Zeffix 5 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Zeffix 100 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje lamivudinum 100 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta)
Potahovaná tableta karamelové barvy, potahovaná, podlouhlého bikonvexního tvaru o rozměrech přibližně 11 mm x 5 mm, na jedné straně vyraženo označení „GX CG5“
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Zeffix je indikován k léčbě dospělých pacientů s chronickou hepatitidou B s:
• kompenzovaným jaterním onemocněním s prokázanými známkami virové replikace, přetrvávajícími zvýšenými hodnotami sérové alaninaminotransferázy (ALT) a histologicky dokumentovaným aktivním zánětem jater a/nebo jaterní fibrózou. Podávání lamivudinu by mělo být zahájeno pouze v případě, kdy není možné nebo přípustné podávat jiné alternativní antivirotikum, u kterého je menší pravděpodobnost vzniku rezistence (viz bod 5.1).
• dekompenzovaným jaterním onemocněním v kombinaci s jiným léčivým přípravkem, který nevykazuje zkříženou rezistenci s lamivudinem (viz bod 4.2).
4.2 Dávkování a způsob podání
Terapii přípravkem Zeffix má zahajovat lékař, který má zkušenosti s léčbou chronické hepatitidy B.
Dávkování
Dospělí
Doporučené dávkování přípravku Zeffix je 100 mg jednou denně.
U pacientů s dekompenzovaným jaterním onemocněním má být lamivudin vždy podáván v kombinaci
s dalším léčivým přípravkem, který nevykazuje zkříženou rezistenci s lamivudinem, aby se snížilo
riziko rezistence a dosáhlo rychlého potlačení replikace viru.
Trvání léčby
Optimální délka léčby není známa. 1
• U HBeAg-negativních pacientů s CHB (infikovaných pre-core mutantou) bez cirhózy má být léčba podávána alespoň, než dojde k HBs sérokonverzi nebo je prokazatelná ztráta účinnosti.
Při prodloužené léčbě se doporučuje pravidelné opakované vyšetření potvrzující, že pokračování ve vybrané léčbě zůstává pro pacienta vhodné.
• U pacientů s dekompenzovanou hepatopatií nebo cirhózou a u příjemců jaterních transplantátů se zastavení léčby nedoporučuje (viz bod 5.1).
Pacienti, u nichž byla aplikace lamivudinu zastavena, mají být pravidelně kontrolováni se zaměřením na známky recidivující hepatitidy (viz bod 4.4).
Klinická rezistence
U pacientů s HBeAg pozitivní nebo HBeAg negativní chronickou hepatitidou B může vznik mutace viru HBV s označením YMDD (tyrozin-methionin-aspartát-aspartát) způsobit snížení terapeutické odpovědi na lamivudin, což se projeví jako zvýšení hladiny HBV DNA a enzymu ALT oproti hodnotám před započetím léčby lamivudinem. Aby se snížilo riziko vzniku rezistence u pacientů léčených monoterapií lamivudinem, má se zvážit přechod na jiný přípravek nebo přidání dalšího přípravku, který nevykazuje zkříženou rezistenci s lamivudinem podle léčebných pokynů, pokud při monoterapii lamivudinem zůstávají hladiny HBV DNA v séru detekovatelné po 24. týdnu léčby (viz bod 5.1).
Pro léčbu pacientů, kteří jsou zároveň nakaženi HIV a léčeni lamivudinem nebo kombinací lamivudinu a zidovudinu, nebo je u nich tato léčba plánována, je nutno zachovat dávku lamivudinu předepsanou pro léčbu infekce HIV (obvykle 150 mg dvakrát denně v kombinaci s dalšími antiretrovirotiky).
Zvláštní skupiny pacientů
Porucha funkce ledvin
U pacientů se středně závažnou až závažnou poruchou funkce ledvin se v důsledku snížené renální clearance zvyšují koncentrace lamivudinu v séru (AUC). Proto má být u pacientů s clearance kreatininu < 50 ml/min sníženo dávkování. Jsou-li potřebné dávky menší než 100 mg, má se aplikovat Zeffix perorální roztok (viz tabulka 1 níže).
Tabulka 1: Dávkování přípravku Zeffix u pacientů se sníženou renální clearance.
|
Clearance kreatininu ml/min |
První dávka přípravku Zeffix perorální roztok1 |
Udržovací dávka jednou denně |
|
30 až < 50 |
20 ml (100 mg) |
10 ml (50 mg) |
|
15 až < 30 |
20 ml (100 mg) |
5 ml (25 mg) |
|
5 až < 15 |
7 ml (35 mg) |
3 ml (15 mg) |
|
< 5 |
7 ml (35 mg) |
2 ml (10 mg) |
* Zeffix perorální roztok obsahuje 5 mg/ml lamivudinu.
Dostupné údaje o pacientech podstupujících intermitentní hemodialýzu (méně než 4 hodiny nebo rovnající se 4 hodinám dialýzy dvakrát až třikrát týdně) ukazují, že po iniciální redukci dávky lamivudinu podle clearance kreatininu po dobu probíhající dialýzy již nejsou další úpravy dávkování nutné.
Porucha funkce jater
Údaje o pacientech s poruchou funkce jater, včetně pacientů s terminální hepatopatií čekajících na transplantaci, ukazují, že porucha funkce jater nemá významný vliv na farmakokinetiku lamivudinu.
Podle těchto údajů se pacientům s poruchou funkce jater, není-li provázena poruchou funkce ledvin, nemusí dávkování upravovat.
Starší pacienti
U starších pacientů nemá stárnutí doprovázené zhoršením funkce ledvin klinicky významný vliv na expozici lamivudinu, s výjimkou pacientů s clearance kreatininu < 50 ml/min.
Pediatrická populace
Bezpečnost a účinnost podávání přípravku Zeffix u kojenců, dětí a dospívajících mladších 18 let nebyly stanoveny. V současnosti dostupné údaje jsou popsány v bodech 4.4 a 5.1, ale na jejich základě nelze doporučit dávkování.
Způsob podání Perorální podání.
Přípravek Zeffix lze užívat jak nalačno, tak s jídlem.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Laktátová acidóza a těžká hepatomegalie se steatózou
Při užívání nukleosidových analogů byl hlášen vznik laktátové acidózy (bez hypoxemie), někdy s fatálním průběhem, obvykle spojený s těžkou hepatomegalií a jaterní steatózou. Jelikož Zeffix je nukleosidovým analogem, toto riziko nelze vyloučit. Léčba nukleosidovými analogy musí být přerušena při rychlém vzestupu hladin aminotransferáz, progresivní hepatomegalii nebo metabolické/laktátové acidóze neznámé etiologie. Benigní zažívací příznaky, jako je nauzea, zvracení a bolest břicha, mohou svědčit o rozvíjející se laktátové acidóze. Závažné případy, někdy s fatálním koncem, byly spojeny s pankreatitidou, jaterním selháním/steatózou jater, renálním selháním a vyššími hladinami sérového laktátu. S opatrností je třeba předepisovat nukleosidové analogy pacientovi (zejména obézním ženám) s hepatomegalií, hepatitidou nebo jinými známými rizikovými faktory vedoucími k onemocnění jater a jaterní steatóze (včetně některých léčivých přípravků a alkoholu). Pacienti koinfikovaní virem hepatitidy C a léčení alfa interferonem a ribavirinem mohou představovat zvláštní riziko. Tito pacienti mají být pečlivě sledováni.
Exacerbace hepatitidy
Exacerbace v průběhu léčby
Spontánní exacerbace jsou u chronické hepatitidy B relativně časté a jsou charakterizované přechodným zvýšením hladin ALT v séru. Po započetí antivirové léčby může u některých pacientů dojít k přechodnému zvýšení hladin ALT v séru v souvislosti s tím, jak klesají v séru hladiny HBV DNA. U pacientů s kompenzovaným jaterním onemocněním tato zvýšení hladin ALT v séru nebyla obvykle doprovázena zvýšením koncentrací sérového bilirubinu ani příznaky jaterní dekompenzace.
Při prodloužené terapii byly identifikovány virové subpopulace HBV se sníženou citlivostí na lamivudin (YMDD-mutace viru HBV). U některých pacientů vede vznik YMDD mutace viru HBV k exacerbaci hepatitidy primárně prokázané zvýšením hodnot ALT v séru a znovu objevením se HBV DNA (viz bod 4.2). U pacientů, kteří mají YMDD mutaci HBV, se má zvážit na základě terapeutických doporučení přechod na jiný léčivý přípravek nebo přidání dalšího léčivého přípravku, který nevykazuje zkříženou rezistenci s lamivudinem (viz bod 5.1).
Exacerbace po ukončení léčby
U pacientů, kteří ukončili léčbu hepatitidy B, byla zaznamenána akutní exacerbace hepatitidy, která se obvykle prokazuje zvýšenou hladinou ALT v séru a znovu objevením se HBV DNA.
V kontrolovaných klinických studiích fáze III s následným sledováním pacientů bez aktivní léčby byla incidence elevací ALT po léčbě (více než 3násobně ve srovnání s výchozími hodnotami) vyšší u pacientů léčených lamivudinem (21 %) ve srovnání s pacienty, kteří dostávali placebo (8 %). Procento pacientů, u kterých došlo po ukončení léčby k elevacím ALT spolu s elevacemi bilirubinu, bylo však nižší a podobné v obou ramenech léčby (viz tabulka 3 v bodu 5.1). U pacientů léčených lamivudinem se většina elevací ALT po léčbě objevila mezi 8. a 12. týdnem po ukončení léčby. Většina případů odezněla spontánně, ale bylo pozorováno i několik fatálních případů. V případě, že se zastaví aplikace přípravku Zeffix, mají být pacienti pravidelně kontrolováni - jak klinicky, tak laboratorním vyšetřováním funkce jater (ALT a hladiny bilirubinu), a to nejméně po dobu čtyř měsíců a dále podle toho, jak je to klinicky indikováno.
Exacerbace u pacientů s dekompenzovanou jaterní cirhózou
Příjemci transplantátů a pacienti s dekompenzovanou jaterní cirhózou jsou více ohroženi aktivní virovou replikací. V důsledku minimální hepatální funkce u těchto pacientů může reaktivace hepatitidy po vysazení lamivudinu nebo snížením účinnosti v průběhu terapie přivodit těžkou i fatální dekompenzaci. Tito pacienti mají být během léčby pravidelně kontrolováni se zaměřením na klinické, virologické a sérologické parametry související s hepatitidou B, na hepatální a renální funkci a na antivirovou terapeutickou odezvu, a to minimálně každý měsíc, a je-li léčba z jakéhokoli důvodu zastavena, pak ještě nejméně 6 měsíců po léčbě. Pravidelně je nutné kontrolovat (minimálně) tyto laboratorní parametry: sérové ALT, bilirubin, albumin, dusík močoviny v krvi (BUN), kreatinin a tyto virologické parametry: antigeny HBV, protilátky proti HBV a pokud možno koncentrace HBV DNA v séru. Pacienti, u kterých se během léčby nebo po jejím zastavení objeví známky hepatální insuficience, mají být kontrolováni v adekvátně kratších časových intervalech.
Není dostatek údajů o tom, zda je u pacientů, u kterých se vyvinou známky rekurentní hepatitidy, prospěšné znovu zahájit terapii lamivudinem.
Mitochondriální dysfunkce
Bylo prokázáno jak in vitro, tak in vivo, že nukleosidové a nukleotidové analogy způsobují poškození mitochondrií různého stupně. Existují hlášení mitochondriální dysfunkce u kojenců vystavených in utero a/nebo postnatálně působení analogů nukleosidů. Hlavními hlášenými nežádoucími účinky jsou hematologické poruchy (anemie, neutropenie), metabolické poruchy (hyperlaktemie, zvýšená hladina lipázy). Byly hlášeny některé neurologické poruchy s pozdním nástupem (hypertonie, křeče, neobvyklé chování). Neurologické poruchy mohou být přechodné nebo stálé. Každé dítě, které bylo in utero vystaveno působení analogů nukleosidů a nukleotidů, musí být klinicky i laboratorně sledováno a v případě relevantních známek nebo příznaků musí projít úplným vyšetřením na možnou mitochondriální dysfunkci.
Pediatričtí pacienti
Lamivudin byl podáván dětem (2 roky a starším) a dospívajícím s kompenzovanou chronickou hepatitidou B. Avšak vzhledem k omezeným údajům se podávání lamivudinu těmto pacientům v současnosti nedoporučuje (viz bod 5.1).
Hepatitida delta nebo hepatitida C
Účinnost lamivudinu u pacientů současně infikovaných virem hepatitidy delta nebo virem hepatitidy C nebyla stanovena a doporučuje se opatrnost.
Imunosupresivní léčba
Údaje o použití lamivudinu u HBeAg negativních pacientů (pre-core mutanty) a u pacientů souběžně léčených imunosupresivními režimy, včetně protinádorové chemoterapie, jsou omezené. Tito pacienti by měli lamivudin užívat s opatrností.
Monitorování
V průběhu léčby přípravkem Zeffix mají být pacienti pravidelně monitorováni. Sérové hladiny ALT a HBV DNA se mají monitorovat ve 3měsíčních intervalech a u HBeAg pozitivních pacientů se mají stanovovat každých 6 měsíců hodnoty HBeAg.
Současná infekce HIV
U pacientů současně infikovaných HIV, kteří jsou současně léčeni lamivudinem nebo kombinací lamivudinu a zidovudinu nebo u nichž se plánuje zahájení takové léčby, má být zachována dávka lamivudinu předepsaná pro terapii infekce HIV (obvykle 150 mg dvakrát denně v kombinaci s jinými antiretrovirotiky). U pacientů současně infikovaných HIV, u nichž není nutná antiretrovirová farmakoterapie, existuje při použití samotného lamivudinu k terapii chronické hepatitidy B riziko mutace HIV.
Přenos hepatitidy B
Nejsou k dispozici informace o přenosu viru hepatitidy B na plod těhotných žen léčených lamivudinem. Je třeba se řídit standardními doporučenými postupy pro imunizaci proti viru hepatitidy B u dětí.
Pacienty je nutné upozornit na to, že nebylo prokázáno, že by terapie lamivudinem snižovala riziko přenosu viru hepatitidy B na jiné osoby, a proto je i nadále nutné dodržovat příslušná preventivní opatření.
Interakce s jinými léčivými přípravky
Zeffix se nemá užívat s jinými přípravky obsahujícími lamivudin, nebo přípravky obsahujícími emtricitabin (viz bod 4.5).
Kombinace lamivudinu s kladribinem se nedoporučuje (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Vzhledem k omezenému rozsahu metabolizace, k omezené míře vazby na plazmatické proteiny a k téměř úplné renální eliminaci intaktního léčiva je pravděpodobnost metabolických interakcí malá.
Lamivudin je eliminován převážně aktivní renální sekrecí organických kationtů. Je třeba brát v úvahu možnost interakcí s jinými souběžně aplikovanými léčivy, zejména těmi, jejichž hlavní eliminační cestou je aktivní renální sekrece prostřednictvím transportního systému pro organické kationty; takovým léčivem je např. trimethoprim. Jiná léčiva (např. ranitidin, cimetidin) jsou tímto mechanismem eliminována jen zčásti a nebylo prokázáno, že by interagovala s lamivudinem.
U léčiv, která jsou převážně eliminována buď aktivní renální sekrecí organických aniontů nebo glomerulární filtrací, nejsou klinicky významné interakce s lamivudinem pravděpodobné. Souběžná aplikace kombinace trimethoprim/sulfamethoxazol v dávkách 160 mg/800 mg zvýšila expozici lamivudinu asi o 40 %. Lamivudin neměl vliv na farmakokinetiku trimethoprimu ani sulfamethoxazolu. Úprava dávkování lamivudinu však není nutná, nejedná-li se o pacienta s poruchou funkce ledvin.
Při souběžné aplikaci lamivudinu se zidovudinem bylo pozorováno mírné (28%) zvýšení Cmax zidovudinu, ale celková expozice tomuto léčivu (AUC) se významně nezměnila. Zidovudin neměl vliv na farmakokinetiku lamivudinu (viz bod 5.2).
Při souběžné aplikaci lamivudinu s alfa-interferonem se nevyskytly farmakokinetické interakce.
U pacientů, kteří užívali lamivudin s běžně používanými imunosupresivy (např. s cyklosporinem A), nebyly pozorovány klinicky významné nežádoucí interakce. Formální interakční studie však nebyly provedeny.
Emtricitabin
Vzhledem k podobnosti se Zeffix nemá podávat současně s jinými analogy cytidinu, jako je např. emtricitabin. Zeffix také nemá být užíván s žádným jiným léčivým přípravkem, který obsahuje lamivudin (viz bod 4.4).
Kladribin
Lamivudin in vitro inhibuje intracelulární fosforylaci kladribinu, což vede k potenciálnímu riziku snížení účinnosti kladribinu v případě léčby kombinací těchto látek. Také některá klinická zjištění ukazují na možnou interakci mezi lamivudinem a kladribinem. Z tohoto důvodu se současné podávání lamivudinu a kladribinu nedoporučuje (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Velké množství dat u těhotných žen (více než 1 000 konečných expozic) neprokázalo malformační toxicitu. Zeffix může být užíván v průběhu těhotenství, pokud je klinicky potřebným.
U pacientek, které jsou léčeny lamivudinem a následně otěhotní, by se mělo uvažovat o možnosti vzniku rekurentní hepatitidy při ukončení léčby lamivudinem.
Kojení
Na základě výsledků pozorování u více než 200 párů matek s dítětem léčených pro infekci virem HIV bylo zjištěno, že sérové koncentrace lamivudinu u dětí kojených matkami, které se léčily pro infekci virem HIV, jsou velmi nízké (méně než 4 % koncentrací naměřených v mateřském séru) a postupně se snižují až na neměřitelné hodnoty v době, kdy kojené děti dosahují 24 týdnů věku. Celkové množství lamivudinu, které se vstřebá u kojených dětí, je velmi nízké, a proto je pravděpodobné, že toto množství lamivudinu má suboptimální antivirové účinky. Infekce hepatitidou B u matky není kontraindikací pro kojení, pokud je u novorozence při porodu adekvátně provedena prevence nákazy infekční hepatitidou B. Nejsou důkazy o tom, že by nízká koncentrace lamivudinu v mateřském mléce vedla ke vzniku nežádoucích reakcí u kojených dětí. Proto je možné uvažovat o kojení dětí matkami, které jsou pro hepatitidu B léčeny lamivudinem, a vzít přitom v úvahu, jaké jsou výhody kojení pro dítě a jaké výhody přináší podávání lamivudinu kojící matce. V případě, že dojde k přenosu viru HBV z matky na dítě i přes adekvátní profylaxi, mělo by se zvážit přerušení kojení, aby se u dítěte snížilo riziko vzniku mutací viru HBV, které by vedly k rezistenci viru vůči lamivudinu.
Fertilita
Reprodukční studie u zvířat neprokázaly žádný vliv na fertilitu u samců ani samic (viz bod 5.3). Mitochondriální dysfunkce
In vitro i in vivo bylo prokázáno, že nukleosidové a nukleotidové analogy způsobují různý stupeň mitochondriálního poškození. Existují hlášení mitochondriální dysfunkce u kojenců vystavených in utero a/nebo postnatálně působení analogů nukleosidů (viz bod 4.4).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacienty je nutno upozornit, že během léčby lamivudinem byly hlášeny malátnost a únava. Při posouzení pacientovy schopnosti řídit nebo obsluhovat stroje je nutno vzít v úvahu klinický stav pacienta a profil nežádoucích účinků lamivudinu.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Výskyt nežádoucích účinků a laboratorních odchylek (s výjimkou zvýšené ALT a CPK, viz níže) byl podobný u pacientů léčených placebem a lamivudinem. Z nežádoucích účinků byly nejčastěji hlášeny: malátnost a únava, infekce respiračního traktu, faryngeální a tonzilární diskomfort, bolest hlavy, abdominální diskomfort a bolest, nevolnost, zvracení a průjem.
Nežádoucí účinky v tabulce
Nežádoucí reakce jsou uvedeny níže podle orgánových systémů a frekvence výskytu. Frekvenční kategorie jsou přiřazeny jen těm nežádoucím reakcím, u kterých je alespoň možné uvažovat o příčinné souvislosti s lamivudinem. Frekvence výskytu je definována jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Kategorie frekvencí přiřazené nežádoucím účinkům jsou založeny převážně na poznatcích z klinických studií zahrnujících celkem 1 171 pacientů s chronickou hepatitidou B, kteří dostávali lamivudin v dávce 100 mg.
|
Poruchy krve a lymfatického systému | |
|
Není známo |
T rombocytopenie |
|
Poruchy imunitního systému | |
|
Vzácné |
Angioedém |
|
Poruchy jater a žlučových cest | |
|
Velmi časté |
Zvýšení ALT (viz bod 4.4) |
|
Exacerbace hepatitidy primárně prokázané zvýšením sérové hladiny ALT byly hlášené při léčbě a po vysazení lamivudinu. Většina případů spontánně odezněla, avšak velmi vzácně došlo k úmrtí (viz bod 4.4). | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Časté |
Zvýšení CPK |
|
Časté |
Svalové poruchy, včetně myalgie a křečí* |
|
Není známo |
Rhabdomyolýza |
|
* Ve fázi III klinickýc |
studií nebyla frekvence rhabdomyolýzy zaznamenaná v léčebné skupině |
s lamivudinem vyšší proti skupině s placebem.
Pediatrická populace
Na základě omezených údajů od dětí ve věku 2 až 17 let nebyly identifikovány nové bezpečnostní signály oproti dospělým.
Další zvláštní populace
U pacientů infikovaných HIV byly hlášeny případy pankreatitidy a periferní neuropatie (nebo parestezie), třebaže jejich souvislost s léčbou lamivudinem nebyla jasně prokázána. U pacientů s chronickou hepatitidou B nebyl pozorován rozdíl ve výskytu těchto projevů mezi pacienty léčenými lamivudinem a pacienty užívajícími placebo.
Při kombinované farmakoterapii nukleosidovými analogy byly u pacientů s HIV hlášeny případy laktátové acidózy, někdy fatální, obvykle spojené s těžkou hepatomegalií a hepatální steatózou.
U pacientů s chronickou hepatitidou B léčených lamivudinem byl vzácně hlášen výskyt laktátové acidózy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Ve studiích akutní toxicity na zvířatech nevedlo podání velmi vysokých dávek lamivudinu k projevům orgánové toxicity. Údaje o následcích užití nadměrných dávek u lidí jsou omezené. Nevyskytly se fatální případy a pacienti se zotavili. Po těchto předávkováních nebyly zjištěny specifické objektivní ani subjektivní příznaky.
Dojde-li k předávkování, má být pacient sledován a v případě potřeby mu má být poskytována standardní podpůrná léčba. Jelikož lamivudin je odstranitelný dialýzou, mohla by se v terapii předávkování použít kontinuální hemodialýza, třebaže to nebylo studováno.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antivirotika pro systémové použití, nukleosidové a nukleotidové inhibitory reverzní transkriptázy, ATC kód: J05AF05.
Lamivudin je antivirotikum, které se ve všech testovaných buněčných liniích a u experimentálně infikovaných zvířat projevilo jako vysoce účinné proti viru hepatitidy B.
Lamivudin je infikovanými i neinfikovanými buňkami metabolizován na lamivudin-trifosfát (TP), který je vlastní účinnou formou výchozí látky. Intracelulární poločas lamivudin-trifosfátu v hepatocytech je in vitro 17 až 19 hodin. Lamivudin-trifosfát působí jako substrát HBV-virové polymerázy.
Inkorporací lamivudin-trifosfátu do řetězce virové DNA s následným ukončením řetězce se znemožní další tvorba virové DNA.
Lamivudin-trifosfát neinterferuje s normálním buněčným metabolismem deoxynukleotidů a je jen slabým inhibitorem savčích DNA-polymeráz alfa a beta. Kromě toho má lamivudin-trifosfát jen malý vliv na obsah DNA v savčích buňkách.
V testech zaměřených na možné účinky léčiva na strukturu mitochondrií a na obsah a funkci mitochondriální DNA neměl lamivudin významné toxické účinky. Lamivudin má velmi nízký potenciál ke snížení obsahu mitochondriální DNA, neinkorporuje se trvale do mitochondriální DNA a nepůsobí jako inhibitor mitochondriální DNA-polymerázy gama.
Klinické zkušenosti
Zkušenosti u pacientů s HBeAg pozitivní CHB a kompenzovaným onemocněním jater Jednoroční terapie lamivudinem v placebem kontrolovaných studiích významně potlačila replikaci HBV DNA [34 až 57 % pacientů mělo hodnoty nižší, než byl detekční limit analýzy (Abbott Genostics hybridizační testovací roztok, LLOD < 1,6 pg/ml)], normalizovala hodnoty ALT (40 až 72 % pacientů), navodila sérokonverzi HBeAg (ztrátu HBeAg a HBeAb s průkazem HBV DNA [s konvenční analýzou] u 16 až 18 % pacientů), zlepšila histologický nález (38 až 52 % pacientů mělo > 2 body poklesu Knodellova skóre - index histologické aktivity [HAI]) a zpomalila progresi fibrózy (u 3 až 17 % pacientů) a progresi jaterní cirhózy.
Trvalá léčba lamivudinem po dobu dalších 2 let u pacientů, kterým selhalo dosažení HBeAg sérokonverze v úvodních jednoročních kontrolovaných studiích, vedla k dalšímu zlepšení přemosťující fibrózy. Z pacientů s YMDD mutací viru HBV vykázalo 41/82 (50 %) pacientů zlepšení jaterního zánětu a 40/56 (71 %) pacientů bez YMDD mutace viru HBV vykázalo zlepšení. Zlepšení přemosťující fibrózy bylo pozorováno u 19/30 (63 %) pacientů bez YMDD mutace a u 22/44 (50 %) pacientů s mutací YMDD. Pět procent (3/56) pacientů bez YMDD mutace a 13 % (11/82) pacientů s YMDD mutací vykázalo zhoršení zánětu jater ve srovnání se stavem před zahájením léčby. Progrese cirhózy byla pozorována u 4/68 (6 %) pacientů s YMDD mutací, zatímco progrese cirhózy nebyla pozorována u pacientů bez mutace.
V rozsáhlé terapeutické studii u asijských pacientů (NUCB3018) byl výskyt HBeAg sérokonverze a normalizace ALT na konci pětiletého léčebného období 48 % (28/58) a 47 % (15/32). HBeAg sérokonverze byla zvýšena u pacientů s elevací hladin ALT; 77 % (20/26) pacientů s ALT před léčbou > 2x ULN sérokonvertovalo. Na konci pěti let měli všichni pacienti hladiny HBV DNA, které nebyly detekovatelné, nebo nižší než hladiny před léčbou.
Další výsledky studie stavu YMDD mutace shrnuje tabulka 2.
Tabulka 2: Výsledky účinnosti po 5 letech podle YMDD stavu (asijská studie) NUCB3018.
|
Předmět % | ||
|
YMDD-mutace HBV |
YMDD1 |
Non-YMDD1 |
|
HBeAg sérokonverze - Všichni pacienti |
38 (15/40) |
72 (13/18) |
|
- Pacienti se základní ALT < 1x ULN2 |
9 (1/11) |
33 (2/6) |
|
- Pacienti se základní ALT > 2x ULN |
60 (9/15) |
100 (11/11) |
|
neměřitelná HBV DNA | ||
|
- Základní3 |
5 (2/40) |
6 (1/18) |
|
- Týden 2604 | ||
|
negativní |
8 (2/25) |
0 |
|
pozitivní < základní |
92 (23/25 |
100 (4/4) |
|
pozitivní > základní |
0 |
0 |
|
normalizované hodnoty ALT | ||
|
- Základní | ||
|
normální |
28 (11/40) |
33 (6/18) |
|
nad normální |
73 (29/40) |
67 (12/18) |
|
- Týden 260 | ||
|
normální |
46 (13/28) |
50 (2/4) |
|
nad normální < základní |
21 (6/28) |
0 |
|
nad normální > základní |
32 (9/28) |
50 (2/4) |
1 Pacienti označení jako pacienti s YMDD mutací byli v > 5 % případů nositeli YMDD mutace viru HBV v kterémkoliv ročním časovém bodu v průběhu pětiletého období. Pacienti zařazení jako bez YMDD mutace byli v > 95 % případů nositeli divoké formy viru HBV ve všech ročních časových bodech v průběhu pětileté studie.
2 Horní hranice normálu.
3 Roztok Abbott Genostics k hybridizačnímu testu (LLOD < 1,6 pg/ml).
4 Chiron Quantiplex assay (LLOD 0,7 Meq/ml).
Srovnávací data podle YMDD charakteru byla rovněž dostupná pro histologické hodnocení, ale pouze do tří let. U pacientů s YMDD mutací HBV 18/39 (46 %) mělo zlepšenou nekroinflamační aktivitu a 9/39 (23 %) vykázalo zhoršení. U pacientů bez mutace mělo 20/27 (74 %) zlepšenou nekroinflamační aktivitu a 2/27 (7 %) měli tuto aktivitu zhoršenou.
V průběhu HBeAg sérokonverze jsou sérologická odpověď a remise klinických příznaků po vysazení lamivudinu obecně neměnné. Avšak mohou se vyskytovat recidivy sérokonverze. V dlouhodobých sledovacích studiích u pacientů, u kterých již došlo k sérokonverzi a ukončení léčby lamivudinem, došlo později k virologickému relapsu u 39 % ze všech sledovaných. Proto by v průběhu HBeAg sérokonverze pacienti měli být pravidelně sledováni, aby se zjistilo, zda je udržována sérologická a klinická odpověď. U pacientů, kteří nemají stálou sérologickou odpověď, by se mělo zvážit znovu podání léčby buď lamivudinem nebo alternativním antivirovým agens k opětovnému nabytí HBV klinické kontroly.
U pacientů sledovaných 16 týdnů po ukončení léčby v jednom roce bylo pozorováno zvýšení ALT častěji u jedinců léčených lamivudinem proti jedincům léčeným placebem. Srovnání vzestupu ALT po léčbě mezi týdny 52 a 68 u pacientů, kteří ukončili léčbu lamivudinem v 52. týdnu, a u pacientů léčených po celou dobu placebem ve stejných studiích, je uvedeno v tabulce 3. Poměr pacientů, u kterých došlo po léčbě k elevaci ALT ve spojitosti se zvýšením hladin bilirubinu, byl nízký a podobný jako u pacientů, kteří obdrželi lamivudin nebo placebo.
Tabulka 3: Zvýšení hladin ALT ve dvou placebem kontrolovaných studiích u dospělých.
|
Patologické výsledky |
Pacienti se zvýšenou hladinou ALT/ pacienti pouze sledovaní* | |
|
Lamivudin |
Placebo | |
|
ALT > 2x základní hodnota |
37/137 (27 %) |
22/116 (19 %) |
|
ALT > 3x základní hodnota^ |
29/137 (21 %) |
9/116 (8 %) |
|
ALT > 2x základní hodnota a absolutní ALT > 500 IU/l |
21/137 (15 %) |
8/116 (7 %) |
|
ALT > 2x základní hodnota; a bilirubin > 2x ULN > základní hodnota |
1/137 (0,7 %) |
1/116 (0,9 %) |
* Jeden pacient může reprezentovat jednu nebo více kategorii. ^ Porovnatelný se stupněm 3 podle WHO kriterií.
ULN = horní hranice normálních hodnot.
Zkušenosti u pacientů s HBeAg negativní CHB
Úvodní data ukazují, že účinnost lamivudinu je u pacientů s HBeAg negativní CHB stejná jako u pacientů s HBeAg pozitivní CHB, kdy má 71 % pacientů HBV DNA sníženou pod detekční limit,
67 % normalizaci ALT a 38 % zlepšení v HAI po jednom roce léčby. Po ukončení léčby lamivudinem došlo u většiny pacientů (70 %) k návratu virové replikace. Údaje jsou získané ze studie prodloužené léčby HBeAg negativních pacientů (NUCAB3017), kteří byli léčeni lamivudinem. Po dvou letech léčby v této studii došlo k normalizaci ALT a nedetekovatelnosti HBV DNA u 30/69 (43 %) a 32/68 (47 %) pacientů a k zlepšení v nekroinflamačním skóre u 18/49 (37 %) pacientů. U pacientů bez YMDD mutace HBV bylo prokázáno u 14/22 (64 %) zlepšení nekroinflamačního skóre a u 1/22 (5 %) pacientů zhoršení ve srovnání s obdobím před léčbou. U pacientů s mutací bylo u 4/26 (15 %) prokázáno zlepšení nekroinflamačního skóre a u 8/26 (31 %) pacientů zhoršení ve srovnání s obdobím před léčbou. Ani v jedné skupině pacientů nedošlo k progresi cirhózy.
Frekvence vzniku YMDD mutace HBV a její vliv na odpověď na léčbu
Monoterapie lamivudinem vede k selekci YMDD mutace HBV přibližně u 24 % pacientů v průběhu jednoho roku léčby se zvýšením na 69 % po 5 letech léčby. Vývoj YMDD mutace HBV je u některých pacientů spojován se snížením léčebné odpovědi dokládané zvýšením hladin HBV DNA a elevací ALT proti předchozím hodnotám při léčbě, progresí příznaků a symptomů hepatitidy a/nebo zhoršením jaterních nekroinflamačních nálezů. Vzhledem k riziku YMDD mutace HBV není udržovací monoterapie lamivudinem vhodná u pacientů s detekovatelnou HBV DNA v séru po 24. týdnu léčby (viz bod 4.4).
Ve dvojitě zaslepené studii u CHB pacientů s YMDD mutací HBV a kompenzovaným jaterním onemocněním (NUC20904) se sníženou virologickou a biochemickou odpovědí na lamivudin (n=95), vedlo přidání adefovir-dipivoxylu 10 mg jednou denně k pokračující léčbě lamivudinem 100 mg po dobu 52 týdnů k střednímu poklesu HBV DNA o 4,6 logJ0 kopií/ml ve srovnání se středním zvýšením o 0,3 logi0 kopií/ml u pacientů léčených monoterapií lamivudinem. K normalizaci hladin ALT došlo u 31 % (14/45) pacientů léčených kombinovanou terapií versus 6 % (3/47) pacientů léčených samotným lamivudinem. Parametry virologické suprese se u pacientů léčených kombinovanou léčbou v pokračovací studii NUC20917 udržely na stejných hodnotách v průběhu druhého roku léčby do 104. týdne léčby. U pacientů docházelo k pokračujícímu zlepšování odpovědi na léčbu, což se projevilo ve výsledcích virologických a biochemických vyšetření.
V retrospektivní studii určené ke zjištění faktorů zodpovědných za nárůst hladiny HBV DNA bylo 159 HBeAg-pozitivních pacientů asijského původu léčeno lamivudinem a sledováno v období mediánu téměř 30 měsíců. U pacientů s hodnotami HBV DNA vyššími než 200 kopií v 1 ml po době 6 měsíců (ve 24. týdnu) léčby lamivudinem byla 60% pravděpodobnost vzniku mutace YMDD viru HBV v porovnání s 8 % pacientů, u kterých byly hodnoty HBV DNA ve 24. týdnu léčby lamivudinem nižší než 200 kopií v 1 ml. Riziko vzniku mutace YMDD bylo 63 % oproti 13 % s mezní hodnotou
1 000 kopií v 1 ml (klinické studie NUCB3009 a NUCB3018).
Zkušenosti u pacientů s dekompenzovaným onemocněním jater
U pacientů s dekompenzovanou hepatopatií nebyly provedeny placebem kontrolované studie. Údaje z nekontrolovaných studií u této populace pacientů, jimž byl lamivudin podáván před transplantací jater, v jejím průběhu a po ní, prokázaly inhibiční účinek na hladiny HBV DNA v séru a normalizaci hodnot aminotransferázy. Pokračovala-li léčba lamivudinem po transplantacích, došlo ke snížení reinfekce HBV, k zvýšené ztrátě HBsAg pozitivity, a jednoleté přežití bylo v rozmezí 76 až 100 %.
Jak se vzhledem k současné imunosupresi očekávalo, byl výskyt YMDD mutace viru HBV po 52 týdnech léčby vyšší (36 % až 64 %) u pacientů po transplantaci jater než u imunokompetentních pacientů s CHB (14 % až 32 %).
40 pacientů (HBeAg negativních nebo HBeAg pozitivních) s dekompenzovaným jaterním onemocněním nebo rekurentní HBV infekcí po jaterní transplantaci a mutací YMDD bylo zařazeno do otevřeného ramene studie s názvem NUC20904. Přidání adefovir-dipivoxilu v dávce 10 mg jednou denně k terapii lamivudinem v dávce 100 mg po dobu 52 týdnů mělo za následek snížení počtu molekul HBV DNA (medián 4,6 logJ0 kopií HBV DNA v 1 ml). Po jednom roce léčby bylo rovněž pozorováno zlepšení funkce jater. Tento stupeň suprese viru zůstával při podávání kombinované léčby během druhého roku léčby stejný do 104. týdne terapie (pokračovací studie NUC20917). U většiny pacientů se zlepšily ukazatele jaterních funkcí a v klinickém obraze byly nadále patrné výhody podávané farmakoterapie.
Zkušenosti u pacientů s CHB s pokročilou fibrózou nebo cirhózou
V placebem kontrolované studii u 651 pacientů s klinicky kompenzovanou chronickou hepatitidou B a histologicky ověřenou fibrózou nebo cirhózou snížila léčba lamivudinem (průměr trvání 32 měsíců) významně celkový rozsah progrese onemocnění (34/436, 7,8 % pro lamivudin versus 38/215, 17,7 % pro placebo, p=0,001), což bylo prokázáno významnou redukcí v poměru pacientů se zvýšeným Child-Pugh skóre (15/436, 3,4 % versus 19/215, 8,8 %, p=0,023) nebo ve vývoji hepatocelulárního karcinomu (17/436, 3,9 % versus 16/215, 7,4 %, p=0,047). Rychlost celkové progrese onemocnění
v lamivudinové skupině byla vyšší pro jednotlivce s detekovatelnou YMDD mutací HBV DNA (23/209, 11 %) ve srovnání s těmi bez detekovatelné YMDD mutace HBV (11/221, 5 %). Avšak progrese onemocnění u jednotlivců s YMDD v lamivudinové skupině byla nižší než progrese onemocnění ve skupině s placebem (23/209, 11 % versus 38/214, 18 %). Potvrzená HBeAg sérokonverze se vyskytla u 47 % (118/252) jednotlivců léčených lamivudinem a 93 % (320/345) jednotlivců léčených lamivudinem se stalo HBV DNA negativními (VERSANT [verze 1], bDNA zkouška LLOD < 0,7 MEq/ml) v průběhu studie.
Zkušenosti u dětí a dospívajících
Lamivudin byl podáván dětem a dospívajícím s kompenzovanou CHB v placebem kontrolované klinické studii u 286 pacientů ve věku od 2 do 17 let. Tato populace sestávala hlavně z dětí s velmi lehkou formou hepatitidy B. Dětem od 2 do 11 let byla podávána dávka 3 mg/kg jedenkrát denně (až do maximální denní dávky 100 mg). Dětem starším než 12 let byla podávána dávka 100 mg jednou denně. Tuto dávku je třeba v budoucnu potvrdit. Rozdíl v sérokonverzi HBeAg (HBeAg a ztráta HBV DNA spolu s detekcí HBeAb) mezi placebem a lamivudinem nebyl v této populaci statisticky významný [po jednom roce 13 % (12/95) pro placebo oproti 22 % (42/191) pro lamivudin; p=0,057].
Výskyt YMDD mutace HBV byl stejný jako u dospělých, v rozsahu od 19 % v 52. týdnu léčby až do 45 % u pacientů léčených kontinuálně 24 měsíců.
5.2 Farmakokinetické vlastnosti
Absorpce
Lamivudin se dobře vstřebává z gastrointestinálního traktu a normální hodnota jeho biologické dostupnosti po perorálním podání je u dospělých mezi 80 a 85 %. Průměrná doba (tmax) dosažení maximálních sérových koncentrací (Cmax) po perorálním podání je kolem jedné hodiny. Po terapeutických dávkách, tj. 100 mg jednou denně, bývají hodnoty Cmax v rozmezí 1,1 až 1,5 pg/ml; nejnižší hladiny na konci dávkovacího intervalu byly 0,015 až 0,020 pg/ml.
Užití lamivudinu spolu s jídlem mělo za následek delší tmax a nižší Cmax (snížení až o 47 %). Celková míra absorpce lamivudinu (hodnocená podle AUC) však nebyla ovlivněna, takže lamivudin lze užívat jak s jídlem, tak nalačno.
Distribuce
Průměrný distribuční objem zjištěný ve studiích po nitrožilním podání je 1,3 l/kg. Lamivudin se v rozmezí terapeutických dávek vyznačuje lineární farmakokinetikou a vykazuje malou míru vazby na plazmatické proteiny (albumin).
Omezené údaje svědčí o tom, že lamivudin proniká do centrálního nervového systému a dostává se do mozkomíšního moku (MMM). Průměrný poměr koncentrace lamivudinu v MMM a v séru 2 až 4 hodiny po perorálním podání byl přibližně 0,12.
Biotransformace
Lamivudin je z organismu odstraňován převážně renální exkrecí v intaktní formě. Pravděpodobnost metabolických lékových interakcí je u lamivudinu malá vzhledem k malému rozsahu (5 až 10 %) jeho hepatální biotransformace a k malé míře jeho vazby na plazmatické proteiny.
Eliminace
Průměrná systémová clearance lamivudinu je přibližně 0,3 l/hod/kg. Pozorovaný eliminační poločas činí 5 až 7 hodin. Převážná část lamivudinu se vylučuje v intaktní formě močí cestou glomerulární filtrace a aktivní sekrece (transportním systémem pro organické kationty). Renální clearance je zodpovědná za eliminaci přibližně 70 % lamivudinu.
Zvláštní skupiny pacientů
Studie u pacientů s poruchou funkce ledvin prokázaly, že porucha funkce ledvin ovlivňuje eliminaci lamivudinu. U pacientů s clearance kreatininu < 50 ml/min je nezbytné snížení dávky (viz bod 4.2).
Porucha funkce jater neovlivňuje farmakokinetiku lamivudinu. Omezené údaje získané u pacientů podstupujících transplantaci jater prokazují, že porucha funkce jater, pokud není provázena poruchou funkce ledvin, nemá významný vliv na farmakokinetiku lamivudinu.
Farmakokinetický profil lamivudinu u starších pacientů nasvědčuje tomu, že normální stárnutí s průvodním poklesem renálních funkcí nemá klinicky významný vliv na expozici lamivudinu, kromě pacientů s clearance kreatininu < 50 ml/min (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
V toxikologických studiích na zvířatech nevyvolalo podání lamivudinu ve vysokých dávkách podstatnou orgánovou toxicitu. Při nejvyšších dávkách byly pozorovány nepříliš intenzivní účinky na indikátory hepatálních a renálních funkcí spolu s občasnou sníženou hmotností jater. Jako efekty, jež by nejspíše mohly mít klinický význam, byly identifikovány poklesy počtu erytrocytů a neutrofilů. Tyto příhody byly zřídka pozorovány v klinických studiích.
Lamivudin nebyl mutagenní v testech na bakteriích, avšak - podobně jako mnohé jiné nukleosidové analogy - vykázal mutagenní účinky in vitro v jednom cytogenetickém testu a v testu myšího lymfomu. In vivo nebyl lamivudin genotoxický ani v dávkách způsobujících plazmatické koncentrace přibližně 60krát až 70krát vyšší, než jsou předpokládané klinické plazmatické hladiny. Protože mutagenní působení lamivudinu in vitro nebylo potvrzeno testy in vivo, usuzuje se, že by lamivudin pro pacienty neměl představovat genotoxické riziko.
Reprodukční studie na zvířatech neprokázaly teratogenitu ani ovlivnění samčí nebo samičí fertility. Při podání březím králičím samicím v expozičních hladinách srovnatelných s hladinami dosaženými u lidí způsobuje lamivudin časnou embryonální letalitu, u potkanů ani při velmi vysoké systémové expozici však tyto účinky nemá.
V dlouhodobých testech na kancerogenitu lamivudinu u potkanů a myší nebyly prokázány žádné známky kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulosa Sodná sůl karboxymethylškrobu Magnesium-stearát
Potahová vrstva tablety Hypromelosa Oxid titaničitý Makrogol 400 Polysorbát 80
Žlutý oxid železitý (syntetický)
Červený oxid železitý (syntetický)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a obsah balení
Krabička obsahující 28 nebo 84 potahovaných tablet v dvoufóliových blistrech, laminovaných polyvinylchloridem.
Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/114/001
EU/1/99/114/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 29. července 1999
Datum posledního prodloužení registrace: 23. června 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Zeffix 5 mg/ml perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml perorálního roztoku obsahuje lamivudinum 5 mg.
Pomocné látky se známým účinkem:
sacharosa 20 % w/v (4 g/20 ml) methylparaben (E218) 1,5 mg/ml propylparaben (E216) 0,18 mg/ml
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok.
Čirý, bezbarvý až světle žlutý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Zeffix je indikován k léčbě dospělých pacientů s chronickou hepatitidou B s:
• kompenzovaným jaterním onemocněním s prokázanými známkami virové replikace, přetrvávajícími zvýšenými hodnotami sérové alaninaminotransferázy (ALT) a histologicky dokumentovaným aktivním zánětem jater a/nebo jaterní fibrózou. Podávání lamivudinu by mělo být zahájeno pouze v případě, kdy není možné nebo přípustné podávat jiné alternativní antivirotikum, u kterého je menší pravděpodobnost vzniku rezistence (viz bod 5.1).
• dekompenzovaným jaterním onemocněním v kombinaci s jiným léčivým přípravkem, který nevykazuje zkříženou rezistenci s lamivudinem (viz bod 4.2).
4.2 Dávkování a způsob podání
Terapii přípravkem Zeffix má zahajovat lékař, který má zkušenosti s léčbou chronické hepatitidy B.
Dávkování
Dospělí
Doporučené dávkování přípravku Zeffix je 100 mg jednou denně.
U pacientů s dekompenzovaným jaterním onemocněním má být lamivudin vždy podáván v kombinaci s dalším léčivým přípravkem, který nevykazuje zkříženou rezistenci s lamivudinem, aby se snížilo riziko rezistence a dosáhlo rychlého potlačení replikace viru.
Trvání léčby
Optimální délka léčby není známa.
• U HBeAg-pozitivních pacientů s chronickou hepatitidou B (CHB) bez cirhózy má léčba trvat alespoň 6-12 měsíců po prokázání sérokonverze HBeAg (ztráty HBeAg a HBV DNA s průkazem HBeAb), aby se omezilo riziko virologického relapsu, nebo do sérokonverze HBsAg, nebo až do ztráty účinnosti (viz bod 4.4). Hladiny sérových ALT a HBV DNA mají být po ukončení léčby pravidelně sledovány pro možnost pozdního virologického relapsu.
• U HBeAg-negativních pacientů s CHB (infikovaných pre-core mutantou) bez cirhózy má být léčba podávána alespoň, než dojde k HBs sérokonverzi nebo je prokazatelná ztráta účinnosti.
Při prodloužené léčbě se doporučuje pravidelné opakované vyšetření potvrzující, že pokračování ve vybrané léčbě zůstává pro pacienta vhodné.
• U pacientů s dekompenzovanou hepatopatií nebo cirhózou a u příjemců jaterních transplantátů se zastavení léčby nedoporučuje (viz bod 5.1).
Pacienti, u nichž byla aplikace lamivudinu zastavena, mají být pravidelně kontrolováni se zaměřením na známky recidivující hepatitidy (viz bod 4.4).
Klinická rezistence
U pacientů s HBeAg pozitivní nebo HBeAg negativní chronickou hepatitidou B může vznik mutace viru HBV s označením YMDD (tyrozin-methionin-aspartát-aspartát) způsobit snížení terapeutické odpovědi na lamivudin, což se projeví jako zvýšení hladiny HBV DNA a enzymu ALT oproti hodnotám před započetím léčby lamivudinem. Aby se snížilo riziko vzniku rezistence u pacientů léčených monoterapií lamivudinem, má se zvážit přechod na jiný přípravek nebo přidání dalšího přípravku, který nevykazuje zkříženou rezistenci s lamivudinem podle léčebných pokynů, pokud při monoterapii lamivudinem zůstávají hladiny HBV DNA v séru detekovatelné po 24. týdnu léčby (viz bod 5.1).
Pro léčbu pacientů, kteří jsou zároveň nakaženi HIV a léčeni lamivudinem nebo kombinací lamivudinu a zidovudinu, nebo je u nich tato léčba plánována, je nutno zachovat dávku lamivudinu předepsanou pro léčbu infekce HIV (obvykle 150 mg dvakrát denně v kombinaci s dalšími antiretrovirotiky).
Zvláštní skupiny pacientů
Porucha funkce ledvin
U pacientů se středně závažnou až závažnou poruchou funkce ledvin se v důsledku snížené renální clearance zvyšují koncentrace lamivudinu v séru (AUC). Proto má být u pacientů s clearance kreatininu < 50 ml/min sníženo dávkování. Jsou-li potřebné dávky menší než 100 mg, má se aplikovat Zeffix perorální roztok (viz tabulka 1 níže).
Tabulka 1: Dávkování přípravku Zeffix u pacientů se sníženou renální clearance.
|
Clearance kreatininu ml/min |
První dávka přípravku Zeffix perorální roztok |
Udržovací dávka jednou denně |
|
30 až < 50 |
20 ml (100 mg) |
10 ml (50 mg) |
|
15 až < 30 |
20 ml (100 mg) |
5 ml (25 mg) |
|
5 až < 15 |
7 ml (35 mg) |
3 ml (15 mg) |
|
< 5 |
7 ml (35 mg) |
2 ml (10 mg) |
Dostupné údaje o pacientech podstupujících intermitentní hemodialýzu (méně než 4 hodiny nebo rovnající se 4 hodinám dialýzy dvakrát až třikrát týdně) ukazují, že po iniciální redukci dávky lamivudinu podle clearance kreatininu po dobu probíhající dialýzy již nejsou další úpravy dávkování nutné.
Porucha funkce jater
Údaje o pacientech s poruchou funkce jater, včetně pacientů s terminální hepatopatií čekajících na transplantaci, ukazují, že porucha funkce jater nemá významný vliv na farmakokinetiku lamivudinu. Podle těchto údajů se pacientům s poruchou funkce jater, není-li provázena poruchou funkce ledvin, nemusí dávkování upravovat.
Starší pacienti
U starších pacientů nemá stárnutí doprovázené zhoršením funkce ledvin klinicky významný vliv na expozici lamivudinu, s výjimkou pacientů s clearance kreatininu < 50 ml/min.
Pediatrická populace
Bezpečnost a účinnost podávání přípravku Zeffix u kojenců, dětí a dospívajících mladších 18 let nebyly stanoveny. V současnosti dostupné údaje jsou popsány v bodech 4.4 a 5.1, ale na jejich základě nelze doporučit dávkování.
Způsob podání Perorální podání.
Přípravek Zeffix lze užívat jak nalačno, tak s jídlem.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Laktátová acidóza a těžká hepatomegalie se steatózou
Při užívání nukleosidových analogů byl hlášen vznik laktátové acidózy (bez hypoxemie), někdy s fatálním průběhem, obvykle spojený s těžkou hepatomegalií a jaterní steatózou. Jelikož Zeffix je nukleosidovým analogem, toto riziko nelze vyloučit. Léčba nukleosidovými analogy musí být přerušena při rychlém vzestupu hladin aminotransferáz, progresivní hepatomegalii nebo metabolické/laktátové acidóze neznámé etiologie. Benigní zažívací příznaky, jako je nauzea, zvracení a bolest břicha, mohou svědčit o rozvíjející se laktátové acidóze. Závažné případy, někdy s fatálním koncem, byly spojeny s pankreatitidou, jaterním selháním/steatózou jater, renálním selháním a vyššími hladinami sérového laktátu. S opatrností je třeba předepisovat nukleosidové analogy pacientovi (zejména obézním ženám) s hepatomegalií, hepatitidou nebo jinými známými rizikovými faktory vedoucími k onemocnění jater a jaterní steatóze (včetně některých léčivých přípravků a alkoholu). Pacienti koinfikovaní virem hepatitidy C a léčení alfa interferonem a ribavirinem mohou představovat zvláštní riziko. Tito pacienti mají být pečlivě sledováni.
Exacerbace hepatitidy
Exacerbace v průběhu léčby
Spontánní exacerbace jsou u chronické hepatitidy B relativně časté a jsou charakterizované přechodným zvýšením hladin ALT v séru. Po započetí antivirové léčby může u některých pacientů dojít k přechodnému zvýšení hladin ALT v séru v souvislosti s tím, jak klesají v séru hladiny HBV DNA. U pacientů s kompenzovaným jaterním onemocněním tato zvýšení hladin ALT v séru nebyla obvykle doprovázena zvýšením koncentrací sérového bilirubinu ani příznaky jaterní dekompenzace.
Při prodloužené terapii byly identifikovány virové subpopulace HBV se sníženou citlivostí na lamivudin (YMDD-mutace viru HBV). U některých pacientů vede vznik YMDD mutace viru HBV k exacerbaci hepatitidy primárně prokázané zvýšením hodnot ALT v séru a znovu objevením se HBV DNA (viz bod 4.2). U pacientů, kteří mají YMDD mutaci HBV, se má zvážit na základě terapeutických doporučení přechod na jiný léčivý přípravek nebo přidání dalšího léčivého přípravku, který nevykazuje zkříženou rezistenci s lamivudinem (viz bod 5.1).
Exacerbace po ukončení léčby
U pacientů, kteří ukončili léčbu hepatitidy B, byla zaznamenána akutní exacerbace hepatitidy, která se obvykle prokazuje zvýšenou hladinou ALT v séru a znovu objevením se HBV DNA.
V kontrolovaných klinických studiích fáze III s následným sledováním pacientů bez aktivní léčby byla incidence elevací ALT po léčbě (více než 3násobně ve srovnání s výchozími hodnotami) vyšší u pacientů léčených lamivudinem (21 %) ve srovnání s pacienty, kteří dostávali placebo (8 %). Procento pacientů, u kterých došlo po ukončení léčby k elevacím ALT spolu s elevacemi bilirubinu, bylo však nižší a podobné v obou ramenech léčby (viz tabulka 3 v bodu 5.1). U pacientů léčených lamivudinem se většina elevací ALT po léčbě objevila mezi 8. a 12. týdnem po ukončení léčby. Většina případů odezněla spontánně, ale bylo pozorováno i několik fatálních případů. V případě, že se zastaví aplikace přípravku Zeffix, mají být pacienti pravidelně kontrolováni - jak klinicky, tak laboratorním vyšetřováním funkce jater (ALT a hladiny bilirubinu), a to nejméně po dobu čtyř měsíců a dále podle toho, jak je to klinicky indikováno.
Exacerbace u pacientů s dekompenzovanou jaterní cirhózou
Příjemci transplantátů a pacienti s dekompenzovanou jaterní cirhózou jsou více ohroženi aktivní virovou replikací. V důsledku minimální hepatální funkce u těchto pacientů může reaktivace hepatitidy po vysazení lamivudinu nebo snížením účinnosti v průběhu terapie přivodit těžkou i fatální dekompenzaci. Tito pacienti mají být během léčby pravidelně kontrolováni se zaměřením na klinické, virologické a sérologické parametry související s hepatitidou B, na hepatální a renální funkci a na antivirovou terapeutickou odezvu, a to minimálně každý měsíc, a je-li léčba z jakéhokoli důvodu zastavena, pak ještě nejméně 6 měsíců po léčbě. Pravidelně je nutné kontrolovat (minimálně) tyto laboratorní parametry: sérové ALT, bilirubin, albumin, dusík močoviny v krvi (BUN), kreatinin a tyto virologické parametry: antigeny HBV, protilátky proti HBV a pokud možno koncentrace HBV DNA v séru. Pacienti, u kterých se během léčby nebo po jejím zastavení objeví známky hepatální insuficience, mají být kontrolováni v adekvátně kratších časových intervalech.
Není dostatek údajů o tom, zda je u pacientů, u kterých se vyvinou známky rekurentní hepatitidy, prospěšné znovu zahájit terapii lamivudinem.
Mitochondriální dysfunkce
Bylo prokázáno jak in vitro, tak in vivo, že nukleosidové a nukleotidové analogy způsobují poškození mitochondrií různého stupně. Existují hlášení mitochondriální dysfunkce u kojenců vystavených in utero a/nebo postnatálně působení analogů nukleosidů. Hlavními hlášenými nežádoucími účinky jsou hematologické poruchy (anemie, neutropenie), metabolické poruchy (hyperlaktemie, zvýšená hladina lipázy). Byly hlášeny některé neurologické poruchy s pozdním nástupem (hypertonie, křeče, neobvyklé chování). Neurologické poruchy mohou být přechodné nebo stálé. Každé dítě, které bylo in utero vystaveno působení analogů nukleosidů a nukleotidů, musí být klinicky i laboratorně sledováno a v případě relevantních známek nebo příznaků musí projít úplným vyšetřením na možnou mitochondriální dysfunkci.
Pediatričtí pacienti
Lamivudin byl podáván dětem (2 roky a starším) a dospívajícím s kompenzovanou chronickou hepatitidou B. Avšak vzhledem k omezeným údajům se podávání lamivudinu těmto pacientům v současnosti nedoporučuje (viz bod 5.1).
Hepatitida delta nebo hepatitida C
Účinnost lamivudinu u pacientů současně infikovaných virem hepatitidy delta nebo virem hepatitidy C nebyla stanovena a doporučuje se opatrnost.
Imunosupresivní léčba
Údaje o použití lamivudinu u HBeAg negativních pacientů (pre-core mutanty) a u pacientů souběžně léčených imunosupresivními režimy, včetně protinádorové chemoterapie, jsou omezené. Tito pacienti by měli lamivudin užívat s opatrností.
Monitorování
V průběhu léčby přípravkem Zeffix mají být pacienti pravidelně monitorováni. Sérové hladiny ALT a HBV DNA se mají monitorovat ve 3měsíčních intervalech a u HBeAg pozitivních pacientů se mají stanovovat každých 6 měsíců hodnoty HBeAg.
Současná infekce HIV
U pacientů současně infikovaných HIV, kteří jsou současně léčeni lamivudinem nebo kombinací lamivudinu a zidovudinu nebo u nichž se plánuje zahájení takové léčby, má být zachována dávka lamivudinu předepsaná pro terapii infekce HIV (obvykle 150 mg dvakrát denně v kombinaci s jinými antiretrovirotiky). U pacientů současně infikovaných HIV, u nichž není nutná antiretrovirová farmakoterapie, existuje při použití samotného lamivudinu k terapii chronické hepatitidy B riziko mutace HIV.
Přenos hepatitidy B
Nejsou k dispozici informace o přenosu viru hepatitidy B na plod těhotných žen léčených lamivudinem. Je třeba se řídit standardními doporučenými postupy pro imunizaci proti viru hepatitidy B u dětí.
Pacienty je nutné upozornit na to, že nebylo prokázáno, že by terapie lamivudinem snižovala riziko přenosu viru hepatitidy B na jiné osoby, a proto je i nadále nutné dodržovat příslušná preventivní opatření.
Interakce s jinými léčivými přípravky
Zeffix se nemá užívat s jinými přípravky obsahujícími lamivudin, nebo přípravky obsahujícími emtricitabin (viz bod 4.5).
Kombinace lamivudinu s kladribinem se nedoporučuje (viz bod 4.5).
Nesnášenlivost pomocných látek
Pacienti se vzácnými vrozenými onemocněními, jako jsou intolerance fruktózy, glukózo-galaktózová malabsorpce a insuficience sacharázy-izomaltázy, by tento přípravek neměli užívat.
Diabetici musí být upozorněni na to, že každá dávka perorálního roztoku (100 mg = 20 ml) obsahuje 4 g sacharózy.
Perorální roztok obsahuje propylparaben a methylparaben. U některých jedinců mohou tyto látky způsobit alergickou reakci. Tato reakce může být opožděná.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Vzhledem k omezenému rozsahu metabolizace, k omezené míře vazby na plazmatické proteiny a k téměř úplné renální eliminaci intaktního léčiva je pravděpodobnost metabolických interakcí malá.
Lamivudin je eliminován převážně aktivní renální sekrecí organických kationtů. Je třeba brát v úvahu možnost interakcí s jinými souběžně aplikovanými léčivy, zejména těmi, jejichž hlavní eliminační cestou je aktivní renální sekrece prostřednictvím transportního systému pro organické kationty; takovým léčivem je např. trimethoprim. Jiná léčiva (např. ranitidin, cimetidin) jsou tímto mechanismem eliminována jen zčásti a nebylo prokázáno, že by interagovala s lamivudinem.
U léčiv, která jsou převážně eliminována buď aktivní renální sekrecí organických aniontů nebo glomerulární filtrací, nejsou klinicky významné interakce s lamivudinem pravděpodobné. Souběžná aplikace kombinace trimethoprim/sulfamethoxazol v dávkách 160 mg/800 mg zvýšila expozici lamivudinu asi o 40 %. Lamivudin neměl vliv na farmakokinetiku trimethoprimu ani sulfamethoxazolu. Úprava dávkování lamivudinu však není nutná, nejedná-li se o pacienta s poruchou funkce ledvin.
Při souběžné aplikaci lamivudinu se zidovudinem bylo pozorováno mírné (28%) zvýšení Cmax zidovudinu, ale celková expozice tomuto léčivu (AUC) se významně nezměnila. Zidovudin neměl vliv na farmakokinetiku lamivudinu (viz bod 5.2).
Při souběžné aplikaci lamivudinu s alfa-interferonem se nevyskytly farmakokinetické interakce.
U pacientů, kteří užívali lamivudin s běžně používanými imunosupresivy (např. s cyklosporinem A), nebyly pozorovány klinicky významné nežádoucí interakce. Formální interakční studie však nebyly provedeny.
Emtricitabin
Vzhledem k podobnosti se Zeffix nemá podávat současně s jinými analogy cytidinu, jako je např. emtricitabin. Zeffix také nemá být užíván s žádným jiným léčivým přípravkem, který obsahuje lamivudin (viz bod 4.4).
Kladribin
Lamivudin in vitro inhibuje intracelulární fosforylaci kladribinu, což vede k potenciálnímu riziku snížení účinnosti kladribinu v případě léčby kombinací těchto látek. Také některá klinická zjištění ukazují na možnou interakci mezi lamivudinem a kladribinem. Z tohoto důvodu se současné podávání lamivudinu a kladribinu nedoporučuje (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Velké množství dat u těhotných žen (více než 1 000 konečných expozic) neprokázalo malformační toxicitu. Zeffix může být užíván v průběhu těhotenství, pokud je klinicky potřebným.
U pacientek, které jsou léčeny lamivudinem a následně otěhotní, by se mělo uvažovat o možnosti vzniku rekurentní hepatitidy při ukončení léčby lamivudinem.
Kojení
Na základě výsledků pozorování u více než 200 párů matek s dítětem léčených pro infekci virem HIV bylo zjištěno, že sérové koncentrace lamivudinu u dětí kojených matkami, které se léčily pro infekci virem HIV, jsou velmi nízké (méně než 4 % koncentrací naměřených v mateřském séru) a postupně se snižují až na neměřitelné hodnoty v době, kdy kojené děti dosahují 24 týdnů věku. Celkové množství lamivudinu, které se vstřebá u kojených dětí, je velmi nízké, a proto je pravděpodobné, že toto množství lamivudinu má suboptimální antivirové účinky. Infekce hepatitidou B u matky není kontraindikací pro kojení, pokud je u novorozence při porodu adekvátně provedena prevence nákazy infekční hepatitidou B. Nejsou důkazy o tom, že by nízká koncentrace lamivudinu v mateřském mléce vedla ke vzniku nežádoucích reakcí u kojených dětí. Proto je možné uvažovat o kojení dětí matkami, které jsou pro hepatitidu B léčeny lamivudinem, a vzít přitom v úvahu, jaké jsou výhody kojení pro dítě a jaké výhody přináší podávání lamivudinu kojící matce. V případě, že dojde k přenosu viru HBV z matky na dítě i přes adekvátní profylaxi, mělo by se zvážit přerušení kojení, aby se u dítěte snížilo riziko vzniku mutací viru HBV, které by vedly k rezistenci viru vůči lamivudinu.
Fertilita
Reprodukční studie u zvířat neprokázaly žádný vliv na fertilitu u samců ani samic (viz bod 5.3). Mitochondriální dysfunkce
In vitro i in vivo bylo prokázáno, že nukleosidové a nukleotidové analogy způsobují různý stupeň mitochondriálního poškození. Existují hlášení mitochondriální dysfunkce u kojenců vystavených in utero a/nebo postnatálně působení analogů nukleosidů (viz bod 4.4).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacienty je nutno upozornit, že během léčby lamivudinem byly hlášeny malátnost a únava. Při posouzení pacientovy schopnosti řídit nebo obsluhovat stroje je nutno vzít v úvahu klinický stav pacienta a profil nežádoucích účinků lamivudinu.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Výskyt nežádoucích účinků a laboratorních odchylek (s výjimkou zvýšené ALT a CPK, viz níže) byl podobný u pacientů léčených placebem a lamivudinem. Z nežádoucích účinků byly nejčastěji hlášeny: malátnost a únava, infekce respiračního traktu, faryngeální a tonzilární diskomfort, bolest hlavy, abdominální diskomfort a bolest, nevolnost, zvracení a průjem.
Nežádoucí účinky v tabulce
Nežádoucí reakce jsou uvedeny níže podle orgánových systémů a frekvence výskytu. Frekvenční kategorie jsou přiřazeny jen těm nežádoucím reakcím, u kterých je alespoň možné uvažovat o příčinné souvislosti s lamivudinem. Frekvence výskytu je definována jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Kategorie frekvencí přiřazené nežádoucím účinkům jsou založeny převážně na poznatcích z klinických studií zahrnujících celkem 1 171 pacientů s chronickou hepatitidou B, kteří dostávali lamivudin v dávce 100 mg.
|
Poruchy krve a lymfatického systému | |
|
Není známo |
T rombocytopenie |
|
Poruchy imunitního systému | |
|
Vzácné |
Angioedém |
|
Poruchy jater a žlučových cest | |
|
Velmi časté |
Zvýšení ALT (viz bod 4.4) |
|
Exacerbace hepatitidy primárně prokázané zvýšením sérové hladiny ALT byly hlášené při léčbě a po vysazení lamivudinu. Většina případů spontánně odezněla, avšak velmi vzácně došlo k úmrtí (viz bod 4.4). | |
|
Poruchy kůže a podkožní tkáně | |
|
Časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Časté |
Zvýšení CPK |
|
Časté |
Svalové poruchy, včetně myalgie a křečí* |
|
Není známo |
Rhabdomyolýza |
|
* Ve fázi III klinickýc |
i studií nebyla frekvence rhabdomyolýzy zaznamenaná v léčebné skupině |
s lamivudinem vyšší proti skupině s placebem.
Pediatrická populace
Na základě omezených údajů od dětí ve věku 2 až 17 let nebyly identifikovány nové bezpečnostní signály oproti dospělým.
Další zvláštní populace
U pacientů infikovaných HIV byly hlášeny případy pankreatitidy a periferní neuropatie (nebo parestezie), třebaže jejich souvislost s léčbou lamivudinem nebyla jasně prokázána. U pacientů s chronickou hepatitidou B nebyl pozorován rozdíl ve výskytu těchto projevů mezi pacienty léčenými lamivudinem a pacienty užívajícími placebo.
Při kombinované farmakoterapii nukleosidovými analogy byly u pacientů s HIV hlášeny případy laktátové acidózy, někdy fatální, obvykle spojené s těžkou hepatomegalií a hepatální steatózou.
U pacientů s chronickou hepatitidou B léčených lamivudinem byl vzácně hlášen výskyt laktátové acidózy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Ve studiích akutní toxicity na zvířatech nevedlo podání velmi vysokých dávek lamivudinu k projevům orgánové toxicity. Údaje o následcích užití nadměrných dávek u lidí jsou omezené. Nevyskytly se fatální případy a pacienti se zotavili. Po těchto předávkováních nebyly zjištěny specifické objektivní ani subjektivní příznaky.
Dojde-li k předávkování, má být pacient sledován a v případě potřeby mu má být poskytována standardní podpůrná léčba. Jelikož lamivudin je odstranitelný dialýzou, mohla by se v terapii předávkování použít kontinuální hemodialýza, třebaže to nebylo studováno.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antivirotika pro systémové použití, nukleosidové a nukleotidové inhibitory reverzní transkriptázy, ATC kód: J05AF05.
Lamivudin je antivirotikum, které se ve všech testovaných buněčných liniích a u experimentálně infikovaných zvířat projevilo jako vysoce účinné proti viru hepatitidy B.
Lamivudin je infikovanými i neinfikovanými buňkami metabolizován na lamivudin-trifosfát (TP), který je vlastní účinnou formou výchozí látky. Intracelulární poločas lamivudin-trifosfátu v hepatocytech je in vitro 17 až 19 hodin. Lamivudin-trifosfát působí jako substrát HBV-virové polymerázy.
Inkorporací lamivudin-trifosfátu do řetězce virové DNA s následným ukončením řetězce se znemožní další tvorba virové DNA.
Lamivudin-trifosfát neinterferuje s normálním buněčným metabolismem deoxynukleotidů a je jen slabým inhibitorem savčích DNA-polymeráz alfa a beta. Kromě toho má lamivudin-trifosfát jen malý vliv na obsah DNA v savčích buňkách.
V testech zaměřených na možné účinky léčiva na strukturu mitochondrií a na obsah a funkci mitochondriální DNA neměl lamivudin významné toxické účinky. Lamivudin má velmi nízký potenciál ke snížení obsahu mitochondriální DNA, neinkorporuje se trvale do mitochondriální DNA a nepůsobí jako inhibitor mitochondriální DNA-polymerázy gama.
Klinické zkušenosti
Zkušenosti u pacientů s HBeAg pozitivní CHB a kompenzovaným onemocněním jater Jednoroční terapie lamivudinem v placebem kontrolovaných studiích významně potlačila replikaci HBV DNA [34 až 57 % pacientů mělo hodnoty nižší, než byl detekční limit analýzy (Abbott Genostics hybridizační testovací roztok, LLOD < 1,6 pg/ml)], normalizovala hodnoty ALT (40 až 72 % pacientů), navodila sérokonverzi HBeAg (ztrátu HBeAg a HBeAb s průkazem HBV DNA [s konvenční analýzou] u 16 až 18 % pacientů), zlepšila histologický nález (38 až 52 % pacientů mělo
> 2 body poklesu Knodellova skóre - index histologické aktivity [HAI]) a zpomalila progresi fibrózy (u 3 až 17 % pacientů) a progresi jaterní cirhózy.
Trvalá léčba lamivudinem po dobu dalších 2 let u pacientů, kterým selhalo dosažení HBeAg sérokonverze v úvodních jednoročních kontrolovaných studiích, vedla k dalšímu zlepšení přemosťující fibrózy. Z pacientů s YMDD mutací viru HBV vykázalo 41/82 (50 %) pacientů zlepšení jaterního zánětu a 40/56 (71 %) pacientů bez YMDD mutace viru HBV vykázalo zlepšení. Zlepšení přemosťující fibrózy bylo pozorováno u 19/30 (63 %) pacientů bez YMDD mutace a u 22/44 (50 %) pacientů s mutací YMDD. Pět procent (3/56) pacientů bez YMDD mutace a 13 % (11/82) pacientů s YMDD mutací vykázalo zhoršení zánětu jater ve srovnání se stavem před zahájením léčby. Progrese cirhózy byla pozorována u 4/68 (6 %) pacientů s YMDD mutací, zatímco progrese cirhózy nebyla pozorována u pacientů bez mutace.
V rozsáhlé terapeutické studii u asijských pacientů (NUCB3018) byl výskyt HBeAg sérokonverze a normalizace Alt na konci pětiletého léčebného období 48 % (28/58) a 47 % (15/32). HBeAg sérokonverze byla zvýšena u pacientů s elevací hladin ALT; 77 % (20/26) pacientů s ALT před léčbou
> 2x ULN sérokonvertovalo. Na konci pěti let měli všichni pacienti hladiny HBV DNA, které nebyly detekovatelné, nebo nižší než hladiny před léčbou.
Další výsledky studie stavu YMDD mutace shrnuje tabulka 2.
Tabulka 2: Výsledky účinnosti po 5 letech podle YMDD stavu (asijská studie) NUCB3018.
|
Předmět % | ||
|
YMDD-mutace HBV |
YMDD1 |
Non-YMDD1 |
|
HBeAg sérokonverze - Všichni pacienti |
38 (15/40) |
72 (13/18) |
|
- Pacienti se základní ALT < 1x ULN2 |
9 (1/11) |
33 (2/6) |
|
- Pacienti se základní ALT > 2x ULN |
60 (9/15) |
100 (11/11) |
|
neměřitelná HBV DNA | ||
|
- Základní3 |
5 (2/40) |
6 (1/18) |
|
- Týden 2604 | ||
|
negativní |
8 (2/25) |
0 |
|
pozitivní < základní |
92 (23/25 |
100 (4/4) |
|
pozitivní > základní |
0 |
0 |
|
normalizované hodnoty ALT | ||
|
- Základní | ||
|
normální |
28 (11/40) |
33 (6/18) |
|
nad normální |
73 (29/40) |
67 (12/18) |
|
- Týden 260 | ||
|
normální |
46 (13/28) |
50 (2/4) |
|
nad normální < základní |
21 (6/28) |
0 |
|
nad normální > základní |
32 (9/28) |
50 (2/4) |
1 Pacienti označení jako pacienti s YMDD mutací byli v > 5 % případů nositeli YMDD mutace viru HBV v kterémkoliv ročním časovém bodu v průběhu pětiletého období. Pacienti zařazení jako bez YMDD mutace byli v > 95 % případů nositeli divoké formy viru HBV ve všech ročních časových bodech v průběhu pětileté studie.
2 Horní hranice normálu.
3 Roztok Abbott Genostics k hybridizačnímu testu (LLOD < 1,6 pg/ml).
4 Chiron Quantiplex assay (LLOD 0,7 Meq/ml).
Srovnávací data podle YMDD charakteru byla rovněž dostupná pro histologické hodnocení, ale pouze do tří let. U pacientů s YMDD mutací HBV 18/39 (46 %) mělo zlepšenou nekroinflamační aktivitu a 9/39 (23 %) vykázalo zhoršení. U pacientů bez mutace mělo 20/27 (74 %) zlepšenou nekroinflamační aktivitu a 2/27 (7 %) měli tuto aktivitu zhoršenou.
V průběhu HBeAg sérokonverze jsou sérologická odpověď a remise klinických příznaků po vysazení lamivudinu obecně neměnné. Avšak mohou se vyskytovat recidivy sérokonverze. V dlouhodobých sledovacích studiích u pacientů, u kterých již došlo k sérokonverzi a ukončení léčby lamivudinem, došlo později k virologickému relapsu u 39 % ze všech sledovaných. Proto by v průběhu HBeAg sérokonverze pacienti měli být pravidelně sledováni, aby se zjistilo, zda je udržována sérologická a klinická odpověď. U pacientů, kteří nemají stálou sérologickou odpověď, by se mělo zvážit znovu podání léčby buď lamivudinem nebo alternativním antivirovým agens k opětovnému nabytí HBV klinické kontroly.
U pacientů sledovaných 16 týdnů po ukončení léčby v jednom roce bylo pozorováno zvýšení ALT častěji u jedinců léčených lamivudinem proti jedincům léčeným placebem. Srovnání vzestupu ALT po léčbě mezi týdny 52 a 68 u pacientů, kteří ukončili léčbu lamivudinem v 52. týdnu, a u pacientů léčených po celou dobu placebem ve stejných studiích, je uvedeno v tabulce 3. Poměr pacientů, u kterých došlo po léčbě k elevaci ALT ve spojitosti se zvýšením hladin bilirubinu, byl nízký a podobný jako u pacientů, kteří obdrželi lamivudin nebo placebo.
Tabulka 3: Zvýšení hladin ALT ve dvou placebem kontrolovaných studiích u dospělých.
|
Patologické výsledky |
Pacienti se zvýšenou hladinou ALT/ pacienti pouze sledovaní* | |
|
Lamivudin |
Placebo | |
|
ALT > 2x základní hodnota |
37/137 (27 %) |
22/116 (19 %) |
|
ALT > 3x základní hodnota T |
29/137 (21 %) |
9/116 (8 %) |
|
ALT > 2x základní hodnota a absolutní ALT > 500 IU/l |
21/137 (15 %) |
8/116 (7 %) |
|
ALT > 2x základní hodnota; a bilirubin > 2x ULN > základní hodnota |
1/137 (0,7 %) |
1/116 (0,9 %) |
* Jeden pacient může reprezentovat jednu nebo více kategorii. ^ Porovnatelný se stupněm 3 podle WHO kriterií.
ULN = horní hranice normálních hodnot.
Zkušenosti u pacientů s HBeAg negativní CHB
Úvodní data ukazují, že účinnost lamivudinu je u pacientů s HBeAg negativní CHB stejná jako u pacientů s HBeAg pozitivní CHB, kdy má 71 % pacientů HBV DNA sníženou pod detekční limit,
67 % normalizaci ALT a 38 % zlepšení v HAI po jednom roce léčby. Po ukončení léčby lamivudinem došlo u většiny pacientů (70 %) k návratu virové replikace. Údaje jsou získané ze studie prodloužené léčby HBeAg negativních pacientů (NUCAB3017), kteří byli léčeni lamivudinem. Po dvou letech léčby v této studii došlo k normalizaci ALT a nedetekovatelnosti HBV DNA u 30/69 (43 %) a 32/68 (47 %) pacientů a k zlepšení v nekroinflamačním skóre u 18/49 (37 %) pacientů. U pacientů bez YMDD mutace HBV bylo prokázáno u 14/22 (64 %) zlepšení nekroinflamačního skóre a u 1/22 (5 %) pacientů zhoršení ve srovnání s obdobím před léčbou. U pacientů s mutací bylo u 4/26 (15 %) prokázáno zlepšení nekroinflamačního skóre a u 8/26 (31 %) pacientů zhoršení ve srovnání s obdobím před léčbou. Ani v jedné skupině pacientů nedošlo k progresi cirhózy.
Frekvence vzniku YMDD mutace HBV a její vliv na odpověď na léčbu
Monoterapie lamivudinem vede k selekci YMDD mutace HBV přibližně u 24 % pacientů v průběhu jednoho roku léčby se zvýšením na 69 % po 5 letech léčby. Vývoj YMDD mutace HBV je u některých pacientů spojován se snížením léčebné odpovědi dokládané zvýšením hladin HBV DNA a elevací ALT proti předchozím hodnotám při léčbě, progresí příznaků a symptomů hepatitidy a/nebo zhoršením jaterních nekroinflamačních nálezů. Vzhledem k riziku YMDD mutace HBV není udržovací monoterapie lamivudinem vhodná u pacientů s detekovatelnou HBV DNA v séru po 24. týdnu léčby (viz bod 4.4).
Ve dvojitě zaslepené studii u CHB pacientů s YMDD mutací HBV a kompenzovaným jaterním onemocněním (NUC20904) se sníženou virologickou a biochemickou odpovědí na lamivudin (n=95), vedlo přidání adefovir-dipivoxylu 10 mg jednou denně k pokračující léčbě lamivudinem 100 mg po dobu 52 týdnů k střednímu poklesu HBV DNA o 4,6 log10 kopií/ml ve srovnání se středním zvýšením
0 0,3 logi0 kopií/ml u pacientů léčených monoterapií lamivudinem. K normalizaci hladin ALT došlo u 31 % (14/45) pacientů léčených kombinovanou terapií versus 6 % (3/47) pacientů léčených samotným lamivudinem. Parametry virologické suprese se u pacientů léčených kombinovanou léčbou v pokračovací studii NUC20917 udržely na stejných hodnotách v průběhu druhého roku léčby do 104. týdne léčby. U pacientů docházelo k pokračujícímu zlepšování odpovědi na léčbu, což se projevilo ve výsledcích virologických a biochemických vyšetření.
V retrospektivní studii určené ke zjištění faktorů zodpovědných za nárůst hladiny HBV DNA bylo 159 HBeAg-pozitivních pacientů asijského původu léčeno lamivudinem a sledováno v období mediánu téměř 30 měsíců. U pacientů s hodnotami HBV DNA vyššími než 200 kopií v 1 ml po době 6 měsíců (ve 24. týdnu) léčby lamivudinem byla 60% pravděpodobnost vzniku mutace YMDD viru HBV v porovnání s 8 % pacientů, u kterých byly hodnoty HBV DNA ve 24. týdnu léčby lamivudinem nižší než 200 kopií v 1 ml. Riziko vzniku mutace YMDD bylo 63 % oproti 13 % s mezní hodnotou
1 000 kopií v 1 ml (klinické studie NUCB3009 a NUCB3018).
Zkušenosti u pacientů s dekompenzovaným onemocněním jater
U pacientů s dekompenzovanou hepatopatií nebyly provedeny placebem kontrolované studie. Údaje z nekontrolovaných studií u této populace pacientů, jimž byl lamivudin podáván před transplantací jater, v jejím průběhu a po ní, prokázaly inhibiční účinek na hladiny HBV DNA v séru a normalizaci hodnot aminotransferázy. Pokračovala-li léčba lamivudinem po transplantacích, došlo ke snížení reinfekce HBV, k zvýšené ztrátě HBsAg pozitivity, a jednoleté přežití bylo v rozmezí 76 až 100 %.
Jak se vzhledem k současné imunosupresi očekávalo, byl výskyt YMDD mutace viru HBV po 52 týdnech léčby vyšší (36 % až 64 %) u pacientů po transplantaci jater než u imunokompetentních pacientů s CHB (14 % až 32 %).
40 pacientů (HBeAg negativních nebo HBeAg pozitivních) s dekompenzovaným jaterním onemocněním nebo rekurentní HBV infekcí po jaterní transplantaci a mutací YMDD bylo zařazeno do otevřeného ramene studie s názvem NUC20904. Přidání adefovir-dipivoxilu v dávce 10 mg jednou denně k terapii lamivudinem v dávce 100 mg po dobu 52 týdnů mělo za následek snížení počtu molekul HBV DNA (medián 4,6 log10 kopií HBV DNA v 1 ml). Po jednom roce léčby bylo rovněž pozorováno zlepšení funkce jater. Tento stupeň suprese viru zůstával při podávání kombinované léčby během druhého roku léčby stejný do 104. týdne terapie (pokračovací studie NUC20917). U většiny pacientů se zlepšily ukazatele jaterních funkcí a v klinickém obraze byly nadále patrné výhody podávané farmakoterapie.
Zkušenosti u pacientů s CHB s pokročilou fibrózou nebo cirhózou
V placebem kontrolované studii u 651 pacientů s klinicky kompenzovanou chronickou hepatitidou B a histologicky ověřenou fibrózou nebo cirhózou snížila léčba lamivudinem (průměr trvání 32 měsíců) významně celkový rozsah progrese onemocnění (34/436, 7,8 % pro lamivudin versus 38/215, 17,7 % pro placebo, p=0,001), což bylo prokázáno významnou redukcí v poměru pacientů se zvýšeným Child-Pugh skóre (15/436, 3,4 % versus 19/215, 8,8 %, p=0,023) nebo ve vývoji hepatocelulárního karcinomu (17/436, 3,9 % versus 16/215, 7,4 %, p=0,047). Rychlost celkové progrese onemocnění
v lamivudinové skupině byla vyšší pro jednotlivce s detekovatelnou YMDD mutací HBV DNA (23/209, 11 %) ve srovnání s těmi bez detekovatelné YMDD mutace HBV (11/221, 5 %). Avšak progrese onemocnění u jednotlivců s YMDD v lamivudinové skupině byla nižší než progrese onemocnění ve skupině s placebem (23/209, 11 % versus 38/214, 18 %). Potvrzená HBeAg sérokonverze se vyskytla u 47 % (118/252) jednotlivců léčených lamivudinem a 93 % (320/345) jednotlivců léčených lamivudinem se stalo HBV DNA negativními (VERSANT [verze 1], bDNA zkouška LLOD < 0,7 MEq/ml) v průběhu studie.
Zkušenosti u dětí a dospívajících
Lamivudin byl podáván dětem a dospívajícím s kompenzovanou CHB v placebem kontrolované klinické studii u 286 pacientů ve věku od 2 do 17 let. Tato populace sestávala hlavně z dětí s velmi lehkou formou hepatitidy B. Dětem od 2 do 11 let byla podávána dávka 3 mg/kg jedenkrát denně (až do maximální denní dávky 100 mg). Dětem starším než 12 let byla podávána dávka 100 mg jednou denně. Tuto dávku je třeba v budoucnu potvrdit. Rozdíl v sérokonverzi HBeAg (HBeAg a ztráta HBV DNA spolu s detekcí HBeAb) mezi placebem a lamivudinem nebyl v této populaci statisticky významný [po jednom roce 13 % (12/95) pro placebo oproti 22 % (42/191) pro lamivudin; p=0,057]. Výskyt YMDD mutace HBV byl stejný jako u dospělých, v rozsahu od 19 % v 52. týdnu léčby až do 45 % u pacientů léčených kontinuálně 24 měsíců.
5.2 Farmakokinetické vlastnosti
Absorpce
Lamivudin se dobře vstřebává z gastrointestinálního traktu a normální hodnota jeho biologické dostupnosti po perorálním podání je u dospělých mezi 80 a 85 %. Průměrná doba (tmax) dosažení maximálních sérových koncentrací (Cmax) po perorálním podání je kolem jedné hodiny. Po terapeutických dávkách, tj. 100 mg jednou denně, bývají hodnoty Cmax v rozmezí 1,1 až 1,5 pg/ml; nejnižší hladiny na konci dávkovacího intervalu byly 0,015 až 0,020 pg/ml.
Užití lamivudinu spolu s jídlem mělo za následek delší tmax a nižší Cmax (snížení až o 47 %). Celková míra absorpce lamivudinu (hodnocená podle AUC) však nebyla ovlivněna, takže lamivudin lze užívat jak s jídlem, tak nalačno.
Distribuce
Průměrný distribuční objem zjištěný ve studiích po nitrožilním podání je 1,3 l/kg. Lamivudin se v rozmezí terapeutických dávek vyznačuje lineární farmakokinetikou a vykazuje malou míru vazby na plazmatické proteiny (albumin).
Omezené údaje svědčí o tom, že lamivudin proniká do centrálního nervového systému a dostává se do mozkomíšního moku (MMM). Průměrný poměr koncentrace lamivudinu v MMM a v séru 2 až 4 hodiny po perorálním podání byl přibližně 0,12.
Biotransformace
Lamivudin je z organismu odstraňován převážně renální exkrecí v intaktní formě. Pravděpodobnost metabolických lékových interakcí je u lamivudinu malá vzhledem k malému rozsahu (5 až 10 %) jeho hepatální biotransformace a k malé míře jeho vazby na plazmatické proteiny.
Eliminace
Průměrná systémová clearance lamivudinu je přibližně 0,3 l/hod/kg. Pozorovaný eliminační poločas činí 5 až 7 hodin. Převážná část lamivudinu se vylučuje v intaktní formě močí cestou glomerulární filtrace a aktivní sekrece (transportním systémem pro organické kationty). Renální clearance je zodpovědná za eliminaci přibližně 70 % lamivudinu.
Zvláštní skupiny pacientů
Studie u pacientů s poruchou funkce ledvin prokázaly, že porucha funkce ledvin ovlivňuje eliminaci lamivudinu. U pacientů s clearance kreatininu < 50 ml/min je nezbytné snížení dávky (viz bod 4.2).
Porucha funkce jater neovlivňuje farmakokinetiku lamivudinu. Omezené údaje získané u pacientů podstupujících transplantaci jater prokazují, že porucha funkce jater, pokud není provázena poruchou funkce ledvin, nemá významný vliv na farmakokinetiku lamivudinu.
Farmakokinetický profil lamivudinu u starších pacientů nasvědčuje tomu, že normální stárnutí s průvodním poklesem renálních funkcí nemá klinicky významný vliv na expozici lamivudinu, kromě pacientů s clearance kreatininu < 50 ml/min (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
V toxikologických studiích na zvířatech nevyvolalo podání lamivudinu ve vysokých dávkách podstatnou orgánovou toxicitu. Při nejvyšších dávkách byly pozorovány nepříliš intenzivní účinky na indikátory hepatálních a renálních funkcí spolu s občasnou sníženou hmotností jater. Jako efekty, jež by nejspíše mohly mít klinický význam, byly identifikovány poklesy počtu erytrocytů a neutrofilů. Tyto příhody byly zřídka pozorovány v klinických studiích.
Lamivudin nebyl mutagenní v testech na bakteriích, avšak - podobně jako mnohé jiné nukleosidové analogy - vykázal mutagenní účinky in vitro v jednom cytogenetickém testu a v testu myšího lymfomu. In vivo nebyl lamivudin genotoxický ani v dávkách způsobujících plazmatické koncentrace přibližně 60krát až 70krát vyšší, než jsou předpokládané klinické plazmatické hladiny. Protože mutagenní působení lamivudinu in vitro nebylo potvrzeno testy in vivo, usuzuje se, že by lamivudin pro pacienty neměl představovat genotoxické riziko.
Reprodukční studie na zvířatech neprokázaly teratogenitu ani ovlivnění samčí nebo samičí fertility. Při podání březím králičím samicím v expozičních hladinách srovnatelných s hladinami dosaženými u lidí způsobuje lamivudin časnou embryonální letalitu, u potkanů ani při velmi vysoké systémové expozici však tyto účinky nemá.
V dlouhodobých testech na kancerogenitu lamivudinu u potkanů a myší nebyly prokázány žádné známky kancerogenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Sacharosa (20%, w/v)
Methylparaben (E218)
Propylparaben (E216)
Kyselina citronová Propylenglykol Dihydrát natrium-citrátu Umělé jahodové aroma Umělé banánové aroma Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Po prvním otevření: 1 měsíc
6.4 Zvláštní opatření pro uchovávání Uchovávejte při teplotě do 25 °C.
6.5 Druh obalu a obsah balení
Krabička obsahující lahvičku z matného bílého polyethylenu vysoké hustoty (HDPE) s polypropylenovým dětským bezpečnostním uzávěrem. Tato lahvička obsahuje 240 ml perorálního roztoku lamivudinu. Součástí balení je také polyethylenový adaptér pro aplikátor a 10ml perorální aplikátor, který se skládá z polypropylenového barelu (s vyznačenou stupnicí v ml) a polyethylenového pístu.
Perorální dávkovací aplikátor je určen k přesnému měření předepsané dávky perorálního roztoku.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/114/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 29. července 1999
Datum posledního prodloužení registrace: 23. června 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
PŘÍLOHA II
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Potahované tablety:
Glaxo Wellcome Operations Priory Street, Ware Hertfordshire SG12 0DJ Velká Británie
nebo
GlaxoSmithKline Pharmaceuticals S.A. ul. Grunwaldzka 189 60-322 Poznaň Polsko
Perorální roztok:
Aspen Bad Oldesloe GmbH Industriestrasse 32-36 23843 Bad Oldesloe Německo
nebo
Glaxo Operations UK Limited (trading as Glaxo Wellcome Operations)
Harmire Road Barnard Castle County Durham DL12 8DT Velká Británie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Neuplatňuje se.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zeffix 100 mg potahované tablety Lamivudinum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna potahovaná tableta obsahuje lamivudinum 100 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 potahovaných tablet 84 potahovaných tablet
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/114/001 28 tablet EU/1/99/114/002 84 tablet
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
zeffix 100 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJĚ UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH 28 tablet, 84 tablet
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zeffix 100 mg tablety Lamivudinum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zeffix 5 mg/ml perorální roztok Lamivudinum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jeden ml perorálního roztoku obsahuje lamivudinum 5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje mimo jiné:
sacharosu, konzervační prostředky: methylparaben (E218) a propylparaben (E216).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Jedna lahvička obsahuje 240 ml perorálního roztoku. Balení obsahuje perorální aplikátor.
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Znehodnoťte po měsíci od prvního otevření.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/114/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
zeffix 5 mg/ml
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Zeffix 5 mg/ml perorální roztok Lamivudinum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jeden ml perorálního roztoku obsahuje lamivudinum 5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje mimo jiné:
sacharosu, konzervační prostředky: methylparaben (E218) a propylparaben (E216).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Jedna lahvička obsahuje 240 ml perorálmho roztoku.
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Znehodnoťte po měsíci od prvního otevření.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/99/114/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE BRAILLOVĚ PÍSMU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Zeffix 100 mg potahované tablety
lamivudinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Zeffix a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zeffix užívat
3. Jak se přípravek Zeffix užívá
4. Možné nežádoucí účinky
5. Jak přípravek Zeffix uchovávat
6. Obsah balení a další informace
1. Co je přípravek Zeffix a k čemu se používá
Léčivou látkou přípravku Zeffix je lamivudin.
B u dospělých.
B a patří do skupiny léčiv
Přípravek Zeffix se používá k léčbě dlouhodobé (chronické) hepatitidy
Přípravek Zeffix je lék proti virové infekci, který potlačuje virus hepatitidy zvaných nukleosidové inhibitory reverzní transkriptázy (NRTI).
Virus hepatitidy B infikuje játra, vyvolává dlouhodobou (chronickou) infekci a může vést k poškození jater. Zeffix mohou užívat lidé, jejichž játra jsou poškozená, ale stále pracují normálně (kompenzované onemocnění jater) a v kombinaci s dalšími léčivými přípravky lidé, jejichž játra jsou poškozená a normálně nepracují (dekompenzované onemocnění jater).
Léčba přípravkem Zeffix sníží množství viru hepatitidy B ve Vašem těle. Tím by se mělo omezit poškození Vašich jater a zlepšit jejich činnost. Odpověď na léčbu přípravkem Zeffix není u všech léčených osob stejná. Váš lékař bude sledovat účinnost Vaší léčby pomocí pravidelných krevních testů.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zeffix užívat Neužívejte přípravek Zeffix
• Jestliže jste alergický(á) na lamivudin nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
^ Pokud se domníváte, že se Vás toto týká, poraďte se se svým lékařem.
Upozornění a opatření
U některých osob užívajících přípravek Zeffix nebo jiné podobné léky existuje vyšší riziko závažných nežádoucích účinků. Je zapotřebí, abyste si byl(a) vědom(a) dalších rizikových faktorů:
• jestli jste někdy prodělal(a) jiné onemocnění jater jako je hepatitida C;
• jestli máte značnou nadváhu (zejména pokud jste žena).
Pokud se Vás cokoli z uvedeného týká, sdělte to svému lékaři. Je možné, že v průběhu léčby budete potřebovat další vyšetření, včetně krevních testů. Další informace o rizicích naleznete v bodě 4.
Nepřestávejte přípravek Zeffix užívat, dokud Vám to nedoporučí Váš lékař, protože jinak by Vám hrozilo nebezpečí zhoršení Vaší hepatitidy. Až skončíte s užíváním přípravku Zeffix, bude Vás lékař ještě minimálně po dobu čtyř měsíců pravidelně kontrolovat, aby se včas odhalily jakékoli případné problémy. Budou Vám odebírány vzorky krve na laboratorní vyšetření zvýšených hodnot jaterních enzymů, které mohou upozornit na poškození jater. Více informací o užívání přípravku Zeffix viz bod 3.
Sledujte, zda se u Vás neobjeví důležité příznaky
U některých osob užívajících léčivé přípravky proti hepatitidě B dochází k rozvoji dalších onemocnění, která mohou být závažná. Je zapotřebí, abyste byl(a) informován(a), jakým důležitým známkám onemocnění a příznakům máte během léčby přípravkem Zeffix věnovat pozornost.
^ Přečtěte si informace „Další možné nežádoucí účinky léčby hepatitidy B“ v bodě 4 této příbalové informace.
Chraňte ostatní osoby
Hepatitida B se přenáší sexuálním kontaktem s někým, kdo je tímto virem infikován nebo přenosem krve, ve které je virus přítomen (např. při používání společných injekčních jehel). Užívání přípravku Zeffix nezabrání přenosu hepatitidy B na další osoby. Abyste ochránil(a) ostatní osoby před nákazou hepatitidou B, je nutné:
• Používat kondom, pokud máte s někým orální nebo penetrativní sexuální styk.
• Neriskovat přenos viru krevní cestou - např. používáním jedné injekční jehly více osobami.
Další léčivé přípravky a přípravek Zeffix
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a přípravků dostupných bez lékařského předpisu.
Nezapomeňte sdělit svému lékaři nebo lékárníkovi, pokud začnete v průběhu léčby přípravkem Zeffix užívat nějaký jiný lék.
Tyto léky nemají být užívány s přípravkem Zeffix:
O jiné léčivé přípravky obsahující lamivudin, užívané k léčbě HIV infekce (onemocnění nazývaného AIDS);
O emtricitabin, užívaný k léčbě HIV nebo hepatitidy B;
O kladribin, užívaný k léčbě vlasatobuněčné leukémie.
^ Pokud jste léčen(a) některým z těchto přípravků, sdělte to svému lékaři.
Těhotenství
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět:
^ poraďte se se svým lékařem o rizicích a přínosech užívání přípravku Zeffix v průběhu těhotenství dříve, než začnete tento přípravek užívat. Nepřestávejte přípravek Zeffix užívat, dokud Vám to nedoporučí Váš lékař.
Kojení
Přípravek Zeffix může přecházet do mateřského mléka. Pokud kojíte, nebo o kojení uvažujete:
^ poraďte se se svým lékařem dříve, než začnete přípravek Zeffix užívat.
Řízení dopravních prostředků a obsluha strojů
Užíváte-li Zeffix, můžete se cítit unavený(á), což může ovlivnit Vaši schopnost řídit nebo obsluhovat stroje.
^ Neřiďte ani neobsluhujte stroje, aniž jste si jistý(á), že nejste ovlivněn(a).
3. Jak se přípravek Zeffix užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Zůstaňte v pravidelném kontaktu se svým lékařem
Přípravek Zeffix pomáhá udržet Vaši hepatitidu B pod kontrolou. Aby mohl udržovat infekci pod kontrolou a zabránit ve zhoršování nemoci, je třeba ho užívat každý den.
Zůstaňte v kontaktu se svým lékařem a nepřestávejte přípravek Zeffix užívat bez toho, abyste se poradil(a) se svým lékařem.
Kolik přípravku se užívá
Obvyklá dávka přípravku Zeffix je jedna tableta (100 mg lamivudinu) jednou denně.
Trpíte-li poruchou činnosti ledvin, Váš lékař Vám předepíše nižší dávku přípravku Zeffix. Pro osoby, které potřebují nižší než obvyklou dávku přípravku nebo nemohou užívat tablety, je k dispozici přípravek Zeffix ve formě perorálního roztoku.
^ Pokud se Vás toto týká, sdělte to svému lékaři.
Pokud již užíváte jiný léčivý přípravek obsahující lamivudin k léčbě infekce virem HIV, bude Váš lékař pokračovat v léčbě vyšší dávkou přípravku (obvykle 150 mg dvakrát denně), protože dávka lamivudinu v přípravku Zeffix (100 mg) není dostačující k léčbě infekce virem HIV. Pokud plánujete změnu Vaší léčby HIV, nejprve si o této změně promluvte se svým lékařem.
Tabletu spolkněte celou a zapijte ji vodou. Přípravek Zeffix může být užíván s jídlem nebo bez jídla. Jestliže jste užil(a) více přípravku Zeffix, než jste měl(a)
Je nepravděpodobné, že by omylem užité velké množství přípravku Zeffix způsobilo nějaké závažné problémy. Pokud nedopatřením užijete vyšší dávku, informujte o tom svého lékaře nebo lékárníka, nebo se poraďte na pohotovostním oddělení nejbližší nemocnice.
Jestliže jste zapomněl(a) užít Zeffix
Zapomenete-li si vzít dávku svého léku, vezměte si ji hned, jakmile si vzpomenete. Potom pokračujte v užívání stejně jako předtím. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Nepřestávejte užívat přípravek Zeffix
Bez porady s lékařem nesmíte ukončit užívání přípravku Zeffix. Riskujete tím zhoršení Vaší hepatitidy (viz bod 2). Přestanete-li Zeffix užívat, lékař Vás bude sledovat alespoň čtyři měsíce. Sledování bude zahrnovat odběry krve ke kontrole zvýšených hladin jaterních enzymů, které mohou ukazovat na poškození jater.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
V průběhu léčby hepatitidy B se, kromě níže uvedených nežádoucích účinků přípravku Zeffix,
mohou vyskytnout i jiné zdravotní potíže.
^ Je důležité, abyste si přečetl(a) informaci „Další možné nežádoucí účinky léčby hepatitidy B“.
Nežádoucími účinky často uváděnými v klinických studiích byly únava, infekce dýchacího ústrojí, bolest v krku, bolest hlavy, žaludeční obtíže a bolest, pocit na zvracení, zvracení a průjem, zvýšení jaterních enzymů a enzymů produkovaných ve svalech (viz níže).
Alergické reakce
Tyto reakce jsou vzácné (mohou se vyskytnout až u 1 z 1 000 osob). Příznaky zahrnují:
• otok očních víček, obličeje nebo rtů,
• obtíže při polykání nebo dýchání.
^ Pokud se u Vás vyskytnou tyto příznaky, neprodleně kontaktujte lékaře. Přestaňte přípravek Zeffix užívat.
Nežádoucí účinky, o kterých se předpokládá, že jsou způsobené přípravkem Zeffix:
Velmi častý nežádoucí účinek (může se vyskytnout u více než 1 osoby z 10), který se může projevit v krevních testech je:
• zvýšená hladina některých jaterních enzymů (transamináz), která může být známkou zánětu nebo poškození jater.
Časté nežádoucí účinky (mohou se vyskytnout až u 1 osoby z 10) jsou:
• křeče a bolesti svalů;
• kožní vyrážka nebo „kopřivka“ kdekoli na těle.
Častý nežádoucí účinek, který se může projevit v krevních testech je:
• zvýšená hladina enzymu tvořeného ve svalech (kreatinfosfokináza), která může být známkou poškození tělesné tkáně.
Jiné nežádoucí účinky
Jiné nežádoucí účinky se mohou vyskytnout u velmi malého počtu pacientů, ale přesná četnost není známa:
• rozpad svalové tkáně;
• zhoršení jaterního onemocnění po ukončení užívání nebo v průběhu užívání přípravku Zeffix, pokud se virus hepatitidy B stane rezistentním (necitlivým) na přípravek Zeffix. Toto může
u některých osob vést k úmrtí.
• laktátová acidóza (viz „Další možné nežádoucí účinky léčby hepatitidy B“ v dalším bodě).
Nežádoucí účinek, který se může projevit v krevních testech:
• snížený počet krevních buněk, které se uplatňují při srážení krve (trombocytopenie).
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků,
^ sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Další možné nežádoucí účinky léčby hepatitidy B
Při léčbě hepatitidy B přípravkem Zeffix a podobnými přípravky (NRTI) může dojít k rozvoji dalších zdravotních potíží.
Laktátová acidóza je vzácný, ale závažný nežádoucí účinek
U některých pacientů, kteří užívají přípravek Zeffix, nebo další podobné přípravky (NRTI), se může rozvinout onemocnění nazývané laktátová acidóza a zvětšení jater.
Laktátová acidóza je způsobená hromaděním kyseliny mléčné v těle. Je vzácná, ale pokud se objeví, je to obvykle v průběhu několika prvních měsíců léčby. Může být život ohrožující, protože může vést k selhání vnitřních orgánů.
Laktátová acidóza se s větší pravděpodobností objeví u osob, které mají onemocnění jater, nebo u obézních osob (s výraznou nadváhou), zejména u žen.
Příznaky laktátové acidózy zahrnují:
• hluboké, zrychlené a obtížné dýchání;
• ospalost;
• pocit snížené citlivosti nebo slabosti končetin;
• nevolnost (pocit na zvracení) a zvracení;
• bolest břicha.
V průběhu léčby bude lékař sledovat, zda se u Vás neobjevují známky laktátové acidózy. Pokud se u Vás objeví jakýkoli z příznaků zmíněných výše, nebo pokud Vás některý z jiných příznaků znepokojuje,
^ navštivte co nejdříve svého lékaře.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zeffix uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru. Uchovávejte při teplotě do 30 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Zeffix obsahuje
Léčivou látkou je lamivudinum. Jedna potahovaná tableta obsahuje lamivudinum 100 mg.
Pomocnými látkami j sou: mikrokrystalická celulosa, sodná sůl karboxymethylškrobu, magnesium-stearát, hypromelosa, oxid titaničitý, makrogol 400, polysorbát 80, žlutý oxid železitý (syntetický) a červený oxid železitý (syntetický).
Jak přípravek Zeffix vypadá a co obsahuje toto balení
Zeffix potahované tablety se dodávají v kartonové krabičce s vnitřním obalem ve formě blistrů. Jedno balení obsahuje 28 nebo 84 tablet. Tablety mají karamelovou barvu, podlouhlý bikonvexní tvar a na jedné straně je vyraženo označení „GX CG5“.
Ve Vaší zemi nemusí být k dispozici všechny velikosti balení.
Vysvětlení cizojazyčných údajů uvedených na obalech přípravku Zeffix:
EXP = Použitelné do Lot = Číslo šarže
Velká Británie
nebo
GlaxoSmithKline Pharmaceuticals S.A. ul. Grunwaldzka 189 60-322 Poznaň Polsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Česká republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111 cz.info@gsk.com
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Eesti
GlaxoSmithKline Eesti OU Tel: + 372 6676 900 estonia@gsk.com
EkXába
GlaxoSmithKline A.E.B.E.
Tqk + 30 210 68 82 100
Espaňa
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700 es-ci@gsk.com
France
Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com
Lietuva
GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Norge
GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no
Osterreich
GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com
Polska
GSK Services Sp. z o.o.
Tel.: + 48 (0)22 576 9000
Portugal
Glaxo Wellcome Farmaceutica, Lda. Tel: + 351 21 412 95 00 FI.PT@gsk.com
|
Hrvatska GlaxoSmithKline d.o.o. Tel: + 385 1 6051 999 |
Románia GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208 |
|
Ireland GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000 |
Slovenija GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com |
|
Ísland Vistor hf. Sími: + 354 535 7000 |
Slovenská republika GlaxoSmithKline Slovakia s. r. o Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com |
|
Italia GlaxoSmithKline S.p.A. Tel: + 39 (0)45 9218 111 |
Suomi/Finland GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com |
|
Kúnpoq GlaxoSmithKline (Cyprus) Ltd TpA.: + 357 22 39 70 00 gskcyprus@gsk.com |
Sverige GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com |
|
Latvija GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com |
United Kingdom GlaxoSmithKline UK Ltd Tel: + 44 (0)800 221441 customercontactuk@gsk.com |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Příbalová informace: informace pro pacienta
Zeffix 5 mg/ml perorální roztok
lamivudinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Zeffix a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zeffix užívat
3. Jak se přípravek Zeffix užívá
4. Možné nežádoucí účinky
5. Jak přípravek Zeffix uchovávat
6. Obsah balení a další informace
1. Co je přípravek Zeffix a k čemu se používá
Léčivou látkou přípravku Zeffix je lamivudin.
Přípravek Zeffix se používá k léčbě dlouhodobé (chronické) hepatitidy B u dospělých.
Přípravek Zeffix je lék proti virové infekci, který potlačuje virus hepatitidy B a patří do skupiny léčiv zvaných nukleosidové inhibitory reverzní transkriptázy (NRTI).
Virus hepatitidy B infikuje játra, vyvolává dlouhodobou (chronickou) infekci a může vést k poškození jater. Zeffix mohou užívat lidé, jejichž játra jsou poškozená, ale stále pracují normálně (kompenzované onemocnění jater) a v kombinaci s dalšími léčivými přípravky lidé, jejichž játra jsou poškozená a normálně nepracují (dekompenzované onemocnění jater).
Léčba přípravkem Zeffix sníží množství viru hepatitidy B ve Vašem těle. Tím by se mělo omezit poškození Vašich jater a zlepšit jejich činnost. Odpověď na léčbu přípravkem Zeffix není u všech léčených osob stejná. Váš lékař bude sledovat účinnost Vaší léčby pomocí pravidelných krevních testů.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zeffix užívat Neužívejte přípravek Zeffix
• Jestliže jste alergický(á) na lamivudin nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
^ Pokud se domníváte, že se Vás toto týká, poraďte se se svým lékařem.
Upozornění a opatření
U některých osob užívajících přípravek Zeffix nebo jiné podobné léky existuje vyšší riziko závažných nežádoucích účinků. Je zapotřebí, abyste si byl(a) vědom(a) dalších rizikových faktorů:
• jestli jste někdy prodělal(a) jiné onemocnění jater jako je hepatitida C;
• jestli máte značnou nadváhu (zejména pokud jste žena).
Pokud se Vás cokoli z uvedeného týká, sdělte to svému lékaři. Je možné, že v průběhu léčby budete potřebovat další vyšetření, včetně krevních testů. Další informace o rizicích naleznete v bodě 4.
Nepřestávejte přípravek Zeffix užívat, dokud Vám to nedoporučí Váš lékař, protože jinak by Vám hrozilo nebezpečí zhoršení Vaší hepatitidy. Až skončíte s užíváním přípravku Zeffix, bude Vás lékař ještě minimálně po dobu čtyř měsíců pravidelně kontrolovat, aby se včas odhalily jakékoli případné problémy. Budou Vám odebírány vzorky krve na laboratorní vyšetření zvýšených hodnot jaterních enzymů, které mohou upozornit na poškození jater. Více informací o užívání přípravku Zeffix viz bod 3.
Sledujte, zda se u Vás neobjeví důležité příznaky
U některých osob užívajících léčivé přípravky proti hepatitidě B dochází k rozvoji dalších onemocnění, která mohou být závažná. Je zapotřebí, abyste byl(a) informován(a), jakým důležitým známkám onemocnění a příznakům máte během léčby přípravkem Zeffix věnovat pozornost.
^ Přečtěte si informace „Další možné nežádoucí účinky léčby hepatitidy B“ v bodě 4 této příbalové informace.
Chraňte ostatní osoby
Hepatitida B se přenáší sexuálním kontaktem s někým, kdo je tímto virem infikován nebo přenosem krve, ve které je virus přítomen (např. při používání společných injekčních jehel). Užívání přípravku Zeffix nezabrání přenosu hepatitidy B na další osoby. Abyste ochránil(a) ostatní osoby před nákazou hepatitidou B, je nutné:
• Používat kondom, pokud máte s někým orální nebo penetrativní sexuální styk.
• Neriskovat přenos viru krevní cestou - např. používáním jedné injekční jehly více osobami.
Další léčivé přípravky a přípravek Zeffix
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a přípravků dostupných bez lékařského předpisu.
Nezapomeňte sdělit svému lékaři nebo lékárníkovi, pokud začnete v průběhu léčby přípravkem Zeffix užívat nějaký jiný lék.
Tyto léky nemají být užívány s přípravkem Zeffix:
O jiné léčivé přípravky obsahující lamivudin, užívané k léčbě HIV infekce (onemocnění nazývaného AIDS);
O emtricitabin, užívaný k léčbě HIV nebo hepatitidy B;
O kladribin, užívaný k léčbě vlasatobuněčné leukémie.
^ Pokud jste léčen(a) některým z těchto přípravků, sdělte to svému lékaři.
Těhotenství
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět:
^ poraďte se se svým lékařem o rizicích a přínosech užívání přípravku Zeffix v průběhu těhotenství dříve, než začnete tento přípravek užívat. Nepřestávejte přípravek Zeffix užívat, dokud Vám to nedoporučí Váš lékař.
Kojení
Přípravek Zeffix může přecházet do mateřského mléka. Pokud kojíte, nebo o kojení uvažujete:
^ poraďte se se svým lékařem dříve, než začnete přípravek Zeffix užívat.
Řízení dopravních prostředků a obsluha strojů
Užíváte-li Zeffix, můžete se cítit unavený(á), což může ovlivnit Vaši schopnost řídit nebo obsluhovat stroje.
^ Neřiďte ani neobsluhujte stroje, aniž jste si jistý(á), že nejste ovlivněn(a).
Přípravek Zeffix obsahuje cukr a konzervační látky
Jestliže jste diabetik, mějte prosím na paměti, že každá dávka přípravku Zeffix (100 mg = 20 ml) obsahuje 4 g sacharózy.
Přípravek Zeffix obsahuje sacharózu. Jestliže Vám Váš lékař sdělil, že trpíte nesnášenlivostí některých cukrů, kontaktujte svého lékaře před zahájením užívání přípravku Zeffix. Sacharóza může mít škodlivý vliv na zuby.
Přípravek Zeffix obsahuje rovněž konzervační látky (parabeny), které mohou způsobovat alergické reakce (někdy opožděné).
3. Jak se přípravek Zeffix užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Zůstaňte v pravidelném kontaktu se svým lékařem
Přípravek Zeffix pomáhá udržet Vaši hepatitidu B pod kontrolou. Aby mohl udržovat infekci pod kontrolou a zabránit ve zhoršování nemoci, je třeba ho užívat každý den.
^ Zůstaňte v kontaktu se svým lékařem a nepřestávejte přípravek Zeffix užívat bez toho, abyste se poradil(a) se svým lékařem.
Kolik přípravku se užívá
Obvyklá dávka přípravku Zeffix perorální roztok je 20 ml (100 mg lamivudinu) jednou denně.
Trpíte-li poruchou činnosti ledvin, Váš lékař Vám předepíše nižší dávku přípravku Zeffix.
^ Pokud se Vás toto týká, sdělte to svému lékaři.
Pokud již užíváte jiný léčivý přípravek obsahující lamivudin k léčbě infekce virem HIV, bude Váš lékař pokračovat v léčbě vyšší dávkou přípravku (obvykle 150 mg dvakrát denně), protože dávka lamivudinu v přípravku Zeffix (100 mg) není dostačující k léčbě infekce virem HIV. Pokud plánujete změnu Vaší léčby HIV, nejprve si o této změně promluvte se svým lékařem.
Přípravek Zeffix může být užíván s jídlem nebo bez jídla.
Návod, jak odměřit a užít dávku léku, je spolu s obrázkem na konci této příbalové informace za bodem 6.
Jestliže jste užil(a) více přípravku Zeffix, než jste měl(a)
Je nepravděpodobné, že by omylem užité velké množství přípravku Zeffix způsobilo nějaké závažné problémy. Pokud nedopatřením užijete vyšší dávku, informujte o tom svého lékaře nebo lékárníka, nebo se poraďte na pohotovostním oddělení nejbližší nemocnice.
Jestliže jste zapomněl(a) užít Zeffix
Zapomenete-li si vzít dávku svého léku, vezměte si ji hned, jakmile si vzpomenete. Potom pokračujte v užívání stejně jako předtím. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Nepřestávejte užívat přípravek Zeffix
Bez porady s lékařem nesmíte ukončit užívání přípravku Zeffix. Riskujete tím zhoršení Vaší hepatitidy (viz bod 2). Přestanete-li Zeffix užívat, lékař Vás bude sledovat alespoň čtyři měsíce. Sledování bude zahrnovat odběry krve ke kontrole zvýšených hladin jaterních enzymů, které mohou ukazovat na poškození jater.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
V průběhu léčby hepatitidy B se, kromě níže uvedených nežádoucích účinků přípravku Zeffix,
mohou vyskytnout i jiné zdravotní potíže.
^ Je důležité, abyste si přečetl(a) informaci „Další možné nežádoucí účinky léčby hepatitidy B“.
Nežádoucími účinky často uváděnými v klinických studiích byly únava, infekce dýchacího ústrojí, bolest v krku, bolest hlavy, žaludeční obtíže a bolest, pocit na zvracení, zvracení a průjem, zvýšení jaterních enzymů a enzymů produkovaných ve svalech (viz níže).
Alergické reakce
Tyto reakce jsou vzácné (mohou se vyskytnout až u 1 z 1 000 osob). Příznaky zahrnují:
• otok očních víček, obličeje nebo rtů,
• obtíže při polykání nebo dýchání.
^ Pokud se u Vás vyskytnou tyto příznaky, neprodleně kontaktujte lékaře. Přestaňte přípravek Zeffix užívat.
Nežádoucí účinky, o kterých se předpokládá, že jsou způsobené přípravkem Zeffix:
Velmi častý nežádoucí účinek (může se vyskytnout u více než 1 osoby z 10), který se může projevit v krevních testech je:
• zvýšená hladina některých jaterních enzymů (transamináz), která může být známkou zánětu nebo poškození jater.
Časté nežádoucí účinky (mohou se vyskytnout až u 1 osoby z 10) jsou:
• křeče a bolesti svalů;
• kožní vyrážka nebo „kopřivka“ kdekoli na těle.
Častý nežádoucí účinek, který se může projevit v krevních testech je:
• zvýšená hladina enzymu tvořeného ve svalech (kreatinfosfokináza), která může být známkou poškození tělesné tkáně.
Jiné nežádoucí účinky
Jiné nežádoucí účinky se mohou vyskytnout u velmi malého počtu pacientů, ale přesná četnost není známa:
• rozpad svalové tkáně;
• zhoršení jaterního onemocnění po ukončení užívání nebo v průběhu užívání přípravku Zeffix, pokud se virus hepatitidy B stane rezistentním (necitlivým) na přípravek Zeffix. Toto může
u některých osob vést k úmrtí.
• laktátová acidóza (viz „Další možné nežádoucí účinky léčby hepatitidy B“ v dalším bodě).
Nežádoucí účinek, který se může projevit v krevních testech:
• snížený počet krevních buněk, které se uplatňují při srážení krve (trombocytopenie).
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků,
^ sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Další možné nežádoucí účinky léčby hepatitidy B
Při léčbě hepatitidy B přípravkem Zeffix a podobnými přípravky (NRTI) může dojít k rozvoji dalších zdravotních potíží.
Laktátová acidóza je vzácný, ale závažný nežádoucí účinek
U některých pacientů, kteří užívají přípravek Zeffix, nebo další podobné přípravky (NRTI), se může rozvinout onemocnění nazývané laktátová acidóza a zvětšení jater.
Laktátová acidóza je způsobená hromaděním kyseliny mléčné v těle. Je vzácná, ale pokud se objeví, je to obvykle v průběhu několika prvních měsíců léčby. Může být život ohrožující, protože může vést k selhání vnitřních orgánů.
Laktátová acidóza se s větší pravděpodobností objeví u osob, které mají onemocnění jater, nebo u obézních osob (s výraznou nadváhou), zejména u žen.
Příznaky laktátové acidózy zahrnují:
• hluboké, zrychlené a obtížné dýchání;
• ospalost;
• pocit snížené citlivosti nebo slabosti končetin;
• nevolnost (pocit na zvracení) a zvracení;
• bolest břicha.
V průběhu léčby bude lékař sledovat, zda se u Vás neobjevují známky laktátové acidózy. Pokud se u Vás objeví jakýkoli z příznaků zmíněných výše, nebo pokud Vás některý z jiných příznaků znepokojuje,
^ navštivte co nejdříve svého lékaře.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Zeffix uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce a krabičce. Uchovávejte při teplotě do 25 °C.
Znehodnoťte po měsíci od prvního otevření.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Zeffix obsahuje
Léčivou látkou je lamivudinum. Jeden ml perorálního roztoku obsahuje lamivudinum 5 mg. Pomocnými látkami jsou:
sacharosa, methylparaben (E218), propylparaben (E216), kyselina citronová, propylenglykol, dihydrát natrium-citrátu, umělé jahodové aroma, umělé banánové aroma, čištěná voda.
Jak přípravek Zeffix vypadá a co obsahuje toto balení
Zeffix perorální roztok se dodává v kartonové krabičce obsahující lahvičku z polyethylenu bílé barvy s dětským bezpečnostním uzávěrem. Roztok je čirý bezbarvý až světle žlutý s jahodově/banánovým aroma. Lahvička obsahuje 240 ml roztoku lamivudinu (5 mg/ml). Součástí balení je také dávkovači perorální aplikátor se stupnicí v ml a adaptér pro aplikátor umožňující jeho nasazení do hrdla lahvičky.
Vysvětlení cizojazyčných údajů uvedených na obalech přípravku Zeffix
EXP = Použitelné do Lot = Číslo šarže
|
Výrobce |
Držitel rozhodnutí o registraci |
|
Aspen Bad Oldesloe GmbH Industriestrasse 32-36 23843 Bad Oldesloe Německo nebo Glaxo Operations UK Limited (trading as Glaxo Wellcome Operations) Harmire Road Barnard Castle County Durham DL12 8DT Velká Británie |
Glaxo Group Ltd. 980 Great West Road Brentford Middlesex TW8 9GS Velká Británie |
|
Další informace o tomto přípravku získáte |
u místního zástupce držitele rozhodnutí o registraci. |
|
Belgie/Belgique/Belgien |
Lietuva |
GlaxoSmithKline Pharmaceuticals s.a./n.v. GlaxoSmithKline Lietuva UAB
Tél/Tel: + 32 (0) 10 85 52 00 Tel: + 370 5 264 90 00
|
EtarapHH EaaKCoCMHTKaaňH EOOfl Tea.: + 359 2 953 10 34 |
Luxembourg/Luxemburg GlaxoSmithKline Pharmaceuticals s.a./n.v. Belgique/Belgien Tél/Tel: + 32 (0) 10 85 52 00 |
|
Česká republika GlaxoSmithKline s.r.o. Tel: + 420 222 001 111 cz.info@gsk.com |
Magyarország GlaxoSmithKline Kft. Tel.: + 36 1 225 5300 |
|
Danmark GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com |
Malta GlaxoSmithKline (Malta) Limited Tel: + 356 21 238131 |
|
Deutschland GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com |
Nederland GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com |
|
Eesti GlaxoSmithKline Eesti OU Tel: + 372 6676 900 estonia@gsk.com |
Norge GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no |
|
EkXúda GlaxoSmithKline A.E.B.E. TpA.: + 30 210 68 82 100 |
Osterreich GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com |
|
Espaňa GlaxoSmithKline, S.A. Tel: + 34 902 202 700 es-ci@gsk.com |
Polska GSK Services Sp. z o.o. Tel.: + 48 (0)22 576 9000 |
|
France Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com |
Portugal Glaxo Wellcome Farmaceutica, Lda. Tel: + 351 21 412 95 00 FI.PT@gsk.com |
|
Hrvatska GlaxoSmithKline d.o.o. Tel: + 385 1 6051 999 |
Románia GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208 |
|
Ireland GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000 |
Slovenija GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com |
|
Ísland Vistor hf. Sími: + 354 535 7000 |
Slovenská republika GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com |
|
Italia GlaxoSmithKline S.p.A. Tel: + 39 (0)45 9218 111 |
Suomi/Finland GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com |
|
Kúnpoq GlaxoSmithKline (Cyprus) Ltd TpA.: + 357 22 39 70 00 gskcyprus@gsk.com |
Sverige GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com |
|
Latvija GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com |
United Kingdom GlaxoSmithKline UK Ltd Tel: + 44 (0)800 221441 customercontactuk@gsk.com |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Jak odměřit a užít dávku léku
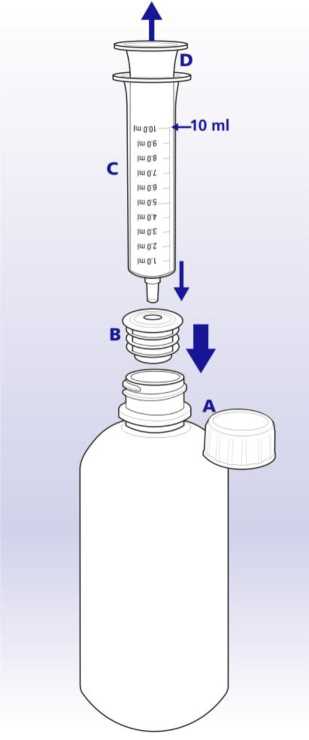
K přesnému odměření své dávky používejte perorální aplikátor, který je součástí dodávaného balení
(viz také bod 3).
Plný aplikátor obsahuje 10 ml roztoku.
1. Sejměte dětský bezpečnostní uzávěr (A). Odložte ho na bezpečné místo.
2. Držte lahvičku. Do jejího hrdla zasuňte adaptér (B), jak nejdále to jde.
3. Do adaptéru upevněte aplikátor (C).
4. Obraťte lahvičku dnem vzhůru.
5. Vytažením pístu (D) natáhněte do aplikátoru první díl celkové dávky.
6. Obraťte lahvičku zpět do polohy hrdlem vzhůru a vyjměte aplikátor z adaptéru.
7. Vložte hrot aplikátoru do úst a nasměrujte ho proti vnitřku tváře. Pomalým stlačením pístu aplikátor vyprázdněte. Při pomalém stlačení pístu máte dost času na spolknutí roztoku. Netlačte na píst příliš silně, rychlé vstříknutí roztoku dozadu do hrdla by mohlo vyvolat dávení nebo dušení.
8. Opakujte kroky 3 až 7 stejným způsobem, dokud neužijete celou dávku. Například, pokud máte předepsánu dávku 20 ml, budete potřebovat 2 plné aplikátory.
9. Po použití vyndejte aplikátor z lahvičky a důkladně umyjte v čisté vodě. Před dalším použitím jej nechte zcela oschnout. Adaptér ponechte v lahvičce.
10. Lahvičku dobře uzavřete pevným utažením uzávěru.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) lamivudinu (chronická hepatitida B) dospěl výbor CHMP k těmto vědeckým závěrům:
Údaje o přípravku Zeffix mají být upraveny tak, aby lépe zohledňovaly současná doporučení pro zvládání rezistence k lamivudinu. Z tohoto důvodu byly výborem PRAC doporučeny změny bodu 4.2 SmPC týkající se přechodu na jiný přípravek nebo přidání dalšího přípravku bez zkřížené rezistence k lamivudinu podle léčebných doporučení. Obdobné změny byly považovány za nutné v bodech 4.4 a 5.1 SmPC, aby se tato informace podpořila. Do příbalové informace nebyly požadovány žádné změny týkající se rezistence k lamivudinu.
Držitel rozhodnutí o registraci využil také příležitosti aktualizovat údaje o přípravku v souladu s nejnovější šablonou QRD v. 10, což bylo přijato.
S ohledem na údaje uvedené v hodnocené PSUR považuje výbor PRAC tyto změny v údajích o přípravcích obsahujících lamivudin za potvrzené.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění doporučující změnu podmínek rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se lamivudinu (chronická hepatitida B) zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících lamivudin (chronická hepatitida B) je příznivý pod podmínkou, že v textech provázejících léčivý přípravek budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu registrace.
59
U HBeAg-pozitivních pacientů s chronickou hepatitidou B (CHB) bez cirhózy má léčba trvat alespoň 6-12 měsíců po prokázání sérokonverze HBeAg (ztráty HBeAg a HBV DNA s průkazem HBeAb), aby se omezilo riziko virologického relapsu, nebo do sérokonverze HBsAg, nebo až do ztráty účinnosti (viz bod 4.4). Hladiny sérových ALT a HBV DNA mají být po ukončení léčby pravidelně sledovány pro možnost pozdního virologického relapsu.