Zinbryta 150 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Zinbryta 150 mg injekční roztok v předplněné injekční stříkačce. Zinbryta 150 mg injekční roztok v předplněném peru.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje daclizumabum 150 mg v 1 ml injekčního roztoku.
Jedno předplněné pero, jehož součástí je předplněná injekční stříkačka, obsahuje daclizumabum 150 mg v 1 ml injekčního roztoku.
Daklizumab je produkován savčí buněčnou linií (NS0) pomocí technologie rekombinantní DNA.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Bezbarvá až nažloutlá, čirá až mírně opalescentní tekutina s pH 6.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Zinbryta je indikován k léčbě dospělých pacientů s relabujícími formami roztroušené sklerózy (RRS) (viz bod 5.1).
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře se zkušenostmi s léčbou roztroušené sklerózy.
Dávkování
Doporučená dávka přípravku Zinbryta je 150 mg podaných subkutánní injekcí jednou měsíčně.
Pokud dojde k vynechání dávky a od data, kdy měla být dávka podána, ještě neuplynuly 2 týdny, pacienti mají být poučeni, aby si neprodleně vynechanou dávku aplikovali a potom pokračovali v původním měsíčním dávkovacím režimu.
Pokud dojde k vynechání dávky a od data, kdy měla být dávka podána, uplynuly více než 2 týdny, pacienti mají vynechanou dávku přeskočit a s aplikací injekce vyčkat až do data podání příští dávky a potom pokračovat v původním měsíčním dávkovacím režimu.
Za vynechanou dávku se má najednou podat pouze jedna dávka přípravku.
Starší populace
V klinických studiích s daklizumabem byla omezená expozice u pacientů starších 55 let. Nebylo zjištěno, zda tito pacienti reagují odlišně v porovnání s mladšími pacienty.
Porucha funkce ledvin
Daklizumab nebyl u pacientů s poruchou funkce ledvin studován. Jelikož exkrece ledvinami není hlavním způsobem eliminace, nepovažuje se za potřebné upravovat dávkování (viz bod 5.2).
Porucha funkce jater
Daklizumab nebyl u pacientů s poruchou funkce jater studován. Jelikož přípravek Zinbryta není metabolizován v játrech, nepovažuje se za potřebné upravovat dávkování u pacientů s poruchou funkce jater (viz bod 5.2). Léčba se nedoporučuje zahajovat u pacientů, u nichž byly již dříve zjištěny elevace alaninaminostransferázy (ALT) nebo aspartátaminotransferázy (AST) na více než dvojnásobek (> 2 krát) horního limitu normálu (ULN) (viz bod 4.4). U pacientů s lehkou či středně těžkou poruchou funkce jater není potřebné dávkování upravovat (viz bod 4.4). Přípravek Zinbryta není vhodný k použití u pacientů s těžkou poruchou funkce jater (třída C klasifikace Child-Pugh) (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost přípravku Zinbryta u dětí a dospívajících mladších 18 let nebyly stanoveny.
K dispozici nejsou žádné údaje.
Způsob podání
Přípravek Zinbryta je určen k subkutánnímu podání.
Doporučuje se, aby byli pacienti zacvičeni ve správné technice samostatného podání podkožní injekce za použití předplněné stříkačky/předplněného pera. Obvyklá místa pro podání podkožní injekce jsou stehno, břicho a zadní strana paže.
Předplněné injekční stříkačky a předplněná pera přípravku Zinbryta jsou dodávány s nasazenou jehlou. Předplněné injekční stříkačky/předplněná pera obsahují pouze jednu dávku a po použití se musí zlikvidovat.
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním Po vyjmutí z chladničky se má přípravek Zinbryta nechat před aplikací zahřát na pokojovou teplotu (20 °C - 30 °C) (přibližně 30 minut). K zahřátí přípravku Zinbryta se nesmí používat externí zdroje tepla, jako je horká voda.
Tento léčivý přípravek se nesmí použít, jestliže:
• je injekční stříkačka/pero prasklá(é) nebo rozbitá(é),
• je roztok zakalený nebo j sou-li v něm vidět plovoucí částice,
• roztok není bezbarvý až nažloutlý,
• pero spadlo na zem nebo je viditelně poškozené.
4.3 Kontraindikace
Přípravek Zinbryta je kontraindikován u pacientů s anamnézou těžké hypersenzitivity (např. anafylaxe nebo anafylaktoidní reakce) na daklizumab nebo na kteroukoli pomocnou látku (viz bod 6.1).
Porucha funkce jater
U pacientů léčených přípravkem Zinbryta bylo zaznamenáno zvýšení hodnot transamináz v séru a těžká porucha funkce jater (viz bod 4.8.).
Před zahájením léčby přípravkem Zinbryta je třeba u pacientů stanovit hodnoty transamináz v séru (ALT a AST) a hladinu bilirubinu. Během léčby a až 4 měsíce po podání poslední dávky přípravku Zinbryta je třeba u pacientů měsíčně sledovat hodnoty transamináz v séru.
Do klinických studií nebyli zařazeni pacienti, u nichž byly před léčbou zjištěny hodnoty ALT nebo AST >
2 krát horní limit normálu (ULN); zahájení léčby se nedoporučuje u pacientů, u nichž byly zjištěny hodnoty ALT nebo AST > 2 krát ULN. U pacientů s již existující lehkou nebo středně těžkou poruchou funkce jater je třeba během podávání přípravku Zinbryta sledovat projevy a příznaky poruchy funkce jater. Léčba přípravkem Zinbryta není vhodná u pacientů s již existující těžkou poruchou funkce jater (třída C klasifikace Child-Pugh). V případě, že se přípravek Zinbryta používá současně s jinými hepatotoxickými léčivými přípravky, je třeba u pacientů sledovat projevy a příznaky poruchy funkce jater.
Souhrn opatření na základě výsledků jaterních testů během léčby přípravkem Zinbryta je uveden v tabulce 1 níže.
Tabulka 1: Souhrn požadovaných opatření podle výsledků jaterních testů
|
Výsledek testu |
Souhrn požadovaných opatření |
|
Potvrzené ALT nebo AST |
Ukončení léčby.* |
|
> 5 krát ULN | |
|
nebo | |
|
Potvrzené ALT nebo AST > 3 krát ULN | |
|
a bilirubin > 2 krát ULN | |
|
ALT nebo AST |
Přerušení léčby a důkladné monitorování stavu |
|
> 3 krát ULN |
pacienta. |
|
Obnovte léčbu, pokud ALT nebo AST dosáhne hodnoty < 2 krát ULN. |
* Opětovné zahájení léčby je možné zvážit v případě, že byla zjištěna jiná příčina zvýšených hodnot jaterních testů, jestliže se hodnoty vrátily do normálu a přínos obnovení léčby pro pacienta převažuj e její rizika.
Pokud se u pacienta vyskytnou klinické projevy nebo příznaky naznačující poruchu funkce jater (např. nauzea neznámého původu, zvracení, bolest břicha, únava, anorexie nebo žloutenka a/nebo tmavá moč), doporučuje se ihned stanovit hodnoty transamináz v séru a podle potřeby přerušit nebo ukončit léčbu přípravkem Zinbryta.
U pacientů s dlouhodobě zvýšenými hodnotami transamináz v séru je vhodné zhodnotit i jiné možné příčiny (např. infekce) a může být potřebné doporučit pacienta na vyšetření ke specialistovi. V rámci klinických studií byla pozorována autoimunitní hepatitida (bez přítomnosti autoprotilátek). Vhodným řešením může být léčba systémovými kortikosteroidy.
Další podrobnosti týkající se Informací pro lékaře a Karty pacienta, které se doporučují používat s tímto přípravkem, jsou uvedeny v části „Odborné poradenství".
Odborné _ poradenství
Všichni lékaři, kteří zamýšlejí přípravek Zinbryta předepisovat, se musí obeznámit s Informacemi pro lékaře pro tento léčivý přípravek.
Lékař musí prodiskutovat s pacienty riziko poškození jater a předat jim Kartu pacienta. Karta informuje pacienty o riziku těžkého poškození jater a možných příznacích, aby uměli rozpoznat situace, ve kterých mají včas kontaktovat zdravotnického pracovníka. Karta pacienta rovněž informuje o nutnosti sledovat funkce jater a poučuje pacienta o důležitosti absolvovat krevní testy každý měsíc.
Kožní reakce
U pacientů léčených přípravkem Zinbryta byly pozorovány kožní reakce, v některých případech závažné (např. exfoliativní vyrážka či dermatitida, toxoalergický exantém). Kožní reakce obecně vymizely při standardní péči, která zahrnovala léčbu lokálními či systémovými steroidy. Pokud se u pacienta objeví difúzní nebo vysoce zánětlivá vyrážka, může být potřebné doporučit pacienta na vyšetření k dermatologovi a ukončit léčbu přípravkem Zinbryta (viz bod 4.8).
Přípravek Zinbryta se má podávat s opatrností pacientům, kteří v minulosti trpěli nebo kteří trpí depresivními poruchami. Pacienti léčení přípravkem Zinbryta mají být poučeni, aby v případě výskytu jakýchkoliv příznaků nové či zhoršující se deprese a/nebo v případě sebevražedných myšlenek o tom informovali svého ošetřujícího lékaře. Pokud se u pacienta rozvine těžká deprese a/nebo sebevražedné myšlenky, je třeba zvážit ukončení léčby přípravkem Zinbryta (viz bod 4.8)
Infekce
U pacientů léčených přípravkem Zinbryta byly hlášeny infekce, v některých případech závažné (např. pneumonie a bronchitida). V případě závažné infekce může být nevyhnutelné přerušit léčbu přípravkem Zinbryta do doby, než infekce odezní.
U pacientů léčených přípravkem Zinbryta byly hlášeny infekce tuberkulózy. U pacientů, kteří měli tuberkulózu nebo žijí v lokalitách s endemickým výskytem tuberkulózy, se má před zahájením léčby provést screening aktivní tuberkulózy a pacienti mají být během léčby monitorováni.
U pacientů s těžkou aktivní infekcí se má zvážit odklad zahájení léčby přípravkem Zinbryta (viz bod 4.8).
Zinbryta nebyla studována u pacientů s příznaky imunodeficience.
Gastrointestinální poruchy
U pacientů léčených přípravkem Zinbryta byla hlášena kolitida. Po ukončení léčby přípravkem Zinbryta a při standardní léčbě kolitida ustoupila. U pacientů s příznaky kolitidy (např. bolest břicha, horečka, přetrvávající průjem) je vhodné doporučit pacienta na vyšetření k specialistovi (viz bod 4.8).
Lymfopenie
Během klinických studií byla u pacientů léčených přípravkem Zinbryta zjištěna lymfopenie, která byla většinou lehká až středně těžká (>500/mm3). Přetrvávající těžká lymfopenie (< 500/mm3) nebyla během klinických studií s přípravkem Zinbryta pozorována. Jako preventivní opatření se však doporučuje sledovat celkový krevní obraz každé 3 měsíce.
Riziko vzniku progresivní multifokální leukoencefalopatie (PML) související s léčbou přípravkem Zinbryta nebylo stanoveno.
Úvahy týkající se pomocných látek
Tento přípravek obsahuje 0,14 mmol sodíku v jedné dávce. To znamená, že v podstatě je “bez sodíku” a přípravek mohou používat pacienti na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nepředpokládá se, že přípravek Zinbryta je metabolizován jatemími enzymy, ani že je vylučován ledvinami. Nepředpokládají se lékové interakce mezi přípravkem Zinbryta a symptomatickou léčbou RS (např. myorelaxancia, fampridin); k dispozici však nejsou dostatečné údaje o souběžném podávaní přípravku Zinbryta a léků k symptomatické léčbě RS.
Imunizace
Bezpečnost imunizace vakcínami, které obsahují živé viry, nebyla během léčby přípravkem Zinbryta studována. Očkování živými vakcínami se během léčby a ještě 4 měsíce po ukončení léčby přípravkem Zinbryta nedoporučuje.
V rámci klinické studie se u pacientů (n=90), kteří byli dlouhodobě léčeni přípravkem Zinbryta, vytvořila odpovídající imunitní odpověď na inaktivovanou trivalentní vakcínu proti sezónní chřipce. Míra imunitní odpovědi na vakcínu proti sezónní chřipce a podíl pacientů s prokázanou sérokonverzí a séroprotekcí byly srovnatelné s výsledky v populaci zdravých dobrovolníků. Pacienti léčení přípravkem Zinbryta mohou být očkováni neživými vakcínami.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání přípravku Zinbryta těhotným ženám jsou omezené. Studie provedené na zvířatech neprokázaly přímé či nepřímé škodlivé účinky z hlediska reprodukční toxicity (viz bod 5.3).
Přípravek Zinbryta se má používat v těhotenství, pouze pokud přínos léčby převyšuje možné riziko plod.
Kojení
Dostupné toxikologické údaje u opic druhu Cynomolgus prokázaly exkreci daklizumabu do mateřského mléka (podrobnosti viz bod 5.3). Není známo, zda se přípravek Zinbryta vylučuje do lidského mateřského mléka. Přestože se lidský IgG vylučuje do lidského mateřského mléka, publikované údaje naznačují, že protilátky obsažené v mateřském mléce nepřecházejí ve větším množství do krevního oběhu novorozenců a kojenců. Riziko pro novorozence/kojence však nelze vyloučit.
Pokud žena chce během léčby přípravkem Zinbryta kojit, je třeba pečlivě zvážit přínos kojení pro dítě a přínos léčby pro pacientku.
Fertilita
Ve studiích na zvířatech nebyl na základě vyhodnocení indexů fertility zaznamenán žádný vliv na fertilitu samců a ani samic (viz bod 5.3). Nejsou k dispozici žádné údaje o účincích přípravku Zinbryta na fertilitu u člověka.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Zinbryta nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
V placebem kontrolované studii (studie SELECT) byl přípravek Zinbryta podáván 417 pacientům (150 mg, n=208; 300 mg, n=209; každé 4 týdny) po dobu 1 roku. V aktivně kontrolované studii (studie DECIDE) byl 919 pacientům podáván přípravek Zinbryta (150 mg každé čtyři týdny) a 922 pacientům podáván intramuskulárně interferon beta-1a (30 mikrogramů týdně) po dobu nejméně dvou a nejdéle tří let.
Nejčastějšími nežádoucími účinky u pacientů léčených přípravkem Zinbryta, které vedly k ukončení léčby, byly jaterní reakce, včetně elevace transamináz v séru (5%), a kožní reakce (4%) (viz bod 4.4).
Nejčastěji hlášenými nežádoucími účinky při léčbě přípravkem Zinbryta byly: kožní vyrážka, zvýšení hladin alaninaminotransferázy (ALT), deprese, nasofaryngitida, infekce horních cest dýchacích, chřipka, orofaryngeální bolest a lymfadenopatie.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky jsou uváděny ve formě MedDRA preferovaných termínů v třídách orgánových systémů MedDRA podle četnosti a výskytu. V každé skupině četnosti jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti. Frekvence nežádoucích účinků jsou vyjádřeny podle následujících kategorií:
• Velmi časté (>1/10)
• Časté (>1/100 až <1/10)
• Méně časté (>1/1 000 až <1/100)
Tabulka 2: Nežádoucí účinky hlášené pro přípravek Zinbryta 150 mg
|
Třídy orgánových systémů |
Nežádoucí účinek |
Četnost |
|
Infekce a infestace |
Infekce horních cest dýchacích f |
Velmi časté |
|
Nasofaryngitida f |
Velmi časté | |
|
Časté | ||
|
Infekce dýchacích cest |
Časté | |
|
Bronchitida |
Časté | |
|
Virová infekce |
Časté | |
|
Chřipka f |
Časté | |
|
Časté | ||
|
Tonzilitida f |
Časté | |
|
Faryngitida |
Časté | |
|
Folikulitida |
Časté | |
|
Rinitida * |
Časté | |
|
Poruchy krve a lymfatického systému |
Lymfadenopatie f |
Časté |
|
Lymfadenitida |
Časté | |
|
Anémie * |
Časté | |
|
Psychické poruchy |
Deprese * |
Časté |
|
Respirační, hrudní a mediastinální poruchy |
Orofaryngeální bolest f |
Časté |
|
Gastrointestinální poruchy |
Časté | |
|
Poruchy kůže a podkožní tkáně |
Časté | |
|
Alergická dermatitida |
Časté | |
|
Ekzém f |
Časté | |
|
Psoriáza |
Časté | |
|
Seboroická dermatitida f |
Časté | |
|
Exfoliace kůže |
Časté | |
|
Vyrážka *f |
Časté | |
|
Makulopapulózní vyrážka |
Časté | |
|
Akné f |
Časté | |
|
Erytém |
Časté | |
|
Pruritus |
Časté | |
|
Suchá kůže |
Časté | |
|
Exfoliativní vyrážka |
Méně časté | |
|
Toxoalergický exantém |
Méně časté | |
|
Numulární ekzém |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Horečka * |
Časté |
|
Vyšetření |
Zvýšení ALT * |
Časté |
|
Zvýšení AST * |
Časté | |
|
Abnormální výsledek testu jaterních funkcí |
Časté | |
|
Zvýšení jaterních enzymů |
Časté | |
|
Snížený počet lymfocytů |
Časté |
* Pozorováno s > 2% vyšším výskytem než u placeba
f Pozorováno s > 2% vyšším výskytem než u interferonu beta-1a (intramuskulárně)
Popis vybraných nežádoucích účinků
Porucha _ funkce _ jater
U pacientů léčených přípravkem Zinbryta bylo zaznamenáno zvýšení hodnot transamináz v séru a těžká porucha funkce jater. U 1 % pacientů byly pozorovány závažné reakce, včetně autoimunitní hepatitidy, hepatitidy a žloutenky. V průběhu klinické studie se vyskytl případ fatální autoimunitní hepatitidy u pacienta, u kterého byla po plánovaném šestiměsíčním přerušení terapie obnovena léčba daklizumabem v dávce 300 mg.
V průběhu klinických studií byly u některých pacientů během léčby a ještě 4 měsíce po podání poslední dávky přípravku Zinbryta pozorovány elevace transamináz v séru. U většiny pacientů se jednalo o asymptomatické elevace, které samovolně odezněly. U pacientů léčených přípravkem Zinbryta byl hlášen častější výskyt zvýšení ALT nebo AST na více než pětinásobek (> 5 krát) ULN v porovnání se skupinou s placebem (4% vs. < 1%) nebo interferonem beta-1a (intramuskulárně) (6% vs. 3%). K ukončení léčby z důvodu jaterních poruch souvisejících s léčbou došlo u 5 % pacientů léčených přípravkem Zinbryta a u 4 % pacientů léčených interferonem beta-1a (intramuskulárně).
Kožní reakce
V klinických studiích vedlo podávání přípravku Zinbryta ke zvýšenému výskytu kožních reakcí [18 % vs. 13 % (placebo); 37 % vs. 19 % (interferon beta-1a intramuskulárně)] a závažných kožních reakcí [<1 % vs. 0 % (placebo); 2 % vs. <1 % (interferon beta-1a intramuskulárně)] v porovnání s placebem a interferonem beta-1a (intramuskulárně).
Nejčastějšími kožními reakcemi byly vyrážka, dermatitida a ekzém. U většiny pacientů byly kožní reakce lehkého nebo středně těžkého stupně. U 4 % pacientů léčených přípravkem Zinbryta došlo k ukončení léčby z důvodu kožních reakcí.
V klinických studiích vedlo podávání přípravku Zinbryta ke zvýšenému výskytu deprese [5 % vs. 1 % (placebo); 8 % vs. 6 % (interferon beta-1a intramuskulárně)]; výskyt závažných depresivních reakcí byl u pacientů léčených přípravkem Zinbryta <1 %.
Infekce
V klinických studiích vedlo podávání přípravku Zinbryta ke zvýšení výskytu infekcí [50 % vs. 44 % (placebo); 65 % vs. 57 % (interferon beta-1a intramuskulárně)] a závažných infekcí [3 % vs. 0 % (placebo); 4 % vs. 2 % (interferon beta-1a intramuskulárně)] v porovnání s placebem a interferonem beta 1-a (intramuskulárně). Nejčastějšími typy infekcí byly infekce horních cest dýchacích a virové infekce. Průměrná doba trvání byla u všech léčebných skupin podobná. Míra infekcí a závažných infekcí se v průběhu času nezvyšovala. Většina pacientů s infekcemi pokračovala v léčbě přípravkem Zinbryta.
K ukončení léčby přípravkem Zinbryta z důvodu infekcí došlo u <1 % pacientů.
Gastrointestinální _ poruchy
V klinických studiích byl hlášen u pacientů léčených přípravkem Zinbryta zvýšený výskyt závažné kolitidy (<1 %).
Lymfadenopatie
V klinických studiích vedlo podávání přípravku Zinbryta ke zvýšenému výskytu lymfadenopatie s nástupem v průběhu léčby. K ukončení léčby z důvodu lymfadenopatie došlo u <1 % pacientů léčených přípravkem Zinbryta. Většina pacientů s lymfoadenopatií pokračovala v léčbě přípravkem Zinbryta; ve většině případů lymfoadenopatie do 3 měsíců odezněla.
Imunogenita
V rámci studie DECIDE (viz bod 5.1) byli pacienti testováni na přítomnost protilátek proti léku (daklizumab) po 4 týdnech léčby a poté každé 3 měsíce. U 19 % (175/913) pacientů zařazených do studie byly pozorovány protilátky proti léku, které se vytvořily během léčby, a u 8 % (71/913) pacientů byly pozorovány neutralizační protilátky. Většina reakcí souvisejících s protilátkami proti léku vznikajícími během léčby byla přechodná (12 % [110/913]), menší část reakcí byla trvalá (7 % [65/913]). U hodnocených pacientů byla většina reakcí souvisejících s neutralizačními protilátkami vznikajícími během léčby přechodná (6 % [56 z 913]), u 2 % pacientů (15 z 913) se vyskytly trvalé reakce. Reakce související s protilátkami proti léku a neutralizačními protilátkami vznikajícími během léčby se většinou objevovaly v prvním roce léčby a jejich četnost se s délkou léčby přípravkem Zinbryta snižovala.
U pacientů s neutralizačními protilátkami se clearance daklizumabu zvýšila v průměru o 19 % (viz bod 5.2). Tvorba protilátek proti léku nebo neutralizačních protilátek se neprojevila žádnou zjevnou klinickou odpovědí, nežádoucími účinky a neovlivnila ani farmakodynamický profil daklizumabu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nahlášené zkušenosti s předávkováním jsou omezené. Bezpečnost dávek nad 300 mg podaných subkutánně a 400 mg podaných intravenózně nebyla hodnocena. Nižší dávky byly dobře snášeny bez známek akutní toxicity. U pacientů s RS se při překročení uvedených hodnot v souladu s bezpečnostním profilem daklizumabu očekávají možné nežádoucí účinky.
Léčba
V případě předávkování může být u pacientů potřebná lékařská péče a má jim být poskytnuta náležitá podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresiva, inhibitory interleukinu, ATC kód: L04AC01 Mechanismus účinku
Daklizumab je humanizovaná monoklonální protilátka IgG1, která se váže na CD25 (IL-2Ra) receptor a zabraňuje tak vazbě IL2 na CD25. Daklizumab moduluje signalizaci IL-2 tím, že blokuje signály CD25-dependentního receptoru IL-2 s vysokou afinitou. V důsledku toho jsou k dispozici vyšší hladiny IL-2 pro signalizaci prostřednictvím IL-2 receptoru se střední afinitou. Mezi klíčové účinky této modulace dráhy IL-2, které potenciálně souvisí s terapeutickými účinky daklizumabu při RS, patří selektivní antagonismus reakcí aktivovaných T-lymfocytů a expanze imunoregulačních CD56bright NK (natural killer) buněk, u kterých se ukázalo, že selektivně snižují počet aktivovaných T-lymfocytů. Předpokládá se, že společně tyto imunomodulační účinky daklizumabu redukují patologické projevy CNS při RS a tím omezují výskyt relapsů a progresi postižení.
Farmakodynamické účinky
V klinických studiích odpovídaly farmakodynamické účinky přípravku Zinbryta podávaného v dávce 150 mg subkutánně každé 4 týdny modulaci signalizace IL-2, což se projevovalo rychlou a trvalou saturací cílových receptorů CD25 na cirkulujících T-lymfocytech a trvalým, přibližně dvojnásobným zvýšením koncentrace IL-2 v séru. Během 2 týdnů od podání první dávky bylo navíc pozorováno zvýšení počtu CD56bright NK buněk a snížení počtu regulačních T-lymfocytů (definovaných jako T-lymfocyty CD4+CD127lowFoxP3+ T-lymfocyty) s trvalým pětinásobným zvýšením počtu CD56bright NK buněk oproti výchozímu stavu a přibližně 60 % snížením počtu regulačních T-lymfocytů v léčebné fázi. Návrat k výchozím hodnotám byl pozorován přibližně po 20-24 týdnech po podání poslední dávky. Během léčby přípravkem Zinbryta zůstávaly průměrné počty buněk pro hlavní imunitní podjednotky (T, B a NK buňky)
v normálních rozmezích; během prvního roku léčby se celkový počet lymfocytů, počet T buněk a B buněk v průměru snížil o <10 % oproti výchozímu stavu. Celkový počet lymfocytů se vrátil k výchozím hodnotám přibližně po 8 - 12 týdnech po podání poslední dávky přípravku Zinbryta (150 mg). Celkový počet lymfocytů <0,8x109 buněk/l ([Všeobecná terminologická kritéria pro nežádoucí účinky - CTCAE] stupeň 2; nejméně jedno měření) byl zjištěn u 4 % pacientů z placebo skupiny a u 5 % pacientů léčených přípravkem Zinbryta v rámci studie SELECT; a u 9 % pacientů léčených intramuskulárně interferonem beta-1a a u 8 % pacientů léčených přípravkem Zinbryta v rámci studie DECIDE. Celkový počet NK buněk se zvýšil přibližně 1,5 krát v důsledku změny počtu CD56bright NK buněk.
Klinická účinnost a bezpečnost
Účinnost přípravku Zinbryta byla prokázána u pacientů s RRS v rámci dvou studií (SELECT a DECIDE). Studie SELECT byla dvojitě zaslepená, randomizovaná, placebem kontrolovaná studie, během níž byl pacientům podáván přípravek Zinbryta buď v dávce 150 mg (n=208) nebo 300 mg (n=209) oproti placebu (n=204) každé 4 týdny po dobu 52 týdnů. Studie DECIDE byla dvojitě zaslepená, randomizovaná, aktivně kontrolovaná studie s paralelními skupinami, během níž byl pacientům podáván buď přípravek Zinbryta v dávce 150 mg každé 4 týdny (n=919), nebo interferon beta-1a (intramuskulárně) v dávce 30 mg týdně (n=922), a to nejméně 2 a nejdéle 3 roky (96 až 144 týdnů). Uspořádání studie a výchozí charakteristiky jsou uvedeny v tabulce 3.
Tabulka 3: Studie SELECT a DECIDE - uspořádání studií a výchozí charakteristiky
|
Název studie |
SELECT |
DECIDE |
|
Uspořádání studie | ||
|
Léčba |
52 týdnů |
96 až 144 týdnů |
|
Historie onemocnění |
Pacienti s RRS, s nejméně 1 relapsem (klinický a/nebo na MRI) v období 1 roku před randomizací a se skóre EDSS 0 až 5,0. V případě studie DECIDE byly požadovány nejméně 2 relapsy (jeden z nich klinický) v období předcházejících 3 let. | |
|
Základní charakteristiky | ||
|
Průměrný věk (roky) |
35,7 |
36,3 |
|
Průměrné trvání nemoci (roky) |
4,1 |
4,2 |
|
Průměrný počet relapsů během 12 měsíců před studií |
1,4 |
1,6 |
|
Medián skóre EDSS |
2,5 |
2,0 |
|
Procento s EDSS > 3,5 |
36 % |
30 % |
|
Procento s > 1 Gd-enhancující lézí (průměr) |
44% (1,8) |
46 % (2,1) |
|
Procento se > 2 relapsy v období 1 roku před studií |
31 % |
46 % |
|
Procento před použitím DMT (%) |
20 % |
41 % |
Výsledky studie SELECT jsou uvedeny v tabulce 4. Léčba přípravkem Zinbryta v dávce 150 mg jednou za 4 týdny v porovnání s placebem významně snížila roční míru relapsů (ARR) a riziko relapsů. Navíc byl u pacientů léčených přípravkem Zinbryta zaznamenán statisticky významný vliv na potvrzenou progresi postižení v průběhu 24 týdnů s mírou rizika 0,24 [95 % CI: 0,09; 0,63]. Dávka 300 mg přípravku Zinbryta nepřinášela další přínos v porovnání s dávkou 150 mg.
Tabulka 4: Studie SELECT: Klinické a MRI výsledky (za 52 týdnů)
|
Placebo |
Zinbryta 150 mg |
Hodnota P | |
|
Klinické závěry | |||
|
Počet pacientů |
196 |
201 | |
|
Roční míra relapsů |
0,458 |
0,211 |
p<0,0001 |
|
Poměr relativních rizik [95 % CI] |
0,461 [0,318; 0,668] | ||
|
Procento pacientů bez relapsu |
64 % |
81 % |
p<0,0001 |
|
Poměr rizik* [95 % CI] |
0,45 [0,30; 0,67] | ||
|
Procento pacientů s potvrzenou progresí postižení za 24 týdnů |
11 % |
2,6 % |
p=0,0037 |
|
Poměr rizik [95 % CI] |
0,24 [0,09; 0,63] | ||
|
Procento pacientů s potvrzenou progresí postižení za 12 týdnů |
13 % |
6 % |
p=0,0211 |
|
Poměr rizik [95 % CI] |
0,43 [0,21; 0,88] | ||
|
Průměrná změna fyzického skóre MSIS-29 |
Zhoršení o 3,0 body |
Zlepšení o 1,0 bod |
p=0,0008 |
|
MRI závěry# | |||
|
Průměrný počet nových nebo nově se zvětšujících T2 hyperintenzních lézí |
8,13 |
2,4 |
p<0,0001 |
|
Průměrný poměr lézí [95 % CI] |
0,30 [0,22; 0,40] | ||
|
Průměrný počet nových T1 Gd-enhancujících lézí mezi týdny 8 a 24 (při měsíčních skenech MRI) |
4,79 |
1,46 |
p<0,0001 |
|
Průměrný poměr lézí [95 % CI] |
0,31 [0,20; 0,48] | ||
* Poměr rizik pro riziko relapsu.
# Při analýze MRI nálezů byl použit hodnotitelný soubor údajů pro každý cílový parametr; T1 Gd-enhancující: populace s intenzivním MRI sledovaním.
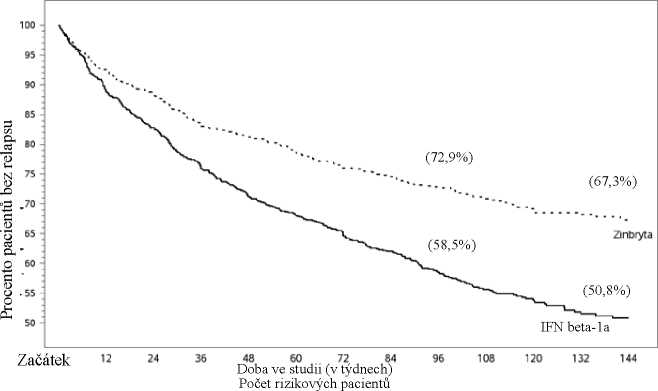
Tabulka 5 a obrázky 1 - 2 ukazují výsledky studie DECIDE. Podávání přípravku Zinbryta významně snížilo ARR a riziko relapsu v porovnání s pacienty léčenými intramuskulárně interferonem beta-1a. Navíc byl u pacientů léčených přípravkem Zinbryta zaznamenán statisticky významný vliv na potvrzenou progresi postižení v průběhu 24 týdnů s mírou rizika 0,73 [95 % CI: 0,55; 0,98]. V 96. týdnu došlo při podávání přípravku Zinbryta ke statisticky významnému snížení počtu nových nebo nově se zvětšujících T2 hyperintenzních lézí, počtu nových T1 Gd-enhancujících lézí a průměrného počtu nových T1 hyperintenzních lézí. Léčba přípravkem Zinbryta navíc vedla ke zmírnění klinicky významného zhoršení fyzického dopadu roztroušené sklerózy podle subjektivního hodnocení pacienta (zhoršení fyzického skóre MSIS-29 o >7,5 bodu oproti výchozímu stavu do 96. týdne) v porovnání s léčbou interferonem beta-1a (intramuskulárně).
Tabulka 5: Klinické a MRI výsledky studie DECIDE (96 až 144 týdnů) (Pokud není uvedeno jinak, hodnoty se vztahují k výsledkům za 96 týdnů.)
|
Interferon beta-1a (intramuskulárně) 30 mikrogramů |
Zinbryta 150 mg |
Hodnota p | |
|
Klinické závěry | |||
|
Počet pacientů |
922 |
919 | |
|
Roční míra relapsů * Poměr relativních rizik* [95 % CI] |
0,393 |
0,216 0,550 [0,469; 0,645] |
p<0,0001 |
|
Procento pacientů bez relapsu |
59 % |
73 % |
p<0,0001 |
|
Poměr rizik # * [95 % CI] |
0,59 [0,50; 0,69] | ||
|
Procento pacientů s potvrzenou progresí postižení za 24 týdnů |
12 % |
9 % |
p=0,03 |
|
Poměr rizik * [95 % CI] |
0,73 [0,55; 0,98] | ||
|
Procento pacientů s potvrzenou progresí postižení za 12 týdnů |
14 % |
12 % |
p=0,16 |
|
Poměr rizik * [95 % CI] |
0,84 [0,66; 1,07] | ||
|
Procento pacientů s klinicky významným zhoršením podle fyzického skóre MSIS-29 (>7,5 bodu) |
23 % |
19 % |
p=0,018 |
|
Poměr pravděpodobnosti [95 % CI] |
0,76 [0,60; 0,95] | ||
|
MRI závěry^ | |||
|
Průměrný počet nových nebo nově se zvětšujících T2-hyperintenzních lézí |
9,44 |
4,31 |
p<0,0001 |
|
Průměrný poměr lézí [95 % CI] |
0,46 [0,39; 0,53] | ||
|
Průměrný počet T1 Gd-enhancujících lézí |
1,0 |
0,4 |
p<0,0001 |
|
Poměr pravděpodobnosti [95 % CI] |
0,25 [0,20; 0,32] | ||
|
Průměrný počet nových T1-hypointenzních lézí |
4,43 |
2,13 |
p<0,0001 |
|
Průměrný poměr lézí [95 % CI] |
0,48 [0,42; 0,55] | ||
* Ukazatele míry a snížení rizika/cílové parametry jsou vypočítány za období léčby do 144 týdnů.
# Poměr rizik pro riziko relapsu.
^ Při analýze MRI nálezů byl použit hodnotitelný soubor údajů pro každý MRI cílový parametr.
Analýzy podskupin studií SELECT a DECIDE prokázaly konzistentní účinek přípravku Zinbryta v porovnání s placebem a interferonem beta-1a (intramuskulárně) napříč podskupinami definovanými demografickými kritérii a kritérii onemocnění RS. V rámci analýzy podskupin studie DECIDE bylo pozorováno v porovnání s pacienty, kterým byl podáván interferon beta-1a (intramuskulárně), statisticky významné snížení ARR a počtu nových nebo nově se zvětšujících T2-hyperintenzních lézí napříč podskupinami (pohlaví, věk, předchozí DMT léčba RS a úrovně aktivity onemocnění).
I když byl vliv na progresi postižení pozorován především u pacientů se vstupní hodnotou EDSS < 3,5, důkazy o účinnosti byly zjevné i u pacientů s relabující sekundárně progresivní RS definovanou pomocí vstupní hodnoty EDSS > 3,5 a alespoň jedním z následujících tří kritérií: 24týdenní potvrzená progrese zhoršení definovaná pomocí EDSS, nebo > 20% pokles při testu chůze na 25 stop (Timed 25-foot Walk, T25FW) nebo > 20% pokles při testu 9-Hole Peg (9-HPT).
Účinnost u pacientů s vysoce aktivním onemocněním
Vysoce aktivní onemocnění bylo definováno následovně:
• pacienti se 2 nebo více relapsy během jednoho roku a s 1 nebo více Gd-enhancujícími lézemi na MRI mozku, nebo
• pacienti, kteří nereagovali na celý a adekvátní cyklus (nejméně 1 rok léčby) předchozí léčby DMT, u kterých došlo v předchozím roce nejméně k 1 relapsu navzdory léčbě a kteří měli nejméně 9 T2 hyperintenzních lézí při kraniální magnetické rezonanci (MRI) nebo nejméně 1 Gd-enhancující lézi, anebo u kterých se nezměnila, případně se zvýšila, míra relapsů v předchozím roce v porovnání s předchozími 2 lety.
Klinické údaje ze studie DECIDE prokázaly konzistentní účinky léčby v podskupině s vysoce aktivním onemocněním. V porovnání s interferonem beta-1a podávaným intramuskulárně (n=440) vedla léčba přípravkem Zinbryta (n=440) ke snížení ARR (poměr relativních rizik 0,52 [95 % CI: 0,42; 0,64], p<0,0001), počtu nových nebo nově se zvětšujících T2-hyperintenzních lézí (průměrný poměr lézí 0,46 [95 % CI: 0,37; 0,57], p<0,0001), a potvrzené progrese postižení za 24 týdnů (poměr rizik 0,60 [95 % CI: 0,40; 0,89], p=0,012).
Obrázek 1: Procento pacientů bez relapsu (studie DECIDE)
922
Zinbryta 919
791
©33
714
7S3
644
711
590
679
551
633
505
599
481
571
445
543
319
413
243
289
174
204
Obrázek 2: Podíl pacientů s potvrzenou progresí postižení za 24 týdnů (studie DECIDE)

Zinbryta 919 897 863 817 796 755 722 688 662 502 361 266 131
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Zinbryta u jedné nebo více podskupin pediatrické populace v léčbě roztroušené sklerózy (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika daklizumabu je dobře popsaná pomocí dvoukompartmentového modelu s absorpcí a eliminačními procesy prvního řádu.
Absorpce
Po subkutánním podávání daklizumabu se medián času dosažení maximální koncentrace v séru (Tmax) pohyboval v rozmezí 5 až 7 dnů. Absolutní biologická dostupnost daklizumabu 150 mg podaného subkutánně byla přibližně 90 % - stanoveno na základě farmakokinetické analýzy subkutánního a intravenózního dávkování napříč studiemi.
Distribuce
Po subkutánním podávání 150 mg daklizumabu každé čtyři týdny bylo dosaženo ustáleného stavu sérové koncentrace daklizumabu po 4. dávce, kdy došlo k jeho akumulaci na přibližně 2,5násobek v porovnání s jednorázovou dávkou. V ustáleném stavu byla průměrná maximální koncentrace (Cmax) daklizumabu v séru přibližně 30 mikrogramů/ml, minimální koncentrace (Cmin) v séru přibližně 15 mikrogramů/ml a plocha pod křivkou sérové koncentrace v čase během dávkovacího intervalu (AUCtau) přibližně 640 den*mikrogramů/ml, přičemž variabilita mezi pacienty (% CV) byla přibližně 40 %.
Na základě populační farmakokinetické analýzy napříč studiemi je distribuční objem daklizumabu v ustáleném stavu 6,34 l u pacienta vážícího 68 kg (přibližný medián hodnocených pacientů). Takto malý distribuční objem naznačuje, že daklizumab je primárně lokalizován ve vaskulárním a intersticiálním prostoru.
Biotransformace
Přesná metabolická dráha pro daklizumab nebyla charakterizována. Předpokládá se, že daklizumab se jako monoklonální protilátka IgG1 pravděpodobně katabolizuje na peptidy a aminokyseliny stejným způsobem jako endogenní IgG. Předpokládá se, že daklizumab není metabolizován jaterními enzymy, jako jsou izoenzymy CYP (viz bod 4.5).
Eliminace
Předpokládá se, že daklizumab se jako monoklonální protilátka IgG1 nevylučuje ledvinami.
Na základě populační farmakokinetické analýzy napříč studiemi je clearance daklizumabu 0,212 l/den s terminálním eliminačním poločasem přibližně 21 dnů. Clearance daklizumabu u pacientů, u nichž se vytvořily neutralizační protilátky, byla přibližně o 19 % vyšší (viz bod 4.8. Imunogenita).
Linearita/nelinearita
Populační farmakokinetická analýza napříč studiemi naznačila (v souladu s výsledky jednotlivých studií), že expozice daklizumabu je vyšší, než je hodnota úměrná dávce v rozmezí od 50 mg do 100 mg při subkutáním podání, a že je úměrná dávce v rozmezí od 100 mg do 300 mg při subkutánním podání.
F armakokinetické/farmakodynamické vztahy
V rámci zkoumaných režimů dávkování u daklizumabu během studií (150 mg a 300 mg subkutánně každé čtyři týdny u pacientů s RS) nebyl zjištěn jednoznačný vztah mezi expozicí daklizumabu a cílovými parametry klinické účinnosti (ARR, T2 léze a Gd-enhancující léze) nebo relevantními bezpečnostními cílovými parametry (závažný infekční stav, středně těžké nebo těžké kožní nežádoucí účinky a zvýšení AST/ALT na > 5 krát ULN).
Zvláštní populace
Porucha _ funkce ledvin nebo _jater
Nebyly provedeny žádné studie, které by hodnotily farmakokinetické vlastnosti daklizumabu u pacientů s poruchou funkce ledvin nebo jater. Nepředpokládá se, že se daklizumab vylučuje ledvinami nebo že je metabolizován jaterními enzymy (viz bod 4.2).
Hmotnost
Na základě populační farmakokinetické analýzy napříč studiemi bylo zjištěno, že tělesná hmotnost se na variabilitě clearance daklizumabu mezi pacienty podílela méně než 40 procenty. V rámci studie DECIDE nebyly pozorovány významné rozdíly v klinické účinnosti nebo bezpečnosti mezi podskupinami pacientů s RS podle hmotnostních kvartilů.
Věk a pohlaví
Na základě populační farmakokinetické analýzy napříč studiemi bylo zjištěno, že farmakokinetické vlastnosti daklizumabu neovlivňuje věk (rozsah: 18 až 66 let; n=1670) a ani pohlaví (n = 567 mužů a 1103 žen).
Rasa
Mezi zdravými dobrovolníky japonské a kavkazské rasy nebyly pozorovány žádné farmakokinetické rozdíly.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické studie bezpečnosti byly provedeny u opic druhu Cynomolgus z důvodu druhové specificky daklizumabu, který se váže pouze na receptor CD25 u člověka nebo primátů.
Kancerogeneze
Studie kancerogenního účinku daklizumabu nebyly provedeny. V průběhu dvou devítiměsíčních studií nebyly u opic pozorovány žádné preneoplastické nebo neoplastické tkáně.
Mutageneze
Studie genotoxicity daklizumabu nebyly provedeny.
Reprodukční toxicita
Daklizumab neovlivnil reprodukční schopnost samic a samců opic druhu Cynomolgus (AUC u samic až 85 krát a u samců až 100 krát vyšší než expozice při klinické dávce). Nebyly zjištěny účinky na vývoj plodu a ani prokázány teratogenní účinky. Daklizumab neměl vliv na perinatální ani postnatální vývoj mláďat od narození až do 6 měsíců věku. Expozice (AUC) se v rámci těchto studií pohybovala v rozmezí od 55 do 140 násobku expozice pozorované při klinické dávce. Daklizumab byl zjištěn v mateřském mléce u 11 ze 14 lakujících opic v množství, která byla <0,122 % hodnot stanovených v séru matky, přičemž u mláďat nebyly pozorované žádné nežádoucí účinky.
Toxikologie
V rámci dvou devítiměsíčních studií provedených u opic druhu Cynomolgus byl daklizumab podáván zvířatům subkutánně každé dva týdny v dávce 10-200 mg/kg.
Chronické podávání daklizumabu ve všech dávkách zvýšilo výskyt kožních nálezů (v porovnání s nálezy pozorovanými u kontrolních zvířat). Tyto nálezy (suché, zarudlé a vystouplé nepravidelné oblasti kůže v porovnání s kontrolami, které na mikroskopické úrovni korelovaly s akantózou/hyperkeratózou a subakutním až chronickým zánětem) byly převážně klasifikovány jako lehké až středně těžké, v jednom případě byly posouzené jako těžké.
V mozku a míše opic léčených dávkou >35 mg/kg, (AUC 27 krát vyšší než při klinické dávce) bylo pozorováno na velikosti dávky závislé zvýšení výskytu mikrogliálních agregátů na pozadí. Po období až 12týdenní rekonvalescence byla pozorovaná reverzibilita. Výskyt nebo závažnost mikrogliálních agregátů u opic se s prodlouženým dávkováním nezvýšily a nesouvisely s neuronálním poškozením nebo neurobehaviorálními účinky. Malý podsoubor mikrogliálních agregátů souvisel s mikrohemoragiemi, ovšem bez zjevných funkčních následků pro organismus opic.
Vyšetřovací studie in vitro naznačují, že mikrogliální agregáty nejsou přímým důsledkem působení daklizumabu na mikrogliální buňky. Spíše je pravděpodobné, že vznikaly z důvodu vyšší lokální biologické dostupnosti IL-2.
Klinický význam mikrogliálních agregátů není znám, u opic nicméně nebyly pozorovány žádné škodlivé neurologické účinky, které by bylo možné přisoudit působení těchto mikroskopických změn.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Natrium-sukcinát Kyselina j antarová Chlorid sodný Polysorbát 80 Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky.
Přípravek Zinbryta může být uchováván při pokojové teplotě (do 30 oC) v původním obalu po dobu 30 dnů. Po zahřátí na pokojovou teplotu nedávejte přípravek Zinbryta zpět do chladničky.
Jestliže byl přípravek Zinbryta uchováván mimo chladničku po dobu delší než celkem 30 dní, nebo pokud si nejste jistý(á), jak dlouho byl přípravek Zinbryta uchováván při pokojové teplotě, přípravek zlikvidujte.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Další informace o uchovávání přípravku při pokojové teplotě jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Předplněná injekční stříkačka vyrobená ze skla (třída 1), uzavřená pryžovou zátkou a pevným termoplastickým krytem jehly, která obsahuje 1 ml roztoku. Na předplněné injekční stříkačce je nasazena injekční jehla velikosti 29 G, o délce 0,5 palce.
Velikosti balení:
- Balení obsahuje jednu 150mg předplněnou injekční stříkačkou.
- Vícečetné balení na 3 měsíce léčby obsahuje tři 150mg předplněné injekční stříkačky (3 krabičky po jedné stříkačce).
Součástí pružinového injekčního pera s názvem Zinbryta Pen je předplněná injekční stříkačka Zinbryta. Uvnitř pera je předplněná injekční stříkačka, vyrobená ze skla (třída I), uzavřená pryžovou zátkou a pevným termoplastickým krytem jehly, která obsahuje 1 ml roztoku. Na předplněné injekční stříkačce je nasazena injekční jehla velikosti 29 G, o délce 0,5 palce.
Velikosti balení:
- Balení obsahuje jedno 150mg předplněné pero.
- Vícečetné balení na 3 měsíce léčby obsahuje tři 150mg předplněná pera (3 krabičky po jednom peru).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
BIOGEN IDEC Limited
Innovation House
70 Norden Road
Maidenhead
Berkshire
SL6 4AY
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/16/1107/001
EU/1/16/1107/002
EU/1/16/1107/003
EU/1/16/1107/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 01. července 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky:
BiogenInc
5000 Davis Drive
Research Triangle Park
North Carolina
27709
USA
Název a adresa výrobce odpovědného za propouštění šarží
Biogen (Denmark) Manufacturing ApS
Biogen Allé 1
Hillered
DK-3400
Dánsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Další opatření k minimalizaci rizik
Informace pro lékaře týkající se zvládání rizika poškození jater, karta pacienta
Před uvedením přípravku Zinbryta na trh v každém členském státě se držitel rozhodnutí o registraci musí spolu s příslušnými národními orgány dohodnout na obsahu a formě edukačního programu, včetně komunikačních médií, způsobu distribuce a veškerých dalších aspektů programu.
Cíl a zdůvodnění:
Informovat pacienty a lékaře o riziku závažného poškození jater a o postupech náležitého zvládání tohoto rizika s cílem minimalizovat jeho výskyt a stupeň závažnosti.
Navrhované opatření:
Příručka o zvládání rizika poškození jater bude obsahovat informace pro lékaře o riziku zvýšení hodnot jaterních enzymů a závažného poškození jater u pacientů léčených přípravkem Zinbryta. Bude rovněž sloužit jako vodítko pro diskuzi mezi lékařem a pacientem o riziku poškození jater a o opatřeních na jeho zvládání. Lékař má s pacientem prodiskutovat riziko poškození jater a má mu poskytnout Kartu pacienta.
Karta pacienta informuje pacienty o riziku závažného poškození jater a o možných příznacích, aby rozpoznali situace, ve kterých je třeba včas kontaktovat lékaře. Karta pacienta rovněž vysvětluje, proč je třeba sledovat jaterní funkce a informuje pacienta o důležitosti podstupování krevních testů každý měsíc.
Karta pacienta je vytvořena tak, aby umožnila lékaři prezentovat pacientovi srozumitelným způsobem informace o přípravku Zinbryta v okamžiku, kdy mu přípravek Zinbryta předepisuje. Bude zaměřená na možnost vzniku závažného poškození jater souvisejícího s léčbou přípravkem Zinbryta a bude rovněž obsahovat informace o příznacích poškození jater a instrukce týkající se kontrolování jaterních funkcí každý měsíc.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zinbryta 150 mg injekční roztok v předplněné injekční stříkačce Zinbryta 150 mg injekční roztok v předplněném peru daclizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje daclizumabum 150 mg v 1 ml. Jedno předplněné pero obsahuje daclizumabum 150 mg v 1 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Natrium-sukcinát, kyselina jantarová, chlorid sodný, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka 1 předplněné pero
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému použití.
Zde otevřít Zde odtrhnout
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Přípravek je možné uchovávat při pokojové teplotě (do 30 °C) nepřerušené po dobu až 30 dní. Po uchovávání při pokojové teplotě nesmí být přípravek vrácen zpět do chladničky.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Biogen Idec Ltd. Innovation House 70 Norden Road Maidenhead Berkshire SL6 4AY Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1107/001
EU/1/16/1107/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Zinbryta
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zinbryta 150 mg injekční roztok v předplněné injekční stříkačce Zinbryta 150 mg injekční roztok v předplněném peru daclizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje daclizumabum 150 mg v 1 ml. Jedno předplněné pero obsahuje daclizumabum 150 mg v 1 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Natrium-sukcinát, kyselina jantarová, chlorid sodný, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Vícečetné balení: 3 (3 balení po 1) předplněné injekční stříkačky.
Vícečetné balení: 3 (3 balení po 1) předplněná pera.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Biogen Idec Ltd. Innovation House 70 Norden Road Maidenhead Berkshire SL6 4AY Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1107/002
EU/1/16/1107/004
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Zinbryta
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VÍCEČETNÉ BALENÍ VNITŘNÍ KRABIČKA (bez blue boxu)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zinbryta 150 mg injekční roztok v předplněné injekční stříkačce Zinbryta 150 mg injekční roztok v předplněném peru daclizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje daclizumabum 150 mg v 1 ml. Jedno předplněné pero obsahuje daclizumabum 150 mg v 1 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Natrium-sukcinát, kyselina jantarová, chlorid sodný, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka. Součást vícečetného balení, nesmí být prodáváno samostatně. 1 předplněné pero. Součást vícečetného balení, nesmí být prodáváno samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému použití.
zde otevřít zde odtrhnout
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Biogen Idec Ltd. Innovation House 70 Norden Road Maidenhead Berkshire SL6 4AY Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1107/002
EU/1/16/1107/004
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Zinbryta
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Zinbryta 150 mg injekce
daclizumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Zinbryta 150 mg injekce
daclizumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Zinbryta 150 mg injekční roztok v předplněné injekční stříkačce Zinbryta 150 mg injekční roztok v předplněném peru daclizumabum
▼ Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
Kromě této příbalové informace obdržíte od svého lékaře i Kartu pacienta. Karta pacienta obsahuje důležité bezpečností informace, které potřebujete znát před zahájením léčby přípravkem Zinbryta a během této léčby.
• Ponechte si příbalovou informaci a Kartu pacienta pro případ, že si je budete potřebovat přečíst znovu. Ponechte si příbalovou informaci a Kartu pacienta u sebe po celou dobu léčby a dále ještě 4 měsíce po poslední dávce přípravku Zinbryta, protože nežádoucí účinky se mohou vyskytnout i po ukončení léčby.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod
4.
Co naleznete v této příbalové informaci
1. Co je přípravek Zinbryta a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zinbryta používat
3. Jak se přípravek Zinbryta používá
4. Možné nežádoucí účinky
5. Jak přípravek Zinbryta uchovávat
6. Obsah balení a další informace
7. Návod na injekční podání přípravku Zinbryta
1. Co je přípravek Zinbryta a k čemu se používá
Léčivou látkou v přípravku Zinbryta je daklizumab. Jedná se o druh přípravku nazývaný monoklonální protilátka.
K čemu se přípravek Zinbryta používá
Přípravek Zinbryta se používá k léčbě relabujících (opětovně se vracejících) forem roztroušené sklerózy (RS) u dospělých.
Při RS imunitní systém způsobuje zánět, který poškozuje ochrannou vrstvu (nazývanou myelin) obklopující nervy centrálního nervového systému (včetně mozku a míchy). Ztráta myelinu se nazývá demyelinizace.
To má za následek, že nervy přestanou správně pracovat.
Pacienti s relabující formou RS mají opakované ataky (relapsy) příznaků postižení nervového systému, které jsou způsobeny tím, že nervy nepracují správně. Tyto příznaky se u jednotlivých pacientů liší, ale obvykle zahrnují potíže při chůzi, problémy se zrakem či udržením rovnováhy.
Příznaky mohou po odeznění relapsu zcela vymizet, ale postupem času může dojít k tomu, že některé problémy přetrvávají i mezi jednotlivými relapsy a mohou zasahovat do každodenních činností.
Jak přípravek Zinbryta působí
Přípravek Zinbryta působí tak, že zabraňuje imunitnímu systému, aby poškozoval mozek a míchu.
To může pomoci snížit počet relapsů, které máte, a zpomalit invalidizující účinky RS. Přestože léčba přípravkem Zinbryta RS nevyléčí, může zabránit zhoršování Vašeho stavu.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zinbryta používat Nepoužívejte přípravek Zinbryta
- jestliže jste v minulosti měl(a) závažnou alergickou reakci na daklizumab, nebo na kteroukoliv další složku tohoto přípravku, uvedenou v bodě 6.
Upozornění a opatření
Než začnete přípravek Zinbryta používat, obraťte se na svého lékaře:
- Jestliže jste někdy měl(a) problémy s játry. Přípravek Zinbryta může způsobit závažné poruchy funkce jater, proto Váš lékař musí rozhodnout, zda je pro Vás přípravek Zinbryta vhodný.
- Jestliže máte nebo jste v minulosti měl(a) depresi.
- Jestliže máte závažnou infekci, jako je například zápal plic.
- Jestliže j ste někdy měl(a) tuberkulózu (nazývanou též TBC) nebo žij ete v oblasti, kde j sou infekce TBC časté, může být riziko TBC u Vás vyšší. Před zahájením léčby přípravkem Zinbryta můžete být testován(a) na TBC a během léčby budete monitorován(a).
Možné poruchy funkce jater
Přípravek Zinbryta může způsobit závažné poruchy funkce jater. Přestože jste v minulosti neměl(a) žádné problémy s játry, Váš lékař Vám odebere vzorek krve, aby vyšetřil funkci jater. Bude nutné provést:
- krevní testy před zahájením léčby,
- měsíčně krevní testy během léčby,
- krevní testy po dobu 4 měsíců po ukončení léčby. Nežádoucí účinky se mohou projevit i po ukončení léčby přípravkem Zinbryta (viz Závažné nežádoucí účinky uvedené v době 4).
Je velice důležité, abyste pravidelně podstupoval(a) krevní testy.
Obdržíte Kartu pacienta, kde naleznete další informace o tom, na co si máte během používání přípravku Zinbryta dávat pozor. Tuto kartu noste během léčby a čtyři měsíce po ukončení léčby při sobě. V případě jakékoliv léčby, i když se nebude jednat o léčbu roztroušené sklerózy, ukažte Kartu pacienta lékaři, lékárníkovi nebo zdravotní sestře.
Pokud se u Vás objeví kterýkoliv z následujících příznaků, okamžitě kontaktujte svého lékaře:
• pocit na zvracení neznámého původu (nevolnost),
• zvracení,
• bolest břicha,
• zvýšená únava,
• ztráta chuti k jídlu,
• zežloutnutí kůže nebo očního bělma,
• tmavé (čajové) zabarvení moči.
Tyto příznaky mohou naznačovat problémy s játry. Jestliže se u Vás vyskytnou problémy s játry, Váš neurolog může léčbu přípravkem Zinbryta přerušit a odeslat Vás k hepatologovi (specialistovi na játra) (viz bod 4. Možné nežádoucí účinky).
Děti a dospívající
Přípravek Zinbryta není určen k použití u dětí a dospívajících mladších 18 let. Bezpečnost a účinnost přípravku Zinbryta nejsou v této věkové skupině známy.
Starší populace
Přípravek Zinbryta byl na pacientech starších 55 let testován pouze v omezené míře. Pokud je Vám více než 55 let, lékař Vám přesto může přípravek Zinbryta předepsat.
Další léčivé přípravky a přípravek Zinbryta
Informujte svého lékaře o všech lécích, vitamínech či rostlinných přípravcích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Očkování
Jestliže se potřebujete nechat očkovat, zeptejte se nejdříve svého lékaře, protože přípravek Zinbryta může mít vliv na účinek očkovací látky. U pacientů léčených přípravkem Zinbryta nebylo prokázáno snížení účinnosti vakcíny proti sezónní chřipce (neaktivní vakcína). Účinek přípravku Zinbryta na jiné vakcíny (živé vakcíny) však není znám.
Těhotenství a kojení
Jelikož údaje o podávání přípravku Zinbryta těhotným ženám jsou omezené, je nutno zvážit přínosy léčby pro matku a riziko pro dítě. Jestliže jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek používat.
Není známo, zda se přípravek Zinbryta vylučuje do mateřského mléka. Lékař Vám poradí, jak se rozhodnout, zda máte ukončit kojení či léčbu přípravkem Zinbryta.
Řízení dopravních prostředků a obsluha strojů
Nepředpokládá se, že přípravek Zinbryta ovlivňuje schopnost řídit a obsluhovat stroje. Lékař Vám sdělí, zda Vám Vaše onemocnění umožňuje bezpečně řídit vozidla a obsluhovat stroje.
Přípravek Zinbryta obsahuje malé množství sodíku
Jedna dávka přípravku Zinbryda obsahuje 0,14 mmol sodíku. To znamená, že v podstatě je “bez sodíku” a přípravek tudíž mohou používat i pacienti na dietě s nízkým obahem sodíku.
3. Jak se přípravek Zinbryta používá
Přípravek Zinbryta Vám předepíše lékař, který má zkušenosti s léčbou RS.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Doporučená dávka
Dávka přípravku Zinbryta je 150 mg jednou za měsíc.
Snažte se podávat si injekci přípravku Zinbryta každý měsíc ve stejný den, abyste si datum podání injekce snadno zapamatoval(a). Podávejte si injekci například první pondělí každého měsíce.
Každý měsíc budete muset rovněž podstoupit krevní testy za účelem kontroly funkce jater. Je velice důležité, abyste tento test nevynechal(a). Zkuste si datum odběru krve pro každý měsíc pevně stanovit. Jestliže se domníváte, že jste krevní odběr vynechal(a), kontaktujte svého lékaře.
Podávání injekce pacientem
Přípravek Zinbryta se podává injekčně pod kůži (subkutánní injekce) do stehna, břicha či zadní strany paže. Podrobné pokyny o podání injekce přípravku Zinbryta jsou uvedeny v bodě 7. Návod na injekční podání přípravku Zinbryta.
Lékař nebo zdravotní sestra Vás mají náležitě zacvičit, abyste si injekci přípravku Zinbryta mohl(a) podávat sám(sama). Přečtěte si a dodržujte pokyny uvedené v návodu v bodě 7. Návod na injekční podání přípravku Zinbryta.
Máte-li se zacházením s injekční stříkačkou/perem problémy, obraťte se svého lékaře nebo zdravotní sestru, kteří Vám s podáním injekce pomohou.
Jak dlouho používat přípravek Zinbryta
Lékař Vám sdělí, jak dlouho budete muset přípravek Zinbryta používat. Pokud Vás o to lékař nepožádá, neprovádějte žádné změny.
Pokud Vám lékař sdělil, abyste přípravek Zinbryta přestal(a) používat, nezačínejte ho používat znovu, dokud Vám k tomu lékař nedá pokyn.
Jestliže jste použil(a) více přípravku Zinbryta, než jste měl(a)
Jestliže jste si podal(a) více injekcí, než je obvyklá dávka, a zaznamenal(a) jste jakékoliv nežádoucí účinky nebo máte obavy, obraťte se na svého lékaře nebo zdravotní sestru. Jsou známy případy, kdy byla pacientům podána dvojnásobná dávka přípravku Zinbryta oproti doporučené dávce a nebyly zaznamenány žádné mimořádné závažné nežádoucí účinky.
Jestliže jste zapomněl(a) použít přípravek Zinbryta
Přípravek Zinbryta se podává injekčně jednou za měsíc. Snažte se podávat si injekci přípravku Zinbryta každý měsíc ve stejný den, abyste si datum podání injekce snadno zapamatoval(a).
• Jestliže j ste si zapomněl(a) dávku podat a od data plánovaného podání injekce neuplynuly ještě více než 2 týdny, podejte si injekci co nejdříve, jak to bude možné. Potom pokračujte jako obvykle a dodržujte zvolený den pro podávání injekce.
• Pokud od vynechání dávky (od data plánovaného podání injekce) uplynuly více než 2 týdny, zapomenutou dávku vynechejte úplně a další dávku si podejte v obvyklý den.
V žádném případě nepoužijte dvě injekce najednou, abyste nahradil(a) vynechanou dávku.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Nesnažte se tyto nežádoucí účinky léčit sám(sama), ale obraťte se na svého lékaře nebo zdravotní sestru. V případě výskytu některých nežádoucích účinků může Váš lékař léčbu přerušit a odeslat Vás ke specialistovi.
Závažné nežádoucí účinky:
Porucha funkce jater:
(časté - mohou postihnout až 1 z 10 lidí)
• pocit na zvracení neznámého původu (nevolnost),
• zvracení,
• zvýšena únava,
• ztráta chuti k jídlu (anorexie),
• zežloutnutí kůže nebo očního bělma,
• tmavé (čajové) zabarvení moči.
Ihned kontaktujte svého lékaře. Může se jednat o příznaky závažné poruchy funkce jater. Více informací o těchto nežádoucích účincích naleznete v Kartě pacienta.
Kožní reakce:
(časté - mohou postihnout až 1 z 10 lidí)
• závažná rozsáhlá vyrážka.
Deprese:
(méně časté - mohou postihnout až 1 ze 100 lidí)
• pocit neobvyklého smutku, beznaděje či negativní pocity vůči sobě samému,
• podrážděnost, lehké rozrušení,
• nervozita, úzkost,
• sebevražedné myšlenky či myšlenky na sebepoškození.
Plicní infekce:
(časté - mohou postihnout až 1 z 10 lidí)
• plicní infekce (např. zápal plic, zánět průdušek)
Zánět zažívacího ústrojí (kolitida):
(méně časté - mohou postihnout až 1 ze 100 lidí)
• přetrvávající průjem,
• bolest břicha,
• horečka,
• krev ve stolici.
Bolest břicha může být rovněž příznakem poruchy funkce jater, viz bod výše Porucha funkce jater.
Nízká hladina určitého typu bílých krvinek (nazývaných lymfocyty):
Přípravek Zinbryta Vám může snížit hladinu těchto bílých krvinek, proto budete každé 3 měsíce absolvovat krevní testy.
Pokud se u Vás vyskytne kterýkoli ze závažných nežádoucích účinků, ihned kontaktujte svého lékaře. Ostatní nežádoucí účinky:
Velmi časté nežádoucí účinky
(mohou postihnout více než 1 z 10 lidí)
• infekce dýchacích cest, např. kašel či nachlazení (zánět sliznice nosohltanu, infekce horních cest dýchacích).
Časté nežádoucí účinky
(mohou postihnout až 1 z 10 lidí)
• chřipka,
• bolest v krku, zánět mandlí (zánět hltanu, zánět hrtanu),
• rýma,
• kožní vyrážka včetně zanícení, podráždění, svědění, odlupování či suchosti pokožky (dermatitida, ekzém, lupénka),
• kožní infekce (folikulitida-zánět vlasového míšku, akné),
• snížení počtu bílých krvinek (prokáže se při krevních testech),
• zvýšení tělesné teploty (horečka),
• zvýšení hladiny jaterních enzymů v krvi (prokáže se při krevních testech),
• zanícení či zvětšení lymfatických uzlin (lymfadenopatie, lymfadenitida-zánět mízních uzlin),
• průjem,
• změny v krvi (anémie), které mohou způsobit únavu či bledost.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nesnažte se nežádoucí účinky léčit sám(sama). Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zinbryta uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a štítku za "EXP". Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Uchovávejte předplněnou injekční stříkačku/pero přípravku Zinbryta v původním obalu, aby byl přípravek chráněn před světlem. Balení otevřete, pouze pokud budete potřebovat novou injekční stříkačku/pero.
• Uchovávejte v chladničce (2 °C - 8 °C).
o Chraňte před mrazem. Jakýkoli náhodně zmrazený přípravek Zinbryda zlikvidujte.
• Pokud není chladnička k dispozici, je možné injekční stříkačky/pera přípravku Zinbryta uchovávat při pokojové teplotě (do 30 oC) v původním obalu po dobu až 30 dní.
o Ujistěte se, že doba, po kterou je přípravek Zinbryta mimo chladničku, není delší než 30 dní. o Jestliže byl přípravek Zinbryta uchováván mimo chladničku do dobu delší než celkem 30 dní, nebo pokud si nejste jistý(á), jak dlouho byl přípravek Zinbryta uchováván při pokojové teplotě, injekční stříkačku/pero zlikvidujte (viz bod 7 Návod na injekční podání přípravku Zinbryta).
• Po zahřátí na pokojovou teplotu nedávejte přípravek Zinbryta zpět do chladničky.
Další informace
Nepoužívejte tento přípravek, pokud si všimnete následujícího:
• Jestliže je injekční stříkačka/pero prasklá(é) nebo rozbitá(é).
• Jestliže je roztok zakalený nebo jsou-li v něm vidět plovoucí částice.
• Jestliže roztok není bezbarvý až nažloutlý.
• Jestliže pero spadlo na zem nebo je viditelně poškozené.
Likvidace
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Zinbryta obsahuje
Léčivou látkou je: daclizumabum.
Jedna předplněná injekční stříkačka obsahuje daclizumabum 150 mg v 1 ml injekčního roztoku.
Jedno předplněné pero obsahuje daclizumabum 150 mg v 1 ml injekčního roztoku.
Dalšími složkami jsou: natrium-sukcinát, kyselina jantarová, chlorid sodný, polysorbát 80, voda na injekci (přečtěte si bod 2 Přípravek Zinbryta obsahuje malé množství sodíku).
Jak přípravek Zinbryta vypadá a co obsahuje toto balení
Zinbryta je bezbarvá až nažloutlá, čirá až opalescentní tekutina v injekční stříkačce/peru.
Velikosti balení: Jedno balení přípravku obsahuje jednu předplněnou skleněnou injekční stříkačku/jedno předplněné pero s nasazenou jehlou připravenou k injekčnímu podání. Dostupné je rovněž i vícečetné balení, které obsahuje tři balení po jedné injekční stříkačce/jednom peru.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Biogen Idec Ltd.
Innovation House 70 Norden Road Maidenhead Berkshire SL6 4AY Velká Británie
Výrobce
Biogen (Denmark) Manufacturing ApS
Biogen Allé 1
Hillerod
DK-3400
Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Biogen Belgium N.V./S.A.
2 +32 2 219 12 18
Btnrapnn
Tn EBOOAPMA
2 +359 2 962 12 00
Česká republika
Biogen (Czech Republic) s.r.o. 2 +420 255 706 200
Danmark
Biogen (Denmark) A/S 2 +45 77 41 57 88
Lietuva
UAB "JOHNSON & JOHNSON"
2 +370 5 278 68 88
Luxembourg/Luxemburg
Biogen Belgium N.V./S.A.
2 +32 2 219 12 18
Magyarország
Biogen Hungary Kft.
2 +36 (1) 899 9883
Malta
Pharma MT limited 2 +356 213 37008/9
Deutschland
Biogen GmbH m +49 (0) 89 99 6170
Eesti
UAB "JOHNSON & JOHNSON" Eesti filiaal m +372 617 7410
EXlába
Genesis Pharma SA m +30 210 8771500
Espaňa
Biogen Spain SL m +34 91 310 7110
France
Biogen France SAS m +33 (0)1 41 37 95 95
Hrvatska
Medis Adria d.o.o. m +385 (0) 1 230 34 46
Ireland
Biogen Idec (Ireland) Ltd. m +353 (0)1 463 7799
Ísland
Icepharma hf m +354 540 8000
Italia
Biogen Italia s.r.l.
m +39 02 584 9901
Kúrcpog
Genesis Pharma Cyprus Ltd m +357 22 769946
Latvija
UAB "JOHNSON & JOHNSON" filiale Latvija m +371 678 93561
Nederland
Biogen Netherlands B.V. m +31 20 542 2000
Norge
Biogen Norway AS m +47 23 40 01 00
Osterreich
Biogen Austria GmbH m +43 1 484 46 13
Polska
Biogen Poland Sp. z o.o. m +48 22 351 51 00
Portugal
Biogen Portugal Sociedade Farmaceutica Unipessoal, Lda m +351 21 318 8450
Románia
Johnson & Johnson Romania S.R.L. m +40 21 207 18 00
Slovenija
Biogen Pharma d.o.o. m +386 1 511 02 90
Slovenská republika
Biogen Slovakia s.r.o. m +421 2 323 340 08
Suomi/Finland
Biogen Finland Oy m +358 207 401 200
Sverige
Biogen Sweden AB m +46 8 594 113 60
United Kingdom
Biogen Idec Limited m +44 (0) 1628 50 1000
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Návod na druhé straně O
7. Návod na injekční podání přípravku Zinbryta
Jak přípravek Zinbryta injekčně podávat
Přečtěte si tento návod k použití dříve, než začnete přípravek Zinbryta používat a pokaždé, když dostanete
nové balení. Nové balení může obsahovat nové informace. Tato informace nenahrazuje konzultaci s Vaším
lékařem nebo zdravotní sestrou o Vašem zdravotním stavu a léčbě.
Poznámka:
• Před prvním použitím předplněné injekční stříkačky přípravku Zinbryta Vám nebo osobě, která o Vás pečuje, musí lékař nebo zdravotní sestra ukázat, jak si připravit a podat injekci pomocí předplněné injekční stříkačky přípravku Zinbryta.
A Nepoužívejte více než jednu předplněnou injekční stříkačku za měsíc.
• Předplněná injekční stříkačka přípravku Zinbryta je určena pouze k podkožnímu podání (subkutánní injekce).
• Každá předplněná injekční stříkačka přípravku Zinbryta je určena pouze pro jedno použití a nelze ji znovu použít. Nedávejte předplněnou injekční stříkačku přípravku Zinbryta žádné jiné osobě.
Co budete k podání injekce přípravku Zinbryta potřebovat
• Předplněnou injekční stříkačku přípravku Zinbryta
/ \
_11 'Ml
Předplněná injekční stříkačka 150 mg Obrázek A
v_I_z
Další pomůcky, které nejsou součástí balení (viz obrázek B):
• tampon napuštěný alkoholem
• gázový polštářek
• adhezivní (samolepicí) obvaz/náplast
Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jak zlikvidovat použité injekční stříkačky.
ALCOHOL
W1PE
Tampon s Gázový Náplast
alkoholem polštářek
Obrázek B
Součásti předplněné injekční stříkačky přípravku Zinbryta (viz obrázek C)
Kryt jeMy Skleněný
Léčiv° válec
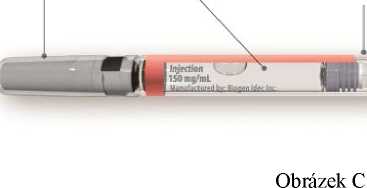
Příruba
Pro , Píst
uchopeni
Příprava na podání injekce Poznámka:
Než si připravíte injekci k podání, vyjměte z chladničky injekční stříkačku a nechte ji zahřát na pokojovou teplotu. To trvá zhruba 30 minut.
Á K zahřátí předplněné injekční stříkačky přípravku Zinbryta nepoužívejte vnější zdroje tepla, jako je horká voda.
Příruba umožní lepší uchopení injekční stříkačky a musí zůstat připojená.
Připravte si pomůcky a umyjte si ruce
Najděte si dobře osvětlenou, čistou, rovnou plochu, jako např. stůl. Připravte si veškeré pomůcky, které budete potřebovat, abyste si injekci mohl(a) sám(sama) podat, nebo aby Vám mohla být podána.
Umyjte si ruce mýdlem a vodou.
2. Zkontrolujte předplněnou injekční stříkačku přípravku Zinbryta
Zkontrolujte dobu použitelnosti uvedenou na předplněné injekční stříkačce přípravku Zinbryta (viz obrázek D).
A Nepoužívejte přeplněnou injekční stříkačku přípravku Zinbryta po _uplynutí doby použitelnosti._
Zkontrolujte, zda je léčivo Zinbryta bezbarvé až nažloutlé (viz obrázek E).
A Nepoužívejte předplněnou injekční stříkačku přípravku Zinbryta, jestliže je tekutina zakalená nebo obsahuje-li plovoucí částice.
o V léčivu Zinbryta můžete vidět
vzduchové bubliny. To je normální a bubliny není třeba před podáním injekce vytlačovat.
Doba
použitelnosti

3. Zvolte a očistěte místo podání injekce
Předplněná injekční stříkačka přípravku Zinbryta je určena k podkožnímu podání (subkutánní injekce).
Předplněná injekční stříkačka přípravku Zinbryta má být aplikována do stehna, břicha nebo zadní strany paže (viz obrázek
F)
A Injekci nepodávejte přímo do pupku.
A Injekci nepodávejte do místa na těle, kde je kůže podrážděná, citlivá, zarudlá, pohmožděná, potetovaná, zanícená či zjizvená.
Zvolte si místo podání injekce a otřete kůži tamponem napuštěným alkoholem.
Před podáním injekce nechte zvolené místo podání oschnout.
Břicho
Zadní strana paže
Obrázek F
a
Před podáním injekce se tohoto místa již nedotýkejte, ani na něj nefoukejte.
4. Odstraňte kryt jehly
A
A
A
Jednou rukou uchopte injekční stříkačku za skleněný válec. Dbejte na to, abyste touto rukou netlačil(a) na přírubu. Druhou rukou pevně uchopte kryt jehly a stáhněte jej z jehly v přímém směru (viz obrázek G).
Při odstraňování krytu jehly postupujte opatrně, abyste se o jehlu neporanil(a).
Nedotýkejte se jehly.
Upozornění - Kryt jehly na předplněnou injekční stříkačku přípravku Zinbryta znovu nenasazujte. Mohl(a) byste se o jehlu poranit.
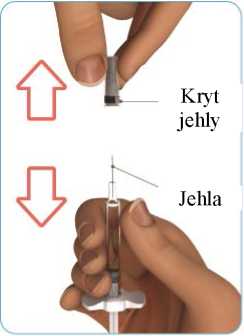
Obrázek G
Palcem a ukazováčkem jemně stiskněte kůži v okolí očištěného místa podání injekce tak, aby vznikla kožní řasa (viz obrázek H).
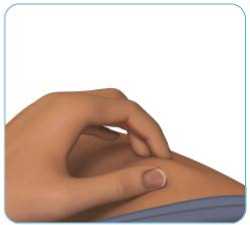
Obrázek H
6. Aplikujte injekci
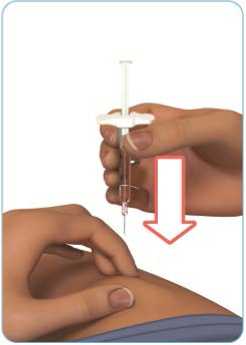
a
Držte předplněnou injekční stříkačku přípravku Zinbryta pod úhlem 45°-90° k místu podání injekce (viz obrázek I). Jehlu rychle vpíchněte přímo do kožního ohybu. Do kůže musí proniknout celá jehla (viz obrázek I.)
Po vpíchnutí celé jehly kůži uvolněte.
Netahejte za píst injekční stříkačky zpětným pohybem.
Obrázek I
A
Pomalu stlačujte píst, dokud se injekční stříkačka zcela nevyprázdní (viz obrázek J).
Dokud nebude píst úplně zatlačen, předplněnou injekční stříkačku přípravku Zinbryta z místa vpichu nevytahujte.
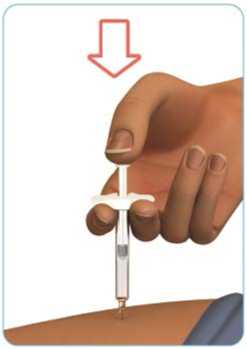
Obrázek J
Vytáhněte jehlu přímým pohybem vzhůru (viz obrázek K).
Upozornění - Kryt jehly na předplněnou injekční stříkačku přípravku Zinbryta znovu nenasazujte. Mohl(a) byste se o jehlu poranit.
Předplněná injekční stříkačka přípravku Zinbryta není určena k opakovanému použití.
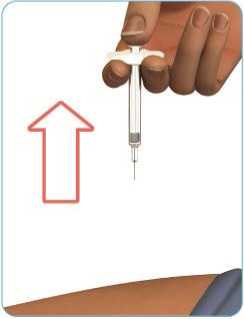
Obrázek K
Po podání injekce
8. Zlikvidujte použitou předplněnou injekční stříkačku přípravku Zinbryta
• Poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou, jak správně zlikvidovat použitou injekční stříkačku.
9. Ošetřete místo podání injekce
• Dle potřeby přiložte na místo podání injekce gázový polštářek, obvaz či náplast.
Všeobecná upozornění
A Předplněná injekční stříkačka přípravku Zinbryta není určena k opakovanému použití.
A Předplněnou injekční stříkačku přípravku Zinbryta nedávejte žádné další osobě.
• Uchovávejte přeplněnou injekční stříkačku přípravku Zinbryta a veškeré ostatní léčivé přípravky mimo dosah a dohled dětí.
Podmínky uchovávání
• Uchovávejte v chladničce při teplotě mezi 2 °C a 8 °C v původním uzavřeném obalu, aby byl přípravek chráněn před světlem.
• Pokud je to nutné, je možné přípravek Zinbryta uchovávat mimo chladničku při teplotě do 30 °C
v původním uzavřeném obalu po dobu až 30 dní.
A Po zahřátí na pokojovou teplotu nedávejte předplněnou injekční stříkačku přípravku Zinbryta zpět do chladničky.
A Chraňte před mrazem a nevystavujte vysokým teplotám.
Návod na injekční podání přípravku Zinbryta
7.
Jak přípravek Zinbryta injekčně podávat
Přečtěte si tento návod k použití dříve, než začnete přípravek Zinbryta používat a pokaždé, když dostanete nové balení. Nové balení může obsahovat nové informace. Tato informace nenahrazuje konzultaci s Vaším lékařem nebo zdravotní sestrou o Vašem zdravotním stavu a léčbě.
Poznámka:

Před prvním použitím pera Vám nebo osobě, která o Vás pečuje, musí lékař nebo zdravotní sestra ukázat, jak si připravit a podat injekci pomocí pera.
Nepoužívejte více než jedno pero za měsíc.
Upozornění - neodstraňujte víčko z pera, dokud nebudete připraven(a) aplikovat si injekci. Nepoužívejte pero, pokud spadlo na zem nebo je-li viditelně poškozené.
Pero je určeno pouze k podkožnímu podání (subkutánní injekce).
Každé pero je určeno pouze pro jedno použití. Nedávejte pero žádné jiné osobě.
Co budete k podání injekce pomocí pera přípravku Zinbryta potřebovat (viz obrázek A):
1 pero přípravku Zinbryta 150 mg
Další pomůcky, které nejsou součástí balení (viz obrázek B):
• tampon napuštěný alkoholem
• gázový polštářek
• adhezivní (samolepicí) obvaz/náplast
Zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jak zlikvidovat použitá pera.

Zobrazují se zelené pruhy
Tělo Okénko zobrazující
| léčivo
1
Víčko
Stavové Před aplikací je viditelné léčivo.
okénko
Obrázek C
Po použití - Součásti pera přípravku Zinbryta (viz obrázek D):
Zobrazují se zelené zaškrtávací symboly
Kryt jehly
,
Tělo Okénko zobrazující léčivo

Po aplikaci je viditelný žlutý píst. Obrázek D
Stavové okénko
Jehla
Poznámka: Jehla je zakrytá krytem jehly a není vidět (viz obrázek D). Nedotýkejte se krytu jehly, ani na něj netlačte, mohl(a) byste se o jehlu poranit.
Příprava na podání injekce
Poznámka:
• Než si připravíte injekci k podání, vyjměte z chladničky pero a nechte jej zahřát na pokojovou teplotu. To trvá zhruba 30 minut.
A K zahřátí pera nepoužívejte vněj ší zdroje tepla, jako je horká voda.
Upozornění - neodstraňujte víčko z pera, dokud nebudete pnpraven(a) aplikovat si injekci.
1. Připravte si pomůcky a umyjte si ruce
• Najděte si dobře osvětlenou, čistou, rovnou plochu, jako např. stůl. Připravte si veškeré pomůcky, které budete potřebovat, abyste si injekci mohl(a) podat, nebo aby Vám mohla být podána.
Umyjte si ruce mýdlem a vodou
Zkontrolujte dobu použitelnosti uvedenou na peru (viz obrázek E).
Zkontrolujte stavové okénko na peru. Ujistěte se, že ve stavovém okénku jsou viditelné zelené pruhy (viz obrázek F).
Na peru zkontrolujte okénko zobrazující léčivo a ujistěte se, že léčivo Zinbryta je bezbarvé až nažloutlé (viz obrázek G). o V okénku zobrazujícím léčivo můžete vidět vzduchové bubliny. To je normální a nemá to vliv na Vaši dávku léčiva.
Nepoužívejte pero, jestliže:
o uplynula doba použitelnosti
(obrázek E).
o ve stavovém okénku nevidíte zelené pruhy (obrázek F). o tekutina je zakalená nebo obsahuje-li plovoucí částice
(obrázek G).
Doba
použitelnosti
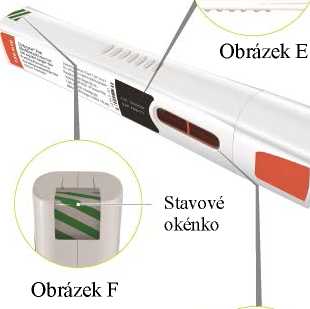
Okénko
zobraz
léčivo
zobrazující
Obrázek G
Podání injekce
3. Zvolte a očistěte místo podání injekce
Pero je učeno pouze k podkožnímu podání (subkutánní injekce).
Pero má být aplikováno do stehna, břicha nebo zadní strany paže (viz vyznačená místa na obrázku H).
Injekci nepodávejte do místa na těle, kde je kůže podrážděná, zarudlá, pohmožděná, potetovaná, zanícená či
zjizvená.
Injekci nepodávejte přímo do pupku, o Jestliže j sou některá místa pro aplikaci
pro Vás obtížně dosažitelná, požádejte o pomoc osobu, která o Vás pečuje a která byla zaškolena.
Zvolte si místo podání injekce a otřete kůži tamponem napuštěným alkoholem.
Před podáním injekce nechte zvolené místo oschnout.
Před podáním injekce se tohoto místa již nedotýkejte, ani na něj nefoukejte.
Obrázek H
Břicho
Zadní strana paže
Stáhněte víčko z pera v přímém směru a dejte je stranou na zlikvidování po aplikaci injekce (viz obrázek I).
o Jehla j e zakrytá krytem j ehly a není vidět (viz obrázek I).
Po odstranění víčka je pero připraveno k podání injekce.
A Upozornění - víčko na pero znovu nenasazujte.
A Upozornění - nedotýkejte se krytu jehly, ani na něj netlačte, mohl(a) byste se o jehlu poranit.
|
o U |
1 j |
|
Víčko | |
|
■ |
_. Kryt |
|
jeMy | |
|
l 1 JI |
\_) |
Obrázek I
5. Umístěte pero přípravku Zinbryta nad místo podání injekce
Umístěte pero nad zvolené místo podání injekce.
o Pero držte kolmo k místu podání injekce.
o Ujistěte se, že ve stavovém okénku vidíte zelené pruhy (viz obrázek J).
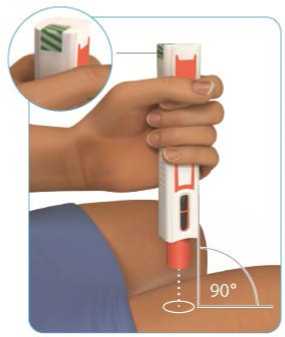
Obrázek J
Pevně zatlačte pero do místa podání injekce a držte jej. Zatlačením pera směrem dolů se vpíchne jehla a automaticky se spustí podání injekce (viz obrázek K).

injekce.
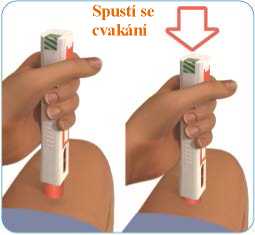
Obrázek K
Až “cvakání” ustane, zkontrolujte stavové okénko. Ujistěte se, že ve stavovém okénku jsou viditelné zelené zaškrtávací symboly (viz obrázek L).
A Upozornění - dokud neustane “cvakání” a dokud ve stavovém okénku neuvidíte zelené zaškrtávací symboly, pero z místa podání injekce nezvedejte.
7-y
Cvaknutí! Cvakání Cvaknutí!
Cvakn nu
..... 171
ustane
Obrázek L
7. Vytáhněte pero přípravku Zinbryta z místa podání injekce
Zvedněte pero z místa podání injekce. Vysune se kryt jehly a jehlu zakryje (viz obrázek M).
Víčko na pero znovu nenasazujte.
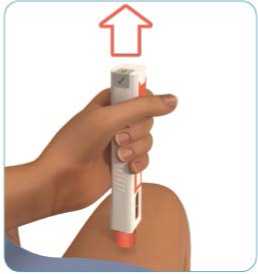
Obrázek M
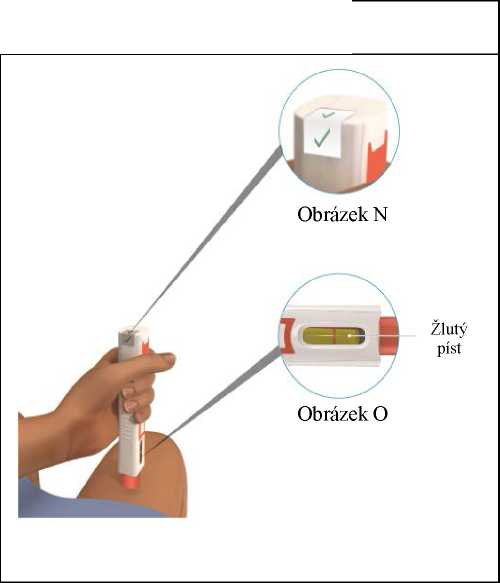
8. Ujistěte se, že jste si podal(a) / byla Vám podána plná dávka přípravku Zinbryta
Zkontrolujte stavové okénko. Ujistěte se, že j sou ve stavovém okénku vidět zelené zaškrtávací symboly (viz obrázek N).
Zkontrolujte okénko zobrazující léčivo. V okénku zobrazujícím léčivo byste měl(a) vidět žlutý píst (viz obrázek O).
Jestliže v místě podání injekce spatříte krev, setřete ji gázovým polštářkem a použijte obvaz nebo místo přelepte náplastí.
|
Po podání injekce | |
|
9. |
Zlikvidujte použité pero přípravku Zinbryta |
|
• |
Poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou, jak správně zlikvidovat použitá |
|
pera. | |
|
10. |
Ošetřete místo podání injekce |
|
• |
Dle potřeby přiložte na místo podání injekce gázový polštářek, obvaz či náplast. |
Podmínky uchovávání
• Uchovávejte v chladničce při teplotě mezi 2 °C a 8 °C v původním uzavřeném obalu, aby byl přípravek chráněn před světlem.
• Pokud je to nutné, je možné přípravek Zinbryta uchovávat mimo chladničku při teplotě do 30 °C
v původním uzavřeném obalu po dobu až 30 dní.
Po zahřátí na pokojovou teplotu nedávejte pero přípravku Zinbryta zpět do chladničky.
A Chraňte před mrazem a nevystavujte vysokým teplotám.
• Uchovávejte pero přípravku Zinbryta a veškeré ostatní léčivé přípravky mimo dosah a dohled dětí.
54