Zelboraf 240 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Zelboraf 240 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje 240 mg vemurafenibum (ve formě ko-precipitátu vemurafenibu a acetátsukcinátu hypromelosy).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Růžovo-bílé až oranžovo-bílé, oválné, bikonvexní potahované tablety o velikosti přibližně 19 mm s vyraženým „VEM“ na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Vemurafenib je indikován v monoterapii k léčbě dospělých pacientů s neresekovatelným nebo metastazujícím melanomem s pozitivní mutací V600 genu BRAF (viz bod 5.1).
4.2 Dávkování a způsob podání
Léčbu vemurafenibem má zahajovat a dohlížet na ni kvalifikovaný lékař se zkušenostmi s podáváním protinádorových léčivých přípravků.
Před zahájením léčby vemurafenibem musí být u pacientů validovaným testem potvrzena pozitivní mutace V600 genu BRAF (viz body 4.4 a 5.1).
Dávkování
Doporučená dávka vemurafenibu je 960 mg (4 tablety po 240 mg) dvakrát denně (odpovídá celkové denní dávce 1920 mg). Vemurafenib může být užíván s jídlem, nebo bez jídla, ale je třeba se vyvarovat podání obou denních dávek na prázdný žaludek (viz bod 5.2).
Trvání léčby
V léčbě vemurafenibem se má pokračovat až do progrese onemocnění nebo do rozvoje nepřijatelné toxicity (viz tabulky 1 a 2 níže).
Vynechané dávky
Pokud dojde k vynechání dávky, je možné ji užít nejpozději 4 hodiny před další dávkou, aby bylo dodrženo dávkovací schéma dvakrát denně. Obě dávky se nemají užít ve stejnou dobu.
V případě zvracení po podání vemurafenibu nemá pacient užívat dodatečnou dávku léčivého přípravku, ale léčba má pokračovat jako obvykle.
Úprava dávkování
Léčba nežádoucích účinků nebo prodloužení QT intervalu může vyžadovat snížení dávky, dočasné přerušení léčby a/nebo její ukončení (viz tabulky 1 a 2). Úprava dávkování, která by vedla k dávkám nižším než 480 mg dvakrát denně, se nedoporučuje.
Pokud u pacienta dojde ke vzniku kožního spinocelulárního karcinomu, doporučuje se pokračovat v léčbě bez úpravy dávky vemurafenibu (viz body 4.4 a 4.8).
Tabulka 1: Schéma úpravy dávkování založené na stupni závažnosti nežádoucích účinků
|
Stupeň (CTC-AE)(a) |
Doporučená úprava dávky |
|
Stupeň 1 nebo stupeň 2 (tolerovatelný) |
Udržení vemurafenibu na dávce 960 mg dvakrát denně. |
|
Stupeň 2 (netolerovatelný) nebo stupeň 3 | |
|
1. výskyt jakéhokoli nežádoucího účinku stupně 2 nebo 3 |
Přerušení léčby až do dosažení stupně 0-1. Znovuzahájení léčby dávkou 720 mg dvakrát denně (nebo 480 mg dvakrát denně, pokud již došlo ke snížení dávky). |
|
2. výskyt jakéhokoli nežádoucího účinku stupně 2 nebo 3 nebo přetrvávání po přerušení léčby |
Přerušení léčby až do dosažení stupně 0-1. Znovuzahájení léčby dávkou 480 mg dvakrát denně (nebo trvalé ukončení léčby, pokud již dávka byla snížena na 480 mg dvakrát denně). |
|
3. výskyt jakéhokoli nežádoucího účinku stupně 2 nebo 3 nebo přetrvávání po druhém snížení dávky |
Trvalé ukončení léčby. |
|
Stupeň 4 | |
|
1. výskyt jakéhokoli nežádoucího účinku stupně 4 |
Trvalé ukončení léčby nebo přerušení léčby vemurafenibem až do dosažení stupně 0-1. Znovuzahájení léčby dávkou 480 mg dvakrát denně (nebo trvalé ukončení léčby, pokud již dávka byla snížena na 480 mg dvakrát denně). |
|
2. výskyt jakéhokoli nežádoucího účinku stupně 4 nebo přetrvávání jakéhokoli nežádoucího účinku stupně 4 po prvním snížení dávky |
Trvalé ukončení léčby. |
(a) Intenzita klinických nežádoucích účinků byla stanovena na základě kritérií CTC-AE (Common Terminology Criteria for Adverse Events) verze 4.0.
Prodloužení QT intervalu závislé na expozici bylo pozorováno v nekontrolované, otevřené studii fáze II u dříve léčených pacientů s metastazujícím melanomem. Léčba prodloužení QTc může vyžadovat specifická opatření týkající se sledování pacienta (viz bod 4.4).
Tabulka 2: Schéma úpravy dávkování založené na prodloužení QT intervalu
|
Hodnota QTc |
Doporučená úprava dávky |
|
QTc > 500 ms v úvodu |
Léčba se nedoporučuje. |
|
Zvýšení QTc dosahuje hodnot > 500 ms a zároveň > 60 ms změny od hodnot před léčbou |
Trvalé ukončení léčby. |
|
1. výskyt QTc > 500 ms během léčby a změna od hodnot před léčbou zůstává < 60 ms |
Dočasné přerušení léčby, dokud QTc nepoklesne pod 500 ms. Viz opatření týkající se sledování pacienta v bodě 4.4. Znovuzahájení léčby dávkou 720 mg dvakrát denně (nebo 480 mg dvakrát denně, pokud dávka již byla snížena). |
|
2. výskyt QTc > 500 ms během léčby a změna od hodnot před léčbou zůstává < 60 ms |
Dočasné přerušení léčby, dokud QTc nepoklesne pod 500 ms. Viz opatření týkající se sledování pacienta v bodě 4.4. Znovuzahájení léčby dávkou 480 mg dvakrát denně (nebo trvalé ukončení léčby, pokud dávka již byla snížena na 480 mg dvakrát denně). |
|
3. výskyt QTc > 500 ms během léčby a změna od hodnot před léčbou zůstává < 60 ms |
Trvalé ukončení léčby. |
Zvláštní populace Starší lidé
U pacientů ve věku > 65 let není nutná žádná zvláštní úprava dávky.
Porucha renálních funkcí
U pacientů s poruchou renálních funkcí jsou k dispozici pouze omezené údaje. Riziko zvýšení expozice u pacientů se závažnou poruchou renálních funkcí nelze vyloučit. Pacienti se závažnou poruchou renálních funkcí mají být pečlivě sledováni (viz body 4.4 a 5.2).
Porucha jaterních funkcí
U pacientů s poruchou jaterních funkcí jsou k dispozici pouze omezené údaje. Vzhledem k tomu, že se vemurafenib vylučuje prostřednictvím jater, může u pacientů se středně závažnou až závažnou poruchou jaterních funkcí dojít ke zvýšení expozice a tito pacienti mají být pečlivě sledováni (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost vemurafenibu u dětí a dospívajících (< 18 let) nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Pacienti jiné než bílé rasy
Bezpečnost a účinnost vemurafenibu u pacientů jiné než bílé rasy nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Tablety vemurafenibu se polykají celé a zapíjejí vodou. Tablety vemurafenibu se nemají žvýkat ani drtit.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Před zahájením léčby vemurafenibem je nutné u pacientů potvrdit pozitivitu mutace V600 genu BRAF validovaným testem. Účinnost a bezpečnost vemurafenibu nebyla přesvědčivě stanovena u pacientů s nádory exprimujícími vzácné mutace V600 genu BRAF, jiné než V600E a V600K (viz bod 5.1). Vemurafenib se nemá používat u pacientů s maligním melanomem s divokým typem genu BRAF.
Reakce přecitlivělosti
V souvislosti s léčbou vemurafenibem byly hlášeny závažné reakce přecitlivělosti včetně anafylaxe (viz body 4.3 a 4.8). Závažné reakce přecitlivělosti mohou zahrnovat Stevens-Johnsonův syndrom, generalizovanou vyrážku, erytém nebo hypotenzi. U pacientů, u kterých dojde k rozvoji závažných reakcí přecitlivělosti, je nutné léčbu vemurafenibem trvale ukončit.
Dermatologické reakce
U pacientů užívajících vemurafenib v klíčové klinické studii byly hlášeny závažné dermatologické reakce, včetně vzácných případů Stevens-Johnsonova syndromu a toxické epidermální nekrolýzy. V post-marketingovém sledování byla v souvislosti s vemurafenibem hlášena poléková reakce s eosinofilií a systémovými příznaky (DRESS) (viz bod 4.8). U pacientů, u kterých dojde k rozvoji závažné dermatologické reakce, je nutné léčbu vemurafenibem trvale ukončit.
Potenciace radiační toxicity
Byly hlášeny případy kožní reakce na ozáření (radiační recall) a radiační senzibilizace u pacientů léčených radiací buď před, v průběhu nebo následně po léčbě vemurafenibem. Ve většině případů se jednalo o kožní reakce obecně, avšak v případech zasažení vnitřních orgánů došlo k úmrtím (viz body
4.5 a 4.8). Vemurafenib je třeba užívat s opatrností, pokud je podáván současně nebo sekvenčně s radiační léčbou.
Prodloužení QT intervalu
V nekontrolované, otevřené studii fáze II u již dříve léčených pacientů s metastazujícím melanomem bylo pozorováno prodloužení QT intervalu závislé na expozici (viz bod 4.8). Prodloužení QT intervalu může vést ke zvýšení rizika ventrikulárních arytmií včetně torsade de pointes. Léčba vemurafenibem se nedoporučuje u pacientů s nekontrolovanými abnormalitami elektrolytové rovnováhy (včetně hořčíku), syndromem dlouhého QT intervalu ani u pacientů, kteří užívají léčivé přípravky, o kterých je známo, že prodlužují QT interval.
Před zahájením léčby vemurafenibem, po jednom měsíci léčby a po úpravě dávek je u všech pacientů nutné sledovat elektrokardiogram (EKG) a hladiny elektrolytů (včetně hořčíku). Další sledování se doporučuje zejména u pacientů se středně závažnou až závažnou poruchou funkce jater a provádí se každý měsíc během prvních 3 měsíců léčby a poté každé 3 měsíce nebo častěji podle klinické indikace. Zahájení léčby vemurafenibem se nedoporučuje u pacientů s QTc intervalem > 500 ms. Pokud během léčby dojde k prodloužení QTc intervalu nad 500 ms, je třeba léčbu vemurafenibem dočasně přerušit, upravit elektrolytovou rovnováhu (včetně hořčíku) a zkontrolovat kardiální rizikové faktory prodloužení QT intervalu (např. městnavé srdeční selhání, bradyarytmie). Jakmile dojde ke zkrácení QTc intervalu pod 500 ms, je možné léčbu znovu zahájit nižšími dávkami, jak je popsáno v tabulce 2. Trvalé ukončení léčby vemurafenibem se doporučuje v případě, kdy prodloužení QTc intervalu dosáhne hodnoty > 500 ms a zároveň změny > 60 ms oproti hodnotám před léčbou.
Oftalmologické reakce
Byly hlášeny závažné oftalmologické reakce, včetně uveitidy, iritidy a uzávěru retinální žíly. Pacienty je nutné rutinně sledovat pro možné oftalmologické reakce.
Kožní spinocelulární karcinom
U pacientů léčených vemurafenibem byly hlášeny případy kožního spinocelulárního karcinomu (které zahrnují nádory klasifikované jako subtyp keratoakantomu nebo smíšeného keratoakantomu) (viz bod 4.8).
Doporučuje se, aby všichni pacienti podstoupili před zahájením léčby dermatologické vyšetření a byli rutinně sledováni i v průběhu léčby. Jakékoli podezřelé kožní léze je nutné excidovat, odeslat na dermatopatologické vyšetření a léčit v souladu s místními standardy péče. Předepisující lékař má během léčby a až šest měsíců po jejím ukončení pacienty každý měsíc vyšetřovat na přítomnost kožního spinocelulámího karcinomu. U pacientů, u kterých dojde k rozvoji kožního spinocelulámího karcinomu, se doporučuje pokračovat v léčbě bez úpravy dávek. Ve sledování je nutné pokračovat po dobu 6 měsíců od ukončení léčby vemurafenibem nebo do zahájení jiné protinádorové léčby. Pacienty je nutné poučit, aby svému lékaři hlásili vznik jakýchkoli kožních změn.
Spinocelulární karcinom v jiné než kožní lokalizaci
V klinických studiích, ve kterých byli pacienti léčeni vemurafenibem, byly hlášeny případy spinocelulámího karcinomu v jiné než kožní lokalizaci.
Pacienti mají před zahájením léčby a dále každé 3 měsíce v průběhu léčby podstupovat vyšetření hlavy a krku, které se skládá minimálně z vyšetření ústní sliznice pohledem a vyšetření lymfatických uzlin pohmatem. Dále pacienti mají před zahájením léčby a poté každých 6 měsíců v průběhu léčby podstupovat vyšetření hrudníku výpočetní tomografií (CT).
Před zahájením léčby a po jejím ukončení, nebo na základně klinických indikací se doporučuje vyšetření konečníku a vyšetření malé pánve (u žen).
Po ukončení léčby vemurafenibem se ve sledování s ohledem na výskyt spinocelulárního karcinomu v jiné než kožní lokalizaci pokračuje po dobu až 6 měsíců nebo do zahájení jiné protinádorové léčby. Abnormální nálezy je třeba zhodnotit v souladu s klinickou praxí.
Nový primární melanom
V klinických studiích byly hlášeny případy nového primárního melanomu. Tyto případy byly řešeny excizí a pacienti pokračovali v léčbě bez úpravy dávkování. Vyšetřování kožních lézí má být prováděno tak, jak je uvedeno výše u kožního spinocelulámího karcinomu.
Další malignity
Na základě mechanismu účinku může vemurafenib způsobit progresi nádoru souvisejícího s RAS mutací (viz bod 4.8). Pečlivě zvažte prospěch a rizika před podáním vemurafenibu pacientům s dřívějším nebo současným nádorem souvisejícím s RAS mutací.
Pankreatitida
Pankreatitida byla hlášena u pacientů léčených vemurafenibem. Bolesti břicha nejasného původu mají být ihned vyšetřeny (včetně měření amylázy a lipázy v séru). Pacienti po epizodě pankreatitidy mají být pečlivě sledováni při znovu nasazení léčby vemurafenibem.
Poškození jater
Při užívání vemurafenibu bylo hlášeno poškození jater, včetně případů závažného poškození jater (viz bod 4.8). Jaterní enzymy (transaminázy a alkalickou fosfatázu) a bilirubin mají být měřeny před zahájením léčby a dále sledovány jednou měsíčně v průběhu léčby nebo podle klinické indikace. Laboratorní abnormality je nutné zvládnout snížením dávky, přerušením léčby nebo jejím ukončením (viz body 4.2 a 4.8).
Renální toxicita
Při užívání vemurafenibu byla hlášena renální toxicita, v rozsahu od zvýšení hladin kreatininu v séru až po akutní intersticiální nefritidu a akutní tubulární nekrózu. Hladina kreatininu v séru má být měřena před zahájením léčby a nadále sledována v průběhu léčby dle klinické indikace (viz body 4.2 a 4.8).
Porucha jaterních funkcí
U pacientů s poruchou jaterních funkcí není nutné upravovat výchozí dávku. Pacienti s mírnou poruchou jaterních funkcí způsobenou jaterními metastázami bez hyperbilirubinemie mohou být sledováni podle všeobecných doporučení. K dispozici jsou pouze velmi omezené údaje týkající se pacientů se středně závažnou až závažnou poruchou jaterních funkcí. U pacientů se středně závažnou až závažnou poruchou jaterních funkcí může docházet ke zvýšení expozice (viz bod 5.2). Proto je nutné pečlivé sledování zejména po prvních několika týdnech léčby, protože k akumulaci může docházet během delšího období (několik týdnů). Během prvních tří měsíců léčby se navíc doporučuje pravidelné sledování EKG jednou měsíčně.
Porucha renálních funkcí
U pacientů s mírnou až středně závažnou poruchou renálních funkcí není nutné upravovat výchozí dávku. K dispozici jsou pouze omezené údaje týkající se pacientů se závažnou poruchou renálních funkcí (viz bod 5.2). Vemurafenib je třeba používat s opatrností u pacientů se závažnou poruchou renálních funkcí a tyto pacienty je třeba pečlivě sledovat.
Fotosenzitivita
U pacientů léčených vemurafenibem v klinických studiích byla hlášena mírná až těžká fotosenzitivita (viz bod 4.8). Všichni pacienti mají být poučeni, aby se během užívání vemurafenibu nevystavovali slunci. Pacienty je nutné poučit, aby během užívání tohoto léčivého přípravku nosili venku ochranné oblečení a užívali opalovací krémy se širokým spektrem ochrany proti UVA/UVB a balzám na rty (SPF > 30), zabrání tak spálení sluncem.
Při fotosenzitivitě 2. stupně (netolerovatelné) nebo vyšší se doporučuje úprava dávkování (viz bod 4.2).
Vliv vemurafenibu na jiné léčivé přípravky
Vemurafenib může zvýšit plazmatickou expozici léčivých přípravků predominantně metabolizovaných CYP1A2 a snížit plazmatickou expozici léků metabolizovaných predominantně CYP3A4, včetně perorální antikoncepce. Před zahájením společného podávání s vemurafenibem má být na základě terapeutického rozpětí zvážena úprava dávky léčivých přípravků, které jsou predominantně metabolizovány CYP1A2 nebo CYP3A4 (viz body 4.5 a 4.6).
Pokud je vemurafenib používán souběžně s warfarinem, je třeba zvýšit opatrnost a zvážit doplňující pravidelné sledování INR (International Normalized Ratio).
Vemurafenib může zvýšit plazmatickou expozici léčivých přípravků, které jsou substráty P-gp. Je třeba zvýšené opatrnosti a zvážení možného snížení dávky a/nebo doplňující sledování hladiny léčiva kvůli úzkému terapeutickému indexu (NTI) léčivých přípravků, které jsou substráty P-gp (např. digoxin, dabigatran-etexilát, aliskiren), pokud jsou tyto léčivé přípravky užívány souběžně s vemurafenibem (viz bod 4.5).
Vliv jiných léčivých přípravků na vemurafenib
Farmakokinetika vemurafenibu může být ovlivněna léčivy, které inhibují nebo ovlivňují P-gp (např. verapamil, klaritromycin, cyklosporin, ritonavir, chinidin, dronedaron, amiodaron, itrakonazol, ranolazin) (viz bod 4.5).
Současné podání silných induktorů P-gp, glukuronidace, CYP3A4 (např. rifampicin, rifabutin, karbamazepin, fenytoin nebo třezalka tečkovaná [hypericin]) má být pokud možno vyloučeno (viz bod 4.5). Pro zachování účinnosti vemurafenibu má být zvážena alternativní léčba s nižším indukčním potenciálem.
Současné podání s ipilimumabem
Ve studii fáze I byla hlášena asymptomatická zvýšení transamináz (ALT/AST > 5x ULN) a bilirubinu (celkový bilirubin > 3x ULN) stupně 3 při současném podání ipilimumabu (3 mg/kg) a vemurafenibu (960 mg 2x denně nebo 720 mg 2x denně). Na základě těchto předběžných údajů není současné podání ipilimumabu a vemurafenibu doporučeno.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vliv vemurafenibu na substráty CYP
Vemurafenib je středně silný inhibitor CYP1A2. Opakované dávky 960 mg vemurafenibu dvakrát denně zvýšily Cmax jednorázové 2 mg dávky tizanidinu (senzitivní substrát CYP1A2) přibližně 2,2krát (geometricky průměrný poměr, v rozsahu 0,7násobně-4,9násobně) a AUCinf přibližně 4,7krát (geometricky průměrný poměr, v rozsahu 0,9násobně-16násobně). V další klinické studii bylo pozorováno po jednorázové dávce kofeinu podané společně po opakovaném dávkování vemurafenibu po dobu 15 dnů průměrné 2,6násobné zvýšení (maximálně až 10násobné) expozice kofeinu v plazmě po léčbě vemurafenibem. Vemurafenib může proto zvýšit plazmatickou expozici látek metabolizovaných predominantně CYP1A2 (např. agomelatin, alosetron, duloxetin, melatonin, ramelteon, takrin, tizanidin, theofylin), a pokud je to klinicky indikováno, měla by být zvážena úprava dávky.
Indukce CYP3A4 byla pozorována v klinických studiích po jednorázové dávce midazolamu podané společně po opakovaném dávkování vemurafenibu po dobu 15 dnů. To vedlo k průměrnému 39 % snížení (maximálně až 80 %) plazmatické expozice midazolamu po léčbě vemurafenibem. Vemurafenib může snížit plazmatickou expozici látek metabolizovaných predominantně CYP3A4. Může tak být snížena účinnost antikoncepčních pilulek metabolizovaných CYP3A4, pokud jsou užívány souběžně s vemurafenibem. Je třeba zvážit úpravu dávky substrátů CYP3A4 s úzkým terapeutickým oknem, pokud je to klinicky indikováno (viz body 4.4 a 4.6).
In vitro byla pozorována mírná indukce CYP2B6 vemurafenibem při koncentracích vemurafenibu 10 pmol. V současné době není známo, zda vemurafenib při plazmatické hladině 100 pmol, pozorované u pacientů v ustáleném stavu (přibližně 50 pg/ml), může snižovat plazmatické koncentrace současně podávaných CYP2B6 substrátů, jako je bupropion.
Pokud se jednorázová dávka warfarinu podala společně po opakovaném dávkování vemurafenibu po dobu 15 dnů, došlo u některých pacientů ke zvýšení expozice warfarinu (průměrně o 18 %) (viz bod 4.4). Při společném podávání vemurafenibu s warfarinem (CYP2C9) u pacientů s melanomem je třeba postupovat s opatrností.
Vemurafenib inhiboval CYP2C8 in vitro. In vivo význam tohoto nálezu není znám, ale riziko pro klinicky relevantní účinek na současně podávané substráty CYP2C8 nemůže být vyloučeno.
Vzhledem k dlouhému poločasu vemurafenibu nelze plný inhibiční účinek vemurafenibu na společně podávané léčivé přípravky pozorovat před 8. dnem léčby vemurafenibem.
Po ukončení léčby vemurafenibem může být nutné vymývací období 8 dní, aby se zabránilo vzájemnému působení s následující léčbou.
Radiační léčba
Byla hlášena potenciace toxicity radiační léčby u pacientů, kterým byl podáván vemurafenib (viz body 4.4 a 4.8). Ve většině případů byly pacientům podávány režimy radioterapie větší nebo rovny 2 Gy/den (hypofrakcionační režimy).
Interakce vemurafenibu s transportními systémy látek
In vitro studie prokázaly, že vemurafenib je inhibitorem efluxních transportérů P-glykoproteinu (P-gp) a proteinu rezistence nádoru prsu (BCRP).
Klinická studie lékové interakce prokázala, že vícenásobná podání perorálních dávek vemurafenibu (960 mg dvakrát denně) zvýšila expozici jednorázové perorální dávky substrátu P-gp digoxinu, což představuje přibližně 1,8násobné zvýšení AUClast digoxinu a 1,5násobné zvýšení Cmax digoxinu. Zvýšené opatrnosti je třeba při podávání vemurafenibu souběžně se substráty P-gp (např. aliskiren, ambrisentan, kolchicin, dabigatran-etexilát, digoxin, everolimus, fexofenadin, lapatinib, maravirok, nilotinib, posakonazol, ranolazin, sirolimus, sitagliptin, talinolol, topotekan) a pokud je to klinicky indikováno, mělo by být zváženo snížení dávky léčivého přípravku podávaného současně. Je třeba zvážit doplňující sledování hladiny léčiva kvůli úzkému terapeutickému indexu (NTI) léčivých přípravků, které jsou substráty P-gp (např. digoxin, dabigatran-etexilát, aliskiren) (viz bod 4.4).
Účinky vemurafenibu na léky, které jsou substráty BCRP nejsou známé. Nelze vyloučit, že vemurafenib může zvyšovat expozici léků transportovaných pomocí BCRP (např. methotrexát, mitoxantron, rosuvastatin).
Mnoho protinádorových léků jsou substráty BCRP a proto je zde teoretické riziko interakcí s vemurafenibem.
Možný vliv vemurafenibu na další transportéry není v současné době znám.
Vliv současně podávaných léků na vemurafenib
In vitro studie naznačují, že za metabolismus vemurafenibu jsou zodpovědné CYP3A4 a glukuronizace. Zdá se, že biliární exkrece je další důležitou cestou eliminace. K dispozici nejsou žádné klinické údaje, které by prokázaly vliv silných induktorů nebo inhibitorů aktivity CYP3A4 a/nebo transportního proteinu na expozici vemurafenibu. Vemurafenib je třeba používat s opatrností v kombinaci se silnými inhibitory CYP3A4, glukuronizace a/nebo transportních proteinů (např. ritonavir, sachinavir, telitromycin, ketokonazol, itrakonazol, vorikonazol, posakonazol, nefazodon, atazanavir).
Společné podávání silných induktorů P-gp, glukuronizace a/nebo CYP3A4 (např. rifampicin, rifabutin, karbamazepin, fenytoin nebo třezalka tečkovaná [Hypericum perforatum]) může vést k suboptimální expozici vemurafenibu a je třeba se mu vyvarovat.
Studie in vitro prokázaly, že vemurafenib je substrátem efluxních transportérů P-gp a BCRP. Vliv induktorů a inhibitorů P-gp a BCRP na expozici vemurafenibu není znám. Nelze vyloučit, že by farmakokinetika vemurafenibu mohla být ovlivněna léky, které ovlivňují P-gp (např. verapamil, cyklosporin, ritonavir, chinidin, itrakonazol) nebo BCRP (např. cyklosporin, gefitinib).
V současné době není známo, zda není vemurafenib rovněž substrátem jiných transportních proteinů.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/Antikoncepce u žen
Ženy ve fertilním věku musí během léčby a po dobu alespoň 6 měsíců po ukončení léčby používat účinnou antikoncepci.
Vemurafenib může snížit účinnost hormonálních antikoncepčních přípravků (viz bod 4.5).
K dispozici nejsou žádné údaje týkající se použití vemurafenibu u těhotných žen.
U vemurafenibu nebyly prokázány žádné důkazy teratogenicity u embryí/plodů potkanů nebo králíků (viz bod 5.3). Ve studiích se zvířaty bylo zjištěno, že vemurafenib prochází placentou. Vemurafenib se nemá podávat těhotným ženám, pokud možný prospěch z léčby pro matku nepřeváží možná rizika pro plod.
Kojení
Není známo, zda se vemurafenib vylučuje do mateřského mléka. Riziko pro novorozence/kojence nelze vyloučit. Při rozhodování, zda přerušit kojení nebo přerušit léčbu vemurafenibem, je nutné vzít v úvahu prospěch z kojení pro dítě a prospěch z léčby pro matku.
Fertilita
U zvířat nebyly provedeny žádné studie, které by specificky hodnotily vliv vemurafenibu na fertilitu. Při studiích, které sledovaly toxicitu opakovaných dávek u potkanů a psů, však nebyly na reprodukčních orgánech patrné žádné histopatologické změny (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Účinky vemurafenibu na schopnost řídit a obsluhovat stroje nebyly studovány. Pacienti si mají být vědomi možné únavy nebo problémů s očima, což může být důvodem, aby neřídili.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nej častější nežádoucí účinky (> 30 %) hlášené v souvislosti s vemurafenibem zahrnují artralgii, únavu, vyrážku, fotosenzitivní reakci, nauzeu, alopecii a pruritus. Velmi často byl hlášen kožní spinocelulární karcinom a nej častěji byl léčen lokální excizí.
Potenciace radiační toxicity: Ze zdrojů po uvedení přípravku na trh byly zaznamenány případy kožní reakce na ozáření (radiační recall) a radiační senzibilizace. Nicméně frekvence těchto nežádoucích
účinků není známá vzhledem k tomu, že informace o radiační léčbě včetně informací o výši dávek radiace nejsou rutinně shromažďovány v rámci spontánních hlášení o bezpečnosti.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky, které byly hlášeny u pacientů s melanomem, jsou shrnuty níže podle MedDRA tříd orgánových systémů, četnosti a stupně závažnosti. Ke stanovení četnosti byla použita následující klasifikace četností:
Velmi časté > 1/10 Časté > 1/100 až < 1/10 Méně časté > 1/1000 až < 1/100 Vzácné > 1/10000 až < 1/1000 Velmi vzácné < 1/10000
V tomto bodě je výskyt nežádoucích účinků založen na výsledcích od 468 pacientů z randomizované otevřené studie fáze III u dospělých pacientů s neresekovatelným melanomem nebo melanomem stádia IV s pozitivní mutací V600 genu BRAF a na výsledcích z jednoramenné studie fáze II u pacientů s melanomem stádia IV s pozitivní mutací V600 genu BRAF, u kterých selhala alespoň jedna předchozí systémová léčba (viz bod 5.1). Dále jsou zahrnuty nežádoucí účinky pocházející
z bezpečnostních hlášení ze všech klinických studií a post-marketingového sledování. Všechny zahrnuté pojmy jsou založené na nejvyšší pozorované četnosti v klinických studiích fáze II a III.
V každé skupině četností jsou nežádoucí účinky řazeny podle klesající závažnosti a ke zhodnocení toxicity byla použita kritéria NCI-CTCEA verze 4.0 (běžná kritéria toxicity).
Tabulka 3: Nežádoucí účinky vyskytující se u pacientů léčených vemurafenibem ve studii fáze II nebo fáze III a příhody* pocházející z bezpečnostních hlášení ze všech klinických studií(1) a postmarketingového sledování(2).
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Folikulitida | |||
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) |
Spinocelulární karcinom kůže(d), seboroická keratóza, kožní papilom |
Bazocelulární karcinom, nový primární melanom(3) |
Spinocelulární karcinom v jiné než kožní lokalizaci1'1'11'3'1 |
Chronická myelomonocy- tická leukemie(2)(4), adenokarcinom pankreatu(5) |
|
Poruchy krve a lymfatického systému |
Neutropenie | |||
|
Poruchy metabolismu a výživy |
Snížení chuti k jídlu | |||
|
Poruchy nervového systému |
Bolest hlavy, dysgeuzie |
Obrna VII. hlavového nervu, závratě |
Periferní neuropatie | |
|
Poruchy oka |
Uveitida |
Uzávěr retinální žíly | ||
|
Cévní poruchy |
Vaskulitida | |||
|
Respirační, hrudní a mediastinální poruchy | ||||
|
Gastrointestinální poruchy |
Pankreatitida(2) |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy jater a žlučových cest |
Poškození jater(1)(2)(s) | |||
|
Poruchy kůže a podkožní tkáně |
Fotosenzitivní reakce, aktinická keratóza, vyrážka, makulopapulózní vyrážka, papulózní vyrážka, pruritus, hyperkeratóza, erytém, alopecie, suchá kůže, spálení sluncem |
Syndrom palmoplantární erytrodysestezie, panikulitida (včetně erythema nodosum), keratosis pilaris |
Toxická epidermální nekrolýza(e), Stevens- Johnsonův syndrom® |
Poléková reakce s eosinofilií a systémovými příznaky(1)(2) |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie, myalgie, bolest končetin, muskuloskeletální bolest, bolest zad |
Artritida | ||
|
Poruchy ledvin a močových cest |
Akutní intersticiální nefritida(1)(2) (h), akutní tubulární nekróza(1)(2) (h) | |||
|
Celkové poruchy a reakce v místě aplikace |
Únava, pyrexie, periferní edém, astenie | |||
|
Vyšetření |
Zvýšení GGT (c) |
Zvýšení ALT (c), zvýšení alkalické fosfatázy(c), zvýšení bilirubinu(c), snížení tělesné hmotnosti, prodloužení QT intervalu, zvýšení kreatininu v krvi(1)(2) (h) |
Zvýšení AST (c) |
11 'Příhody pocházející z bezpečnostních hlášení ze všech klinických studií.
(2) Příhody pocházející z postmarketingového sledování.
(3) Příčinná souvislost mezi léčivým přípravkem a nežádoucí příhodou je možná.
(4) Progrese dřívější chronické myelomonocytické leukemie s NRAS mutací.
(5) Progrese dřívějšího adenokarcinomu pankreatu s KRAS mutací.
Popis vybraných nežádoucích účinků
Zvýšení jaterních enzymů
Abnormality jaterních enzymů hlášené v klinické studii fáze III jsou vyjádřeny níže jako poměr pacientů, u kterých došlo ke změně od výchozích hodnot k abnormalitám jaterních enzymů stupně 3 nebo 4.
• Velmi časté: GGT
• Časté: ALT, alkalická fosfatáza, bilirubin
• Méně časté: AST
Zvýšení stupně 4 nebyla u ALT, alkalické fosfatázy ani bilirubinu zaznamenána.
Poškození jater (g)
Na základě kritérií na posouzení přípravkem způsobeného poškození jater, která byla vyvinuta mezinárodní expertní pracovní skupinou lékařů a vědců, bylo poškození jater definováno jako jedna z následujících abnormálních hodnot laboratorních testů:
• > 5x horní limit normy ALT
• > 2x horní limit normy ALP (bez další příčiny pro zvýšení ALP)
• > 3x horní limit normy ALT se současným zvýšením koncentrace bilirubinu > 2x horní limit normy
Kožní spinocelulární karcinom (d)
U pacientů léčených vemurafenibem byly hlášeny případy kožního spinocelulárního karcinomu. Incidence kožního spinocelulárního karcinomu u pacientů léčených vemurafenibem v klinických studiích byla přibližně 20 %. Většina excidovaných lézí hodnocených nezávislou centrální dermatopatologickou laboratoří byla klasifikována jako spinocelulární karcinom - subtyp keratoakantomu nebo s rysy smíšeného keratoakantomu (52 %). Většinu lézí klasifikovaných jako “jiné” (43 %) tvořily benigní kožní léze (např. verucca vulgaris, aktinická keratóza, benigní keratóza, cysta/benigní cysta). Kožní spinocelulární karcinom se obvykle objevuje časně v průběhu léčby s mediánem doby do prvního výskytu 7 až 8 týdnů. Asi u 33 % pacientů, u kterých vznikl kožní spinocelulární karcinom, byl zaznamenán > 1 výskyt s mediánem doby mezi jednotlivými výskyty 6 týdnů. Případy kožního spinocelulárního karcinomu byly typicky zvládnuty jednoduchou excizí a pacienti obvykle pokračovali v léčbě bez úpravy dávkování (viz body 4.2 a 4.4).
Spinocelulární karcinom v jiné než kožní lokalizaci
Případy spinocelulárního karcinomu v jiné než kožní lokalizaci byly hlášeny u pacientů dostávajících vemurafenib při zařazení do klinických studií. Sledování spinocelulárního karcinomu v jiné než kožní lokalizaci se má provádět, jak je uvedeno v bodě 4.4.
Nový primární melanom
V klinických studiích byly hlášeny případy nového primárního melanomu. Tyto případy byly řešeny excizí a pacienti pokračovali v léčbě bez úpravy dávky. Kožní léze mají být sledovány tak, jak je uvedeno v bodě 4.4.
Potenciace radiační toxicity
Hlášené případy zahrnují recall fenomén, radiační poškození kůže, radiační pneumonitidu, radiační ezofagitidu, radiační proktitidu, radiační hepatitidu, radiační cystitidu a radiační nekrózu.
Reakce přecitlivělosti(e)
V souvislosti s léčbou vemurafenibem byly hlášeny závažné reakce přecitlivělosti včetně anafylaxe. Závažné reakce přecitlivělosti mohou zahrnovat Stevens-Johnsonův syndrom, generalizovanou vyrážku, erytém nebo hypotenzi. U pacientů, u kterých dojde k rozvoji závažných reakcí přecitlivělosti, je nutné léčbu vemurafenibem trvale ukončit (viz bod 4.4).
Dermatologické reakce (f)
U pacientů léčených vemurafenibem v klíčové klinické studii byly hlášeny závažné dermatologické reakce, včetně vzácných případů Stevens-Johnsonova syndromu a toxické epidermální nekrolýzy. U pacientů, u kterých dojde k rozvoji závažné dermatologické reakce, je nutné léčbu vemurafenibem trvale ukončit.
Prodloužení QT intervalu
Analýza centralizovaných EKG údajů z otevřené nekontrolované substudie fáze II hodnotící QT interval u 132 pacientů léčených vemurafenibem v dávce 960 mg dvakrát denně (studie NP22657) prokázala prodloužení QTc v závislosti na expozici. Průměrný účinek na QTc zůstával stabilní v rozmezí mezi 12-15 ms po prvním měsíci léčby, s nejdelším průměrným prodloužením QTc (15,1 ms; horní 95% interval spolehlivosti: 17,7 ms) pozorovaným během prvních 6 měsíců (n=90 pacientů). U dvou pacientů (1,5 %) došlo k rozvoji léčbou vyvolaných absolutních hodnot QTc > 500 ms (CTC stupeň 3) a pouze u jednoho pacienta (0,8 %) došlo ke změně QTc o > 60 ms od výchozích hodnot (viz bod 4.4).
Akutní poškození ledvin (h>
Při užívání vemurafenibu byly hlášeny případy renální toxicity v rozsahu od zvýšení hladin kreatininu až po akutní intersticiální nefritidu a akutní tubulární nekrózu, některé pozorované příhody byly stanovené při dehydrataci. Elevace kreatininu v séru byly většinou mírné (>1-1,5x ULN) až středně závažné (>1,5-3x ULN) a bylo pozorováno, že jsou reverzibilní povahy (viz tabulka 4).
Tabulka 4: Změny hladin kreatininu od počáteční hodnoty ve studii fáze III
|
vemurafenib (%) |
dakarbazin (%) | |
|
Změny o > 1 stupeň od počáteční hodnoty u všech stupňů |
27,9 |
6,1 |
|
Změny o > 1 stupeň od počáteční hodnoty se zvýšením na stupeň 3 nebo vyšší |
1,2 |
1,1 |
|
• až na stupeň 3 |
0,3 |
0,4 |
|
• až na stupeň 4 |
0,9 |
0,8 |
Tabulka 5: Případy akutního poškození ledvin ve studii fáze III
|
vemurafenib (%) |
dakarbazin (%) | |
|
Případy akutního poškození ledvin* |
10,0 |
1,4 |
|
Případy akutního poškození ledvin ve spojitosti s dehydratací |
5,5 |
1,0 |
|
Úprava dávky z důvodu akutního poškození ledvin |
2,1 |
0 |
Všechny procentní podíly jsou vyjádřeny jako případy z celkového počtu pacientů vystavených jednotlivým léčivým přípravkům.
* Zahrnuje akutní poškození ledvin, poruchu funkce ledvin a laboratorní změny v souladu s akutním poškozením ledvin.
Zvláštní populace
Starší lidé
Ve studii fáze III bylo 94 (28 %) z 336 pacientů s neresekovatelným nebo metastazujícím melanomem léčených vemurafenibem ve věku > 65 let. Starší pacienti (> 65 let) mohou být náchylnější k výskytu nežádoucích účinků, včetně kožního spinocelulárního karcinomu, snížení chuti k jídlu a kardiálních poruch.
Pohlaví
Nežádoucí účinky stupně 3, hlášené během klinických studií s vemurafenibem častěji u žen než mužů, byly vyrážka, artralgie a fotosenzitivita.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pro případ předávkování vemurafenibem není k dispozici žádné specifické antidotum. Pacienti, u kterých dojde k rozvoji nežádoucích účinků, mají dostat vhodnou symptomatickou léčbu. V klinických studiích nebyly pozorovány žádné případy předávkování vemurafenibem. V případě podezření na předávkování je třeba vemurafenib vysadit a zahájit podpůrnou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Protinádorové léky, inhibitory protein kinázy, ATC kód: L01XE15 Mechanismus účinku a farmakodynamické účinky
Vemurafenib je inhibitor serin-threoninové kinázy BRAF. Mutace genu BRAF vede ke konstituční aktivaci proteinů BRAF, což může vést k buněčné proliferaci při absenci růstových faktorů. Preklinické údaje dle biochemických stanovení prokázaly, že vemurafenib může účinně blokovat kinázy BRAF s aktivující mutací v kodonu 600 (tabulka 6).
Tabulka 6: Kinázová inhibiční aktivita vemurafenibu proti různým kinázám BRAF
|
Kináza |
Předpokládaná frekvence u melanomu s pozitivitou mutace V600(t) |
Inhibiční koncentrace 50 (nmol) |
|
brafV600E |
87,3 % |
10 |
|
brafV600K |
7,9 % |
7 |
|
brafV600R |
1 % |
9 |
|
brafV600D |
<0,2 % |
7 |
|
brafV600G |
<0,1 % |
8 |
|
brafV600M |
<0,1 % |
7 |
|
brafV600A |
<0,1 % |
14 |
|
brafwt |
N/A |
39 |
(t) Odhadnuta na základě 16403 melanomů se známou mutací kodonu 600 BRAF ve veřejné databázi COSMIC, vydání 71 (listopad 2014).
Tento inhibiční účinek byl potvrzen na dostupných buněčných liniích melanomu s mutací V600 BRAF při stanovení fosforylace ERK a buněčných antiproliferačních analýzách. V buněčných antiproliferačních analýzách s inhibiční koncentrací 50 (IC50) proti buněčným liniím s mutací V600 (buněčné linie s mutací V600E, V600R, V600D a V600K) se pohyboval v rozmezí od 0,016 do 1,131 pmol, kdy IC 50 proti buněčným liniím divokého typu genu BRAF byla 12,06 resp. 14,32 pmol.
Určení stavu BRAF mutace
Před zahájením léčby vemurafenibem je nutné, aby pacienti měli validovaným testem potvrzenou pozitivní mutaci V600 genu BRAF. V klinických studiích fáze II a fáze III byli vhodní pacienti identifikováni za použití polymerázové řetězové reakce v reálném čase (cobas 4800 BRAF V600 Mutation Test). Tento test má CE značku a používá se ke stanovení mutací BRAF na DNA izolované z nádorové tkáně fixované formalinem a zalité do parafinu. Byl sestrojen tak, aby s vysokou senzitivitou detekoval predominantní mutace BRAF V600E (až k 5 % sekvencí V600E na pozadí sekvence „divokého“ typu v DNA izolované z nádorové tkáně fixované formalinem a zalité do parafinu). Neklinické a klinické studie s retrospektivní sekvenční analýzou prokázaly, že tento test detekuje s nižší senzitivitou rovněž méně obvyklé mutace BRAF V600D a V600K. Z dalších vzorků dostupných z neklinických a klinických studií (n=920), ve kterých byla pozitivita mutace stanovená pomocí testu cobas a které byly následně analyzovány sekvencováním, nebyly žádné vzorky identifikovány jako divoký typ genu pomocí sekvencování dle Sangera ani 454 sekvencování.
Klinická účinnost a bezpečnost
Účinnost vemurafenibu byla hodnocena u 336 pacientů v klinické studii fáze III (NO25026) a u 278 pacientů ve dvou klinických studiích fáze II (NP22657 a MO25743). U všech pacientů bylo vyžadováno, aby měli pokročilou formu melanomu s pozitivní mutací V600 genu BRAF stanovenou pomocí testu cobas 4800 BRAF V600 Mutation Test.
Výsledky ze studie fáze III (studie NO25026) u dříve neléčených pacientů
Otevřená, multicentrická, mezinárodní randomizovaná studie fáze III podpořila použití vemurafenibu u dříve neléčených pacientů s neresekovatelným nebo metastazujícím melanomem s pozitivní mutací V600E genu BRAF. Pacienti byli randomizováni k léčbě vemurafenibem (960 mg dvakrát denně) nebo dakarbazinem (1000 mg/m2 v den 1 každé 3 týdny).
K léčbě vemurafenibem (n=337) nebo dakarbazinem (n=338) bylo randomizováno celkem 675 pacientů. Většina pacientů byli muži (56 %) a běloši (99 %) s mediánem věku 54 let (24 % bylo > 65 let), všichni pacienti měli stav výkonnosti (PS) dle ECOG 0 nebo 1 a většina pacientů měla stupeň onemocnění M1c (65 %). Ko-primární cílové parametry účinnosti studie zahrnovaly celkové přežití a dobu přežití bez progrese.
V předdefinované interim analýze s ukončením sběru údajů ke dni 30. prosince 2010 bylo významné zlepšení pozorováno u ko-primárních cílových parametrů celkové přežití (p<0,0001) a doba přežití bez progrese (p<0,0001) (nestratifikovaný log-rank test). Podle doporučení DSMB (Data Safety Monitoring Board), jehož výsledky byly uveřejněny v lednu 2011, byla studie modifikovaná a pacientům užívajícím dakarbazin bylo umožněno přejít na léčbu vemurafenibem. Poté byly provedeny post-hoc analýzy přežití, jak je popsáno v tabulce 7.
Tabulka 7: Celkové přežití u dříve neléčených pacientů s melanomem s pozitivní mutací V600 genu BRAF do data ukončení sběru údajů ze studie (N=338 dakarbazin, N=337 vemurafenib)
|
Data ukončení sběru údajů |
Léčba |
Počet úmrtí (%) |
Poměr rizik (95% CI) |
Počet pacientů, kteří změnili léčbu (%) |
|
30. prosinec 2010 |
dakarbazin |
75 (22) |
0,37 (0,26; 0,55) |
0 (NA) |
|
vemurafenib |
43 (13) | |||
|
31. březen 2011 |
dakarbazin |
122 (36) |
0,44 (0,33; 0,59) (w) |
50 (15 %) |
|
vemurafenib |
78 (23) | |||
|
3. říjen 2011 |
dakarbazin |
175 (52) |
0,62 (0,49; 0,77) (w) |
81 (24%) |
|
vemurafenib |
159 (47) | |||
|
1. únor 2012 |
dakarbazin |
200 (59) |
0,70 (0,57; 0,87) (w) |
83(25 %) |
|
vemurafenib |
199 (59) | |||
|
20. prosinec 2012 |
dakarbazin |
236 (70) |
0,78 (0,64; 0,94) (w) |
84 (25 %) |
|
vemurafenib |
242 (72) |
(w) Cenzurované výsledky v době změny léčby
Necenzurované výsledky v době změny léčby: 31. březen 2011: HR (95% CI) = 0,47 (0,35; 0,62); 3. říjen 2011: HR (95% CI) = 0,67 (0,54; 0,84); 1. únor 2012: HR (95% CI) = 0,76 (0,63; 0,93); 20. prosinec 2012: HR (95% CI) = 0,79 (0,66; 0,95)
Obrázek 1: Kaplan-Meierovy křivky celkového přežití - dříve neléčení pacienti (ukončení sběru údajů ke dni 20. prosince 2012)
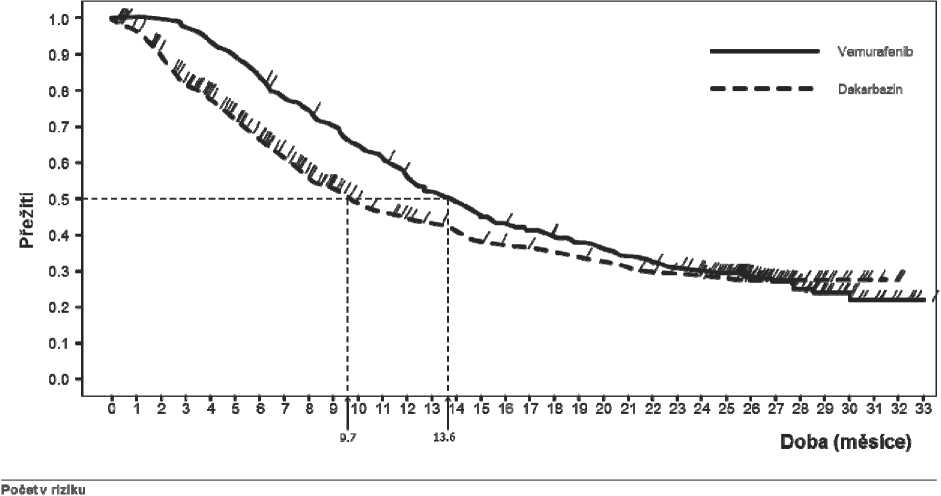
Dakarbazin 338 306 276 243 217 193172 154126110 97 91 82 79 76 68 66 63 60 68 66 61 48 46 41 36 28 20 17 11 8 4 0 0
Vemurabnib 337336336 326314 303281 260 248 232214 203183171161 148 140136 129123 117110104 98 91 81 66 43 30 17 13 8 4 1
Tabulka 8 znázorňuje účinek léčby pro všechny předdefinované stratifikační proměnné, které byly stanoveny jako prognostické faktory.
Tabulka 8: Celkové přežití u dříve neléčených pacientů s melanomem s pozitivní mutací V600 genu BRAF podle LDH, stadia tumoru a ECOG statusu (post-hoc analýza, ukončení sběru údajů ke dni 20. prosince 2012, cenzurované výsledky v době změny léčby)
|
Stratifikační proměnná |
N |
Poměr rizik |
95% Interval spolehlivosti |
|
Normální LDH |
391 |
0,88 |
0,67; 1,16 |
|
LDH >ULN |
284 |
0,57 |
0,44; 0,76 |
|
Stadium IIIc/M1A/M1B |
234 |
1,05 |
0,73; 1,52 |
|
Stadium MIC |
441 |
0,64 |
0,51; 0,81 |
|
ECOG PS=0 |
459 |
0,86 |
0,67; 1,1 |
|
ECOG PS=1 |
216 |
0,58 |
0,42; 0,9 |
LDH: laktát dehydrogenáza, ECOG PS: Stav výkonnosti (performance status) dle Východní kooperativní onkologické skupiny (Eastern Cooperative Oncology Group).
Tabulka 9 ukazuje výskyt celkové odpovědi na léčbu a dobu přežití bez progrese u dříve neléčených pacientů s melanomem s pozitivní mutací V600 genu BRAF.
Tabulka 9: Výskyt celkové odpovědi na léčbu a doba přežití bez progrese u dříve neléčených pacientů s melanomem s pozitivní mutací V600 genu BRAF
|
vemurafenib |
dakarbazin |
p-hodnota (x) | |
|
Ukončení sběru údajů ke dni 30. prosince 2010 (y) | |||
|
Výskyt celkové odpovědi na léčbu (95% interval spolehlivosti) |
48,4 % (41,6 %; 55,2 %) |
5,5 % (2,8 %; 9,3 %) |
<0,0001 |
|
Doba přežití bez progrese Poměr rizik (95% interval spolehlivosti) |
0,26 (0,20; 0,33) |
<0,0001 | |
|
Počet příhod (%) |
104 (38 %) |
182 (66%) | |
|
Medián doby přežití bez progrese (měsíce) (95% interval spolehlivosti) |
5,32 (4,86; 6,57) |
1,61 (1,58; 1,74) | |
|
Ukončení sběru údajů ke dni 1. února 2012 (z) | |||
|
Doba přežití bez progrese Poměr rizik (95% interval spolehlivosti) |
0,38 (0,32; 0,46) |
<0,0001 | |
|
Počet příhod (%) |
277(82 %) |
273 (81 %) | |
|
Medián doby přežití bez progrese (měsíce) (95% interval spolehlivosti) |
6,87 (6,14; 6,97) |
1,64 (1,58; 2,07) | |
(x) Nestratifikovaný log-rank test pro dobu přežití bez progrese a chí-kvadrát test pro výskyt celkové odpovědi na léčbu.
(y) Ke dni 30. prosince 2010 celkem 549 pacientů bylo hodnotitelných s ohledem na dobu přežití bez progrese a 439 pacientů bylo hodnotitelných s ohledem na výskyt celkové odpovědi na léčbu.
(z) Ke dni 1. února 2012 celkem 675 pacientů bylo hodnotitelných v následné analýze údajů doby přežití bez progrese.
Celkem 57 pacientů z 673 pacientů ve studii NO25026, u kterých byly tumory analyzovány retrospektivně pomocí sekvencování, mělo melanom s pozitivní mutací V600K genu BRAF. Analýzy účinnosti mezi těmito pacienty s V600K pozitivními tumory, ačkoli byly limitovány nízkým počtem pacientů, naznačovaly podobný prospěch léčby vemurafenibem, pokud jde o celkové přežití, dobu přežití bez progrese a potvrzenou nejlepší celkovou odpověď na léčbu. Pro pacienty se vzácnou mutací V600 genu BRAF jinou než V600E a V600K nejsou k dispozici žádné údaje.
Výsledky studie fáze II (studie NP22657) u pacientů, u kterých selhala alespoň jedna předchozí léčba
Jednoramenná, multicentrická, mezinárodní studie fáze II byla provedena u 132 pacientů s metastazujícím melanomem s pozitivitou mutace V600E genu BRAF stanovenou pomocí testu cobas 4800 BRAF V600 Mutation Test, kteří podstoupili alespoň jednu předchozí léčbu. Medián věku byl 52 let, 19 % pacientů bylo starších 65 let. Většina pacientů byli muži (61 %), běloši (99 %) a měli onemocnění stádia M1c (61 %). U 49 % pacientů selhaly > 2 předchozí léčby. Při mediánu následného sledování 12,9 měsíců (rozmezí 0,6 až 20,1) byl primární cílový parametr účinnosti potvrzeného výskytu nejlepší celkové odpovědi (úplná odpověď + částečná odpověď), jak byl stanoven nezávislou hodnotící komisí (IRC) 53 % (95% CI: 44 %, 62 %). Medián celkového přežití byl 15,9 měsíců (95% CI: 11,6; 18,3). Výskyt celkového přežití v 6 měsících byl 77 % (95% CI: 70 %, 85 %) a ve 12 měsících 58 % (95% CI: 49 %, 67 %).
Devět ze 132 pacientů zařazených do studie NP22657 mělo dle retrospektivního sekvencování (Sanger) nádory s mutací V600K. Z těchto pacientů 3 měli částečnou odpověď, 3 měli stabilizaci onemocnění, 2 měli progresi nemoci a jeden nebyl hodnotitelný.
Výsledky studie fáze II (MO25743) u pacientů s mozkovými metastázami
Byla provedena jednoramenná, multicentrická studie (N = 146) s vemurafenibem u dospělých pacientů s histologicky potvrzeným metastazujícím melanomem, u nichž je přítomna mutace BRAF V600 (dle cobas 4800 BRAF V600 Mutation Test) a mozkové metastázy. Ve studii byly dvě kohorty souběžně zařazující pacienty:
- 1. kohorta s dosud neléčenými pacienty (N = 90): Pacienti, u nichž dosud mozkové metastázy nebyly léčeny; předchozí systémová léčba metastazujícího melanomu byla povolena, nezahrnují se BRAF inhibitory a MEK inhibitory.
- 2. kohorta s dříve léčenými pacienty (N = 56): Pacienti, u nichž byly mozkové metastázy dříve léčeny, a u nichž po této léčbě došlo k progresi. U pacientů, kteří byli léčeni stereotaktickou radioterapií (SRT) nebo chirurgicky, muselo po této předchozí léčbě dojít k rozvoji nové mozkové léze hodnotitelné podle kritérií RECIST.
Bylo zařazeno celkem 146 pacientů. Většina pacientů byli muži (61,6 %), běloši (92,5 %), medián věku byl 54 let (rozmezí 26 až 83 let), kteří byli obdobně rozděleni do obou kohort. Při zahájení byl v obou kohortách střední počet mozkových cílových lézí 2 (rozmezí 1 až 5).
Primárním cílem účinnosti studie byla četnost nejlepší celkové odpovědi (BORR) v mozku pacientů s metastazujícím melanomem s dosud neléčenými mozkovými metastázami na základě posouzení nezávislé hodnotící komise (IRC).
Sekundární cíle zahrnovaly posouzení účinnosti vemurafenibu podle BORR v mozku již léčených pacientů, trvání odpovědi (DOR), přežití bez progrese (PFS) a celkové přežití (OS) u pacientů s melanomem metastazujícím do mozku (viz tabulka 10).
Tabulka 10:
Účinnost vemurafenibu u pacientů s mozkovými metastázami
|
1. kohorta Bez předchozí léčby n = 90 |
2. kohorta Dříve léčení n = 56 |
Celkem n = 146 | |
|
BORRa v mozku Pacienti s odpovědí n (%) (95% CI)b |
16 (17,8 %) (10,5; 27,3) |
10 (17,9 %) (8,9; 30,4) |
26 (17,8 %) (12,0; 25,0) |
|
DORc v mozku (n) Medián (v měsících) (95% CI)d |
(n = 16) 4,6 (2,9; 6,2) |
(n = 10) 6,6 (2,8; 10,7) |
(n = 26) 5,0 (3,7; 6,6) |
|
BORR extra-kraniální n (%)a |
26 (32,9 %) |
9 (22,5 %) |
35 (29,4 %) |
|
PFS - celkově Medián (v měsících)e (95% CI)d |
3,7 (3,6; 3,7) |
3,7 (3,6; 5,5) |
3,7 (3,6; 3,7) |
|
PFS - pouze u pacientů s metastázami v mozku Medián (v měsících)e (95% CI)d |
3,7 (3,6; 4,0) |
4,0 (3,6; 5,5) |
3,7 (3,6; 4,2) |
|
OS Medián (v měsících) (95% CI)d |
8,9 (6,1; 11,5) |
9,6 (6,4; 13,9) |
9,6 (6,9; 11,5) |
a Četnost nejlepší potvrzené celkové odpovědi posouzená nezávislou hodnotící komisí, počet pacientů s odpovědí n (%)
b Oboustranný 95% Clopper-Pearsonův interval spolehlivosti (CI) c Trvání odpovědi posouzené nezávislou hodnotící komisí d Kaplan-Meierův odhad
e
Posouzené zkoušejícím lékařem
5.2 Farmakokinetické vlastnosti
Vemurafenib je látka skupiny IV (nízká rozpustnost a permeabilita) dle kritérií popsaných v Klasifikačním systému biofarmaceutik. Farmakokinetické parametry vemurafenibu byly stanoveny za použití non-kompartmentální analýzy ve studiích fáze I a fáze III (20 pacientů po 15 dnech léčby dávkou 960 mg dvakrát denně a 204 pacientů v ustáleném stavu 22. den) a rovněž v populační analýze farmakokinetiky za použití souhrnných údajů od 458 pacientů. Z tohoto počtu bylo 457 pacientů bílé rasy.
Absorpce
Absolutní biologická dostupnost tablet vemurafenibu 240 mg není známa. Vemurafenib je absorbován s mediánem Tmax přibližně 4 hodiny po podání jedné dávky 960 mg (čtyři 240 mg tablety). Vemurafenib vykazuje vysokou variabilitu mezi jednotlivými pacienty. Ve studii fáze II byly v den 1 AUC0-8h 22,1 ± 12,7 pg.h/ml a Cmax 4,1 ± 2,3 pg/ml. Po několikadenním dávkování vemurafenibu dvakrát denně dochází k akumulaci. V non-kompartmentální analýze při dávkování vemurafenibu dvakrát denně 960 mg se poměr den 15/den 1 pohyboval v rozmezí 15- až 17násobku pro AUC a 13-až 14násobku pro Cmax, když v podmínkách ustáleného stavu hodnota AUC0-8h dosahovala 380,2 ± 143,6 pg.h/ml a Cmax 56,7 ± 21,8 pg/ml.
Jídlo (s vysokým obsahem tuku) zvyšuje relativní biologickou dostupnost jednorázové dávky 960 mg vemurafenibu. Geometricky průměrné poměry mezi stavy po jídle a nalačno byly pro Cmax 2,5- a pro AUC 4,6 až 5,1-násobkem. Medián Tmax byl zvýšen ze 4 na 7,5 hodiny, pokud jednorázová dávka vemurafenibu byla užita s jídlem.
Vliv jídla na expozici vemurafenibu v ustáleném stavu není v současné době znám. Podání vemurafenibu na prázdný žaludek může vést k významně nižší expozici v ustáleném stavu než podání vemurafenibu s jídlem, nebo v krátké době po jídle. Při příležitostném podání vemurafenibu na prázdný žaludek se očekává, že má omezený vliv na expozici v ustáleném stavu z důvodu vysoké akumulace vemurafenibu v ustáleném stavu. Údaje o bezpečnosti a účinnosti z klíčových studií byly shromážděny od pacientů užívajících vemurafenib s jídlem, nebo bez jídla.
Z důvodu rozdílů v obsahu gastrointestinálních tekutin, objemu, pH, motility, času přenosu a složení žluči může také docházet k variabilitě v expozici.
V ustáleném stavu je průměrná expozice vemurafenibu v plazmě během 24hodinového období stabilní, jak ukazuje průměrný poměr 1,13 mezi plazmatickou koncentrací před ranní dávkou a 2-4 hodiny po ranní dávce.
Po perorálním podání je konstanta rychlosti absorpce u populace pacientů s metastazujícím melanomem stanovena na 0,19 h-1 (se 101 % variabilitou mezi pacienty).
Distribuce
Populační zdánlivý distribuční objem vemurafenibu u pacientů s metastazujícím melanomem je stanoven na 91 l (se 64,8 % variabilitou mezi pacienty). Vemurafenib je in vitro vysoce vázaný na bílkoviny lidské plazmy (> 99 %).
Biotransformace
Relativní poměr vemurafenibu a jeho metabolitů byl popsán v metabolické bilanční studii u člověka při jednorázové dávce vemurafenibu značeného 14C podaného perorálně. Primárním enzymem odpovědným za metabolismus vemurafenibu in vitro je CYP3A4. U člověka byla rovněž identifikována konjugace metabolitů (glukuronizace a glykosylace). Nicméně mateřská složka byla hlavní složkou (95 %) v plazmě. Ačkoli se nezdá, že by metabolismus vedl k relevantním hladinám metabolitů v plazmě, nelze vyloučit význam metabolismu pro exkreci.
Eliminace
Populační zdánlivá clearance vemurafenibu u pacientů s metastazujícím melanomem je odhadnuta na 29,3 l/den (s 31,9 % variabilitou mezi pacienty). Populační eliminační poločas odhadnutý dle populační analýzy farmakokinetiky pro vemurafenib je 51,6 hodin (5. a 95. percentil rozptylu individuálních eliminačních poločasů je 29,8 až 119,5 hodin).
V metabolické bilanční studii u člověka s vemurafenibem podávaným perorálně bylo v průměru 95 % dávky vyloučeno během 18 dní. Většina látek souvisejících s vemurafenibem (94 %) byla vyloučena ve stolici a <1 % v moči. Důležitou cestou eliminace může být i biliární exkrece nezměněné složky. Vzhledem k tomu, že není známa absolutní biologická dostupnost, je však význam jaterní a renální exkrece pro clearanci vemurafenibu nejistý. Vemurafenib je in vitro substrátem a inhibitorem P-gp.
Zvláštní populace
Starší lidé
Na základě populační analýzy farmakokinetiky nemá věk žádný statisticky významný vliv na farmakokinetiku vemurafenibu.
Pohlaví
V populační farmakokinetické analýze byla prokázána o 17 % vyšší zdánlivá clearance (CL/F) a o 48 % vyšší zdánlivý distribuční objem (V/F) u mužů než u žen. Není zřejmé, zda se jedná o vliv pohlaví nebo tělesného povrchu. Rozdíly v expozici však nejsou natolik veliké, aby bylo nutné na základě tělesného povrchu nebo pohlaví upravovat dávku.
Porucha funkce ledvin
V populační farmakokinetické analýze za použití údajů z klinických studií u pacientů s metastazujícím melanomem neovlivňovala mírná a středně závažná porucha funkce ledvin zdánlivou clearance vemurafenibu (clearance kreatininu >40 ml/min). U pacientů se závažnou poruchou renálních funkcí nejsou k dispozici žádné údaje (viz body 4.2 a 4.4).
Porucha jaterních funkcí
Na základě preklinických údajů a metabolické bilanční studie u člověka se většina vemurafenibu vylučuje játry. V populační farmakokinetické analýze za použití údajů z klinických studií u pacientů s metastazujícím melanomem nemělo zvýšení AST a ALT až k trojnásobku horní hranice normálu vliv na zdánlivou clearance vemurafenibu. Ke stanovení vlivu metabolické poruchy nebo poruchy exkreční funkce jater na farmakokinetiku vemurafenibu není k dispozici dostatek údajů (viz body 4.2 a 4.4).
Pediatrická populace
Žádné studie hodnotící farmakokinetiku vemurafenibu u pediatrických pacientů nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Preklinický bezpečnostní profil vemurafenibu byl hodnocen u potkanů, psů a králíků.
Studie toxicity po opakovaném podávání rozpoznaly jako cílové orgány u psů játra a kostní dřeň. Reverzibilní toxické účinky (hepatocelulární nekróza a degenerace) v játrech při expozicích nižších, než je předpokládaná klinická expozice (na základě porovnání AUC), byly zaznamenány v 13týdenní studii se psy. V předčasně ukončené 39týdenní studii se psy s dávkováním dvakrát denně při expozicích podobných, jako jsou očekávané klinické expozice (na základě porovnání AUC), byly u jednoho psa zaznamenány fokální nekrózy kostní dřeně. V in vitro studii hodnotící cytotoxicitu v kostní dřeni byla při klinicky relevantních koncentracích pozorována mírná cytotoxicita u některých buněčných populací lymfo-hematopoetických buněk potkana, psa a člověka.
Bylo prokázáno, že vemurafenib je fototoxický in vitro, na kulturách myších fibroblastů po ozáření UVA zářením, ale nebylo to prokázáno ve studii s potkany in vivo při dávkách až 450 mg/kg/den (při expozicích nižších než jsou předpokládané klinické expozice (na základě porovnání AUC)).
Žádné zvláštní studie hodnotící účinky vemurafenibu na fertilitu nebyly u zvířat provedeny. Ve studiích toxicity po opakovaném podávání však nebyly zaznamenány žádné histopatologické nálezy na reprodukčních orgánech u samců a samic potkanů a psů při dávkách až 450 mg/kg/den (při expozicích nižších než jsou předpokládané klinické expozice na základě porovnání AUC). Ve studiích vývoje embryí a plodů u potkanů při dávkách až 250 mg/kg/den a u králíků při dávkách až 450 mg/kg/den, což jsou dávky vedoucí k expozicím nižším, než jsou předpokládané klinické expozice (na základě porovnání AUC), nebyla pozorována teratogenita. Expozice ve studiích vývoje embryí a plodů byly však nižší než klinické expozice na základě porovnání AUC, proto je obtížné definovat, do jaké míry lze tyto výsledky extrapolovat na člověka. Proto vliv vemurafenibu na plod nelze vyloučit. Nebyly provedeny žádné studie hodnotící pre- a postnatální vývoj.
Ve studiích in vitro (bakteriální mutace [Amesův test], aberace chromozomů lidských lymfocytů) ani v in vivo mikrojadérkovém testu kostní dřeně u potkanů prováděných s vemurafenibem nebyly prokázány žádné známky genotoxicity.
Studie kancerogenity nebyly s vemurafenibem provedeny.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Jádro
Sodná sůl kroskarmelosy Koloidní bezvodý oxid křemičitý Magnesium-stearát Hyprolosa
Potahová vrstva Polyvinylalkohol Oxid titaničitý (E171)
Makrogol 3350 Mastek
Červený oxid železitý (E172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Al/Al perforovaný jednodávkový blistr
Velikost balení: 56 x 1potahovaná tableta (7 blistrů po 8 x 1 tabletě)
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/751/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. února 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zelboraf 240 mg potahované tablety vemurafenibum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje 240 mg vemurafenibum (ve formě ko-precipitátu vemurafenibu a acetátsukcinátu hypromelosy).
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
56 x 1 potahovaná tableta
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/12/751/001
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
zelboraf
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem.>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH PERFOROVANÝ BLISTR JEDNODÁVKOVÝ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zelboraf 240 mg tablety vemurafenibum
2 NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Zelboraf 240 mg potahované tablety
vemurafenibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je psáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Zelboraf a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zelboraf užívat
3. Jak se přípravek Zelboraf užívá
4. Možné nežádoucí účinky
5. Jak přípravek Zelboraf uchovávat
6. Obsah balení a další informace
1. Co je přípravek Zelboraf a k čemu se používá
Přípravek Zelboraf je protinádorový lék, který obsahuje léčivou látku vemurafenib. Používá se k léčbě dospělých pacientů s melanomem, který se rozšířil do dalších částí těla nebo který nelze odstranit chirurgicky.
Lze ho použít pouze u pacientů, jejichž nádor má změnu (mutaci) genu „BRAF“. Tato změna může vést k rozvoji melanomu.
Přípravek Zelboraf se zaměřuje na bílkoviny, které jsou produkovány tímto modifikovaným genem, a tak zpomaluje nebo zastavuje rozvoj rakoviny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zelboraf užívat Neužívejte přípravek Zelboraf:
• Jestliže jste alergický(á) na vemurafenib nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6 této příbalové informace). K příznakům alergických reakcí může patřit otok obličeje, rtů nebo jazyka, obtíže s dýcháním, vyrážka nebo pocit na omdlení.
Upozornění a opatření
Před užitím přípravku Zelboraf se poraďte se svým lékařem.
Alergické reakce
• Při užívání přípravku Zelboraf může dojít k alergickým reakcím, které mohou být závažné.
Přestaňte užívat přípravek Zelboraf a neprodleně vyhledejte lékařskou pomoc, pokud pocítíte některý z příznaků alergické reakce, jako jsou otok obličeje, rtů nebo jazyka, obtíže s dýcháním, vyrážka nebo pocit na omdlení.
Závažné kožní reakce
• Při užívání přípravku Zelboraf může dojít ke vzniku závažných kožních reakcí. Pokud se u Vás objeví kožní vyrážka spolu s kterýmkoli z následujících příznaků: puchýře na kůži, puchýře nebo vředy v ústech, olupování kůže, horečka, zarudnutí nebo otok obličeje, rukou nebo chodidel nohou, přestaňte užívat přípravek Zelboraf a neprodleně se poraďte se svým lékařem.
Dřívější výskyt nádoru
• Informujte svého lékaře, pokud se již u Vás vyskytl jiný typ nádoru než melanom, protože přípravek Zelboraf může způsobit rozvoj určitých typů nádorů.
Reakce na radiační terapii
• Informujte svého lékaře, pokud jste podstoupil(a) nebo podstupujete radioterapii, neboť přípravek Zelboraf může zhoršit nežádoucí účinky radiační léčby.
Problémy se srdcem
• Sdělte svému lékaři, pokud máte problémy se srdcem, jako je porucha převodu elektrického signálu nazývaná „prodloužení QT intervalu“. Váš lékař provede testy ke kontrole správné srdeční činnosti před zahájením léčby přípravkem Zelboraf i v jejím průběhu. Pokud to bude nutné, může lékař rozhodnout o dočasném přerušení léčby nebo o jejím úplném ukončení.
Oční problémy
• Při užívání přípravku Zelboraf je potřeba, aby Vám lékař pravidelně kontroloval Vaše oči.
Pokud se u Vás během léčby objeví bolest očí, otok, zarudnutí, rozmazané vidění nebo jiné změny zraku, neprodleně to sdělte svému lékaři.
Vyšetření kůže před zahájením léčby, v jejím průběhu a po jejím ukončení
• Pokud při užívání tohoto léku zaznamenáte jakékoli změny na kůži, prosím, sdělte to svému lékaři co nejdříve.
• Pravidelně během léčby a po dobu až 6 měsíců po ukončení léčby bude Váš lékař provádět kontroly Vaší kůže s ohledem na nádor zvaný „kožní spinocelulární (dlaždicobuněčný) karcinom“.
• Tyto léze obvykle vypadají jako poškození kůže sluncem, zůstávají na jednom místě a lze je léčit chirurgickým odstraněním.
• Pokud Váš lékař prokáže tento typ kožního nádoru, bude jej léčit nebo Vás odešle k jinému lékaři k následné léčbě.
• Dále bude nutné, aby Vám lékař vyšetřil hlavu, krk, ústa, mízní (lymfatické) uzliny a abyste pravidelně podstupoval(a) CT vyšetření. Toto jsou preventivní opatření prováděná pro případ, kdyby došlo k rozvoji spinocelulárního (dlaždicobuněčného) karcinomu uvnitř Vašeho těla. Před zahájením léčby a po jejím ukončení se doporučuje také gynekologické vyšetření (u žen) a vyšetření konečníku.
• Při užívání přípravku Zelboraf může dojít ke vzniku nových ložisek melanomu. Tyto léze se obvykle odstraňují chirurgicky a pacienti pokračují ve své léčbě. Kontrola těchto lézí je shodná s kontrolami kožního spinocelulárního (dlaždicobuněčného) karcinomu, jak je uvedeno výše.
Problémy s ledvinami nebo játry
• Sdělte svému lékaři, pokud máte problémy s ledvinami nebo játry. Může to ovlivnit účinnost přípravku Zelboraf. Váš lékař bude také provádět určité krevní testy ke kontrole funkce jater a funkce ledvin před zahájením užívání přípravku Zelboraf a v průběhu léčby.
Ochrana před sluncem
• Pokud užíváte přípravek Zelboraf, můžete být citlivější ke slunečnímu záření a snadněji se spálit, což může být závažné. Během léčby se vyvarujte vystavování kůže přímému slunečnímu záření.
• Pokud plánujete jít ven na slunce:
• oblečte si oblečení, které bude chránit Vaši kůži, včetně hlavy a obličeje, paží a nohou;
• použijte balzám na rty a širokospektrý opalovací krém (s ochranným faktorem minimálně 30, který se aplikuje opakovaně každé 2 až 3 hodiny).
• Toto pomůže předcházet spálení kůže způsobenému sluncem.
Děti a dospívající
Přípravek Zelboraf se nedoporučuje u dětí a dospívajících. Účinky přípravku Zelboraf u osob mladších 18 let nejsou známy.
Další léčivé přípravky a přípravek Zelboraf
Prosím, informujte před zahájením léčby svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat (včetně těch, které jste si koupil(a) v lékárně, supermarketu nebo zdravotnickém zařízení). Toto je velmi důležité, protože užívání více než jednoho léku ve stejný čas může zesílit nebo oslabit účinky léků.
Zejména sdělte svému lékaři, pokud užíváte:
• Léky, které jsou známy, že ovlivňují srdeční tep:
• léky k léčbě poruch srdečního rytmu (např. chinidin, amiodaron)
• léky k léčbě deprese (např. amitriptylin, imipramin)
• léky k léčbě bakteriálních infekcí (např. azitromycin, klaritromycin)
• léky k léčbě nevolnosti (pocitu na zvracení) a zvracení (např. ondansetron, domperidon).
• Léky, které jsou odbourávány především prostřednictvím bílkoviny nazývané CYP1A2 (např. kofein, olanzapin, teofylin), CYP3A4 (např. některé perorální (užívané ústy) antikoncepční přípravky) nebo nazývané CYP2C8.
• Léky, které ovlivňují bílkovinu nazývanou P-gp nebo BCRP (např. verapamil, cyklosporin, ritonavir, chinidin, itrakonazol, gefitinib).
• Léčivé přípravky, které mohou být ovlivněny bílkovinou nazývanou P-gp (např. aliskiren, kolchicin, digoxin, everolimus, fexofenadin) nebo bílkovinou nazývanou BCRP (např. methotrexát, mitoxantron, rosuvastatin).
• Léky, které stimulují bílkovinu nazývanou CYP3A4 nebo metabolizační proces nazývaný glukuronizace (např. rifampicin, rifabutin, karbamazepin, fenytoin nebo třezalka tečkovaná).
• Lék užívaný k prevenci tvorby krevních sraženin nazývaný warfarin
• Lék nazývaný YERVOY (ipilimumab, další lék používaný k léčbě melanomu). Není doporučena kombinace tohoto léku s přípravkem Zelboraf, z důvodu zvýšené jaterní toxicity.
Pokud užíváte jakýkoli z těchto léků (nebo pokud si nejste jistý(á)), prosím, sdělte to svému lékaři dříve, než začnete užívat přípravek Zelboraf.
Těhotenství a kojení
• Během léčby a po dobu alespoň 6 měsíců po ukončení léčby používejte vhodnou metodu antikoncepce. Přípravek Zelboraf může snižovat účinnost některých druhů perorálních antikoncepčních přípravků. Pokud užíváte perorální antikoncepci, prosím, sdělte to svému lékaři.
• Přípravek Zelboraf se nedoporučuje užívat během těhotenství, pokud se Váš lékař nedomnívá, že prospěch pro matku převyšuje riziko pro dítě. K dispozici nejsou žádné informace týkající se bezpečnosti přípravku Zelboraf u těhotných žen. Pokud jste těhotná, nebo těhotenství plánujete, sdělte to svému lékaři.
• Není známo, zda složky přípravku Zelboraf přecházejí do mateřského mléka. Během léčby přípravkem Zelboraf se proto kojení nedoporučuje.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Není známo, zda přípravek Zelboraf bude ovlivňovat schopnost řídit nebo obsluhovat stroje. Pozor na únavu a problémy s očima, které mohou být důvodem proč neřídit.
3. Jak se přípravek Zelboraf užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Kolik tablet je třeba užívat
• Doporučená dávka je 4 tablety dvakrát denně (celkem 8 tablet).
• Vezměte si 4 tablety ráno. Poté si vezměte 4 tablety večer.
• Pokud se u Vás objeví nežádoucí účinky, může Váš lékař rozhodnout pokračovat v léčbě, ale dávku snížit. Vždy užívejte přípravek Zelboraf přesně podle pokynů svého lékaře.
• V případě zvracení pokračujte v užívání přípravku Zelboraf jako obvykle a neberte si doplňující dávku.
Užívání tablet
• Neužívejte přípravek Zelboraf pravidelně na prázdný žaludek.
• Tablety spolkněte celé a zapijte sklenicí vody.
Jestliže jste užil(a) více přípravku Zelboraf, než jste měl(a)
Jestliže jste užil(a) více přípravku Zelboraf, než jste měl(a), sdělte to okamžitě svému lékaři. Užívání přílišného množství přípravku Zelboraf může vést k vyšší pravděpodobnosti výskytu nežádoucích účinků, nebo jejich větší závažnosti. Nebyly pozorovány žádné případy předávkování přípravkem Zelboraf.
Jestliže jste zapomněl(a) užít přípravek Zelboraf
• Jestliže jste zapomněl(a) užít dávku a do další dávky zbývají více než 4 hodiny, užijte dávku, jakmile si vzpomenete. Poté užijte následující dávku v obvyklý čas.
• Pokud již do další dávky zbývají méně než 4 hodiny, zapomenutou dávku vynechejte. Poté užijte následující dávku v obvyklý čas.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Zelboraf
Je důležité pokračovat v léčbě přípravkem Zelboraf tak dlouho, jak Vám jej předepsal Váš lékař. Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i přípravek Zelboraf nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Závažné alergické reakce
Pokud se u Vás objeví jakýkoli z těchto příznaků:
• Otok obličeje, rtů nebo jazyka
• Obtíže s dýcháním
• Vyrážka
• Pocit na omdlení.
Zavolejte okamžitě lékaře. Neužívejte dál přípravek Zelboraf, dokud si nepromluvíte s lékařem.
U pacientů léčených radioterapií před, v průběhu nebo po léčbě přípravkem Zelboraf se může vyskytnout zhoršení nežádoucích účinků spojených s radiační léčbou. Ty se mohou objevit v místech ozáření, jako například na kůži, jícnu, močovém měchýři, játrech, konečníku a plicích.
Informujte ihned svého lékaře, pokud se u Vás vyskytne jakýkoli z následujících příznaků:
• kožní vyrážka, puchýře, odlupování kůže nebo změna barvy kůže
• dušnost, což může být doprovázeno kašlem, horečkou nebo zimnicí (pneumonitida)
• potíže nebo bolest při polykání, bolest na hrudi, pálení žáhy nebo reflux kyseliny (ezofagitida).
Prosím, kontaktujte svého lékaře co nejdříve, jakmile zaznamenáte jakékoli změny na kůži.
Nežádoucí účinky jsou uvedeny níže podle frekvence jejich výskytu:
Velmi časté: mohou postihnout více než 1 osobu z 10
• Vyrážka, svědění, suchá nebo šupinatá kůže
• Kožní problémy včetně bradavic
• Určitý typ kožního nádoru (kožní dlaždicobuněčný karcinom)
• Spálení kůže od slunce, zvýšená citlivost na sluneční záření
• Ztráta chuti k jídlu
• Bolest hlavy
• Změny vnímání chuti
• Průjem
• Zácpa
• Pocit na zvracení (nevolnost), zvracení
• Vypadávání vlasů
• Bolest kloubů nebo svalů, bolest svalů a kostí
• Bolest končetin
• Bolest zad
• Pocit unavenosti (únava)
• Horečka
• Otok obvykle na nohou (periferní otok)
• Změny výsledků jaterních testů (zvýšení GGT)
• Kašel.
Časté: mohou postihnout až 1 z 10 osob
• Typy kožních nádorů (bazocelulární karcinom, nový primární melanom)
• Palmoplantární syndrom (tj. zčervenání, olupování nebo tvorba puchýřů na kůži rukou a chodidel)
• Zánět oka (zánět duhovky)
• Obrna lícního nervu (forma lícní obrny, která je často vratná)
• Pocity brnění a pálení v rukou a nohou
• Zánět kloubů
• Zánět vlasových míšků (kořínků)
• Pokles hmotnosti
• Změny výsledků jaterních testů (zvýšení ALT, alkalické fosfatázy a bilirubinu)
• Závratě
• Změny elektrické aktivity srdce (prodloužení QT intervalu)
• Zánět podkožní tukové tkáně
• Abnormální výsledky krevních testů ledvin (zvýšená hladina kreatininu).
Méně časté: mohou postihnout až 1 ze 100 osob
• Alergické reakce, které mohou zahrnovat otok obličeje a obtíže s dýcháním
• Blokáda průtoku krve v části oka (uzávěr žíly v sítnici)
• Problémy s nervy, které vedou k pocitům bolesti, ztrátě vnímání a/nebo svalové slabosti (periferní neuropatie)
• Zánět krevních cév
• Zánět slinivky břišní
• Změny výsledků jatemích laboratorních testů nebo poškození jater, včetně závazného poškození jater, kdy játra jsou poškozena do té míry, že nejsou schopna plně vykonávat svou funkci
• Typ nádoru (spinocelulární karcinom v jiné než kožní lokalizaci)
• Snížený počet bílých krvinek (neutropenie).
Vzácné: mohou postihnout až 1 z 1000 osob
• Rozvoj určitého typu již existujícího nádorového onemocnění s RAS mutací (chronická myelomonocytická leukemie, adenokarcinom pankreatu)
• Typ závažné kožní reakce charakterizované vyrážkou doprovázenou horečkou a zánětem vnitřních orgánů, např. jater a ledvin
• Typy poškození ledvin charakterizované zánětem (akutní intersticiální nefritida) nebo poškozením tubulů ledvin (akutní tubulární nekróza).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zelboraf uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Přípravek Zelboraf nepoužívejte po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“ a na blistru za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Zelboraf obsahuje
• Léčivou látkou je vemurafenibum. Jedna potahovaná tableta obsahuje 240 miligramů (mg) vemurafenibu (ve formě ko-precipitátu vemurafenibu a acetátsukcinátu hypromelosy).
• Dalšími složkami jsou:
• Koloidní bezvodý oxid křemičitý, sodná sůl kroskarmelosy, hyprolosa a magnesium-stearát.
• Potahová vrstva: Červený oxid železitý, makrogol 3350, polyvinylalkohol, mastek a oxid titaničitý.
Jak přípravek Zelboraf vypadá a co obsahuje toto balení
Zelboraf 240 mg potahované tablety jsou růžovo-bílé až oranžovo-bílé. Jsou oválné s vyraženým „VEM“ na jedné straně.
Jsou dostupné v hliníkovém perforovaném jednodávkovém blistru v balení po 56 x 1 tabletě.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639
Grenzach-Wyhlen
Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
Bt^rapnn Pom Etnrapua EOOfl Tea: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Česká republika Roche s. r. o. Tel: +420 - 2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (ara Renju Unit) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 - 6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
ELLába Roche (Hellas) A.E. Ttfk: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kónpoq r.A.Exapáxn? & Eia AxS. Ttfk: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 - 6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Tato příbalová informace byla naposledy revidována <{MM/RRRR}>.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
39