetření.
Kontaktujte okamžitě svého lékaře a stomatologa, pokud se u Vás objeví jakékoli obtíže v ústní dutině nebo zubní potíže, jako je padání zubů, bolest nebo otoky, nebo nehojící se vřídky nebo výtok, protože může jít o příznaky stavu zvaného osteonekróza čelistí.
Pacienti, kteří podstupují chemoterapii a/nebo radioterapii, pacienti užívající kortikosteroidy, pacienti, kteří absolvovali zubní zákrok, kteří nepodstupují pravidelnou zubní péči, kteří mají potíže s dásněmi, kuřáci nebo pacienti, kteří byli dříve léčeni bisfosfonáty (užívanými k léčbě nebo prevenci kostních onemocnění) mohou mít vyšší riziko vzniku osteonekrózy čelisti.
U pacientů léčených zoledronovou kyselinou byly hlášeny snížené hladiny vápníku v krvi (hypokalcemie), které někdy vedou ke svalovým křečím, suché kůži a pocitům pálení. Jako sekundární (druhotné) příznaky při závažné hypokalcemii byly hlášeny nepravidelný srdeční tep (srdeční arytmie), záchvaty, křeče a svalové záškuby (tetanie). V některých případech může být hypokalcemie život ohrožující. Pokud se některý z výše uvedených příznaků u Vás objeví, okamžitě informujte svého lékaře. Pokud jste před zahájením léčby trpěl(a) hypokalcémií, musí být hypokalcemie před podáním první dávky Zoledronic Acid upravena. Bude vám podáváno přiměřené množství vápníku a vitamínu D.
Další léčivé přípravky a přípravek Zoledronic Acid Hospira
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Především je důležité, abyste svému lékaři sdělil(a), zda též užíváte:
- aminoglykosidy (léčivé přípravky používané k léčbě závažných infekcí), kalcitonin (druh léku používaného k léčbě postmenopauzálníl osteoporózy a hyperkalcémie), kličková (loop) diuretika (druh léku určeného k léčbě vysokého krevního tlaku nebo edému) nebo jiné léky snižující hladinu vápníku, neboť kombinace těchto přípravků, s bisfosfonáty může způsobit příliš velké snížení hladiny vápníku v krvi.
- thalidomid (léčivý přípravek užívaný k léčbě určitých typů rakoviny krve postihující kost) nebo jiné léčivé přípravky, které mohou poškodit ledviny,
- jakékoli jiné léčivé přípravky, které obsahují kyselinu zoledronovou a používají se k léčbě osteoporózy a jiných onemocnění kostí nenádorového původu nebo jiný bisfosfonát, protože kombinovaný účinek těchto léků užívaných současně s přípravkem Zoledronic Acid Hospira není znám,
- antiangiogenní léčivé přípravky (používané k léčbě rakoviny), neboť jejich kombinace s kyselinou zoledronovou je spojována se zvýšeným rizikem výskytu osteonekrózy čelisti (OČ).
Pacienti ve věku 65 let a starší
Přípravek Zoledronic Acid Hospira k infuzi může být podáván lidem ve věku 65 a více let. Neexistují žádné důkazy, které by svědčily o tom, že jsou nutná jakákoli zvláštní opatření.
Děti a dospívající
Přípravek Zoledronic Acid Hospira se nedoporučuje podávat dospívajícím a dětem mladším 18 let. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, nesmíte používat přípravek Zoledronic Acid Hospira.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Ve velmi vzácných případech byla při použití přípravku Zoledronic Acid Hospira zaznamenána mátožnost a ospalost. Při řízení dopravních prostředků, obsluze strojů nebo vykonávání jiné činnosti, která vyžaduje plnou pozornost, byste proto měl(a) být opatrný/opatrná.
Přípravek Zoledronic Acid Hospira obsahuje sodík
Tento léčivý přípravek obsahuje 16 mmol (neboli 360 mg) sodíku v jedné dávce. To je třeba vzít v úvahu u pacientů na dietě s nízkým příjmem sodíku.
3. Jak se přípravek Zoledronic Acid Hospira používá
- Přípravek Zoledronic Acid Hospira smí podávat pouze zdravotničtí pracovníci zaškolení v intravenózním podávání bisfosfonátů, tj. do žíly.
- Lékař Vám doporučí, abyste před každou léčbou vypil(a) dostatečné množství vody, aby se zabránilo dehydrataci.
- Pečlivě dodržujte všechny další pokyny, které Vám dal Váš lékař, zdravotní sestra nebo lékárník.
Jaké množství přípravku Zoledronic Acid Hospira se podává
- Obvyklá jednorázová dávka je 4 mg.
- Jestliže trpíte onemocněním ledvin, lékař Vám podle závažnosti Vašeho onemocnění podá nižší dávku.
Jak často se přípravek Zoledronic Acid Hospira podává
- Pokud jste léčen(a) za účelem prevence kostních komplikací způsobených metastázami v kostech, bude Vám podávána jedna infuze přípravku Zoledronic Acid Hospira jednou za tři až čtyři týdny.
- Pokud jste léčen(a) za účelem snížení množství vápníku v krvi, bude Vám zpravidla podána pouze jedna infuze přípravku Zoledronic Acid Hospira.
Jak se přípravek Zoledronic Acid Hospira podává
- Přípravek Zoledronic Acid Hospira se podává formou kapačky (infuze) do žíly, která musí trvat nejméně 15 minut a musí být podávána jako samostatný nitrožilní roztok oddělenou infuzní linkou.
Pacientům, jejichž hladina vápníku v krvi není příliš vysoká, budou též předepsány doplňkové denní dávky vápníku a vitamínu D.
Jestliže jste užil(a) více přípravku Zoledronic Acid Hospira, než jste měl(a)
Pokud Vám byly podány vyšší než doporučené dávky, musíte být pečlivě sledován(a) lékařem. Je tomu tak proto, že může dojít k abnormálním změnám v hladině elektrolytů v séru (např. abnormální hladina vápníku, fosforu a hořčíku) a/nebo ke změnám funkce ledvin, včetně těžkého poškození ledvin. Jestliže se Vaše hladina vápníku příliš sníží, může Vám být vápník doplněn infuzí.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Nejčastější nežádoucí účinky jsou obvykle mírné a po krátké době pravděpodobně vymizí.
Pokud se u Vás projeví následující závažné nežádoucí účinky, okamžitě informujte svého lékaře:
Časté (postihují 1 až 10 uživatelů ze 100):
- Těžké poškození ledvin (obvykle je zjistí Váš lékař určitými speciálními krevními testy).
- Nízká hladina vápníku v krvi.
Méně časté (postihují 1 až 10 uživatelů z 1 000):
- Bolest v ústech, bolest zubů a/nebo čelisti, otok nebo nehojící se vředy v ústech nebo na dásních, necitlivost nebo pocit ztěžknutí čelisti nebo viklání zubu. Mohlo by jít o známky kostního poškození čelisti (osteonekróza). Pokud se u Vás projeví tyto příznaky během léčby přípravkem Zoledronic Acid Hospira nebo po ukončení léčby, okamžitě to sdělte svému lékaři a zubnímu lékaři.
- U pacientek, které užívaly kyselinu zoledronovou k léčbě postmenopauzální osteoporózy, byl pozorován nepravidelný srdeční rytmus (fibrilace síní). V současné době není jasné, zda kyselina zoledronová tento nepravidelný srdeční rytmus způsobuje, ale pokud se u Vás tyto příznaky po podání kyseliny zoledronové projeví, informujte svého lékaře.
- Závažné alergické reakce: dušnost, otoky, zejména otok obličeje a krku.
Vzácné (mohou postihnout až 1 z 1000 lidí):
- jako následek nízkých hodnot vápníku: nepravidelný srdeční tep (srdeční arytmie;sekundárně po hypokalcemii).
Velmi vzácné (mohou postihnout až 1 z 10 000 lidí):
- Jako následek nízkých hodnot vápníku: křeče, pocit necitlivosti a tetanie (sekundárně po hypokalcemii).
- Poraďte se se svým lékařem, pokud máte bolest ucha, výtok z ucha a/nebo infekci ucha. Mohlo by se jednat o známky poškození kosti v uchu.
Pokud se u Vás vyskytne kterýkoli z následujících nežádoucích účinků, co nejdříve to sdělte svému lékaři:
Velmi časté (postihují více než 1 uživatele z 10):
- Nízká hladina fosfátů v krvi.
Časté (postihují 1 až 10 uživatelů ze 100):
- Bolest hlavy a chřipkové příznaky, jako je horečka, únava, slabost, mátožnost, zimnice, bolesti kostí, kloubů a/nebo svalů. Ve většině případů není třeba žádná specifická léčba a příznaky po krátké době vymizí (po několika hodinách nebo dnech).
- Zažívací potíže, jako je např. nevolnost, zvracení a ztráta chuti k jídlu.
- Zánět spojivek.
- Nízký počet červených krvinek (anemie).
Méně časté (postihují 1 až 10 uživatelů z 1 000):
- Hypersenzitivní reakce.
- Nízký krevní tlak.
- Bolest na hrudi.
- Kožní reakce (zarudnutí a otok) v místě zavedení infuze, vyrážka, svědění.
- Vysoký krevní tlak, dušnost, závrať, úzkost, poruchy spánku, třes, brnění nebo necitlivost rukou nebo nohou, průjem, zácpa, bolest břicha, sucho v ústech.
- Nízký počet bílých krvinek a krevních destiček.
- Nízká hladina hořčíku a draslíku v krvi. Lékař bude tyto hladiny sledovat a učiní nezbytná opatření.
- Zvýšení tělesné hmotnosti.
- Zvýšení pocení.
- Ospalost.
- Rozmazané vidění, slzení očí, citlivost očí na světlo.
- Náhlý pocit chladu a mdloby, ochablost nebo kolaps.
- Dýchací obtíže se sípáním nebo kašlem.
- Kopřivka.
Vzácné (postihují 1 až 10 uživatelů z 10 000):
- Pomalý rytmus srdce.
- Zmatenost.
- Vzácně může vzniknout neobvyklá zlomenina stehenní kosti zvláště u pacientů dlouhodobě léčených pro osteoporózu. Jestliže se u Vás projeví bolest, slabost nebo máte nepříjemný pocit v oblasti stehna,
kyčle nebo třísla, obraťte se na svého lékaře, neboť se může jednat o časný příznak možné zlomeniny stehenní kosti.
- Intersticiální plicní choroba (zánět tkáně v okolí plicních sklípků v plicích).
- Příznaky podobné chřipce, včetně artritidy a otoky kloubů.
- Bolestivé zarudnutí a/nebo otok očí.
Velmi vzácné (postihují méně než 1 uživatele z 10 000):
- Mdloba způsobená nízkým krevním tlakem.
- Silná bolest kostí, svalů a/nebo kloubů, občas zneschopňující.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zoledronic Acid Hospira uchovávat
Váš lékař, zdravotní sestra nebo lékárník vědí, jak se přípravek Zoledronic Acid Hospira správně uchovává (viz bod 6).
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu za EXP.
6. Obsah balení a další informace Co přípravek Zoledronic Acid Hospira obsahuje
- Léčivou látkou je acidum zoledronicum. Jeden vak o objemu 100 ml obsahuje acidum zoledronicum 4 mg (ve formě acidum zoledronicum monohydricum).
Jeden ml roztoku obsahuje acidum zoledronicum 0,04 mg (ve formě acidum zoledronicum monohydricum).
- Dalšími složkami jsou: mannitol, dihydrát natrium-citrát, chlorid sodný a voda na injekci.
Jak přípravek Zoledronic Acid Hospira vypadá a co obsahuje toto balení
Přípravek Zoledronic Acid Hospira je čirý bezbarvý roztok. Dodává se ve 100ml plastových vacích jako infuzní roztok k přímému použití. Každé balení obsahuje jeden vak, který obsahuje acidum zoledronicum 4 mg. což odpovídá acidum zoledronicum monohydricum 4,264 mg.
Držitel rozhodnutí o registraci
Hospira UK Limited
Horizon
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Velká Británie
Výrobce
Hospira Enterprises B.V.
Randstad 22-11 1316 BN Almere Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
AT
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1521 15-0
BG/CZ/EE/EL/HU/LT/LV/ MT / PL / RO / SI / SK / UK
Hospira UK Limited Tel: + 44 (0) 1628 515500
DE
Pfizer Pharma PFE GmbH Tel: + 49 (0)800 8535555
ES
Pfizer GEP, S.L.
Tel: +34 91 490 99 00
HR
Alvogen d.o.o.
Tél/Tel: + 385 1 6641 830
BE/LU/NL Pfizer S.A. / N.V.
Tél/Tel: +32 2 554 62 11
CY
Name: N.Karoullas Pharmaceutical Trading Co Ltd 33, Artemidos avenue, 6025 Larnaca Tel: 24656165/ Mob.: 99403969 Email: n.karoullas@ptc-ltd.com
DK / FI / IS / NO / SE Hospira Nordic AB Tel: + 46 (0) 8 672 85 00
FR
Hospira France
Tél: + 33 (0) 1 40 83 82 00
IE
Hospira Ireland Sales Limited Tel: 1800 633 363 (toll free)
+44 (0) 1304 616161
PT
Hospira Portugal Lda (+351) 21 423 55 00
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Jak připravit a podávat přípravek Zoledronic Acid Hospira
- Přípravek Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok obsahuje 4 mg kyseliny zoledronové ve 100 ml infuzního roztoku k okamžitému použití u pacientů s normální funkcí ledvin.
- Určeno pouze k jednorázovému použití. Veškerý nepoužitý roztok zlikvidujte. Smí se použít pouze čirý roztok bez obsahu částic a zabarvení. Během přípravy infuze musí být dodržen aseptický postup.
- Z mikrobiologického hlediska má být přípravek použit okamžitě. Pokud není použit okamžitě, jsou doba a podmínky uchovávání před použitím v zodpovědnosti uživatele a obvykle by doba uchovávání neměla být delší než 24 hodin při teplotě 2 °C - 8 °C. Chlazený roztok musí být před podáním vytemperován na pokojovou teplotu.
- Roztok obsahující kyselinu zoledronovou se nesmí dále ředit ani mísit s jinými infuzními roztoky. Podává se jako jednorázová 15minutová nitrožilní infuze v samostatné infuzní lince. Před podáním kyseliny zoledronové a poté musí být posouzena hydratace pacienta, aby bylo zajištěno jeho odpovídající zavodnění.
- Přípravek Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok lze použít okamžitě bez další přípravy u pacientů s normální funkcí ledvin. U pacientů s mírnou až středně závažnou poruchou funkce ledvin je nutné připravit snížené dávky podle níže uvedených pokynů.
Při přípravě snížených dávek pro pacienty s výchozí CLcr < 60 ml/min se řiďte níže uvedenou tabulkou 1. Před podáním odstraňte z infuzního vaku uvedený objem roztoku přípravku Zoledronic Acid Hospira.
Tabulka 1: Příprava snížených dávek přípravku Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok
|
Výchozí clearance kreatininu (ml/min) |
Odstraňte tento objem přípravku Zoledronic Acid Hospira 4 mg/100 ml infuzní roztok (ml) |
Upravená dávka (mg kyseliny zoledronové) * |
|
50-60 |
12,0 |
3,5 |
|
40-49 |
18,0 |
3,3 |
|
30-39 |
25,0 |
3,0 |
*Dávky byly vypočítány za předpokladu cílové hodnoty AUC 0,66 (mg^h/l) (CLcr = 75 ml/min). Předpokládá se, že snížené dávky u pacientů s poruchou funkce ledvin dosáhnou stejné hodnoty AUC, jakou lze pozorovat u pacientů s clearancí kreatininu 75 ml/min.
- Studie s několika druhy infuzních linek vyrobených z polyvinylchloridu, polyetylenu a polypropylenu neprokázaly inkompatibilitu s kyselinou zoledronovou.
- Vzhledem k tomu, že nejsou k dispozici žádné údaje o kompatibilitě kyseliny zoledronové s dalšími nitrožilně podávanými látkami, nesmí se přípravek Zoledronic Acid Hospira mísit s jinými léčivými přípravky/látkami a vždy je nutné jej podávat samostatnou infuzní linkou.
Jak přípravek Zoledronic Acid Hospira uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu EXP.
- Vak nevyžaduje žádné zvláštní podmínky uchovávání.
Zoledronic Acid Hospira 5 mg/100 ml infuzní roztok
Acidum zoledronicum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Zoledronic Acid Hospira a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zoledronic Acid Hospira používat
3. Jak se přípravek Zoledronic Acid Hospira používá
4. Možné nežádoucí účinky
5. Jak přípravek Zoledronic Acid Hospira uchovávat
6. Obsah balení a další informace
1. Co je Zoledronic Acid Hospira a k čemu se používá
Přípravek Zoledronic Acid Hospira obsahuje léčivou látku kyselinu zoledronovou. Patří do skupiny léčivých přípravků zvaných bisfosfonáty a používá se k léčbě Pagetovy choroby kostí u dospělých.
Je normální, že se stará kostní tkáň odstraňuje a nahrazuje novou. Tento proces se nazývá remodelace. U Pagetovy choroby je remodelace kostní tkáně příliš rychlá a nová kost se vytváří neuspořádaně, což způsobuje, že je slabší než normální kost. Pokud se tato choroba neléčí, mohou se kosti deformovat a bolet a mohou se zlomit. Přípravek Zoledronic Acid Hospira účinkuje tak, že vrací proces remodelace k normálu, čímž zajišťuje tvorbu normální kostní tkáně a obnovuje tak sílu kosti.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zoledronic Acid Hospira používat
Před tím, než Vám bude přípravek Zoledronic Acid Hospira podán, se pečlivě řiďte všemi pokyny Vašeho lékaře, lékárníka nebo zdravotní sestry.
Nepoužívejte přípravek Zoledronic Acid Hospira:
- jestliže jste alergický(á) na kyselinu zoledronovou, jiné bisfosfonáty nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6),
- jestliže trpíte hypokalcemií (to znamená, že máte příliš nízkou hladinu vápníku v krvi),
- jestliže trpíte závažnými potížemi s ledvinami,
- jestliže jste těhotná,
- jestliže kojíte.
Upozornění a opatření
Před použitím přípravku Zoledronic Acid Hospira se poraďte se svým lékařem:
- jestliže jste léčen(a) jakýmkoli jiným bisfosfonátovým lékem, neboť kombinované účinky těchto léků užívaných společně s přípravkem Zoledronic Acid Hospira nejsou známy. Patří sem např. přípravek Zometa nebo Aclasta (léčivé přípravky, které také obsahují kyselinu zoledronovou a používají se k léčbě stejného onemocnění nebo k léčbě rakoviny kostí),
- jestliže máte problémy s ledvinami, nebo jste nějaké měl(a) v minulosti,
- jestliže nejste schopen (schopna) užívat denně doplňky vápníku,
- jestliže jste podstoupil(a) částečné nebo úplné chirurgické odstranění příštítných tělísek ve Vašem krku,
- jestliže Vám byly chirurgicky odstraněny části střeva.
Nežádoucí účinek zvaný osteonekróza čelisti (OČ) (kostní poškození čelisti) byl hlášen v postmarketingovém období u pacientů užívajících kyselinu zoledronovou k léčbě osteoporózy. OČ se může objevit také po ukončení léčby.
Je důležité pokusit se zabránit vzniku OČ, protože jde o bolestivý stav, který se může obtížně léčit. Abyste snížil(a) riziko vzniku osteonekrózy čelisti, můžete provést některá opatření.
Před zahájením léčby přípravkem Zoledronic Acid Hospira informujte svého lékaře, lékárníka nebo zdravotní sestru pokud
- máte jakékoli potíže v dutině ústní nebo se zuby, například špatné zdraví chrupu, onemocnění dásní nebo plánované extrakce zubů;
- pokud si nenecháváte pravidelně ošetřovat chrup nebo Vám nebylo po dlouhou dobu provedeno vyšetření chrupu;
- pokud kouříte (protože může dojít ke zvýšení rizika dentálních potíží);
- pokud jste byli dříve léčeni bisfosfonáty (užívané k léčbě nebo prevenci kostních potíží);
- pokud užíváte léky nazývané kortikosteroidy (jako je prednizolon nebo dexamethason);
- pokud máte rakovinu.
Před zahájením léčby přípravkem Zoledronic Acid Hospira Vás může lékař požádat, abyste prodělal(a) vyšetření zubů.
Během léčby přípravkem Zoledronic Acid Hospira byste měl(a) udržovat kvalitní ústní hygienu (včetně pravidelného čištění zubů) a podstoupit pravidelné zubní vyšetření. Pokud máte zubní protézu, měl(a) byste se ujistit, že je nasazená správně. Před zubním ošetřením nebo zubním chirurgickým zákrokem (např. extrakce zubů) informujte Vašeho lékaře o zubním ošetření a informujte svého dentistu, že j ste léčen přípravkem Zoledronic Acid Hospira. Kontaktujte svého lékaře a dentistu, pokud se u Vás projeví onemocnění úst nebo zubní potíže, jako je padání zubů, bolesti nebo otoky, nebo nehojící se vřídky nebo výtok, protože může jít o příznaky osteonekrózy čelistí.
Sledování
Váš lékař by Vám měl před každou dávkou přípravku Zoledronic Acid Hospira provést krevní testy, aby zkontroloval funkci ledvin (hladina kreatininu). Je pro Vás důležité, abyste vypil(a) nejméně 2 sklenice tekutiny (např. vody) během několika hodin před podáním přípravku Zoledronic Acid Hospira, jak Vám poradil Váš lékař.
Děti a dospívající
Podávání přípravku Zoledronic Acid Hospira jedincům mladším 18 let se nedoporučuje. Použití přípravku Zoledronic Acid Hospira u dětí a dospívajících nebylo studováno.
Další léčivé přípravky a přípravek Zoledronic Acid Hospira
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Je obzvlášť důležité, aby Váš lékař věděl o všech lécích, které užíváte, zejména pokud užíváte léky, o nichž je známo, že mohou poškozovat ledviny (např. aminoglykosidy), nebo diuretika („pilulky na odvodnění“), která mohou způsobit dehydrataci.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, nesmíte používat přípravek Zoledronic Acid Hospira.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Přípravek Zoledronic Acid Hospira nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Pokud při užívání přípravku Zoledronic Acid Hospira pociťujete závratě, neřiďte dopravní prostředky ani neobsluhujte stroje, dokud se nebudete cítit lépe.
Přípravek Zoledronic Acid Hospira obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. je prakticky „bez sodíku“.
3. Jak se přípravek Zoledronic Acid Hospira používá
Pečlivě dodržujte všechny pokyny, které Vám dal Váš lékař nebo zdravotní sestra. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo zdravotní sestrou.
Obvyklá dávka je 5 mg podaných jako jedna jednorázová infuze do žíly Vaším lékařem nebo zdravotní sestrou. Infuze bude trvat nejméně 15 minut. Přípravek Zoledronic Acid Hospira může účinkovat déle než jeden rok a Váš lékař Vám sdělí, zda je třeba, abyste byl(a) znovu léčen(a).
Váš lékař Vám může doporučit, abyste užíval(a) doplňky vápníku a vitamínu D (např. tablety) alespoň během prvních deseti dnů po podání přípravku Zoledronic Acid Hospira. Je důležité, abyste se tímto doporučením pečlivě řídil(a), aby u Vás nedošlo v období po podání infuze k přílišnému poklesu hladiny vápníku v krvi. Váš lékař Vás bude informovat o příznacích spojených s hypokalcemií.
Přípravek Zoledronic Acid Hospira s jídlem a pitím
Před zahájením a po léčbě přípravkem Zoledronic Acid Hospira musíte vypít dostatečné množství tekutin (nejméně jednu až dvě sklenice) tak, jak Vám lékař doporučil. Tím pomůžete předejít dehydrataci (nedostatku tekutin). V den, kdy Vám bude podán přípravek Zoledronic Acid Hospira, můžete normálně jíst. To je důležité především u pacientů, kteří užívají diuretika („pilulky na odvodnění“), a u starších pacientů.
Jestliže jste vynechal(a) dávku přípravku Zoledronic Acid Hospira
Co nejdříve se obraťte na svého lékaře nebo nemocnici, abyste se přeobjednal(a).
Před ukončením léčby přípravkem Zoledronic Acid Hospira
Pokud uvažujete o ukončení léčby přípravkem Zoledronic Acid Hospira, dostavte se prosím ke své další návštěvě u lékaře a poraďte se o tom se svým lékařem. Váš lékař Vám poradí a rozhodne, jak dlouho byste se měl(a) léčit přípravkem Zoledronic Acid Hospira.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky související s podáním první infuze jsou velmi časté (vyskytují se u více než 30 % pacientů), avšak po podání následujících infuzí jsou již méně časté. Většina nežádoucích účinků, jako je horečka a zimnice, bolest svalů nebo kloubů a bolest hlavy, se objevuje během prvních tří dnů po podání dávky přípravku Zoledronic Acid Hospira. Tyto příznaky jsou obvykle mírné až středně závažné a do tří dnů vymizí. Váš lékař Vám může doporučit mírný přípravek na úlevu od bolesti, jako je ibuprofen nebo paracetamol, aby se tyto nežádoucí účinky omezily. Pravděpodobnost výskytu těchto nežádoucích účinků se při dalších dávkách přípravku Zoledronic Acid Hospira snižuje.
Časté (mohou postihnout až 1 osobu z 10)
U pacientek s postmenopauzální osteoporózou, které jsou léčeny kyselinou zoledronovou, byl zaznamenán nepravidelný srdeční rytmus (fibrilace síní). Doposud není jasné, zda tyto nepravidelnosti srdečního rytmu způsobuje kyselina zoledronová, avšak jestliže tyto příznaky po podání přípravku Zoledronic Acid Hospira zaznamenáte, je třeba to oznámit Vašemu lékaři.
Mohou se objevit otoky a/nebo bolest v místě podání infuze.
Méně časté (mohou postihnout až 1 osobu ze 100)
Kožní reakce, jako je zarudnutí.
Otoky, zarudnutí, bolest a svědění očí nebo citlivost očí na světlo.
Velmi vzácné (mohou postihnout až 1 osobu z 10 000)
Poraďte se se svým lékařem, pokud máte bolest ucha, výtok z ucha a/nebo infekci ucha. Mohlo by se jednat o známky poškození kosti v uchu.
Není známo (z dostupných údajů nelze určit)
Bolest v ústech a/nebo čelisti, otoky nebo nehojící se vřídky v ústech nebo na dásních, snížená citlivost nebo pocit těžké čelisti nebo uvolnění zubů, může jít o příznaky poškození kosti v čelisti (osteonekróza). Pokud se u Vás během léčby nebo po ukončení léčby přípravkem Zoledronic Acid Hospira objeví tyto příznaky, okamžitě kontaktujte svého lékaře a zubního lékaře.
Může dojít k poruchám funkce ledvin (např. snížený výdej moči). Váš lékař by Vám měl před každou dávkou přípravku Zoledronic Acid Hospira provést krevní test, aby zkontroloval funkci ledvin. Je pro Vás důležité, abyste vypil(a) nejméně 2 sklenice tekutiny (např. vody) během několika hodin před podáním přípravku Zoledronic Acid Hospira, jak Vám poradil Váš lékař.
Pokud se objeví některý z výše uvedených nežádoucích účinků, okamžitě kontaktujte svého lékaře.
Přípravek Zoledronic Acid Hospira může také způsobit další nežádoucí účinky
Velmi časté ( mohou postihnout více než 1 osobu z 10)
Časté (mohou postihnout až 1 osobu z 10)
Bolest hlavy, závrať, nevolnost, zvracení, průjem, bolest svalů, bolest kostí a/nebo kloubů, bolest zad, paží nebo nohou, chřipkovité příznaky (např. únava, zimnice, bolest kloubů a svalů), zimnice, pocit únavy a ztráty zájmu, slabost, bolest, pocit, že se necítíte dobře.
U pacientů s Pagetovou chorobou byly hlášeny příznaky způsobené nízkou hladinou vápníku v krvi, jako jsou svalové křeče nebo necitlivost či brnění zejména v oblasti okolo úst.
Méně časté (mohou postihnout až 1 osobu ze 100)
Chřipka, infekce horních cest dýchacích, snížený počet červených krvinek, nechutenství, nespavost, ospalost, která může zahrnovat sníženou bdělost a pozornost, brnění nebo necitlivost, extrémní únava, třes, dočasná ztráta vědomí, infekce nebo podráždění oka s bolestí a zčervenáním, točení hlavy, zvýšený krevní tlak, návaly, kašel, dušnost, žaludeční nevolnost, bolest břicha, zácpa, sucho v ústech, pálení žáhy, kožní vyrážka, nadměrné pocení, svědění, zčervenání kůže, bolest krku, ztuhlost svalů, kostí a/nebo kloubů, otok kloubů, svalové křeče, bolest ramene, bolest svalů hrudníku a hrudního koše, zánět kloubů, svalová slabost, abnormální výsledky renálních testů, abnormálně časté močení, otok rukou, kotníků nebo chodidel, žízeň, bolest zubů, poruchy vnímání chuti.
Vzácné (mohou postihnout až 1 osobu z 1 000)
Vzácně se může objevit neobvyklá zlomenina stehenní kosti, zvláště u pacientů dlouhodobě léčených pro osteoporózu (řídnutí kostí).
Pokud se u Vás objeví bolest, slabost nebo nepříjemné pocity v oblasti stehna, kyčle nebo třísla, obraťte se na svého lékaře, protože to mohou být časné příznaky možné zlomeniny stehenní kosti.
Není známo (z dostupných údajů nelze určit)
Závažné alergické reakce včetně závratě a dechových potíží, otok zejména obličeje a hrdla, snížený krevní tlak, dehydratace v důsledku příznaků po podání dávky, jako je horečka, zvracení a průjem.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zoledronic Acid Hospira uchovávat
Váš lékař, lékárník nebo zdravotní sestra vědí, jak se přípravek Zoledronic Acid Hospira správně uchovává.
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu za EXP.
- Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6. Obsah balení a další informace
Co přípravek Zoledronic Acid Hospira 5 mg/100 ml infuzní roztok obsahuje
- Léčivou látkou je acidum zoledronicum. Jeden vak s roztokem o objemu 100 ml obsahuje acidum zoledronicum anhydricum 5 mg (ve formě acidum zoledronicum monohydricum).
Jeden ml roztoku obsahuje acidum zoledronicum 0,05 mg (ve formě acidum zoledronicum monohydricum).
- Dalšími složkami jsou: mannitol, dihydrát natrium-citrát a voda na injekci.
Jak přípravek Zoledronic Acid Hospira vypadá a co obsahuje toto balení
Přípravek Zoledronic Acid Hospira je čirý bezbarvý roztok. Dodává se ve 100ml plastových vacích jako infuzní roztok k přímému použití. Každé balení obsahuje jeden vak.
Držitel rozhodnutí o registraci
Hospira UK Limited
Horizon
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Velká Británie
Výrobce
Hospira Enterprises B.V.
Randstad 22-11 1316 BN Almere Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
AT
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1521 15-0
BG / CZ / EE / EL / HU / LT / LV / MT / PL / RO / SI / SK / UK
Hospira UK Limited Tel: + 44 (0) 1628 515500
DE
Pfizer Pharma PFE GmbH Tel: + 49 (0)800 8535555
ES
Pfizer GEP, S.L.
Tel: +34 91 490 99 00
HR
Alvogen d.o.o.
Tél/Tel: + 385 1 6641 830
BE/LU/NL Pfizer S.A. / N.V.
Tél/Tel: +32 2 554 62 11
CY
Name: N.Karoullas Pharmaceutical Trading Co Ltd 33, Artemidos avenue, 6025 Larnaca Tel: 24656165/ Mob.: 99403969 Email: n.karoullas@ptc-ltd.com
DK / FI / IS / NO / SE Hospira Nordic AB Tel: + 46 (0) 8 672 85 00
FR
Hospira France
Tél: + 33 (0) 1 40 83 82 00
IE
Hospira Ireland Sales Limited Tel: 1800 633 363 (toll free)
+44 (0) 1304 616161
PT
Hospira Portugal Lda (+351) 21 423 55 00
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Jak připravit a podávat přípravek Zoledronic Acid Hospira
- Přípravek Zoledronic Acid Hospira je připraven k okamžitému použití.
Určeno pouze k jednorázovému použití. Veškerý nepoužitý roztok zlikvidujte. Smí se použít pouze čirý roztok bez obsahu částic a zabarvení. Přípravek Zoledronic Acid Hospira se nesmí mísit nebo podávat intravenózně s jakýmkoli jiným léčivým přípravkem a musí se podávat oddělenou infuzní linkou se zavzdušněním stálou rychlostí infuze. Doba infuze musí být nejméně 15 minut. Přípravek Zoledronic Acid Hospira nesmí přijít do styku s roztoky obsahujícími vápník. Pokud je přípravek uchováván v chladničce, nechte chlazený roztok před podáním vytemperovat na pokojovou teplotu. Při přípravě infuze musí být dodržena aseptická technika. Infuze se musí provádět v souladu se standardním lékařským postupem.
Jak přípravek Zoledronic Acid Hospira uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu za EXP.
- Neotevřený vak nevyžaduje žádné zvláštní podmínky uchovávání.
- Po otevření vaku má být přípravek okamžitě použit, aby se zabránilo mikrobiální kontaminaci. Pokud není použit okamžitě, zodpovídá za délku a podmínky jeho uchovávání po otevření před použitím uživatel; tato doba by neměla obvykle překročit 24 hodin při teplotě 2 °C - 8 °C. Pokud je přípravek uchováván v chladničce, nechte chlazený roztok před podáním vytemperovat na pokojovou teplotu.
Zahrnuje všechny kostní příhody, celkový počet i dobu do každé příhody během studie.
NR Nedosaženo