Zarzio 48 Mu/0,5 Ml
1. název přípravku
Zarzio 30 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce
2. kvalitativní a kvantitativní složení
Zarzio 30 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce
Jeden ml roztoku obsahuje filgrastimum* 60 milionů jednotek (MU) (odpovídá 600 mikrogramům
([ůg)]).
1 předplněná injekční stříkačka obsahuje filgrastimum 30 milionů jednotek (MU) (odpovídá 300 ^g) v 0,5 ml.
Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce
Jeden ml roztoku obsahuje filgrastimum* 96 milionů jednotek (MU) (odpovídá 960 mikrogramům
[ůg]).
1 předplněná injekční stříkačka obsahuje filgrastimum 48 milionů jednotek (MU) (odpovídá 480 ^g) v 0,5 ml.
* rekombinantní methionylovaný humánní faktor stimulující kolonie granulocytů (G-CSF, granulocyte-colony stimulating factor) produkovaný v E. coli rekombinantní DNA-technologií.
Pomocná látka se známým účinkem:
Jeden ml roztoku obsahuje 50 mg sorbitolu (E420).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční nebo infuzní roztok v předplněné injekční stříkačce (injekce nebo infuze) Čirý, bezbarvý až slabě nažloutlý roztok.
4. klinické údaje
4.1 Terapeutické indikace
- Zkrácení trvání neutropenie a snížení incidence febrilní neutropenie u pacientů léčených obvyklými cytotoxickými chemoterapeutiky pro malignitu (s výjimkou chronické myeloidní leukémie a myelodysplastických syndromů) a zkrácení doby trvání neutropenie u pacientů podstupujících myeloablativní terapii následovanou transplantací kostní dřeně, u nichž se předpokládá riziko prodloužené těžké neutropenie.
U dospělých a dětí léčených cytotoxickými chemoterapeutiky je bezpečnost a účinnost filgrastimu podobná.
- Mobilizace periferních progenitorových kmenových buněk (PBPCs, peripheral blood progenitor cells).
- U pacientů, dětí nebo dospělých, s těžkou kongenitální, cyklickou nebo idiopatickou neutropenií s absolutním počtem neutrofilů (ANC, absolute neutrophil count) < 0,5 x 109/l, a s těžkými anebo opakovanými infekcemi v anamnéze, je indikováno dlouhodobé podávání filgrastimu, aby se zvýšily počty neutrofilů a aby se snížila incidence a zkrátilo trvání příhod souvisejících
s infekcí.
- Léčba perzistující neutropenie (ANC < 1,0 x 109/l) u pacientů s pokročilou infekcí HIV, aby se snížilo riziko bakteriálních infekcí, pokud není vhodné zvolit jiné možnosti léčby neutropenie.
4.2 Dávkování a způsob podání
Terapie filgrastimem se provádí pouze ve spolupráci s onkologickým centrem, které má zkušenosti s léčbou G-CSF a s hematologií a které má potřebné diagnostické vybavení. Procedury mobilizace a aferézy je třeba provádět ve spolupráci s onkologicko-hematologickým centrem, které má v tomto směru přijatelné zkušenosti a kde je možné přesně provádět monitorování hemopoetických progenitorových kmenových buněk.
Obvykle používaná cytotoxická chemoterapie
Dávkovaní
Doporučená dávka filgrastimu je 0,5 MU/kg/den (5 ^g/kg/den). První dávka filgrastimu se podává nejméně 24 hodin po cytotoxické chemoterapii. V randomizovaných klinických hodnoceních byla použita subkutánní dávka 230 ^g/m2/den (4,0 až 8,4 ^g/kg/den).
Každodenní podávání filgrastimu má pokračovat, dokud není překročena doba očekávaného nejhlubšího poklesu počtu neutrofilů a dokud se počet neutrofilů nevrátí do normálního rozsahu. Po obvykle používané chemoterapii solidních tumorů, lymfomů a lymfoidní leukémie je očekávaná doba trvání léčby, potřebná pro splnění těchto kritérií, až 14 dní. Po indukčním a konsolidačním léčení akutní myeloidní leukémie může být trvání léčby podstatně delší (až 38 dní) v závislosti na typu, dávce a schématu podávání použitého cytotoxického chemoterapeutika.
U pacientů léčených cytotoxickou chemoterapií je v typickém případě vidět přechodný vzestup počtu neutrofilů za 1-2 dny po zahájení terapie filgrastimem. Pro trvalou terapeutickou odpověď se však terapie filgrastimem nemá ukončit dříve, než byla překročena očekávaná doba nejhlubšího poklesu a než se počet neutrofilů se opět vrátil do normálního rozmezí. Předčasné přerušení terapie filgrastimem, před dobou očekávaného nejhlubšího poklesu počtu neutrofilů, se nedoporučuje.
Způsob podání
Filgrastim naředěný v 5% roztoku glukózy se může podávat jako každodenní subkutánní injekce nebo jako každodenní intravenózní infuze trvající alespoň 30 minut (viz bod 6.6). Ve většině případů se dává přednost subkutánní aplikaci. Ze studie sledující jednodávkovou aplikaci jsou určité náznaky, že při intravenózním podávání se může zkrátit trvání účinku. Klinický význam tohoto nálezu pro opakované podávání není jasný. Výběr způsobu podání by měl záležet na individuálních klinických okolnostech.
U pacienů léčených myeloablativní terapií následovanou transplantací kostní dřeně
Dávkování
Doporučená dávka filgrastimu je 1,0 MU/kg/den (10 ^g/kg/den). První dávka filgrastimu se má podat nejméně 24 hodin po cytotoxické chemoterapii a nejméně 24 hodin po transfuzi kostní dřeně.
Jakmile byl překonán nejnižší pokles počtu neutrofilů, vytitruje se každodenní dávka filgrastimu v závislosti na odpovědi neutrofilů takto:
|
Počet neutrofilů |
Úprava dávky filgrastimu |
|
> 1,0 x 109/l po 3 po sobě následující dny |
Snížit na 0,5 MU/kg denně (5 ^g/kg/den) |
|
Pak, jestliže ANC zůstává > 1,0 x 109/l po 3 další po sobě následující dny |
Přerušit léčbu filgrastimem |
|
Jestliže se ANC sníží na < 1,0 x 109/l v průběhu léč uvedených údajů. |
ry, zahájí se opět dávkování filgrastimu podle výše |
|
ANC = absolutní počet neutrofilů | |
Způsob podání
Filgrastim se může podávat jako 30minutová nebo 24hodinová intravenózní infuze nebo jako kontinuální 24hodinová subkutánní infuze. Filgrastim by se měl naředit ve 20 ml 5 % roztoku glukózy (viz bod 6.6).
K mobilizaci PBPC u pacientů podstupujících myelosupresivní nebo myeloablativní léčbu následovanou autologní PBPC transplantací
Dávkování
Doporučená dávka filgrastimu k mobilizaci PBPC, pokud se používá samotný, je 1,0 MU/kg/den (10 gg/kg/den) po 5-7 po sobě následujících dní. Časový rozvrh leukaferézy: Často postačují 1 nebo 2 leukaferézy pátý a šestý den. Za jiných okolností mohou být nutné další leukaferézy. Dávkování filgrastimu je třeba provádět až do poslední leukaferézy.
Doporučená dávka filgrastimu k mobilizaci PBPC po myelosupresivní chemoterapii je 0,5 MU/kg/den (5 gg/kg/den), podávaná od prvního dne po ukončení chemoterapie až do doby, kdy již je překročeno očekávané nejnižší snížení počtu neutrofilů a kdy se počty neutrofilů navrátily do normálního rozmezí. Leukaferézu je třeba provádět v období, kdy se ANC zvýšil z < 0,5 x 109/l na > 5,0 x 109/l. U pacientů, kteří nepodstoupili extenzivní chemoterapii, často dostačuje jediná leukaferéza. Za jiných okolností se doporučují další leukaferézy.
Způsob podání
Filgrastim k mobilizaci PBPC, pokud se používá samotný:
Filgrastim se může podávat jako 24hodinová kontinuální subkutánní infuze nebo subkutánní injekce. Pro potřeby infuze se filgrastim naředí ve 20 ml 5 % roztoku glukózy (viz bod 6.6).
Filgrastim k mobilizaci PBPC po myelosupresivní chemoterapii:
Filgrastim by se měl podávat subkutánní injekcí.
K mobilizaci PBPC u normálních dárců před alogenní transplantací PBPC
Dávkování
K mobilizaci PBPC u normálních dárců se filgrastim podává v dávce 1,0 MU/kg/den (10 gg/kg/den) po dobu 4-5 po sobě následujících dní. Leukaferáza se má zahájit 5. dne a má pokračovat do 6. dne, pokud je to nutné k odběru 4 x 106 buněk CD34+/kg tělesné hmotnosti příjemce.
Způsob podání
Filgrastim by se měl podávat subkutánní injekcí.
U pacientů s těžkou chronickou neutropenií (SCN, Severe chronic neutropenia)
Dávkování
Kongenitální neutropenie:
Doporučená zahajovací dávka je 1,2 MU/kg/den (12 gg/kg/den) jako jednorázová injekce anebo v rozdělených dávkách.
Idiopatická nebo cyklická neutropenie:
Doporučená zahajovací dávka je 0,5 MU/kg/den (5 gg/kg/den) jako jednorázová injekce anebo v rozdělených dávkách.
Úprava dávkování:
Filgrastim je třeba podávat denně subkutánní injekcí, dokud počet neutrofilů nedosáhl hodnoty více než 1,5 x 109/l a dokud na těchto hodnotách nemůže být udržen. Po dosažení odpovědi je třeba stanovit nejmenší účinnou dávku k udržení této hladiny účinku. Pro udržení přiměřeného počtu neutrofilů je nutné dlouhodobé každodenní podávání. Po 1-2 týdnech léčby se zahajovací dávka může zdvojnásobit anebo snížit na polovinu v závislosti na pacientově odpovědi. Pak se dávka může individuálně upravovat každé 1-2 týdny tak, aby se udržel průměrný počet neutrofilů mezi 1,5 x 109/l a 10 x 109/l. Rychlejší postup zvyšování dávky je možné zvážit u pacientů s těžkými infekcemi.
V klinických hodnoceních 97 % pacientů, kteří na léčbu reagovali, dosáhlo kompletní odpovědi při dávkách < 24 ^g/kg/den. Dlouhodobá bezpečnost podávání filgrastimu ve větších dávkách než 24 ^g/kg/den u pacientů s SCN nebyla stanovena.
Způsob podání
Kongenitální, idiopatická nebo cyklická neutropenie: Filgrastim by se měl podávat subkutánní injekcí. U pacientů s infekcí HIV
Dávkování
Ke zvratu neutropenie:
Doporučená zahajovací dávka filgrastimu je 0,1 MU/kg/den (1 ^g/kg/den), podávaná s titrací až do maxima 0,4 MU/kg/den (4 ^g/kg/den), dokud není dosaženo normálního počtu neutrofilů a dokud není možné jej udržovat (ANC > 2,0 x 109/l). V klinických studiích bylo dosaženo při těchto dávkách odpovědi u > 90 % těchto pacientů a zvratu neutropenie bylo dosaženo v průměru po 2 dnech.
U malého počtu pacientů (< 10 %) byly k dosažení zvratu neutropenie nutné dávky až do 1,0 MU/kg/den (10 ^g/kg/den).
K udržení normálních počtů neutrofilů:
Když bylo dosaženo zvratu neutropenie, je třeba stanovit minimální účinnou dávku pro udržení normálního počtu neutrofilů. Doporučuje se úprava zahajovacího dávkování na střídavé podávání 30 MU/den (300 ^g/den). V závislosti na pacientově ANC může být nutná další úprava dávky k udržení počtu neutrofilů na > 2,0 x 109/l. V klinických studiích bylo nutné dávkování 30 MU/den (300 ^g/den) 1-7 dní v týdnu, aby se ANC udržel na > 2,0 x 109/l, s mediánem frekvence dávkování 3 dny v týdnu. Pro udržení ANC > 2,0 x 109/l může být nutné dlouhodobé podávání.
Způsob podání
Zvrat neutropenie nebo udržení normálních počtů neutrofilů: Filgrastim by se měl podávat subkutánní injekcí.
Starší osoby
Do klinických studií s filgrastimem byl zařazen malý počet pacientů vyššího věku. Pro tuto populaci pacientů ale nebyly provedeny žádné speciální studie, a proto není možné pro tyto pacienty vydat specifická doporučení pro dávkování.
Porucha funkce ledvin
Studie filgrastimu u pacientů s těžkou poruchou renálních nebo jaterních funkcí ukázaly, že filgrastim vykazuje podobný farmakokinetický a farmakodynamický profil jako u normálních jedinců. Za těchto okolností není úprava dávek nutná.
Použití u pediatrických pacientů při SCN a nádorovém onemocnění
Šedesát pět procentpacientů hodnocených v programu studie SCN bylo ve věku do 18 let. V této věkové skupině pacientů, která většinou zahrnovala pacienty s kongenitální neutropenií, byla účinnost léčby jasná. Nebyly zaznamenány rozdíly v bezpečnostním profilu u pediatrických pacientů léčených pro SCN.
Údaje z klinických studií u pediatrických pacientů naznačují, že bezpečnost a účinnost filgrastimu jsou podobné jak u dospělých, tak u dětí léčených cytotoxickými chemoterapeutiky.
Dávkovači doporučení u pediatrických pacientů jsou stejná jako u dospělých, léčených myelosupresivní cytotoxickou chemoterapií.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Zvláštní upozornění
Filgrastim se nesmí použít ke zvýšení dávky cytotoxického chemoterapeutika nad stanovené dávkovací režimy (viz níže).
Filgrastim se nesmí podávat pacientům s těžkou kongenitální neutropenií, u kterých se vyvine leukémie nebo kteří mají průkaz vývoje leukémie.
U pacientů léčených filgrastimem byla hlášena hypersenzitivita, včetně anafylaktických reakcí, vyskytující se při úvodní nebo následné léčbě. U pacientů s klinicky významnou hypersenzitivitou ukončete trvale podávání přípravku Zarzio. Nepodávejte přípravek Zarzio pacientům s hypersenzitivitou vůči filgrastimu nebo pegfilgrastimu v anamnéze.
Podobně jako u všech terapeutických proteinů existuje i zde možnost imunogenicity. Míra tvorby protilátek proti filgrastimu je obecně nízká. Objevují se vazebné protilátky, jak lze očekávat u všech biologických přípravků, nicméně v současnosti nejsou spojeny s neutralizační aktivitou.
Stanovená cytotoxická chemoterapie
Růst maligních buněk
G-CSF může podpořit růst myeloidních buněk in vitro a podobné účinky je možné pozorovat u některých myeloidních buněk in vitro.
Bezpečnost a účinnost podávání filgrastimu u pacientů s myelodysplastickým syndromem nebo s chronickou myeloidní leukémií nebyly stanoveny. Filgrastim není pro použití v těchto případech indikován. Zvláštní péči je třeba věnovat diagnostickému rozlišení blastické transformace chronické myeloidní leukémie od akutní myeloidní leukémie (AML).
Protože údaje o bezpečnosti a účinnosti u pacientů se sekundární AML jsou omezené, je třeba podávat filgrastim opatrně.
Bezpečnost a účinnost podávání filgrastimu u pacientů s de novo AML, ve věku < 55 let s dobrými cytogenetickými parametry [t(8; 21), t(15;17), a inv(16)] nebyly stanoveny.
Zvláštní upozornění u pacientů s nádorovým onemocněním
Po podání filgrastimu byly méně často hlášeny případy splenomegalie a ruptury sleziny. Některé případy ruptury sleziny skončily fatálně. Je třeba proto kontrolovat velikost sleziny a na možnost ruptury sleziny pomýšlet u pacientů léčených filgrastimem, kteří si stěžují na bolesti v levém nadbřišku a/nebo v horní části ramena.
Leukocytóza
Počty bílých krvinek 100 x 109/l nebo vyšší byly pozorovány u méně než 5 % pacientů léčených filgrastimem v dávkách vyšších než 0,3 MU/kg/den (3 ^g/kg/den). Nebyly hlášeny žádné nežádoucí účinky, které by se daly přisoudit přímo tomuto stupni leukocytózy. Se zřetelem k možným rizikům spojeným se závažnou leukocytózou je však třeba v průběhu terapie filgrastimem počet leukocytů v pravidelných intervalech sledovat. Jestliže počty leukocytů převýší hodnotu 50 x 109/l po očekávaném nejhlubším snížení, je třeba léčbu filgrastimem okamžitě přerušit. V průběhu podávání filgrastimu k mobilizaci PBPC je však třeba léčbu přerušit anebo snížit dávkování, jestliže počty leukocytů vystoupí nad 70 x 109/l.
Rizika spojená se zvýšenými dávkami chemoterapeutik
Zvláštní opatrnosti je třeba při léčení pacientů vysokými dávkami chemoterapeutik, protože zlepšení výsledků léčení tumoru nebylo zjištěno a zvýšené dávky chemoterapeutik mohou vést ke zvýšeným projevům toxicity včetně účinků na srdce, plíce, účinků neurologických a dermatologických (viz „Souhrn údajů o přípravku“ použitých specifických chemoterapeutik).
Terapie samotným filgrastimem nevylučuje vznik trombocytopenie a anémie vyvolané myelosupresivní chemoterapií. V případě možné léčby vyššími dávkami chemoterapeutik (např. plnými dávkami při předepsaném léčebném postupu) může být u pacienta zvýšené riziko trombocytopenie a anémie. Doporučuje se pravidelné monitorování počtu krevních destiček a hematokritu. Zvláštní opatrnosti je třeba při podávání jednoho chemoterapeutika nebo kombinace chemoterapeutik, o nichž je známo, že vyvolávají závažnou trombocytopenii.
Ukázalo se, že použití filgrastimem mobilizovaných PBPC snižuje stupeň a dobu trvání trombocytopenie po myelosupresivní nebo myeloablativní chemoterapii.
Jiná zvláštní opatření
Účinky filgrastimu u pacientů s podstatně sníženými počty myeloidních kmenových buněk nebyly studovány. Filgrastim působí zejména na prekurzory neutrofilů, a tak se jeho účinek projeví na zvýšení počtu neutrofilů. Proto u pacientů se sníženým počtem prekurzorů může být odpověď neutrofilů slabší (jak u pacientů léčených extenzivní radioterapií nebo chemoterapií, tak u pacientů s kostní dření infiltrovanou tumorem).
Příležitostně byly u pacientů podstupujících vysokodávkovou chemoterapii po transplantaci hlášeny cévní poruchy, včetně venookluzivní choroby a poruchy objemu tekutin.
U pacientů, kteří dostávali G-CSF po alogenní transplantaci kostní dřeně, byly hlášené reakce štepu proti hostiteli (Graft versus Host Disease, GvHD) a případy úmrtí (viz bod 4.8 a 5.1).
Zvýšená hematopoetická aktivita kostní dřeně jako reakce na léčbu růstovým faktorem souvisela s přechodně abnormálními snímky kostí. To by mělo být zváženo při interpretaci výsledků zobrazování kostí.
Mobilizace PBPC
Nejsou k dispozici žádná prospektivní randomizovaná srovnání dvou doporučených metod mobilizace (filgrastim v monoterapii nebo v kombinaci s myelosupresivní chemoterapií) u stejné populace pacientů. Stupeň variability mezi jednotlivými pacienty a mezi laboratorními testy CD34+ buněk znamená, že přímé srovnání mezi různými studiemi je obtížné. Je proto těžké doporučit optimální metodu. Volba mobilizační metody by měla být zvážena s ohledem na celkové cíle léčby u každého pacienta.
Předchozí expozice cytotoxickým látkám
U pacientů, kteří podstoupili velmi extenzivní myelosupresivní terapii nemusí být mobilizace PBPC dostatečná k dosažení doporučovaného minimálního množství (> 2,0 x 106 CD34+ buněk/kg) ani ke zrychlení obnovy krevních destiček na stejnou úroveň.
Některé cytotoxické látky vykazují obzvláštní toxicitu vůči hemopoetickým kmenovým buňkám a mohou nepříznivě ovlivnit mobilizaci kmenových buněk. Látky, jako např. melfalan, karmustin (BCNU) a karboplatina, podávány dlouhodobě anebo před pokusy o mobilizaci kmenových buněk, mohou výtěžek kmenových buněk snížit. Ukázalo se však, že podávání melfalanu, karboplatiny anebo BCNU spolu s filgrastimem na mobilizaci kmenových buněk působí příznivě. Jestliže se uvažuje o transplantaci PBPC, doporučuje se plánovat proceduru mobilizace kmenových buněk v časné fázi léčebného rozvrhu pacienta. Zvláštní pozornost je potřeba věnovat počtu kmenových buněk, mobilizovaných u těchto pacientů před nasazením vysokých dávek chemoterapeutik. Jestliže výtěžky nejsou přiměřené podle kritérií uvedených výše, je třeba uvažovat o alternativních způsobech léčby, nevyžadujících podporu kmenových buněk.
Stanovení výtěžku kmenových buněk
Při stanovení počtu kmenových buněk získaných u pacientů léčených filgrastimem je třeba věnovat zvláštní pozornost metodě kvantifikace. Výsledky průtokové cytometrie počtu CD34+ buněk jsou rozdílné v závislosti na přesnosti použité metodologie, a doporučení o množství, vycházející ze studií v jiných laboratořích, je třeba interpretovat opatrně.
Statistická analýza vztahu mezi počtem reinfundovaných buněk CD34+ a rychlostí obnovy krevních destiček po vysokých dávkách chemoterapeutik naznačuje komplikovaný, ale kontinuální vztah.
Doporučení minimálního výtěžku > 2,0 x 106 CD34+ buněk/kg vychází z publikovaných zkušeností, kdy bylo dosaženo přiměřené hematologické úpravy. Ukazuje se, že výtěžky přesahující tento minimální výtěžek korelují s rychlejší obnovou, výtěžky nižší korelují s obnovou pomalejší.
Normální dárci podstupující mobilizaci PBPC
Mobilizace PBPC nepřináší normálnímu dárci přímý klinický prospěch a má se o něm uvažovat pouze pro potřebu alogenní transplantace kmenových buněk.
O mobilizaci PBPC se má uvažovat pouze u dárců, kteří vykazují normální klinická a laboratorní kritéria vhodnosti pro dárcovství kmenových buněk. Zvláštní pozornost je třeba věnovat hematologickým hodnotám a infekčním nemocem.
Bezpečnost a účinnost filgrastimu nebyla stanovena u normálních dárců < 16 let nebo > 60 let.
U pacientů léčených filgrastimem byla velmi často hlášena trombocytopenie. Je nezbytné pečlivě sledovat počet krevních destiček.
Přechodná trombocytopenie (počet trombocytů < 100 x 109/l) po podávání filgrastimu a po leukaferéze byla pozorována u 35 % studovaných osob. Mezi nimi byly uvedeny dva případy počtu destiček < 50 x 109/l, o kterých se předpokládá, že jsou z důvodu leukaferézy.
Jestliže je zapotřebí více než jedna leukaferéza, je třeba věnovat zvláštní pozornost dárcům s počtem destiček < 100 x 109/l před leukaferézou; všeobecně se aferéza nesmí provádět při počtu destiček < 75 x 109/l.
Leukaferéza se nesmí provádět u dárců, kteří užívají antikoagulancia, nebo kteří mají poruchy hemostázy.
Podávání filgrastimu se musí přerušit nebo se musí snížit dávkování, jestliže se počty leukocytů zvýší na > 70 x 109/l.
Dárce, kteří dostávají G-CSF k mobilizaci PBPC, je třeba monitorovat, dokud se hematologické ukazatele nevrátí na normální úroveň.
U zdravých dárců byly v souvislosti s podáváním G-CSF pozorovány přechodné cytogenetické abnormality. Význam těchto změn není znám.
Riziko vzniku maligního myeloidního klonu nelze zcela vyloučit. Doporučuje se proto, aby centra provádějící aferézu systematicky sledovala dárce kmenových buněk po dobu alespoň 10 let, aby tak bylo zajištěno sledování dlouhodobé bezpečnosti přípravku.
Časté, ale obvykle asymptomatické případy splenomegalie a méně časté případy ruptury sleziny byly hlášeny u zdravých dárců (a u pacientů) po podávání G-CSF. Některé případy ruptury sleziny byly fatální. Proto je třeba pečlivě monitorovat velikost sleziny (např. klinickým vyšetřením, ultrazvukem). Je třeba uvažovat o diagnóze ruptury sleziny u dárců a/nebo pacientů, kteří udávají bolest v levém nadbřišku nebo v horní části ramena.
U normálních dárců byla často hlášena dyspnoe a méně často další nežádoucí reakce plic (hemoptýza, krvácení do plic, plicní infiltráty a hypoxie). V případě, že jsou nežádoucí reakce plic pravděpodobné nebo již potvrzené, mělo by se uvažovat o přerušení léčby přípravkem a zahájení vhodné lékařské péče.
Příjemci alogenních PBPC mobilizovaných filgrastimem
Průběžné údaje naznačují, že imunologické interakce mezi alogenním štěpem PBPC a příjemcem mohou být spojeny se zvýšeným rizikem akutní a chronické GvHD ve srovnání s transplantací kostní dřeně.
SCN
Hodnoty krevního obrazu
U pacientů léčených filgrastimem byla často hlášena trombocytopenie. Počty destiček je třeba monitorovat v krátkých intervalech, zejména v průběhu několika prvních týdnů terapie filgrastimem.
Je třeba uvážit intermitentní přerušování nebo snížení dávky filgrastimu u pacientů, u nichž se vyvinula trombocytopenie, tj. počty destiček jsou trvale < 100 000/mm3.
V krevním obrazu se mohou vyskytnout i další změny, včetně anémie a přechodného zvýšení myeloidních kmenových buněk, které vyžadují časté monitorování počtu buněk.
Transformace na leukémii nebo myelodysplastický syndrom
Je třeba věnovat zvláštní pozornost při rozlišování diagnózy SCN od ostatních hemopoetických poruch jako je aplastická anémie, myelodysplazie a myeloidní leukémie. Před zahájením léčby je třeba provést kompletní krevní obraz s diferenciálním krevním obrazem a určením počtu krevních destiček, musí se také vyhodnotit morfologie kostní dřeně a karyotyp.
V klinickém hodnocení pacientů s SCN léčených filgrastimem se s nízkou frekvencí (přibližně 3 %) vyvinuly myelodysplastické syndromy (MDS) nebo leukémie. To bylo pozorováno pouze u pacientů s kongenitální neutropenií. MDS a leukémie jsou přirozené komplikace základního onemocnění a jejich vztah k terapii filgrastimem je nejistý. U podskupiny přibližně 12 % pacientů, jejichž cytogenetická hodnocení byla za výchozí situace normální, byly později při rutinním opakovaném hodnocení nalezeny abnormality včetně monosomie 7. V současné době není jasné, zda dlouhodobá terapie pacientů s SCN predisponuje pacienty k cytogenetickým abnormalitám, k transformaci MDS nebo leukémii. Doporučuje se provádět morfologická a cytogenetická vyšetření kostní dřeně u pacientů v pravidelných intervalech (přibližně každých 12 měsíců).
Další zvláštní opatření
Je třeba vyloučit případy přechodné neutropenie např. při virových infekcích.
Po podání filgrastimu byly velmi často hlášeny případy splenomegalie a často případy ruptury sleziny. Je třeba proto kontrolovat velikost sleziny a na možnost ruptury sleziny pomýšlet u pacientů léčených filgrastimem, kteří si stěžují na bolesti v levém nadbřišku a/nebo v horní části ramena.
Přímý účinek terapie filgrastimem je splenomegalie. U třiceti jednoho procenta (31 %) pacientů ve studiích bylo dokumentováno, že mají hmatnou splenomegalii. Zvětšení objemu, měřené radiograficky, se zjistilo hned na začátku terapie filgrastimem a postupně se stabilizovalo. Bylo zjištěno, že snížení dávek zpomaluje nebo zastaví progresi zvětšování sleziny a u 3 % pacientů byla nutná splenektomie. Velikost sleziny je třeba pravidelně hodnotit. Palpační vyšetření břicha by mělo dostačovat pro zjištění abnormálního zvětšení objemu sleziny.
U malého počtu pacientů se objevila často hematurie a proteinurie. Je třeba provádět pravidelné vyšetření moči pro sledování těchto změn.
Bezpečnost a účinnost přípravku u novorozenců a u pacientů s autoimunitní neutropenií nebyla stanovena.
HIV infekce
Po podání filgrastimu byly často hlášeny případy splenomegalie. Je třeba proto kontrolovat velikost sleziny a na možnost ruptury sleziny pomýšlet u pacientů léčených filgrastimem, kteří si stěžují na bolesti v levém nadbřišku a/nebo v horní části ramena.
Hodnoty krevního obrazu
Je třeba v krátkých intervalech sledovat absolutní počet neutrofilů (ANC), zejména v průběhu prvních několika týdnů terapie filgrastimem. Někteří pacienti mohou na zahajovací dávku filgrastimu reagovat velmi rychle a značným zvýšením počtu neutrofilů. Doporučuje se stanovovat ANC denně během 2-3 dnů podávání filgrastimu. Potom se doporučuje stanovovat ANC minimálně dvakrát týdně během prvních dvou týdnů a dále jednou týdně nebo jednou za dva týdny v průběhu udržovací léčby. V průběhu intermitentního dávkování 30 MU/den (300 pg/den) filgrastimu se mohou objevit značné výkyvy v hodnotách ANC u pacientů. Aby se zjistila maximální i minimální hodnota ANC pacienta, doporučuje se odebírat vzorky krve pro určení ANC bezprostředně před plánovanou dávkou filgrastimu.
Riziko spojené se zvýšenými dávkami myelosupresivních léčivých přípravků
Léčba filgrastimem samotným nevylučuje vznik trombocytopenie a anémie vyvolané myelosupresivní léčbou. Následkem případného zvýšení dávek nebo většího počtu těchto léčivých přípravků při terapii filgrastimem může být pro pacienta vyšší riziko vývoje trombocytopenie a anémie. Doporučuje se pravidelné monitorování krevního obrazu (viz výše).
Infekce a maligní onemocnění způsobující myelosupresi
Neutropenie může být vyvolána náhodnými infekcemi postihujícími kostní dřeň, jako je například komplexMycobacterium avium anebo malignitami, například lymfomem. U pacientů se známým postižením kostní dřeně infekcemi nebo malignitou je potřeba kromě podávání filgrastimu pro léčbu neutropenie zvážit vhodnou léčbu základního onemocnění. Účinky filgrastimu na neutropenii vyvolanou infiltrací kostní dřeně nebo malignitou nebyly dostatečně prokázány.
Nosičství genu pro hemoglobin S a srpkovitá anémie
Krize srpkovité anémie, v některých případech fatální, byly hlášeny při použití filgrastimu u pacientů s nosičstvím genu pro hemoglobin S nebo se srpkovitou anémií. Lékaři by měli při předepisování filgrastimu pacientům s nosičstvím genu pro hemoglobin S nebo se srpkovitou anémii postupovat opatrně.
Jiná zvláštní opatření
Po podávání G-CSF byly hlášeny nežádoucí reakce plic, zejména intersticiální plicní onemocnění.
U pacientů s nedávnou anamnézou plicních infiltrátů anebo pneumonie může být riziko vyšší. Nástup známek postižení plic, jako je kašel, horečka a dušnost, ve spojení s radiologicky zjištěnými plicními infiltráty a zhoršením plicních funkcí mohou představovat první známky syndromu akutní dechové tísně (ARDS, Acute Respiratory Distress Syndrome). V těchto případech je třeba léčbu filgrastimem přerušit a zahájit vhodnou terapii.
Monitorování kostní denzity může být indikováno u pacientů s osteoporotickými chorobami kostí, kteří jsou kontinuálně léčeni filgrastimem déle než 6 měsíců.
Po podání faktoru stimulujícího granulocytové kolonie byl hlášen syndrom zvýšené permeability kapilár, který se vyznačuje hypotenzí, hypoalbuminemií, edémem a hemokoncentrací. Pacienti, u kterých se vyskytnou příznaky syndromu zvýšené permeability kapilár, mají být pečlivě sledováni a mají dostávat standardní symptomatickou léčbu, která může zahrnovat i intenzivní péči (viz bod 4.8).
U pacientů léčených filgrastimem nebo pegfilgrastimem byla hlášena glomerulonefritida. Případy glomerulonefritidy obvykle odezněly po snížení dávky nebo vysazení filgrastimu nebo pegfilgrastimu. Doporučuje se sledování rozboru moči.
Jedinci citliví na latex
Snímatelný kryt jehly předplněné injekční stříkačky obsahuje derivát přírodního kaučukového latexu. Ve snímatelném krytu jehly nebyl dosud detekován žádný přírodní kaučukový latex. Použití přípravku Zarzio injekční roztok v předplněné injekční stříkačce u jedinců citlivých na latex však nebylo studováno, a tudíž u nich existuje možné riziko hypersenzitivních reakcí, které nelze zcela vyloučit.
Pomocné látky
Zarzio obsahuje sorbitol (E420). Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy by neměli tento přípravek užívat.
Za účelem lepší vysledovatelnosti faktorů stimulujících růst kolonií granulocytů (G-CSFs) je nutno do zdravotních záznamů pacienta zřetelně uvést obchodní název podávaného přípravku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Bezpečnost a účinnost filgrastimu podávaného v tentýž den jako myelosupresivní cytotoxická chemoterapeutika nebyla definitivně stanovena. Se zřetelem k citlivosti rychle se dělících myeloidních buněk na myelosupresivní cytotoxická chemoterapeutika se nedoporučuje použití filgrastimu v době od 24 hodin před chemoterapií do 24 hodin po ní. Předběžné nálezy u malého počtu pacientů léčených současně filgrastimem a 5-fluorouracilem naznačují, že závažnost neutropenie může být zvýšena.
Možné interakce s jinými hemopoetickými růstovými faktory a cytokiny dosud nebyly v klinických hodnoceních sledovány.
Protože litium zvyšuje uvolňování neutrofilů, je pravděpodobné, že litium zesiluje účinek filgrastimu. Ačkoli tato interakce nebyla formálně studována, není k dispozici důkaz, že by byla škodlivá.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání filgrastimu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu. Zvýšená incidence potratů byla pozorována u králíků při mnohonásobcích klinické expozice a za přítomnosti toxicity pro matku (viz bod 5.3). Existují literární údaje, ve kterých byl prokázán transplacentární průchod filgrastimu u těhotných žen.
Podávání přípravku Zarzio se v těhotenství nedoporučuje.
Kojení
Není známo, zda se filgrastim/metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit podávání přípravku Zarzio.
Fertilita
Filgrastim neovlivnil reprodukční schopnost ani fertilitu u samců či samic potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie účinků na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
V klinických studiích u pacientů s nádorovým onemocněním byla nejčastějším nežádoucím účinkem mírná až středně silná muskuloskeletální bolest vyskytující se u 10 % pacientů a závažná u 3 % pacientů.
Také byla hlášena reakce štěpu proti hostiteli (GvHD) (viz níže).
U PBPC mobilizace u normálních dárců byla nejčastěji hlášeným nežádoucím účinkem muskuloskeletální bolest. Leukocytóza byla pozorována u dárců a trombocytopenie po filgrastimu a
leukaferéze byla též pozorována u dárců. Také byla hlášena splenomegalie a ruptura sleziny. Některé případy ruptury sleziny byly fatální.
U pacientů se SCN byly nejčastější nežádoucí účinky související s léčbou filgrastimem bolest kostí, obecná muskuloskeletální bolest a splenomegalie. Myelodysplastické syndromy (MDS) nebo leukémie se vyvinuly u pacientů s vrozenou neutropenií léčených filgrastimem (viz bod 4.4).
Syndrom zvýšené permeability kapilár, který může být život ohrožující v případě opožděné léčby, byl hlášen méně často (> 1/1 000 až < 1/100) u pacientů s nádorem podstupujících chemoterapii a zdravých dárců podstupujících mobilizace PBPC po podání faktoru stimulujícího granulocytové kolonie; viz níže a bod 4.4.
V klinických studiích u pacientů s HIV byly jedinými nežádoucími účinky, které byly shodně považovány za související s podáváním filgrastimu, muskuloskeletální bolest, bolest kostí a myalgie.
Tabulkový seznam nežádoucích účinků
Data v tabulkách níže popisují nežádoucí účinky hlášené z klinických studií a spontánního hlášení.
V každé skupině frekvence jsou nežádoucí účinky uvedeny v pořadí s klesající závažností. Data jsou uvedena samostatně pro pacienty s nádorovým onemocněním, u PBPC mobilizace u normálních dárců, pacientů se SCN a u pacientů s HIV, což odráží různé profily nežádoucích účinků u těchto populací.
Tab.1: Pacienti s nádorovým onemocněním
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky | ||||
|
Velmi časté (>i/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>i/ioooo až <1/1000) |
Velmi vzácné (<1/10000) | |
|
Poruchy krve a lymfatického systému |
Ruptura slezinya Splenomegaliea,e Krize srpkovité anémiea | ||||
|
Poruchy imunitního systému |
Hypersenzitivita na léka |
Reakce štěpu proti hostitelib | |||
|
Poruchy metabolismu a výživy |
Zvýšení hladiny kyseliny močové Zvýšení hladiny laktátu v krvi Snížená chuť k jídlua |
Pseudodnaa | |||
|
Poruchy nervového systému | |||||
|
Cévní poruchy |
Venookluzivní chorobad Poruchy objemu tekutin Syndrom zvýšené permeability kapilára | ||||
|
Respirační, hrudní a mediastinální poruchy |
Orofaryngeální bolesta |
Hemoptýzae |
Syndrom akutní respirační tísněa Respirační selhánía Plicní edéma Intersticiální plicní chorobaa Plicní infiltrátya Plicní krvácení | ||
|
Gastrointestinální poruchy |
Zácpaa | ||||
Viz níže
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky | ||||
|
Velmi časté (>i/io) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>i/ioooo až <1/1000) |
Velmi vzácné (<1/10000) | |
|
Poruchy jater a žlučových cest |
Zvýšení hladiny gamaglutamyl transferázy Zvýšení hladiny alkalické fosfatázy v krvi | ||||
|
Poruchy kůže a podkožní tkáně |
Alopeciea |
Sweetův syndrom Kožní vaskulitidaa | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletální bolestc |
Exacerbace revmatoidní artritidy | |||
|
Poruchy ledvin a močových cest |
Abnormality moči Glomerulonefritida | ||||
|
Celkové poruchy a reakce v místě aplikace |
Asteniea Únavaa Zánět sliznica Bolesta | ||||
a
b
Vyskytly se zprávy o GvHD a fatální případy u pacientů léčených pomocí G-CSF po alogenní transplantaci kostní dřeně (viz níže)
c
d
e
Patří sem bolest kostí, zad, artralgie, myalgie, bolest končetin, muskuloskeletální bolest, muskuloskeletální bolest hrudníku, bolest krku
Po uvedení přípravku na trh byly pozorovány případy u pacientů podstupujících transplantaci kostní dřeně nebo mobilizaci periferních progenitorových kmenových buněk (PBPC)
Byly pozorovány případy v klinických studiích
Tab.2: Mobilizace PBPC u normálních dárců
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky | ||||
|
Velmi časté (>i/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>i/ioooo až <1/1000 ) |
Velmi vzácné (<i/ioooo ) | |
|
Poruchy krve a lymfatického systému |
Trombocytopeni ea Leukocytózaa |
Splenomegaliea |
Ruptura slezinya Krize srpkovité anémiea | ||
|
Poruchy imunitního systému |
Anafylaktická reakce | ||||
|
Poruchy metabolismu a výživy |
Zvýšení hladiny laktátdehydrogenáz y v krvi |
Hyperurikémie (zvýšení hladiny kyseliny močové) | |||
|
Poruchy nervového systému | |||||
|
Cévní poruchy |
Syndrom zvýšené permeability kapilára | ||||
|
Respirační, hrudní a mediastináln í poruchy |
Plicní krvácení Hemoptýza Plicní infiltráty Hypoxie | ||||
|
Poruchy jater a žlučových cest |
Zvýšení hladiny alkalické fosfatázy v krvi |
Zvýšení hladiny aspartátaminotransferáz _y_ | |||
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky | ||||
|
Velmi časté (>i/io) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>i/ioooo až <1/1000 ) |
Velmi vzácné (<1/10000 ) | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletál ní bolestb |
Zhoršení revmatoidní artritidy | |||
|
Poruchy ledvin a močových cest |
Glomerulonefritida | ||||
Viz níže
Patří sem bolest kostí, bolest zad, artralgie, myalgie, bolest končetin, muskuloskeletální bolest, muskuloskeletální bolest hrudníku, bolest krku
Tab.3: Pacienti se SCN
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky | ||||
|
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>i/ioooo až <1/1000) |
Velmi vzácné (<i/ioooo) | |
|
Poruchy krve a lymfatického systému |
Splenomegaliea |
Ruptura slezinya Trombocytopeniea |
Krize srpkovité | ||
|
Poruchy metabolismu a výživy |
Hyperurikémie Snížení hladiny krevního cukru Zvýšení hladiny laktátdehydrogenázy v krvi | ||||
|
Poruchy nervového systému | |||||
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe | ||||
|
Gastrointestinální poruchy | |||||
|
Poruchy jater a žlučových cest |
Hepatomegalie Zvýšení hladiny alkalické fosfatázy v krvi | ||||
|
Poruchy kůže a podkožní tkáně |
Kožní vaskulitida Alopecie | ||||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletální bolestb Artralgie |
Osteoporóza | |||
|
Poruchy ledvin a močových cest |
Hematurie Glomerulonefritida |
Proteinurie | |||
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě aplikace injekce | ||||
Viz níže
Patří sem bolest kostí, bolest zad, artralgie, myalgie, bolest končetin, muskuloskeletální bolest, muskuloskeletální bolest hrudníku, bolest krku
|
Třídy orgánových systémů podle MedDRA |
Nežádoucí účinky | ||||
|
Velmi časté (>i/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1000 až <1/100) |
Vzácné (>i/ioooo až <1/1000) |
Velmi vzácné (<1/10000) | |
|
Poruchy krve a lymfatického systému |
Splenomegaliea |
Krize srpkovité | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletální bolestb | ||||
|
Poruchy ledvin a močových cest |
Glomerulonefritida | ||||
Viz níže
Patří sem bolest kostí, bolest zad, artralgie, myalgie, bolest končetin, muskuloskeletální bolest, muskuloskeletální bolest hrudníku, bolest krku
Popis vybraných nežádoucích účinků
Vyskytly se zprávy o GvHD a fatální případy u pacientů léčených pomocí G-CSF po alogenní transplantaci kostní dřeně (viz bod 4.4 a 5.1).
V postmarketingovém sledování byly hlášeny případy syndromu zvýšené permeability kapilár při použití faktoru stimulujícího granulocytové kolonie. Obecně se vyskytly u pacientů s pokročilým stádiem zhoubného onemocnění, se sepsí, užívajících vícesložkovou chemoterapii nebo podstupujících aferézu (viz bod 4.4).
Pacienti s nádorovým onemocněním
V randomizovaných, placebem kontrolovaných klinických studiích nezvyšoval filgrastim incidenci nežádoucích účinků spojených s cytotoxickou chemoterapií. Nežádoucí účinky uváděné se stejnou frekvencí u pacientů léčených kombinací filgrastim/chemoterapie a placebo/chemoterapie byly nevolnost a zvracení, alopecie, průjem, únava, anorexie, zánět sliznic, bolesti hlavy, kašel, vyrážka, bolest na hrudi, astenie, faryngolaryngeální bolest a zácpa.
Po uvedení přípravku na trh byla u pacientů léčených filgrastimem hlášena kožní vaskulitida. Mechanismus vaskulitidy u pacientů užívajících filgrastin není znám. Frekvence z klinických studií se odhaduje jako méně častá.
Po uvedení přípravku na trh byly hlášeny případy Sweetova syndromu (akutní febrilní dermatóza). Frekvence z klinických studií se odhaduje jako méně častá.
V klinických studiích a po uvedení přípravku na trh byly hlášeny plicní nežádoucí účinky, včetně intersticiální plicní choroby, plicního edému a plicních infiltrátů, v některých případech s následným rozvojem respiračního selhání nebo syndromu akutní respirační tísně (ARDS), který může být fatální (viz bod 4.4).
Po podání filgrastimu byly méně často hlášeny případy splenomegalie a ruptury sleziny. Některé případy ruptury sleziny skončily fatálně (viz bod 4.4).
V klinických studiích a po uvedení přípravku na trh byly hlášeny hypersenzitivní reakce, včetně anafylaxe, vyrážky, kopřivky, angioedému, dyspnoe a hypotenze, vyskytující se při úvodní nebo následné léčbě. Celkově byla hlášení častější po intravenózním podání. V některých případech se příznaky znovu vyskytly při opětovném zahájení léčby, což naznačuje příčinnou souvislost. Léčba filgrastimem by měla být trvale ukončena u pacientů s anamnézou prodělané závažné alergické reakce.
Po uvedení přípravku na trh byly u pacientů s nosičstvím genu pro hemoglobin S nebo se srpkovitou anémií hlášeny izolované případy srpkovité anémie s krizí (viz bod 4.4). Frekvence z klinických studií se odhaduje jako méně častá.
U pacientů s nádorovým onemocněním léčených filgrastimem byla hlášena pseudodna. Frekvence z klinických studií se odhaduje jako méně častá.
Mobilizace periferních progenitorových kmenových buněk (PBPC) u normálních dárců U zdravých dárců a u pacientů po podání filgrastimu byly hlášeny časté, ale obecně asymptomatické případy splenomegalie a měně časté případy ruptury sleziny. Některé případy ruptury sleziny skončily fatálně (viz bod 4.4).
Byly hlášeny plicní nežádoucí účinky (hemoptýza, plicní krvácení, plicní infiltráty, dyspnoe a hypoxie) (viz bod 4.4).
Méně často byla pozorována exacerbace příznaků artritidy.
Leukocytóza (WBC >50x109/l) byla pozorována u 41 % dárců a přechodná trombocytopenie (trombocyty <100x109/l) po podání filgrastimu a leukaferéze byla pozorována u 35 % dárců (viz bod 4.4).
Pacienti se SCN
Pozorované nežádoucí účinky zahrnují splenomegalii, která může být u menšího počtu případů progresivní, rupturu sleziny a trombocytopenii (viz bod 4.4).
Nežádoucí účinky, které mohou souviset s léčbou filgrastimem a které se typicky objevují u <2 % pacientů se SCN, byly reakce v místě aplikace injekce, bolest hlavy, hepatomegalie, artralgie, alopecie, osteoporóza a vyrážka.
Během dlouhodobého užívání byla u 2 % pacientů se SCN hlášena kožní vaskulitida.
U pacientů s HIV
Splenomegalie byla hlášena v souvislosti s léčbou filgrastimem u <3 % pacientů. Ve všech případech se jednalo o mírnou až středně závažnou formu při tělesném vyšetření a klinický průběh byl benigní; žádní pacienti neměli diagnózu hypersplenismu a žádný pacient nemusel podstoupit splenektomii. Jelikož splenomegalie je běžným nálezem u pacientů s HIV infekcí a vyskytuje se v různém stupni u většiny pacientů s AIDS, je souvislost s léčbou filgrastimem nejasná (viz bod 4.4).
Pediatrická populace
Data z klinických studií u pediatrických pacientů naznačují, že bezpečnost a účinnost filgrastimu jsou podobné u dospělých a dětí léčených cytotoxickou chemoterapií, což naznačuje, že se nevyskytují žádné věkem podmíněné rozdíly ve farmakokinetice filgrastimu. Jediným shodně hlášeným nežádoucím účinkem byla muskuloskeletální bolest, která se neliší od dospělé populace.
Údaje pro další hodnocení filgrastimu u pediatrických pacientů nejsou dostatečné.
Další zvláštní skupiny populací
Geriatrické použití
U pacientů nad 65 let věku nebyly ve srovnání s mladšími dospělými (>18 let věku) užívajícími cytotoxickou chemoterapii pozorovány žádné celkové rozdíly v bezpečnosti nebo účinnosti a klinická zkušenost neidentifikovala rozdíly v odpovědi mezi staršími a mladšími pacienty. Údaje pro další hodnocení filgrastimu u geriatrických pacientů u dalších schválených indikací filgrastimu nejsou dostatečné.
Pediatričtí pacienti se SCN
U pediatrických pacientů se závažnou chronickou neutropenií, kteří dostávali chronickou léčbu filgrastimem, byly hlášeny případy snížené kostní density a osteoporózy. Frekvence z klinických studií se odhaduje jako „častá“.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkovánf
Účinky předávkování filgrastimu nebyly stanoveny. Ukončení léčby filgrastimem vede obvykle k 50 % snížení cirkulujících neutrofilů během 1 až 2 dnů s návratem k normálním hodnotám za 1 až 7 dnů.
5. farmakologické vlastnosti
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunostimulancia. Faktory stimulující kolonie, ATC kód: L03AA02
Zarzio je tzv. podobným biologickým léčivým přípravkem („biosimilar“). Podrobné informace jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Lidský G-CSF je glykoprotein, který reguluje tvorbu a uvolňování funkčních neutrofilů z kostní dřeně. Zarzio, obsahující r-metHuG-CSF (filgrastim) vyvolává během 24 hodin výrazné zvýšení počtu neutrofilů v periferní krvi a mírné zvýšení monocytů. U některých pacientů s SCN může filgrastim také vyvolat mírné zvýšení počtu cirkulujících eosinofilů a bazofilů nad výchozí hodnoty; někteří z těchto pacientů mohou mít eosinofilii nebo bazofilii již před léčením. Zvýšení počtu neutrofilů je při doporučeném dávkování závislé na dávce. Neutrofily vytvářené jako odpověď na filgrastim mají funkci normální nebo zvýšenou, jak se ukázalo v testech chemotaktických a fakocytárních funkcí. Po ukončení terapie filgrastimem se počty cirkulujících neutrofilů sníží o 50 % během 1-2 dní a dále potom k normálním hladinám v průběhu 1-7 dní.
Použití filgrastimu u pacientů léčených cytotoxickými chemoterapeutiky významně snižuje incidenci, závažnost a trvání neutropenie a febrilní neutropenie. Terapie filgrastimem významně snižuje trvání febrilní neutropenie, nutnou dobu podávání antibiotik a hospitalizace po úvodní chemoterapii pro akutní myeloidní leukémii nebo po myeloablativní léčbě s následnou transplantací kostní dřeně. Incidence horečky a doložených infekcí nebyla snížena v žádné skupině. Trvání horečky nebylo zkráceno u pacientů s myeloablativní terapií s následnou transplantací kostní dřeně.
Použití filgrastimu, ať již samotného anebo po chemoterapii, mobilizuje hemopoetické kmenové buňky v periferní krvi. Tyto autologní PBPC je možno odebrat a infundovat zpět po terapii vysokými dávkami cytotoxických látek, a to buď namísto transplantace kostní dřeně anebo jako její doplněk. Infúze PBPC urychluje obnovu krvetvorby a zkracuje tak období rizika hemoragických komplikací a snižuje potřebu transfuzí krevních destiček.
Příjemci alogenních PBPC mobilizovaných filgrastimem měli významně rychlejší hematologické zotavení, což vede k významnému snížení doby do nepodporovaného zotavení trombocytů při srovnání s alogenní transplantací kostní dřeně.
Jedna retrospektivní evropská studie hodnotící užívání G-CSF po transplantaci alogenní kostní dřeně pacientům s akutní leukémií naznačila zvýšené riziko reakce štěpu proti hostiteli (GvHD), mortalitu související s léčbou (TRM, treatment related mortality) a mortalitu při podání G-CSF. V jiné mezinárodní retrospektivní studii u pacientů s akutní a chronickou myelogenní leukémií nebyl prokázán žádný efekt na riziko GvHD, TRM ani na mortalitu. Metaanalýza studií o alogenní transplantaci, včetně výsledků 9 prospektivních randomizovaných zkoušek, 8 retrospektivních studií a 1 studie s kontrolovanými případy, nezjistila žádný efekt na rizika akutní GvHD, chronické GvHD ani na časnou mortalitu související s léčbou.
|
Relativní riziko (95 % Cl) GvHD a TRM Následná léčba s G-CSF po transplantaci kostní dřeně | |||||
|
Studie |
Délka studie |
n |
Akutní stupeň II - IV GvHD |
Chronická GvHD |
trm |
|
Metaanalýza (2003) |
1986 - 2001a |
1198 |
1,08 (0,87, 1,33) |
1,02 (0,82, 1.26) |
0,70 (0,38, 1,31) |
|
Evropská retrospektivní studie (2004) |
1992 - 2002b |
1789 |
1,33 (1,08, 1,64) |
1,29 (1,02, 1,61) |
1,73 (1,30, 2,32) |
|
Mezinárodní retrospektivní studie (2006) |
1995 - 2000b |
2110 |
1,11 (0,86, 1,42) |
1,10 (0,86, 1,39) |
1,26 (0,95, 1,67) |
a
b
Analýza obsahuje studie zahrnující transplantaci kostní dřeně během tohoto období, některé studie použily GM-CSF
Analýza zahrnuje pacienty, kteří dostávali kostní dřeň během tohoto období
Použití filgrastimu pro mobilizaci PBPC u normálních dárců před transplantací alogenních PBPC U normálních dárců dávka 1 MU/kg/den (10 pg/kg tělesné hmotnosti/den) podávaná subkutánně v průběhu 4-5 po sobě následujících dnů, umožní získat > 4 x 106 CD34+ buněk/kg příjemcovy tělesné hmotnosti od většiny dárců po dvou leukaferezách.
Použití filgrastimu u pacientů, dětí a dospělých, se SCN (těžkou kongenitální, cyklickou a idiopatickou neutropenií) vyvolá trvalé zvýšení ANC v periferní krvi a snížení počtu infekcí a s tím souvisejících stavů.
Použití filgrastimu u pacientů s infekcí HIV udrží normální počty neutrofilů, takže umožňuje podávání antivirových a/nebo jiných myelosupresivních léčiv podle plánovaného rozpisu. Nejsou důkazy o tom, že by u pacientů s infekcí HIV léčených filgrastimem byla zvýšena replikace HIV.
Tak jako jiné hematopoetické růstové faktory, také G-CSF in vitro vykázal stimulační vlastnosti na lidské endoteliální buňky.
5.2 Farmakokinetické vlastnosti
Randomizované dvojitě zaslepené křížové studie s jednorázovou a vícenásobnou dávkou, prováděné u 204 zdravých dobrovolníků prokázaly, že farmakokinetický profil přípravku Zarzio je srovnatelný s profilem referenčního přípravku po subkutánním a intravenózním podání.
Absorpce
Jedna subkutánní dávka 0,5 MU/kg (5 pg/kg) vedla k maximálním koncentracím v séru po tmax 4,5 ± 0,9 hodin (průměr ± SD).
Distribuce
Distribuční objem v krvi je přibližně 150 ml/kg. Po subkutánním podání doporučených dávek se sérové koncentrace držely nad 10 ng/ml pro 8 - 16 hodin. Mezi dávkou a sérovou koncentrací filgrastimu je kladná lineární korelace, bez ohledu na to, zda byly podány intravenózně nebo subkutánně.
Eliminace
Průměrný eliminační poločas (tJ/2) filgrastimu v séru po jednorázových subkutánních dávkách kolísal od 2,7 hodin (1,0 MU/kg, 10 pg/kg) do 5,7 hodin (0,25 MU/kg, 2,5 pg/kg) a po 7 dnech léčby se prodloužil na 8,5 resp. 14 hodin.
Kontinuální infoze přípravku filgrastim po dobu až 28 dní u pacientů zotavujících se po autologní transplantaci kostní dřeně neměla za následek výskyt akumulace léku ani srovnatelných poločasů eliminace.
5.3 Předklinické údaje vztahující se k bezpečnosti
Filgrastim byl studován ve studiích toxicity po opakovaném podávání trvajících až 1 rok, které ukázaly změny, jež lze přičíst očekávanému farmakologickému působení a jež zahrnují zvýšení leukocytů, myeloidní hyperplazii v kostní dřeni, extramedulární granulopoézu a zvětšení sleziny. Všechny tyto změny po ukončení léčby vymizely.
Účinky filgrastimu na prenatální vývoj byly studovány u potkanů a králíků. Intravenózní (80 ^g/kg/den) podávání filgrastimu králíkům během období organogeneze bylo toxické pro matku a byla pozorována zvýšená míra spontánních potratů, postimplantačních ztrát a snížený průměrný počet živých zvířat ve vrhu a snížená fetální hmotnost.
Dle hlášených údajů pro jiný přípravek s filgrastimem podobný referenčnímu přípravku s filgrastimem byly pozorovány srovnatelné nálezy a zvýšené fetální malformace při dávce 100 ^g/kg/den toxické pro matku, která odpovídala systémové expozici rovnající se přibližně 50-90násobku expozic pozorovaných u pacientů léčených klinickou dávkou 5 ^g/kg/den. Dávka, při níž byly pozorovány nežádoucí účinky embryofetální toxicity, v této studii činila 10 ^g/kg/den, což odpovídalo systémové expozici rovnající se přibližně 3-5násobku expozic pozorovaných u pacientů léčených klinickou dávkou.
U březích potkanů nebyla pozorována žádná toxicita pro matku ani fetální toxicita při dávkách až 575 ^g/kg/den. Mláďata potkanů, kterým byl podán filgrastim během perinatálního a laktačního období, vykazovala opoždění v zevní diferenciaci a růstovou retardaci (>20 ^g/kg/den) a mírně sníženou míru přežití (100 ^g/kg/den).
U filgrastimu nebyl pozorován žádný účinek na fertilitu samců či samic potkanů.
6. farmaceutické údaje
6.1 Seznam pomocných látek
Kyselina glutamová Sorbitol (E420)
Polysorbát 80 Voda na injekci
6.2 Inkompatibility
Zarzio se nesmí ředit roztokem, obsahujícím chlorid sodný.
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
Zředěný filgrastim se může absorbovat na sklo a plastové materiály, pokud není naředěn v 5 %
(50 mg/ml) roztoku glukózy (viz bod 6.6).
6.3 Doba použitelnosti
36 měsíců.
Po zředění: Chemická a fyzikální stabilita již zředěného infuzního roztoku byla prokázána pro dobu 24 hodiny při 2 °C až 8 °C. Z mikrobiologického hlediska je třeba použít roztok ihned. Pokud se nepoužije ihned, jsou doba do použití i podmínky před vlastním použitím na odpovědnosti uživatele a za normálních okolností ne delší než 24 hodiny při 2 °C až 8 °C, pokud se naředění neprovedlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku ve vnějším obalu, aby byl přípravek chráněn před světlem.
Během doby použitelnosti a pro účely ambulantního použití může pacient jednorázově vyndat přípravek z chladničky a uchovávat ho při pokojové teplotě (do 25 °C) až 72 hodin. Po uplynutí této doby přípravek nesmí být přípravek znovu vrácen do chladničky, ale musí být zlikvidován.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Předplněné injekční stříkačky (sklo typu I) s injekční jehlou (nerezavějící ocel) s bezpečnostním krytem jehly anebo bez něho, obsahující 0,5 ml roztoku.
Velikost balení: 1, 3, 5 nebo 10 předplněných injekčních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před použitím je třeba roztok prohlédnout. Mohou se použít jen čiré roztoky bez částic.
Náhodná expozice teplotám pod bodem mrazu nemá nepříznivý vliv na stabilitu filgrastimu.
Zarzio neobsahuje žádné konzervační látky. Vzhledem k možnému riziku mikrobiální kontaminace jsou stříkačky Zarzio určeny pouze k jednorázovému použití.
Vnitřek krytu jehly injekční stříkačky může obsahovat suchou pryž (latex). Osoby citlivé na latex by měly při používání přípravku Zarzio postupovat se zvláštní opatrností (viz bod 4.4).
Ředění před podáním (není nutné)
Pokud je třeba, může se Zarzio ředit 5 % (50 mg/ml) roztokem glukózy.
Rozhodně se nedoporučuje ředit na konečnou koncentraci < 0,2 MU/ml (2 ^g/ml).
Pro pacienty léčené filgrastimem zředěným na koncentrace < 1,5 MU/ml (15 ^g/ml) je nutné přidat lidský sérový albumin (HAS, Human Serum Albumin) k dosažení koncentrace 2 mg/ml.
Příklad: Konečný objem 20 ml celkové dávky filgrastimu nižší než 30 MU (300 ^g) se má použít až po přidání 0,2 ml 20 % (200 mg/ml) roztoku lidského albuminu Ph. Eur.
Pokud je naředěn v5 % roztoku glukózy (50 mg/ml), je filgrastim kompatibilní se sklem a s různými plastickými materiály včetně polyvinylchloridu, polyolefinu (kopolymeru polypropylenu a polyethylenu) a polypropylenu.
Použití předplněné injekční stříkačky s bezpečnostním krytem jehly
Bezpečnostní kryt jehly překrývá jehlu po injekci, aby zabránil zranění píchnutím jehlou. Tím nijak neruší normální zacházení se stříkačkou. Tlačte na píst pomalu a až do té doby, kdy byla podána celá dávka a kdy píst se již nedá dále stlačit. Zatímco udržujete tlak na píst, vytáhněte stříkačku s jehlou z pacientova těla. Bezpečnostní kryt zakryje jehlu, jakmile povolíte tlak na píst.
Použití předplněné injekční stříkačky bez ochranného krytu jehly
Podejte dávku standardním způsobem.
Likvidace
Všechen nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. držitel rozhodnutí o registraci
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Rakousko
8. registrační čísla
Zarzio 30 MU/0,5 ml, injekční nebo infíizní roztok v předplněné injekční stříkačce
EU/1/08/495/001
EU/1/08/495/002
EU/1/08/495/003
EU/1/08/495/004
EU/1/08/495/009
EU/1/08/495/010
EU/1/08/495/011
EU/1/08/495/012
Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce
EU/1/08/495/005
EU/1/08/495/006
EU/1/08/495/007
EU/1/08/495/008
EU/1/08/495/013
EU/1/08/495/014
EU/1/08/495/015
EU/1/08/495/016
9. datum první registrace/prodloužení registrace
Datum první registrace: 6. února 2009
Datum posledního prodloužení registrace: 13. listopadu 2013
io. datum revize textu
<{MM.RRRR}>
příloha ii
a. výrobce biologické léčivé látky a výrobce odpovědný za propouštění šarží
b. podmínky nebo omezení výdeje a použití
c. další podmínky a požadavky registrace
d. podmínky nebo omezení s ohledem na bezpečné a účinné používání léčivého přípravku
a. výrobce biologické léčivé látky a výrobce odpovědný za propouštění šarží
Název a adresa vvrobce/vvrobců biologické léčivé látky
Sandoz GmbH Biochemiestrasse 10 6250 Kundl Rakousko
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Sandoz GmbH Biochemiestrasse 10 AT-6250 Kundl Rakousko
b. podmínky nebo omezení výdeje a použití
Výdej léčivého přípravku je vázán na lékařský předpis (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
c. další podmínky a požadavky registrace
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
d. podmínky nebo omezení s ohledem na bezpečné a účinné používání léčivého přípravku
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• Na žádost Evropské agentury pro léčivé přípravky,
• Při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
příloha iii
označení na obalu a příbalová informace
Zarzio 30 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce Filgrastimum
Jedna předplněná injekční stříkačka obsahuje filgrastimum 30 milionů jednotek (odpovídá 300 mikrogramům) v 0,5 ml (60 MU/ml).
Pomocné látky: kyselina glutamová, polysorbát 80, voda na injekci a sorbitol (E420).Další informace jsou uvedeny v příbalové informaci.
Injekční nebo infuzní roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka bez ochranného krytu injekční jehly 3 předplněné injekční stříkačky bez ochranného krytu injekční jehly 5 předplněných injekčních stříkaček bez ochranného krytu injekční jehly 10 předplněných injekčních stříkaček bez ochranného krytu injekční jehly
Pouze k jednorázovému užití. Před použitím si přečtěte příbalovou informaci. Subkutánní nebo intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku ve vnějším obalu, aby byl přípravek chráněn před světlem.
io. zvláštní opatření pro likvidaci nepoužitých léčivých přípravků nebo odpadu z nich, pokud je to vhodné
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Rakousko
EU/1/08/495/009
EU/1/08/495/010
EU/1/08/495/011
EU/1/08/495/012
č.š.
Zarzio 30 MU/0,5 ml
Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce Filgrastimum
Jedna předplněná injekční stříkačka obsahuje filgrastimum 48 milionů jednotek (odpovídá 480 mikrogramům) v 0,5 ml (96 MU/ml).
Pomocné látky: kyselina glutamová, polysorbát 80, voda na injekci a sorbitol (E420). Další informace jsou uvedeny v příbalové informaci.
Injekční nebo infuzní roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka bez ochranného krytu injekční jehly 3 předplněné injekční stříkačky bez ochranného krytu injekční jehly 5 předplněných injekčních stříkaček bez ochranného krytu injekční jehly 10 předplněných injekčních stříkaček bez ochranného krytu injekční jehly
Pouze k jednorázovému užití. Před použitím si přečtěte příbalovou informaci. Subkutánní nebo intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Po naředění užijte do 24 hodin.
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku ve vnějším obalu, aby byl přípravek chráněn před světlem.
io. zvláštní opatření pro likvidaci nepoužitých léčivých přípravků nebo odpadu z nich, pokud je to vhodné
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Rakousko
EU/1/08/495/013
EU/1/08/495/014
EU/1/08/495/015
EU/1/08/495/016
č.š.
Zarzio 48 MU/0,5 ml
Zarzio 30 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce Filgrastimum
Jedna předplněná injekční stříkačka obsahuje filgrastimum 30 milionů jednotek (odpovídá 300 mikrogramům) v 0,5 ml (60 MU/ml).
Pomocné látky: kyselina glutamová, polysorbát 80, voda na injekci a sorbitol (E420). Další informace jsou uvedeny v příbalové informaci.
Injekční nebo infuzní roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s bezpečnostním krytem jehly 3 předplněné injekční stříkačky s bezpečnostním krytem jehly 5 předplněných injekčních stříkaček s bezpečnostním krytem jehly 10 předplněných injekčních stříkaček s bezpečnostním krytem jehly
Pouze k jednorázovému užití. Před použitím si přečtěte příbalovou informaci. Subkutánní nebo intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku ve vnějším obalu, aby byl přípravek chráněn před světlem.
io. zvláštní opatření pro likvidaci nepoužitých léčivých přípravků nebo odpadu z nich, pokud je to vhodné
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Rakousko
EU/1/08/495/001
EU/1/08/495/002
EU/1/08/495/003
EU/1/08/495/004
č.š.
Zarzio 30 MU/0,5 ml
Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce Filgrastimum
Jedna předplněná injekční stříkačka obsahuje filgrastimum 48 milionů jednotek (odpovídá 480 mikrogramům) v 0,5 ml (96 MU/ml).
Pomocné látky: kyselina glutamová, polysorbát 80, voda na injekci a sorbitol (E420). Další informace jsou uvedeny v příbalové informaci.
Injekční nebo infuzní roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s bezpečnostním krytem jehly 3 předplněné injekční stříkačky s bezpečnostním krytem jehly 5 předplněných injekčních stříkaček s bezpečnostním krytem jehly 10 předplněných injekčních stříkaček s bezpečnostním krytem jehly
Pouze k jednorázovému užití. Před použitím si přečtěte příbalovou informaci. Subkutánní nebo intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku ve vnějším obalu, aby byl přípravek chráněn před světlem.
io. zvláštní opatření pro likvidaci nepoužitých léčivých přípravků nebo odpadu z nich, pokud je to vhodné
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Rakousko
EU/1/08/495/005
EU/1/08/495/006
EU/1/08/495/007
EU/1/08/495/008
č.š.
Zarzio 48 MU/0,5 ml
předplněná injekční stříkačka / předplněná injekční stříkačka s bezpečnostním krytem jehly
i. název léčivého přípravku a cesta/cesty podání
Zarzio 30 MU/0,5 ml, injekce nebo infuze
Filgrastimum
s.c./i.v.
2. způsob podání
3. použitelnost
EXP
4. číslo šarže
Lot
5. obsah udaný jako hmotnost, objem nebo počet
6. jiné
předplněná injekční stříkačka / předplněná injekční stříkačka s bezpečnostním krytem jehly
i. název léčivého přípravku a cesta/cesty podání
Zarzio 48 MU/0,5 ml, injekce nebo infuze
Filgrastimum
s.c./i.v.
2. způsob podání
3. použitelnost
EXP
4. číslo šarže
Lot
5. obsah udaný jako hmotnost, objem nebo počet
6. jiné
Zarzio 30 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce
Filgrastimum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Zarzio a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Zarzio používat
3. Jak se přípravek Zarzio používá
4. Možné nežádoucí účinky
5. Jak přípravek Zarzio uchovávat
6. Obsah balení a další informace
1. Co je přípravek Zarzio a k čemu se používá
Zarzio je růstový faktor bílých krvinek (faktor stimulující kolonie granulocytů) a patří do skupiny bílkovin nazývaných cytokiny. Růstové faktory jsou bílkoviny, které jsou vytvářeny přirozeně v těle, ale mohou být také vyrobeny biotechnologicky pro použití jako lék. Přípravek Zarzio povzbuzuje kostní dřeň, aby tvořila více bílých krvinek.
Snížení počtu bílých krvinek (neutropenie) může nastat z různých důvodů a způsobí, že Vaše tělo má nižší schopnost boje s infekcí. Přípravek Zarzio stimuluje kostní dřeň, aby tvořila rychleji nové bílé krvinky.
Přípravek Zarzio může být použit:
• Pro zvýšení počtu bílých krvinek po chemoterapii s cílem zabránit infekcím.
• Pro zvýšení počtu bílých krvinek po transplantaci kostní dřeně s cílem zabránit infekcím.
• Před vysokou dávkou chemoterapeutik pro povzbuzení kostní dřeně, aby tvořila více
kmenových buněk, které mohou být odebrány a vráceny zpět po Vaší léčbě. Tyto kmenové buňky mohou být odebrány od Vás nebo od dárce. Kmenové buňky pak budou vráceny do kostní dřeně a budou vytvářet krevní buňky.
• Pro zvýšení počtu bílých krvinek, pokud trpíte těžkou chronickou neutropenií s cílem zabránit
infekcím.
• U pacientů s pokročilou HIV infekcí, což pomůže snížit riziko infekcí.
2. Čemu musíte věnovat pozornost, než začnete přípravek Zarzio používat Nepoužívejte přípravek Zarzio
- jestliže jste alergický(á) na filgrastim nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Zarzio se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Zvláštní opatrnosti při použití přípravku Zarzio je zapotřebí, pokud jste někdy měl(a) alergickou reakci na latex.
Před zahájením léčby prosím informujte svého lékaře, pokud máte:
- osteoporózu (onemocnění kostí),
- srpkovitou anémii, protože přípravek Zarzio může způsobit krizi srpkovité anémie.
Informujte, prosím, ihned svého lékaře během léčby přípravkem Zarzio, pokud:
- máte bolest v levém nadbřišku, pod levým žeberním obloukem nebo v horní části ramena [může se jednat o příznaky zvětšené sleziny (splenomegalie) nebo možné prasknutí sleziny],
- si všimnete neobvyklého krvácení nebo tvorby podlitin [může se jednat o příznaky snížení počtu krevních destiček (trombocytopenie) se sníženou srážlivostí krve],
- máte náhlé známky alergie, jako je vyrážka, svědění nebo kopřivka na kůži, otok obličeje, rtů, jazyka nebo jiných částí těla, dýchavičnost, sípot nebo potíže s dechem, neboť by se mohlo jednat o známky závažné alergické reakce.
- pokud budete mít otok tváře nebo kotníků, krev v moči nebo hnědě zbarvenou moč nebo si povšimnete, že močíte méně než obvykle.
Ztráta odpovědi na filgrastim
Jestliže u Vás dojde ke ztrátě odpovědi nebo nemožnosti udržet si odpověď na léčbu filgrastimem,
Váš lékař vyšetří důvody tohoto stavu včetně toho, zda u Vás nedošlo ke vzniku protilátek, které neutralizují působení filgrastimu.
Váš lékař Vás bude možná chtít pečlivě sledovat, viz bod 4.4 příbalové informace.
Pokud jste pacient s těžkou chronickou neutropenií, můžete mít riziko, že se u Vás objeví rakovina krve (leukémie, myelodysplastický syndrom [MDS]). Měl(a) byste prodiskutovat se svým lékařem jaká jsou Vaše rizika vývoje rakoviny krve a jaká vyšetření by měla být provedena. Pokud se u Vás objeví leukémie nebo je pravděpodobné, že může dojít k jejímu rozvoji, neměl(a) byste přípravek Zarzio užívat, pokud nedostanete od svého lékaře jiné instrukce.
Jestliže jste dárce kmenových buněk, musíte být ve věku mezi 16 a 60 lety.
Zvláštní opatrnosti při užívání jiných přípravků stimulujících bílé krvinky je zapotřebí
Přípravek Zarzio patří do skupiny přípravků, které stimulují tvorbu bílých krvinek. Zdravotnický personál, který se o Vás stará, by měl vždy přesně zaznamenat název přípravku, který je Vám podáván.
Další léčivé přípravky a přípravek Zarzio
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Přípravek Zarzio nebyl hodnocen u těhotných nebo kojících žen.
Je důležité, abyste sdělila svému lékaři, jestliže:
• jste těhotná,
• domníváte se, že můžete být těhotná, nebo
• plánujete otěhotnět.
Pokud během léčby přípravkem Zarzio otěhotníte, sdělte to, prosím, svému lékaři.
Pokud Váš lékař neurčí jinak, musíte během používání přípravku Zarzio přestat kojit.
Řízení dopravních prostředků a obsluha strojů
Přípravek Zarzio by neměl mít žádný vliv na Vaši schopnost řídit nebo obsluhovat stroje. Doporučuje se však počkat a zjistit, jak se cítíte po užití přípravku Zarzio před tím, než začnete řídit nebo obsluhovat stroje.
Přípravek Zarzio obsahuje sorbitol (E420). Jestliže Vám Váš lékař řekl, že máte reakci na některé cukry, spojte se se svým lékařem, než začnete tento léčivý přípravek používat.
3. Jak se přípravek Zarzio používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jak a jaké množství přípravku Zarzio se podává?
Přípravek Zarzio se obvykle podává denně v injekci do tkáně těsně pod kůží (označuje se jako podkožní injekce). Může se podávat denně pomalou injekcí do žíly (označuje se jako nitrožilní infuze). Obvyklá dávka se mění v závislosti na Vašem onemocnění a tělesné hmotnosti. Váš lékař Vám řekne, jaké množství přípravku Zarzio byste měl(a) užívat.
Pacienti s transplantací kostní dřeně po chemoterapii:
Obvykle dostanete první dávku přípravku Zarzio nejméně 24 hodin po chemoterapii a nejméně 24 hodin po transplantaci kostní dřeně.
Vy nebo lidé, kteří o Vás pečují, se mohou naučit podávat podkožní injekce, abyste mohl(a) ve své léčbě pokračovat doma. Neměl(a) byste se o to však pokoušet, pokud Vás v tom nejprve patřičně nezaškolil Váš poskytovatel zdravotní péče.
Jak dlouho budu muset přípravek Zarzio užívat?
Bude nutné, abyste užíval(a) přípravek Zarzio, dokud nebude počet Vašich bílých krvinek normální. Budou prováděna pravidelná vyšetření krve pro sledování počtu bílých krvinek ve Vašem těle.
Váš lékař Vám řekne, jak dlouho bude nutné užívat přípravek Zarzio.
Použití u dětí
Přípravek Zarzio se používá k léčbě dětí, které jsou léčeny chemoterapií nebo které mají závažný nízký počet bílých krvinek (neutropenie). Dávky pro děti, které dostávají chemoterapii, jsou stejné jako pro dospělé.
Jestliže jste použil(a) více přípravku Zarzio, než jste měl(a)
Nezvyšujte si dávku, kterou Vám lékař předepsal. Jestliže si myslíte, že jste si injekčně podal(a) více přípravku Zarzio, než jste měl(a), spojte se co možná nejdříve se svým lékařem.
Jestliže jste zapomněl(a) použít přípravek Zarzio
Jestliže jste zapomněl(a) podat injekci přípravku Zarzio, nebo si podal(a) příliš malé množství, spojte se co možná nejdříve se svým lékařem. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Prosím, sdělte svému lékaři ihned během léčby:
• jestliže se u Vás objeví alergická reakce, včetně slabosti, poklesu krevního tlaku, problémy
s dýcháním, otok obličeje (anafylaxe), kožní vyrážka, svědivá vyrážka (kopřivka), otok obličeje, rtů, úst, jazyka nebo krku (angioedém) a dechová nedostatečnost (dušnost). Přecitlivělost je častá u pacientů s rakovinou.
• jestliže se u Vás objeví kašel, horečka a dýchací obtíže (dušnost), protože to může být známka
syndromu akutní respirační tísně (ARDS). ARDS je méně častý u pacientů s rakovinou.
• jestliže dostanete bolesti v levém nadbřišku, bolest pod levým žeberním obloukem nebo bolest
v horní části ramene, protože to může souviset s problémy s Vaší slezinou [zvětšení sleziny (splenomegalie) nebo prasknutí sleziny].
• jestliže jste léčen(a) pro těžkou chronickou neutropenii a máte krev v moči (hematurie).
Váš lékař může pravidelně vyšetřovat Vaši moč, pokud se u Vás objeví tento nežádoucí účinek nebo pokud se ve Vaší moči objeví bílkovina (proteinurie).
• jestliže máte některý z následujících nežádoucích účinků nebo kombinaci následujících
nežádoucích účinků:
- otok nebo opuchlina, které mohou být spojeny s méně častým močením, dušnost, otoky břicha a pocit plnosti a celkový pocit únavy. Tyto příznaky mají obvykle rychlý nástup. Mohou to být příznaky méně častého stavu (může postihnout až 1 ze 100 pacientů) nazývaného „syndrom zvýšené permeability kapilár“, který způsobuje prosakování krve z malých cév do Vašeho těla a vyžaduje okamžitou lékařskou pomoc.
• jestliže budete mít poruchu funkce ledvin (glomerulonefritidu). Porucha funkce ledvin byla u
pacientů léčených filgrastimem pozorována. Pokud budete mít otok tváře nebo kotníků, krev v moči nebo hnědě zbarvenou moč nebo si povšimnete, že močíte méně než obvykle, volejte okamžitě svého lékaře.
Velmi častý nežádoucí účinek užívání filgrastimu je bolest svalů a kostí (muskuloskeletální bolest), kterou je možné snížit užíváním běžných léků proti bolesti (analgetika). U pacientů podstupujících transplantaci kmenových buněk nebo kostní dřeně se může objevit reakce štěpu proti hostiteli (GvHD), to je reakce buněk dárce proti pacientovi, který je příjemcem transplantátu; známky a příznaky zahrnují vyrážku na dlaních nebo chodidlech a vřed a bolavá místa v ústech, střevech, játrech, na kůži, ve Vašich očích, v plících, v pochvě a v kloubech. Velmi často se u normálních dárců kmenových buněk pozoruje zvýšení počtu bílých krvinek (leukocytóza) a snížení počtu krevních destiček, což snižuje schopnost krve se srážet (trombocytopenie). Tyto nežádoucí účinky budou sledovány Vaším lékařem.
Velmi časté nežádoucí účinky (nežádoucí účinky se mohou vyskytnout u více než 1 z 10 osob užívajících přípravek Zarzio)
u pacientů s nádorovým onemocněním
• změny hladiny chemických látek v krvi
• zvýšení hladiny určitých enzymů v krvi
• snížená chuť k jídlu
• bolest hlavy
• bolest v ústech a hrdle (orofaryngeální bolest)
• kašel
• průjem
• zvracení
• zácpa
• nevolnost
• kožní vyrážka
• neobvyklé vypadávání nebo řídnutí vlasů (alopecie)
• bolest svalů nebo kostí (muskuloskeletální bolest)
• celková slabost (astenie)
• únava
• bolest a otok výstelky trávicího traktu od dutiny ústní až po konečník (zánět sliznice)
• dušnost (dyspnoe) • bolest
u normálních dárců kmenových buněk
• snížení počtu krevních destiček, což snižuje schopnost krve se srážet (trombocytopenie)
• zvýšení počtu bílých krvinek (leukocytóza)
• bolest hlavy
• bolest svalů nebo kostí (muskuloskeletální bolest)
u pacientů s těžkou chronickou neutropenií
• zvětšení sleziny (splenomegalie)
• nízký počet červených krvinek (anémie)
• změny hladiny chemických látek v krvi
• zvýšení hladiny určitých enzymů v krvi
• bolest hlavy
• krvácení z nosu (epistaxe)
• průjem
• zvětšení jater (hepatomegalie)
• kožní vyrážka
• bolest svalů nebo kostí (muskuloskeletální bolest)
• bolest kloubů (artralgie)
u HIV pacientů
• bolest svalů nebo kostí (muskuloskeletální bolest)
Časté nežádoucí účinky (nežádoucí účinky se mohou vyskytnout až u 1 ze 10 osob užívajících přípravek Zarzio)
u pacientů s nádorovým onemocněním
• alergická reakce (přecitlivělost na lék)
• nízký krevní tlak (hypotenze)
• bolest při močení (dysurie)
• bolest na hrudi
• vykašlávání krve (hemoptýza)
u normálních dárců kmenových buněk
• zvýšení hladiny určitých enzymů v krvi
• dušnost (dyspnoe)
• zvětšení sleziny (splenomegalie)
u pacientů s těžkou chronickou neutropenií
• prasknutí sleziny
• snížení počtu krevních destiček, což snižuje schopnost krve se srážet (trombocytopenie)
• změny hladiny chemických látek v krvi
• zánět krevních cév v kůži (kožní vaskulitida)
• neobvyklé vypadávání vlasů nebo řídnutí vlasů (alopecie)
• onemocnění, které způsobuje, že kosti ztrácejí hustotu, jsou slabší, křehčí a mohou se lámat
(osteoporóza)
• krev v moči (hematurie)
• bolest v místě aplikace injekce
• poškození malých filtračních struktur v ledvinách (glomerulonefritida) u HIV pacientů
• zvětšení sleziny (splenomegalie)
Méně časté nežádoucí účinky (nežádoucí účinky se mohou vyskytnout až u 1 z 100 osob užívajících přípravek Zarzio)
u pacientů s nádorovým onemocněním
• prasknutí sleziny
• zvětšení sleziny (splenomegalie)
• silná bolest kostí, hrudníku, střev nebo kloubů (krize srpkovité anémie)
• odmítnutí transplantované kostní dřeně (reakce štěpu proti hostiteli)
• bolest a otok kloubů podobný dně (pseudodna)
• těžký zánět plic způsobující obtíže při dýchání (syndrom akutní respirační tísně)
• plíce nefungují tak, jak by měly, což způsobuje dušnost (respirační selhání)
• otok a/nebo tekutina v plících (plicní edém)
• zánět plic (intersticiální plicní choroba)
• abnormální rentgenový snímek plic (plicní infiltrát)
• švestkovitě zbarvené, vyvýšené, bolavé plochy na končetinách a někdy na obličeji a krku
s horečkou (Sweetův syndrom)
• zánět krevních cév v kůži (kožní vaskulitida)
• zhoršení revmatoidní artritidy
• neobvyklá změna moči
• bolest
• poškození jater způsobené ucpáním malých cév v játrech (venookluzivní choroba)
• krvácení z plic (plicní hemorhagie)
• změna regulace tekutin v těle, což může vést k opuchlosti
• poškození malých filtračních struktur v ledvinách (glomerulonefritida)
u normálních dárců kmenových buněk
• prasknutí sleziny
• silná bolest kostí, hrudníku, střev nebo kloubů (krize srpkovité anémie)
• náhlá život ohrožující alergická reakce (anafylaktická reakce)
• změny hladin chemických látek v krvi
• krvácení z plic (plicní hemorhagie)
• vykašlávání krve (hemoptýza)
• abnormální rentgenový snímek plic (plicní infiltrát)
• snížené vstřebávání kyslíku v plících (hypoxie)
• zvýšení hladiny určitých enzymů v krvi
• zhoršení revmatoidní artritidy
• poškození malých filtračních struktur v ledvinách (glomerulonefritida)
u pacientů s těžkou chronickou neutropenií
• silná bolest kostí, hrudníku, střev nebo kloubů (krize srpkovité anémie)
• nadbytek bílkoviny v moči (proteinurie)
u HIV pacientů
• silná bolest kostí, hrudníku, střev nebo kloubů (krize srpkovité anémie)
Neznámé nežádoucí účinky (frekvenci nelze odhadnout z dostupných údajů)
• poškození malých filtračních struktur v ledvinách (glomerulonefritida)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a injekční stříkačce za Použitelné do:/EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Uchovávejte předplněnou injekční stříkačku ve vnějším obalu, aby byl přípravek chráněn před světlem.
Neúmyslné zmrazení přípravek Zarzio nepoškodí.
Předplněná injekční stříkačka může být vyndána z chladničky a ponechána při pokojové teplotě maximálně jedenkrát na dobu maximálně 72 hodin (při teplotě ne vyšší než 25 °C). Po uplynutí této doby nesmí být přípravek znovu vrácen do chladničky, ale musí být zlikvidován.
Nepoužívejte tento přípravek, pokud si všimnete změny barvy, zákalu nebo částic. Měla by to být čirá, bezbarvá až slabě nažloutlá tekutina.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Zarzio obsahuje
- Léčivou látkou je filgrastimum.
Zarzio 30 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce: 1 předplněná injekční stříkačka obsahuje filgrastimum 30 MU v 0,5 ml, odpovídající 60 MU/ml.
Zarzio 48 MU/0,5 ml, injekční nebo infuzní roztok v předplněné injekční stříkačce: 1 předplněná injekční stříkačka obsahuje filgrastimum 48 MU v 0,5 ml, odpovídající 96 MU/ml.
- Pomocnými látkami jsou kyselina glutamová, sorbitol (E420), polysorbát 80, voda na injekci.
- Kryt jehly injekční stříkačky může obsahovat suchou pryž (latex).
Jak přípravek Zarzio vypadá a co obsahuje toto balení
Zarzio je čirý, bezbarvý nebo slabě nažloutlý injekční nebo infuzní roztok v předplněné injekční stříkačce.
Zarzio je k dispozici v baleních obsahujících 1, 3, 5 nebo 10 předplněných injekčních stříkaček s bezpečnostním krytem injekční jehly anebo bez něho.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Rakousko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci: Belgie/Belgique/Belgien Lietuva
Sandoz nv-sa Sandoz Pharmaceuticals d.d filialas
Tél/Tel: +32 2 722 97 97
Tel: +370 5 2636 037
|
Btarapna TtproBCKO npegcraBHTencTBO CaHgo3 g.g. Ten.: +359 2 970 47 47 |
Luxembourg/Luxemburg Sandoz GmbH Tel: +43 5338 200 |
|
Česká republika Sandoz s.r.o. Tel: +420 221 421 611 |
Magyarország Sandoz Hungária Kft. Tel.: +36 1 430 2890 |
|
Danmark/Norge/ísland/Sverige Sandoz A/S Tlf/Sími /Tel: +45 63 95 10 00 |
Malta Sandoz GmbH Tel: +43 5338 200 |
|
Deutschland Hexal AG Tel: +49 8024 908 0 |
Nederland Sandoz B.V. Tel: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel: +372 665 2400 |
Osterreich Sandoz GmbH Tel: +43 5338 2000 |
|
EXXáSa Sandoz Pharmaceuticals d.d, Tn^: +30 216 600 5000 n DEMO Avróvu^og Bio^nxaviKq Kai E^nopiKq Exaipsía Ten.: +30 210 816 18 02 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
Espaňa Sandoz Farmacéutica, S.A. Tlf: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel: +351 21 924 19 11 |
|
France Sandoz SAS Tél: +33 1 49 64 48 00 |
Románia S.C. Sandoz Pharma Services S.R.L Tel: +40 21 4075160 |
|
Hrvatska Sandoz d.o.o. Tel: +385 1 23 53 111 |
Slovenija Lek farmacevtska družba d.d. Tel: +386 1 580 21 11 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenská republika Sandoz d.d. - organizačná zložka Tel: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel: +358 10 6133 415 |
|
Kúrcpog P.T.Hadjigeorgiou co ltd Tel: +357 25372425 Latvija Sandoz d.d. Parstavnieciba Latvija Tel: +371 67 892 006 |
United Kingdom Sandoz Ltd Tel: +44 1276 69 8020 |
Tato přfbalová informace byla naposledy revidována {MM.RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky (EMA): http://www.ema.europa.eu/.
Instrukce, jak si sám (sama) podáte injekci
Tento oddíl obsahuje informace, jak si sám/sama podáte injekci přípravku Zarzio. Je důležité, abyste se nesnažil(a) si injekci sám/sama podat, pokud to s Vámi Váš lékař nebo zdravotní sestra zvlášť nenatrénovali. Přípravek Zarzio je anebo není vybaven ochranným krytem jehly a Váš lékař nebo ošetřovatelka Vám ukáží, jak jej používat. Jestliže si nejste jistí, jak si injekci podávat anebo máte-li jakékoli další otázky, prosím, požádejte svého lékaře anebo zdravotní sestru o pomoc.
1. Umyjte si ruce.
2. Vyjměte jednu injekční stříkačku z balení a odstraňte ochranný kryt z injekční jehly. Injekční stříkačky mají vyražené kroužky označující stupnici, aby bylo možné částečné použití v případě potřeby. Každý kroužek stupnice odpovídá objemu 0,1 ml. Pokud je nutné částečné použití, odstraňte před aplikací injekce nepotřebný roztok.
3. Očistěte kůži v místě aplikace injekce pomocí tampónu namočeného v alkoholu.
4. Vytvořte záhyb kůže sevřením kůže mezi palcem a ukazovákem.
5. Jehlu rychlým a pevným pohybem zaveďte do kožního záhybu. Injikujte roztok přípravku Zarzio tak, jak Vám to ukázal Váš lékař. Pokud si nejste jistý(á), zeptejte se svého lékaře nebo lékárníka.
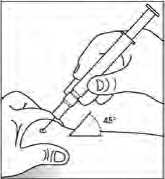
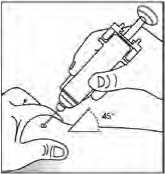
Předplněná stříkačka bez ochranného krytu injekční jehly
6. Stále držte kůži zřasenou mezi palcem a ukazováčkem a tlačte na píst pomalu a stejnoměrně.
7. Po injekci tekutiny vytáhněte injekční jehlu a uvolněte kůži.
8. Použitou stříkačku vložte do kontejneru pro odpad. Každou stříkačku použijte pouze pro jednu injekci.
Předplněná stříkačka s ochranným krytem injekční jehly
6. Stále držte kůži zřasenou mezi palcem a ukazováčkem a tlačte na píst pomalu a stejnoměrně, dokud jste nepodal(a) celou dávku
a dokud píst se již nedá dále zatlačit. Neuvolňujte tlak na píst!
7. Po ukončené injekci tekutiny vytáhněte injekční jehlu za stálého tlaku na píst, a potom uvolněte kůži.
8. Uvolněte píst. Chránič injekční jehly se rychle vysune a přikryje injekční jehlu.
9. Zlikvidujte veškerý nepoužitý přípravek nebo odpadní materiál. Každou stříkačku použijte pouze pro jednu injekci.
Následující informace je určena pouze pro zdravotnické pracovníky:
Před použitím je třeba roztok vizuálně zkontrolovat. Mohou se použít jen čiré roztoky bez částic. Náhodná expozice mrazu nemá nepříznivý vliv na stabilitu přípravku Zarzio.
Zarzio neobsahuje žádné konzervační látky: Vzhledem k možnému riziku mikrobiální kontaminace jsou stříkačky Zarzio určeny pouze k jednorázovému použití.
Kryt jehly injekční stříkačky může obsahovat suchou pryž (latex), a proto by s ním neměly manipulovat osoby citlivé na tuto látku.
Ředění před podáním (není nutné)
Je-li nutné, může být přípravek Zarzio naředěn 5% (50 mg/ml) roztokem glukózy. Zarzio se nesmí ředit žádným roztokem s obsahem chloridu sodného.
V žádném případě se nedoporučuje ředit do konečné koncentrace < 0,2 MU/ml (2 ^g/ml).
U pacientů léčených filgrastimem naředěným na koncentrace < 1,5 MU/ml (15 ^g/ml), je nutné přidat lidský sérový albumin (HSA, Human Serum Albumin) k dosažení koncentrace albuminu 2 mg/ml.
Příklad: Konečný objem 20 ml celkové dávky filgrastimu nižší než 30 MU (300 ^g) se má použít až po přidání 0,2 ml 20% (200 mg/ml) roztoku lidského albuminu Ph. Eur.
Pokud je Zarzio naředěn v 5% roztoku glukózy (50 mg/ml), je kompatibilní se sklem a s různými plastickými materiály včetně polyvinylchloridu, polyolefinu (kopolymeru polypropylenu a polyethylenu) a polypropylenu.
Po naředění: chemická a fyzikální stabilita v užívání naředěného infuzního roztoku byla prokázána po dobu 24 hodin při 2 °C až 8 °C. Z mikrobiologického hlediska se má přípravek užít ihned. Není-li užit okamžitě, odpovídá uživatel za podmínky a dobu uchovávání před užitím, ty by neměly překročit 24 hodin a 2 °C až 8 °C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
Použití předplněné injekční stříkačky s bezpečnostním krytem jehly
Bezpečnostní kryt jehly překrývá jehlu po injekci, aby zabránil zranění píchnutím jehlou. Tím nijak neruší normální zacházení se stříkačkou. Tlačte na píst pomalu a až do té doby, kdy byla podána celá dávka a kdy píst se již nedá dále stlačit. Zatímco udržujete tlak na píst, vytáhněte stříkačku s jehlou z pacientova těla. Bezpečnostní kryt zakryje jehlu, jakmile povolíte tlak na píst.
Použití předplněné injekční stříkačky bez ochranného krytu jehly
Podejte dávku standardním způsobem.
Likvidace
Všechen nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
46