Xyrem 500 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Xyrem 500 mg/ml perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje natrii oxybutyras 500 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok
Perorální roztok je čirý až mírně opalizující.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba narkolepsie s kataplexií u dospělých pacientů.
4.2 Dávkování a způsob podání
Léčba má být zahájena a zůstávat pod vedením lékaře se zkušeností s léčbou poruch spánku. Dávkování
Doporučená zahajovací dávka je 4,5 g natrium-oxybutyrátu denně a je rozdělená do dvou stejných dávek po 2,25 g. Dávka se má titrovat na účinnou dávku podle účinnosti a snášenlivosti (viz bod 4.4) až na maximální dávku 9 g denně, rozdělenou do dvou stejných dávek po 4,5 g. Dávka se nastavuje oběma směry o 1,5 g denně (tj. po 0,75 g/dávku). Mezi jednotlivými vzestupy dávky se doporučuje interval minimálně 1-2 týdny. Z důvodu možného výskytu závažných příznaků při dávce 18 g/den nebo vyšší (viz bod 4.4) nemá být dávka 9 g denně překročena.
Jednotlivá dávka 4,5 g se nemá užívat bez předchozí titrace pacienta na tuto dávku.
Jestliže se užívá současně natrium-oxybutyrát a valproát (viz bod 4.5), doporučuje se snížení dávky natrium-oxybutyrátu o 20 %. Doporučená zahajovací dávka natrium-oxybutyrátu, pokud je užíván současně s valproátem, je 3,6 g za noc, podávaná perorálně ve dvou stejných dávkách, asi 1,8 g. Pokud je současné užívání nutné, je třeba sledovat odpověď pacienta na léčbu a její snášenlivost, a dávku upravit příslušným způsobem (viz bod 4.4).
Vysazení Xyremu
Účinky vysazení natrium-oxybutyrátu nebyly v kontrolovaných klinických studiích systematicky hodnoceny (viz bod 4.4).
Pokud pacient přeruší užívání přípravku na déle než 14 po sobě následujících dní, musí být znovu zahájena titrace od nejnižší dávky.
Zvláštní populace Starší pacienti
Starší pacienti musí být při užívání natrium-oxybutyrátu pečlivě monitorováni z hlediska zhoršení motorických a/nebo kognitivních funkcí (viz bod 4.4).
Porucha funkce jater
U všech pacientů s poruchou funkce jater se zahajovací dávka sníží na polovinu a je nutné pečlivě monitorovat odpověď na zvyšování dávek přípravku (viz bod 4.4).
Porucha funkce ledvin
U všech pacientů s poruchou funkce ledvin je nutné vzít v úvahu dietní doporučení, aby byl při užívání natrium-oxybutyrátu snížen příjem sodíku (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost natrium-oxybutyrátu u dětí a dospívajících ve věku 0-18 let nebyla ještě stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Xyrem se užívá perorálně po ulehnutí do postele a druhá dávka s odstupem 2,5-4 hodin. Doporučuje se připravit si obě dávky Xyremu najednou před ulehnutím do postele.
Balení léčivého přípravku Xyrem obsahuje kalibrovanou odměrnou stříkačku a dvě 90ml dávkovací nádobky s dětským bezpečnostním uzávěrem. Každá odměřená dávka Xyremu musí být před užitím připravena do dávkovací nádobky a naředěna přidáním 60 ml vody.
Jelikož jídlo významně snižuje biologickou dostupnost natrium-oxybutyrátu, pacienti mají jíst alespoň několik hodin (2-3) před užitím první dávky přípravku Xyrem v čase, kdy chodí spát. Pacienti mají vždy dodržet stejnou dobu odstupu užití dávky přípravku od jídla. Dávky přípravku mají být užity do 24 hodin po naředění, jinak musí být zlikvidovány.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Pacienti se závažnou depresí.
Pacienti s deficitem sukcinát-semialdehyd dehydrogenázy.
Pacienti léčení opioidy nebo barbituráty.
4.4 Zvláštní upozornění a opatření pro použití
Xyrem může navodit respirační depresi.
Respirační deprese a deprese CNS
Natrium-oxybutyrát může také navodit dechový útlum. U zdravého dobrovolníka byla pozorována apnoe a respirační deprese po jednotlivé dávce 4,5 g nalačno (dvojnásobek doporučené počáteční dávky). Pacienti mají být dotázáni na případné příznaky útlumu centrálního nervového systému (CNS) nebo dechový útlum. Zvláštní opatrnosti je třeba u pacientů s poruchou dýchání. Vzhledem k vyššímu riziku spánkové apnoe musí být pacienti s BMI > 40 kg/m2 při užívání natrium-oxybutyrátu pečlivě monitorováni.
Přibližně 80% pacientů, kteří užívali natrium-oxybutyrát v klinických studiích, pokračovalo v užívání stimulancií CNS. Není známo, zda toto užívání ovlivnilo dýchání v noci. Před zvýšením dávky natrium-oxybutyrátu (viz bod 4.2) si musí být předepisující lékař vědom, že až u 50 % pacientů s narkolepsií se vyskytuje spánková apnoe.
• Benzodiazepiny
Vzhledem k možnosti zvýšení rizika útlumu dýchání je třeba se vyhnout současnému užívání benzodiazepinů a natrium-oxybutyrátu.
• Alkohol a látky s tlumivým účinkem na CNS
Současné užití alkoholu či jiného přípravku s tlumivým účinkem na CNS spolu s natrium-oxybutyrátem může vést k zesílení tlumivých účinků natrium-oxybutyrátu na CNS, stejně jako ke zvýšení rizika útlumu dechu. Proto musí být pacienti upozorněni na to, aby nepožívali alkohol spolu s natrium-oxybutyrátem.
• Inhibitory dehydrogenázy kyseliny gama-hydroxymáselné (gamma hydroxybutyrate,
GHB)
Opatrnost je nutná u pacientů, kteří jsou současně léčeni valproátem nebo jiným inhibitorem GHB dehydrogenázy, protože byly pozorovány farmakokinetické a farmakodynamické interakce, pokud je natrium-oxybutyrát podáván spolu s valproátem (viz bod 4.5). Pokud je současné podávání nutné, je třeba zvážit úpravu dávkování (viz bod 4.2). Vedle toho je nutno pečlivě sledovat odpověď pacienta na léčbu a její snášenlivost a dávku upravit příslušným způsobem.
• Topiramát
Po současném podávání natrium-oxybutyrátu a topiramátu bylo klinicky pozorováno kóma a zvýšené plazmatické koncentrace GHB. Pacienti by tudíž měli být varováni před současným užíváním topiramátu a natrium-oxybutyrátu (viz bod 4.5).
Možnost abúzu a závislosti
Natrium-oxybutyrát, což je sodná sůl gama-hydroxybutyrátu (GHB), je látka s tlumivým účinkem na CNS s dobře známým potenciálem pro zneužívání. Před zahájením léčby musí lékaři u pacientů vyhodnotit anamnézu zneužívání léků nebo náchylnost k takovému zneužívání. Pacienti musí být rutinně monitorováni a v případě podezření na abúzus musí být léčba natrium-oxybutyrátem přerušena.
Existují kazuistiky závislosti po ilegálním užití GHB v častých opakovaných dávkách (18250 g denně) převyšujících rozsah terapeutických dávek. I když nemáme jednoznačný důkaz o vzniku závislosti u pacientů užívajících natrium-oxybutyrát v terapeutických dávkách, tuto možnost nelze vyloučit.
Pacienti s porfVrií
Natrium-oxybutyrát je považován za nebezpečný u pacientů s porfyrií, protože se ukázalo, že je u zvířat a v in vitro systémech porfyriogenní.
Neuropsychiatrické příhody
Během léčby natrium-oxybutyrátem se může u pacientů vyskytnout zmatenost. Pokud k tomu dojde, je zapotřebí kompletní vyšetření a podle individuálního stavu učinit příslušná opatření.
K dalším neuropsychiatrickým příhodám patří anxieta, psychóza, paranoia, halucinace a agitovanost. Vznik poruch myšlení a/nebo abnormálního chování při léčbě natrium-oxybutyrátem vyžaduje pečlivé a okamžité vyšetření.
Vznik deprese při léčbě natrium-oxybutyrátem vyžaduje pečlivé a okamžité vyšetření. Pacienty s anamnézou deprese a/nebo pokusu o sebevraždu je nutné při léčbě natrium-oxybutyrátem mimořádně pečlivě sledovat z hlediska možnosti vzniku depresivních symptomů. U těžké deprese je použití Xyremu kontraindikováno (viz bod 4.3).
Pokud se během léčby přípravkem vyskytne inkontinence moče nebo stolice, musí předepisující lékař zvážit vyšetření k vyloučení etiologie této poruchy.
U pacientů léčených natrium-oxybutyrátem byla v klinických studiích popsána náměsíčnost. Není jasné, zda některé nebo všechny epizody odpovídaly pravému somnambulismu (parasomnie vyskytující se během non-REM spánku) nebo jiné specifické zdravotní poruše.
U každého pacienta s náměsíčností je nutné myslet na riziko poranění nebo sebepoškození. Proto je nutné stavy náměsíčnosti kompletně vyšetřit a zvážit příslušnou intervenci.
Příjem sodíku
Pacienti užívající natrium-oxybutyrát budou mít další denní příjem sodíku v rozmezí od 0,82 g (při dávce Xyremu 4,5 g denně) do 1,6 g (při dávce 9 g denně). Při léčbě pacientů se srdečním selháním, hypertenzí nebo poruchou funkce ledvin by měla být pečlivě zvážena dietní opatření ke snížení příjmu sodíku (viz bod 4.2).
Starší pacienti
S podáváním přípravku starším pacientům jsou pouze velmi omezené zkušenosti. Ti musí být proto pečlivě monitorováni z hlediska potenciálního zhoršení motorických a/nebo kognitivních funkcí.
Pacienti s epilepsií
U pacientů léčených natrium-oxybutyrátem byly pozorovány záchvaty. U pacientů s epilepsií nebyla jeho bezpečnost a účinnost stanovena, proto se podávání přípravku nedoporučuje.
Rebound efekt a abstinenční syndrom
Účinky vysazení přípravku nebyly v kontrolovaných klinických studiích systematicky hodnoceny. U některých pacientů se může po ukončení léčby vrátit kataplexie ve vyšší frekvenci, může to však být i z důvodů normální variability této choroby. I když zkušenosti z klinických studií u pacientů s narkolepsií/kataplexií léčených terapeutickými dávkami natrium-oxybutyrátu neprokazují jednoznačně abstinenční syndrom, byly ve vzácných případech pozorovány po vysazení GHB stavy jako nespavost, bolest hlavy, úzkost, závratě, poruchy spánku, somnolence, halucinace a psychotické poruchy.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Současné požití alkoholu a natrium-oxybutyrátu může vést k zesílení jeho tlumivých účinků na centrální nervový systém (CNS). Pacienty je nutné upozornit, aby nepili v době léčby natrium-oxybutyrátem alkoholické nápoje.
Natrium-oxybutyrát se nesmí používat v kombinaci se sedativními hypnotiky nebo jinými přípravky, které mají tlumivý účinek na CNS.
Sedativní hypnotika
Studie lékových interakcí s natrium-oxybutyrátem (jednotlivá dávka 2,25 g), lorazepamem (jednotlivá dávka 2 mg) a zolpidem-tartarátem (jednotlivá dávka 5 mg) u zdravých dospělých osob neprokázaly farmakokinetické interakce. Zvýšená ospalost byla pozorována po souběžném podání natrium-oxybutyrátu (2,25 g) a lorazepamu (2 mg). Farmakodynamická interakce se zolpidemem nebyla hodnocena. Jestliže budou kombinovány vyšší dávky natrium-oxybutyrátu až 9 g/d s vyššími dávkami hypnotik (v rámci doporučeného dávkového rozmezí), nelze vyloučit farmakodynamické interakce spojené se symptomy útlumu CNS a/nebo útlumu dýchání (viz bod 4.3).
Tramadol
V jedné studii lékových interakcí s natrium-oxybutyrátem (jednotlivá dávka 2,25 g) a tramadolem (jednotlivá dávka 100 mg) u zdravých dospělých nebyla prokázána farmakokinetická/farmakodynamická interakce. Jestliže budou kombinovány vyšší dávky natrium-oxybutyrátu až 9 g/d s vyššími dávkami opioidů (v rámci doporučeného dávkového rozmezí), nelze vyloučit farmakodynamické interakce spojené se symptomy útlumu CNS a/nebo útlumu dýchání (viz bod 4.3).
Antidepresiva
Studie lékových interakcí u zdravých dospělých neprokázaly žádné interakce mezi natrium-oxybutyrátem (jednotlivá dávka 2,25 g), antidepresivem protriptylin-hydrochloridem (jednotlivá dávka 10 mg) a duloxetinem (60 mg v ustáleném stavu). Žádné další účinky na ospalost nebyly pozorovány při porovnání jednotlivé dávky samotného natrium-oxybutyrátu (2,25 g) a natrium-oxybutyrátu (2,25 g) v kombinaci s duloxetinem (60 mg v ustáleném stavu). Antidepresiva byla použita k léčbě kataplexie. Možný aditivní účinek antidepresiv a natrium-oxybutyrátu nelze vyloučit. Výskyt nežádoucích účinků se zvýšil při souběžném podávání natrium-oxybutyrátu s tricyklickými antidepresivy.
Modafinil
V jedné studii lékových interakcí u zdravých dospělých nebyly prokázány farmakokinetické interakce mezi natrium-oxybutyrátem (jednotlivá dávka 4,5 g) a modafinilem (jednotlivá dávka 200 mg). Natrium-oxybutyrát byl podáván současně s látkami stimulujícími CNS přibližně u 80 % pacientů v klinických studiích při narkolepsii. Není známo, zda bylo ovlivněno dýchání v noci.
Omeprazol
Současné podávání spolu s omeprazolem nemá žádný klinicky významný účinek na farmakokinetiku natrium-oxybutyrátu. Dávku natrium-oxybutyrátu proto není nutno upravovat při současném podávání s inhibitory protonové pumpy.
Ibuprofen
Studie lékových interakcí u zdravých dospělých neprokázaly žádné farmakokinetické interakce mezi natrium-oxybutyrátem a ibuprofenem.
Diklofenak
Studie lékových interakcí u zdravých dospělých neprokázaly žádné farmakokinetické interakce mezi natrium-oxybutyrátem a diklofenakem. Současné podávání natrium-oxybutyrátu a diklofenaku u zdravých dobrovolníků vedlo ke snížení poruch pozornosti způsobených podáváním samotného Xyremu při hodnocení psychometrickými testy.
Inhibitory GHB dehydrogenázy
Jelikož je natrium-oxybutyrát metabolizován GHB dehydrogenázou, existuje potenciální riziko interakce s přípravky stimulujícími nebo inhibujícími tento enzym (např. valproát, fenytoin, nebo ethosuximid) (viz bod 4.4).
Současné podávání natrium-oxybutyrátu (6 g denně) spolu s valproátem (1250 mg denně) vedlo ke zvýšení systémové expozice vůči natrium-oxybutyrátu přibližně o 25 % a nevyvolalo žádnou významnou změnu Cmax. Nebyl pozorován žádný vliv na farmakokinetiku valproátu. Výsledné farmakodynamické účinky, a to včetně zvýšeného narušení kognitivních funkcí a ospalosti, byly výraznější při současném podávání obou léků než účinky pozorované při podávání těchto léků jednotlivě. Pokud stav pacienta vyžaduje současné podávání obou léků, je třeba monitorovat odpověď pacienta na léčbu a její snášenlivost, a pokud je to nutné, je třeba provést úpravu dávky (viz bod 4.2).
Topiramát
Nelze vyloučit možné farmakodynamické a farmakokinetické interakce, když je natrium-oxybutyrát užíván současně s topiramátem. Klinicky bylo pozorováno kóma a zvýšené plazmatické koncentrace GHB u pacientů, kteří současně užívali natrium-oxybutyrát a topiramát (viz bod 4.4).
Studie in vitro s lidskými jaterními mikrozomy prokázaly, že natrium-oxybutyrát neinhibuje významně aktivity lidských izoenzymů (viz bod 5.2).
4.6 Fertilita, těhotenství a kojení
Studie na zvířatech neprokázaly teratogenitu, ale ve studiích u potkanů i králíků byla pozorována embryoletalita (viz bod 5.3).
Údaje od omezeného počtu těhotných žen exponovaných v prvním trimestru svědčí o možném zvýšení rizika spontánních potratů. Dosud nejsou k dispozici jiná relevantní epidemiologická data. Omezené údaje od těhotných pacientek užívajících natrium-oxybutyrát ve druhém a třetím trimestru těhotenství nenaznačují možnost vzniku malformací ani fetální/neonatální toxicitu natrium-oxybutyrátu.
Užívání natrium-oxybutyrátu v těhotenství se nedoporučuje.
Kojení
Není známo, zda se natrium-oxybutyrát a/nebo jeho metabolity vylučují do mateřského mléka. Při léčbě natrium-oxybutyrátem se nedoporučuje kojit.
Fertilita
Nejsou k dispozici žádné údaje o vlivu natrium-oxybutyrátu na fertilitu. Studie u samců a samic potkanů neprokázaly při dávkách GHB až 1000 mg/kg/den žádný nežádoucí vliv na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Natrium-oxybutyrát má výrazný vliv na schopnost řídit nebo obsluhovat stroje.
Po dobu nejméně 6 hodin po požití natrium-oxybutyrátu nesmí pacienti provádět aktivity, které vyžadují úplnou duševní bdělost nebo motorickou koordinaci, jako je např. obsluha strojů nebo řízení.
Pokud pacient zahajuje terapii, pak do doby, než bude vědět, zda tento léčivý přípravek nemá nějaké přetrvávající účinky následující den, musí být mimořádně opatrný při řízení vozidla, ovládání těžkých strojů nebo provádění jiných úkolů, které by mohly být nebezpečné nebo které vyžadují úplnou duševní čilost.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Nejčastěji hlášenými nežádoucími účinky jsou závratě, nauzea a bolest hlavy, které se vyskytují u 10 %-20 % pacientů. Nejzávažnější nežádoucí účinky jsou sebevražedný pokus, psychózy, respirační deprese a konvulze.
Bezpečnost a účinnost natrium-oxybutyrátu při léčbě příznaků narkolepsie byla stanovena ve čtyřech multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných paralelních skupinových studiích u pacientů s narkolepsií s kataplexií, s výjimkou jedné studie, kde kataplexie nebyla požadována k zařazení. Dvě studie fáze 3 a jedna studie fáze 2 dvojitě zaslepených, paralelních, placebem kontrolovaných studií byly provedeny k posouzení indikace natrium-oxybutyrátu pro fibromyalgii. Navíc randomizované, dvojitě zaslepené, placebem kontrolované, zkřížené studie lékových interakcí s ibuprofenem, diklofenakem a valproátem byly provedeny u zdravých subjektů a shrnuty v bodě 4.5.
K nežádoucím účinkům hlášených z klinických studií byly navíc hlášeny nežádoucí účinky po uvedení přípravku na trh. Není vždy možné spolehlivě odhadnout frekvenci jejich výskytu u léčené populace.
Přehled nežádoucích účinků v tabulce
Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů MedDRA.
Odhad frekvence: Velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Infekce a infestace
Časté: nazofaryngitida, sinusitida.
Poruchy imunitního systému Méně časté: hypersenzitivita.
Poruchy metabolismu a výživy Časté: anorexie, snížená chuť k jídlu Není známo: dehydratace.
Psychiatrické poruchy
Časté: deprese, kataplexie, anxieta, abnormální sny, stav zmatenosti, dezorientace, noční můry, náměsíčnost, poruchy spánku, nespavost, střední insomnie, nervozita Méně časté: sebevražedný pokus, psychóza, paranoia, halucinace, abnormální myšlení, agitovanost, časná insomnie
Není známo: sebevražedné představy, euforie, syndrom poruchy spánku s nočním jedlictvím, panická ataka, mánie/bipolární porucha, blud, bruxismus.
Poruchy nervového systému Velmi časté: závratě, bolest hlavy
Časté: spánková obrna, somnolence, třes, poruchy rovnováhy, poruchy pozornosti, hypestezie, parestezie, sedace, dysgeuzie
Méně časté: myoklonus, amnézie, syndrom neklidných nohou Není známo: konvulze, ztráta vědomí.
Poruchy oka
Časté: rozmazané vidění.
Poruchy ucha a labyrintu Časté: vertigo.
Není známo: tinitus
Srdeční poruchy Časté: palpitace.
Cévní poruchy Časté: hypertenze.
Respirační, hrudní a mediastinální poruchy Časté: dyspnoe, chrápání, nazální kongesce Není známo: respirační deprese, spánková apnoe.
Gastrointestinální poruchy
Velmi časté: nauzea (frekvence je vyšší u žen než u mužů)
Časté: zvracení, průjem, bolest v horní části břicha Méně časté: inkontinence stolice.
Není známo: sucho v ústech
Poruchy kůže a podkožní tkáně Časté: hyperhidróza, vyrážka Není známo: urtikarie, angioedém.
Poruchy svalové a kosterní soustavy a pojivové tkáně Časté: artralgie, svalové spasmy, bolest zad.
Poruchy ledvin a močových cest
Časté: enuresis nocturna, inkontinence moči
Není známo: polakisurie, nutkání k močení.
Celkové poruchy a reakce v místě aplikace Časté: astenie, únava, pocit opilosti, periferní otoky.
Vyšetření
Časté: zvýšení krevního tlaku, snížení tělesné hmotnosti.
Poranění, otravy a procedurální komplikace
Popis vybraných nežádoucích účinků
U některých pacientů se může po ukončení léčby natrium-oxybutyrátem vrátit kataplexie ve vyšší frekvenci, může to však být i z důvodů normální variability této choroby. I když zkušenosti z klinických studií s terapeutickými dávkami natrium-oxybutyrátu u pacientů s narkolepsií/kataplexií neprokazují jednoznačně abstinenční syndrom, byly pozorovány ve vzácných případech po vysazení GHB nežádoucí účinky jako nespavost, bolesti hlavy, úzkost, závratě, poruchy spánku, somnolence, halucinace a psychotické poruchy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Informace o příznacích a projevech předávkování natrium-oxybutyrátem jsou omezené. Většina údajů pochází z ilegálního používání gama-hydroxybutyrátu (GHB), protože natrium-oxybutyrát je sodná sůl GHB. Příhody související s abstinenčním syndromem byly pozorovány u dávek mimo terapeutické rozmezí natrium-oxybutyrátu.
Příznaky
U pacientů se vyskytl různý stupeň útlumu vědomí, který může rychle kolísat mezi zmateností, agitovaným bojovným stavem s ataxií a kómatem. Může se vyskytnout zvracení (i při porušeném vědomí), nadměrné pocení, bolest hlavy a porucha psychomotorických dovedností. Popsáno bylo rozmazané vidění. Při vyšších dávkách bylo pozorováno prohloubení komatu. Byly hlášeny myoklonus a tonicko-klonické křeče. Existují hlášení o porušení rychlosti a hloubky dýchání a život ohrožujícím útlumu dechu, který vyžaduje intubaci a ventilaci. Byly pozorovány Cheyne-Stokesovo dýchání a apnoe. Bezvědomí může být provázeno bradykardií, hypotermií a svalovou hypotonií, šlachové reflexy ale zůstávají intaktní. Bradykardie reaguje na intravenózní podání atropinu.
Léčba
Pokud je podezření i na další požité látky, je třeba zvážit výplach žaludku. Jelikož se zvracení může vyskytovat při porušeném vědomí, musí se pacient uložit do příslušné polohy (vleže na levém boku) a musí být zajištěna ochrana dýchacích cest intubací. I když u pacientů v hlubokém kómatu může chybět dávivý reflex, i pacienti v bezvědomí se mohou bránit intubaci a je třeba zvážit rychlou sekvenční indukci (bez použití sedativa).
Po podání flumazenilu nelze předpokládat zvrat centrálních tlumivých účinků natrium-oxybutyrátu. Pro doporučení naloxonu v léčbě předávkování GHB není dostatek důkazů.
Použití hemodialýzy a dalších forem mimotělního odstranění nebylo při předávkování natrium-oxybutyrátem předmětem klinických studií. Vzhledem k rychlému metabolismu přípravku není ale přijetí takových opatření odůvodněné.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva nervového systému, ATC kód N07XX04.
Natrium-oxybutyrát je látka s tlumivým účinkem na centrální nervový systém, která snižuje excesivní ospalost v průběhu dne a kataplexii u pacientů s narkolepsií a modifikuje spánkovou architekturu redukcí fragmentace nočního spánku. Není znám přesný mechanismus, kterým
látka účinkuje. Předpokládá se, že vyvolává podporu spánku s pomalými (delta) vlnami a konsolidaci nočního spánku. Natrium-oxybutyrát podaný před nočním spánkem zvyšuje spánek stadia 3 a 4 a spánkovou latenci, ale snižuje frekvenci nástupu spánkových period REM (SOREMP). Na účinku přípravku se mohou podílet i další mechanismy, které dosud nebyly objasněny. Více než 80 % pacientů v databázi klinických studií užívalo současně stimulancia.
Účinnost natrium-oxybutyrátu v léčbě příznaků narkolepsie byla stanovena ve 4 multicentrických randomizovaných dvojitě zaslepených placebem kontrolovaných studiích s paralelními skupinami (studie 1, 2, 3 a 4) u pacientů s narkolepsií s kataplexií (s výjimkou studie 2, u které nebyla kataplexie k zařazení vyžadována). Současné užívání stimulancií bylo povoleno ve všech studiích (s výjimkou fáze aktivní léčby u studie 2); antidepresiva byla vysazena před nasazením účinné léčby ve všech studiích mimo studii 2. V každé studii byla denní dávka rozdělena do 2 stejných dílčích dávek. První dávka byla užita každý večer před ulehnutím, druhá dávka o 2,5-4 hodiny později.
Tabulka 1 Souhrn klinických studií s natrium-oxybutyrátem v léčbě narkolepsie
|
Studie |
Primární parametry účinnosti |
N |
Sekundární parametry účinnosti |
Trvání |
Aktivní léčba a dávka (g/d) |
|
Studie 1 |
EDS (ESS); CGIc |
246 |
MWT/spánková architektura/ kataplexie/Naps/FOSQ |
8 týdnů |
Xyrem 4,5-9 |
|
Studie 2 |
EDS (MWT) |
231 |
Spánková architektura/ ESS/CGIc/Naps |
8 týdnů |
Xyrem 6-9 Modafinil 200-600 mg |
|
Studie 3 |
Kataplexie |
136 |
EDS (ESS)/CGIc/Naps |
4 týdny |
Xyrem 3-9 |
|
Studie 4 |
Kataplexie |
55 |
žádné |
4 týdny |
Xyrem 3-9 |
EDS - excesivní denní spavost; ESS - Epworthská spánková škála; MWT - Test udržení bdělosti; Naps - počet nechtěných denních usnutí; CGIc - Celkový klinický dojem změny; FOSQ - funkční výsledky spánkového dotazníku
Do studie 1 bylo zařazeno 246 pacientů s narkolepsií. Studie 1 obsahovala periodu titrace v trvání 1 týdne. Primárním parametrem účinnosti byly změny v nadměrné denní spavosti měřené Epworthskou spánkovou škálou a změna celkové tíže příznaků narkolepsie hodnocená zkoušejícím pomocí škály Celkového klinického dojmu změny (CGI-c).
Tabulka 2 Souhrn výsledků ESS ve studii 1
Epworthská spánková škála (ESS; rozsah 0-24)
|
Skupina s dávkou [g/d (n)] |
Počáteční stav |
Cílový parametr |
Medián změny od počátečního stavu |
Změna proti počátečnímu stavu v porovnání s placebem (p-hodnota) |
|
Placebo (60) |
17,3 |
16,7 |
-0,5 |
- |
|
4,5 (68) |
17,5 |
15,7 |
-1,0 |
0,119 |
|
6 (63) |
17,9 |
15,3 |
-2,0 |
0,001 |
|
9 (55) |
17,9 |
13,1 |
-2,0 |
<0,001 |
|
Celkový klinický dojem změny (CGI-c) | ||
|
Skupina s dávkou [g/d (n)] |
Respondéři* N (%) |
Změna proti počátečnímu stavu v porovnání s placebem (p-hodnota) |
|
Placebo (60) |
13 (21,7) |
- |
|
4,5 (68) |
32 (47,1) |
0,002 |
|
6 (63) |
30 (47,6) |
<0,001 |
|
9 (55) |
30 (54,4) |
<0,001 |
* Data CGI-c byla analyzována s definováním respondérů jako pacientů, kteří vykázali velmi výrazné nebo výrazné zlepšení.
Studie 2 porovnávala účinek perorálně užívaného natrium-oxybutyrátu, modafinilu a natrium-oxybutyrátu + modafinilu s placebem v léčbě denní ospalosti u narkolepsie. Během 8 týdnů trvající dvojitě zaslepené periody užívali pacienti modafinil v jejich obvyklé dávce nebo placebo. Natrium-oxybutyrát nebo placebo užívali v dávce 6 g/den první 4 týdny a v dávce 9 g/den po zbylé 4 týdny. Primárním parametrem účinnosti byla excesivní denní spavost měřená objektivní odpovědí v MWT.
Tabulka 4 Souhrn výsledků MWT ve studii 2
|
STUDIE 2 | ||||
|
Skupina s dávkou |
Počáteční stav |
Cílový parametr |
Průměrná změna proti počátečnímu stavu |
Cílový parametr v porovnání s placebem |
|
Placebo (56) |
9,9 |
6,9 |
-2,7 |
- |
|
Natrium-oxybutyrát (55) |
11,5 |
11,3 |
0,16 |
<0,001 |
|
Modafinil (63) |
10,5 |
9,8 |
-0,6 |
0,004 |
|
Natrium-oxybutyrát + Modafinil (57) |
10,4 |
12,7 |
2,3 |
<0,001 |
Do studie 3 bylo zařazeno 136 pacientů s narkolepsií se střední až těžkou kataplexií (medián 21 kataplektických záchvatů za týden) v počátečním stavu. Primárním parametrem účinnosti byla frekvence záchvatů kataplexie.
Tabulka 5 Souhrn výsledků ve studii 3
|
Dávka |
Počet subjektů |
Záchvaty kataplexie | ||
|
Studie 3 |
Počáteční stav |
Medián změny od počátečního stavu |
Změna proti počátečnímu stavu v porovnání s placebem (p-hodnota) | |
|
Medián záchvatů/týden | ||||
|
Placebo |
33 |
20,5 |
-4 |
- |
|
3,0 g/den |
33 |
20,0 |
-7 |
0,5235 |
|
6,0 g/den |
31 |
23,0 |
-10 |
0,0529 |
|
9,0 g/den |
33 |
23,5 |
-16 |
0,0008 |
Do studie 4 bylo zařazeno 55 pacientů s narkolepsií, kteří užívali natrium-oxybutyrát v otevřeném uspořádání po dobu 7-44 měsíců. Pacienti byli randomizováni k pokračování v léčbě v jejich obvyklé dávce nebo k podávání placeba. Studie 4 byla uspořádána specificky
k zhodnocení kontinuální účinnosti natrium-oxybutyrátu po dlouhodobé léčbě. Primárním parametrem účinnosti byla frekvence záchvatů kataplexie.
Tabulka 6 Souhrn výsledků ve studii 4
|
Skupina |
Počet subjektů |
Záchvaty kataplexie | ||
|
Studie 4 |
Počáteční stav |
Medián změny od počátečního stavu |
Změna proti počátečnímu stavu v porovnání s placebem (p-hodnota) | |
|
Medián záchvatů/2 týdny | ||||
|
Placebo |
29 |
4,0 |
21,0 |
- |
|
Natrium-oxybutyrát |
26 |
1,9 |
0 |
p <0,001 |
Ve studii 4 byla odpověď numericky podobná u pacientů léčených dávkou 6-9 g/den, ale nebyl pozorován žádný účinek u dávek nižších než 6 g/den.
5.2 Farmakokinetické vlastnosti
Natrium-oxybutyrát se po perorálním podání rychle a téměř úplně absorbuje. Absorpce je prodloužena a snížena po velmi tučném jídle. Vylučuje se hlavně metabolismem s poločasem 0,5-1 hodina.
Farmakokinetika je nelineární s plochou pod křivkou plazmatických koncentrací (AUC) v porovnání s časovou křivkou, která se zvýší 3,8x při zdvojnásobení dávky přípravku ze 4,5 g na 9 g. Farmakokinetika se nemění po opakovaných dávkách přípravku.
Absorpce
Natrium-oxybutyrát se po perorálním podání rychle absorbuje s absolutní biologickou dostupností přibližně 88 %. Průměrné vrcholové plazmatické koncentrace (1. a 2. vrchol) po podání denní dávky 9 g rozdělené do dvou stejných dávek podaných v rozmezí 4 hodin, byly 78, resp. 142 pg/ml. Průměrná doba k dosažení vrcholové plazmatické koncentrace (Tmax) se v osmi farmakokinetických studiích pohybovala od 0,5 do 2 hodin. Po perorálním podání látky stoupají plazmatické hladiny s její rostoucí dávkou více než proporcionálně. Jednotlivé dávky nad 4,5 g nebyly studovány. Podání natrium-oxybutyrátu bezprostředně po jídle s vysokým obsahem tuků vedlo k opožděné absorpci (průměrná Tmax stoupla z 0,75 hodiny na 2,0 hodiny) a ke snížení vrcholové plazmatické hladiny (Cmax) průměrně o 58 % a systémové expozice (AUC) o 37 %.
Distribuce
Natrium-oxybutyrát je hydrofilní sloučenina se zdánlivým distribučním objemem v průměru 190-384 ml/kg. Při koncentracích v rozmezí 3-300 pg/ml se méně než 1 % látky váže na plazmatické bílkoviny.
Biotransformace
Studie na zvířatech ukazují, že metabolismus je hlavním způsobem eliminace látky. Látka se metabolizuje na oxid uhličitý a vodu cyklem trikarboxylových kyselin (Krebsův cyklus) a sekundárně P-oxidací. Cytosolový enzym spojený s NADP+, GHB dehydrogenáza, katalyzuje primárně přeměnu výchozího substrátu na sukcinát semialdehyd, který je pak biotransformací enzymem sukcinit semialdehyd dehydrogenázou přeměněn na kyselinu jantarovou. Ta vstupuje do Krebsova cyklu a je metabolizována na oxid uhličitý a vodu. Druhý mitochondriální oxidoredukční enzym transhydrogenáza také katalyzuje v přítomnosti a-ketoglutarátu přeměnu natrium-oxybutyrátu na sukcinát semialdehyd. Alternativní biotransformací je P-oxidace přes 3,4-dihydroxybutyrát na acetyl-CoA, který se také následně přeměňuje v cyklu kyseliny citrónové na oxid uhličitý a vodu. Aktivní metabolit natrium-oxybutyrátu nebyl zjištěn.
Studie in vitro s lidskými jatemími mikrozomy prokázaly, že natrium-oxybutyrát neinhibuje významně až do koncentrace 3 mM (378 pg/ml) aktivitu lidských izoenzymů CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 a CYP3A. Uvedené hladiny byly výrazně vyšší než hladiny dosažené dávkami terapeutickými.
Eliminace
Odstranění natrium-oxybutyrátu z organismu probíhá z velké části biotransformací na oxid uhličitý, který je pak eliminován dýcháním. Méně než 5 % nezměněného přípravku se vylučuje močí do 6-8 hodin po podání. Vylučování stolicí je zanedbatelné.
Starší pacienti
U omezeného počtu pacientů starších než 65 let nebyl rozdíl ve farmakokinetice natrium-oxybutyrátu ve srovnání s pacienty mladšími než 65 let.
Pediatrická populace
Farmakokinetika natrium-oxybutyrátu u pediatrických pacientů do 18 let nebyla studována. Porucha funkce ledvin
Ledviny nehrají významnou úlohu ve vylučování látky, proto nebyla provedena farmakokinetická studie u pacientů s poruchou funkce ledvin. Možnost ovlivnění farmakokinetiky natrium-oxybutyrátu u pacientů s poruchou funkce ledvin se nepředpokládá.
Porucha funkce jater
Natrium-oxybutyrát se výrazně metabolizuje během prvního průchodu játry. Po jednotlivé perorální dávce 25 mg/kg byly hodnoty AUC u pacientů s cirhózou dvojnásobné, zdánlivá perorální clearance se snížila z 9,1 ml/min/kg u zdravých dospělých osob na 4,5 ml/min/kg u pacientů ve třídě A (bez ascitu) a na 4,1 ml/min/kg u pacientů třídy C (s ascitem). Eliminační poločas byl u pacientů ve třídě C a třídě A významně delší než u osob v kontrolní skupině (průměrný tJ/2 59 minut u pacientů ve třídě C a 32 minut u pacientů ve třídě A proti 22 minutám u osob na placebu). U pacientů s poruchou jaterní funkce má být zahajovací dávka přípravku snížena na polovinu a odpověď na zvýšení dávky je třeba pečlivě monitorovat (viz bod 4.2).
Etnický původ
Vliv etnického původu na metabolismus natrium-oxybutyrátu nebyl zkoumán.
5.3 Předklinické údaje vztahující se k bezpečnosti
Opakované podávání natrium-oxybutyrátu potkanům (90 dnů a 26 týdnů) a psům (52 týdnů) nevedlo k žádnému významnému nálezu při biochemickém vyšetření, makroskopické i mikroskopické patologii. Klinické příznaky vztahující se k podávání přípravku souvisely se sedací, sníženou spotřebou potravy a druhotnými změnami tělesné hmotnosti, nárůstem tělesné hmotnosti a hmotnosti orgánů. Expozice psů a potkanů k dosažení NOEL byla nižší (~50%) než u lidí. Natrium-oxybutyrát nebyl v in vivo a in vitro testech mutagenní ani klastogenní.
Gamabutyrolakton (GBL), prekurzor látky GHB, byl testován v dávkách 1,21-1,64krát vyšších než dávky terapeutické a byl klasifikován jako nekancerogenní u potkanů a neprůkazně kancerogenní u myší (došlo k mírnému zvýšení počtu feochromocytomů). Tento výsledek byl vzhledem k vysoké mortalitě skupiny s vysokou dávkou GBL obtížně interpretovatelný. Ve studii kancerogenity u potkanů byly také identifikovány nádory bez vztahu k podané látce.
Látka GHB nevykazovala žádný účinek na páření, obecnou fertilitu nebo parametry spermatu a nepůsobila ani embryofetální toxicitu u potkanů exponovaných dávkám do 1000 mg/kg/den (1,64x vyšší expozice v porovnání s člověkem stanovená výpočtem pro zvířata, která nejsou březí). Během laktace byla také po této vysoké dávce zvýšena u F1 zvířat perinatální mortalita a snížena průměrná hmotnost mláďat. Souvislost těchto vývojových defektů s maternální toxicitou nemohla být stanovena. U králíků byla pozorována mírná fetotoxicita.
Lékové diskriminační studie ukázaly, že GHB produkuje specifický diskriminační stimul, který je v některých ohledech podobný alkoholu, morfinu a některým GABA agonistům. Studie u potkanů, myší a opic, ve kterých si zvířata sama mohla podávat přípravek, vedly k rozporuplným výsledkům. U hlodavců však byla jasně prokázána tolerance k GHB i zkřížená tolerance mezi GHB a alkoholem a baklofenem.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Čištěná voda
Kyselina jablečná (k úpravě pH)
Hydroxid sodný (k úpravě pH)
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti 5 let
Po prvním otevření: 40 dní.
Po naředění v dávkovacích nádobkách by se měl přípravek použít do 24 hodin.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3. Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a velikost balení a zvláštní vybavení pro použití
180 ml roztoku v oválné 240 ml PET lahvi jantarově hnědé barvy s plastovou fólií a dětským bezpečnostním uzávěrem z HDPE/polypropylenu a s dřevolepenkovou vnitřní vložkou.
Papírová krabička obsahuje jednu lahev, zatlačovací adaptér lahve sestávající z LDPE vložky, ventilu z Silastic Biomedical ETR elastomeur, zpětného ventilu z akrylonitryl butadien styrenového terpolymeru a z LDPE trubičky, kalibrovanou odměrku (polypropylenová stříkačka) a dvě polypropylenové dávkovací nádobky se dvěma HDPE závitovými dětskými bezpečnostními uzávěry.
6.6 Zvláštní požadavky pro likvidaci přípravku
Žádné zvláštní požadavky
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
UCB Pharma Ltd 208 Bath Road Slough Berkshire SL1 3WE Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/312/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. října 2005
Datum posledního prodloužení registrace: 8. září 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
UCB Pharma Ltd 208 Bath Road Slough
Berkshire SL1 3WE Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci zavede edukační program pro Xyrem k zajištění toho, že lékaři, u kterých se předpokládá, že budou předepisovat Xyrem, budou seznámeni s dávkováním Xyremu a důležitými riziky při jeho užívání. Edukační program obsahuje 4 následující položky:
• Kontrolní seznam pro zdravotnické pracovníky (např. formulář k zahájení léčby): pro připomenutí lékařům kontrolovat kontraindikace, upozornění a opatření v SmPC, se zvláštním vyznačením, že Xyrem může navodit respirační depresi a depresi CNS, že alkohol může zesilovat depresi CNS a že Xyrem má potenciál k abúzu.
• Často kladené otázky, informační brožura pro pacienta (určena pro předání pacientovi): poskytne pacientovi odpovědi na některé otázky, které mohou mít pacienti ohledně užívání Xyremu.
• Příručka pacienta (určena pro předání pacientovi): poskytne pacientovi informace ohledně užívání Xyremu.
• Kartička pro pacienta (určena pro předání pacientovi): k připomenutí pacientům, lékařům a/nebo farmaceutům důležité bezpečnostní informace při užívání Xyremu
Držitel rozhodnutí o registraci zavede kontrolovaný distribuční program, který zvýší existující kontroly pro Xyrem a umožní dosažení zamýšlené populace pacientů s narkolepsií a zároveň minimalizuje riziko Xyremu vyřazením těch, kteří by ho chtěli zneužít.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU A VNITRNÍM OBALU Krabička a lahev
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xyrem 500 mg/ml perorální roztok natrii oxybutyras
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml roztoku obsahuje natrii oxybutyras 500 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 lahev se 180 ml perorálního roztoku
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Tento léčivý přípravek by měl být spotřebován do 40 dní po prvním otevření. Po naředění v dávkovacích nádobkách by se měl přípravek použít do 24 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma Ltd 208 Bath Road Slough Berkshire SL1 3WE Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/312/001
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Xyrem 500 mg/ml (po uze krabička)
B. PŘÍBALOVÁ INFORMACE
Xyrem 500 mg/ml perorální roztok
natrii oxybutyras
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Xyrem a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Xyrem užívat
3. Jak se Xyrem užívá
4. Možné nežádoucí účinky
5. Jak Xyrem uchovávat
6. Obsah balení a další informace
1. Co je Xyrem a k čemu se používá
Xyrem obsahuje léčivou látku natrium-oxybutyrát. Xyrem účinkuje tak, že upravuje noční spánek, i když přesný mechanismus účinku není znám.
Xyrem se používá k léčbě narkolepsie s kataplexií u dospělých.
Narkolepsie je spánková porucha, při které se mohou vyskytovat záchvaty spánku během normálních hodin bdění a dále kataplexie, spánková obrna, halucinace a špatný spánek. Kataplexie je nástup náhlé svalové slabosti nebo ochrnutí bez ztráty vědomí jako odpověď na náhlou emoční reakci, jako je zlost, strach, štěstí, smích nebo překvapení.
2. Čemu musíte věnovat pozornost, než začnete Xyrem užívat
Neužívejte Xyrem:
- jestliže jste alergický(á) na natrium-oxybutyrát nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže máte nedostatek sukcinát semialdehyd dehydrogenázy (vzácná metabolická porucha)
- jestliže trpíte těžkou depresí
- jestliže jste léčen(a) opiáty nebo barbituráty.
Upozornění a opatření
Před užitím Xyremu se poraďte se svým lékařem nebo lékárníkem.
- jestliže máte dechové nebo plicní problémy (zvláště jste-li obézní), protože Xyrem může způsobit dýchací problémy
- jestliže máte nebo jste v minulosti měl(a) depresi
- jestliže máte srdeční selhání, hypertenzi (vysoký krevní tlak), problémy s játry nebo ledvinami, může být zapotřebí dávku Xyremu upravit
- jestliže jste v minulosti zneužíval(a) léky
- jestliže máte epilepsii, protože užívání Xyremu se v tomto případě nedoporučuje
- jestliže máte porfyrii (málo častá metabolická porucha).
Pokud se Vás něco z toho týká, oznamte to svému lékaři ještě předtím, než začnete užívat Xyrem.
Pokud při užívání Xyremu zpozorujete, že máte vlhké lůžko a inkontinenci (neudržíte moč nebo stolici), jste zmatený(á), máte halucinace, jste náměsíčný(á) nebo máte abnormální myšlení, sdělte to ihned svému lékaři. I když tyto nežádoucí účinky jsou málo časté, pokud se vyskytnou, jsou obvykle mírné až středně závažné.
Jestliže jste starší, lékař bude pečlivě sledovat Váš stav a kontrolovat, zda má Xyrem požadované účinky.
Xyrem má dobře známý potenciál pro zneužívání. Případy závislosti se vyskytly po nedovoleném užívání natrium-oxybutyrátu.
Lékař se Vás bude před začátkem užívání Xyremu ptát, zda u Vás někdy došlo ke zneužívání léků. Rovněž se Vás bude ptát v průběhu užívání.
Děti a dospívající
Nepodávejte tento lék dětem a dospívajícím.
Další léčivé přípravky a Xyrem
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Xyrem zvláště nesmí být užíván společně s léky, které navozují spánek a které mají tlumivý účinek na centrální nervový systém (pojem centrální nervový systém zahrnuje mozek a míchu).
Informujte také svého lékaře nebo lékárníka, pokud užíváte některý z následujících léků:
• léky, které zvyšují aktivitu centrálního nervového systému, a antidepresiva
• léky, které mohou být v těle podobně metabolizovány (např. valproát, fenytoin nebo ethosuximid, které se používají k léčbě záchvatů)
• topiramát (používaný k léčbě epilepsie)
• Jestliže užíváte valproát, bude třeba upravit Vaši denní dávka Xyremu (viz bod 3), protože to může vést k interakcím.
Xyrem s jídlem, pitím a alkoholem
Při užívání Xyremu nesmíte pít alkohol, protože jeho účinky mohou být zvýšeny.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Existuje velmi málo žen, které užívaly Xyrem nějakou dobu během svého těhotenství, a několik z nich spontánně potratilo. Riziko užívání Xyremu v těhotenství není známo, a proto se užívání Xyremu u těhotných žen nebo u žen, které plánují otěhotnět, nedoporučuje.
Není známo, zda se Xyrem vylučuje do mateřského mléka. Pacientky užívající Xyrem mají přerušit kojení.
Řízení dopravních prostředků a obsluha strojů
Xyrem může narušit schopnost řídit nebo obsluhovat stroje a zařízení. Po dobu nejméně 6 hodin po požití Xyremu neřiďte dopravní prostředky, neobsluhujte žádné přístroje nebo stroje ani neprovádějte žádné činnosti, které jsou nebezpečné nebo vyžadují duševní bdělost. Když začnete poprvé užívat Xyrem, buďte extrémně opatrný(á) při řízení vozidla, obsluze strojů či vykonávání jakékoli činnosti, která může být nebezpečná nebo vyžaduje plnou duševní bdělost, dokud nebudete vědět, zda nejste následující den ospalý(á).
Xyrem obsahuje sodík
Měl(a) byste sledovat množství soli, které přijímáte, protože Xyrem obsahuje sodík (ten se nachází v kuchyňské soli). To může ovlivnit Váš zdravotní stav, pokud jste měl(a) v minulosti problémy s vysokým krevním tlakem, se srdcem nebo ledvinami. Pokud užíváte dvě dávky natrium-oxybutyrátu po 2,25 g každou noc, přijímáte zároveň 0,82 g sodíku. Pokud užíváte dvě dávky natrium-oxybutyrátu po 4,5 g každou noc, přijímáte zároveň 1,6 g sodíku. Může být tedy třeba snížit množství soli, které přijímáte.
3. Jak se Xyrem užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená zahajovací dávka je 4,5 g denně, rozdělená do dvou stejných dávek po 2,25 g. Lékař Vám může postupně zvyšovat dávku až na 9 g denně, rozdělené do dvou stejných dávek po 4,5 g.
Užívejte Xyrem perorálně (ústy) dvakrát každou noc. První dávku si vezměte po ulehnutí do postele a druhou dávku za 2,5-4 hodiny. Možná bude nutné, abyste si nařídil(a) budík, aby bylo jisté, že se vzbudíte na druhou dávku. Jídlo snižuje množství Xyremu, které se vstřebá do organismu. Proto je nejlepší si vzít Xyrem v určenou dobu dvě až tři hodiny po jídle. Obě dávky si připravte před spaním. Obě dávky užijte do 24 hodin po přípravě.
Pokud užíváte valproát společně s Xyremem, lékař upraví dávku Xyremu. Doporučená zahajovací dávka Xyremu, pokud užíváte valproát, je 3,6 g denně, rozdělená do dvou stejných dávek po 1,8 g. První dávku si vezměte po ulehnutí do postele a druhou dávku za 2,5-4 hodiny.
Jestliže máte problém s ledvinami, měl(a) byste zvážit dietní opatření, abyste snížil(a) příjem sodíku.
Jestliže máte problém s játry, zahajovací dávka Vám bude snížena na polovinu. Lékař Vám může pak dávku postupně zvyšovat.
Návod na ředění Xyremu
V níže uvedených pokynech je vysvětleno, jak se Xyrem připravuje. Přečtěte si prosím tyto pokyny pečlivě a postupujte podle nich krok za krokem.
Pro Vaši informaci, krabička s Xyremem obsahuje 1 lahev s přípravkem, odměrnou stříkačku a dvě dávkovací nádobky s dětským bezpečnostním uzávěrem.
1. Odstraňte uzávěr lahve zatlačením a otáčením uzávěru proti směru hodinových ručiček (doleva). Po odstranění uzávěru postavte lahev na desku stolu. Hrdlo lahve je pokryto těsnící plastovou fólií, která se musí před prvním použitím odstranit. Držte lahev ve svislé poloze a zasuňte zatlačovací adaptér lahve do jejího hrdla. To je zapotřebí udělat pouze při prvním otevření lahve. Adaptér se pak ponechá v lahvi pro všechna další použití.
2. Zasuňte hrot odměrné stříkačky do středu otvoru lahve a pevně ji zatlačte (viz obrázek 1).
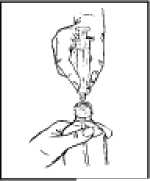
Figure 1
3. Držte lahev a stříkačku jednou rukou, natáhněte předepsanou dávku druhou rukou
tažením za píst. POZNÁMKA: Lék nepoteče do stříkačky, pokud nebudete držet lahev rovně ve svislé poloze (viz obrázek 2).
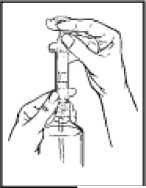
Figure 2
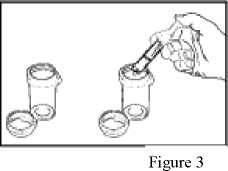
4. Vyjměte stříkačku ze středu otvoru lahve. Stlačením pístu stříkačky vyprázdněte lék do jedné dávkovači nádobky (viz obrázek 3). Zopakujte tento postup pro druhou dávkovači nádobku. Poté přidejte do každé dávkovací nádobky 60 ml vody (60 ml jsou asi 4 polévkové lžíce).
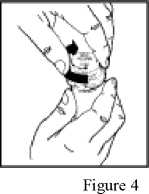
5. Dejte uzávěry na dávkovací nádobky a otočte ve směru hodinových ručiček (doprava), až uslyšíte zaklapnuti a uzávěr se zajistí v poloze chránící před otevřením dětmi (viz obrázek 4). Stříkačku vypláchněte vodou.
6. Těsně před tím, než půjdete spát, si dejte druhou dávku k posteli. Měl(a) byste si nařídit budík, abyste se vzbudil(a) pro druhou dávku nejdříve za 2,5 hodiny a nejpozději za
4 hodiny po první dávce. Odstraňte uzávěr z první dávkovací nádobky zatlačením na dětský bezpečnostní uzávěr a otočte uzávěr proti směru hodinových ručiček (doleva) Vsedě na posteli vypijte celou první dávku, poté zavřete víčko na nádobce a lehněte si do postele.
7. Když se za 2,5 až 4 hodiny probudíte, odstraňte uzávěr z druhé dávkovači nádobky. Vsedě na posteli vypijte čelou druhou dávku těsně předtím, než si lehnete do postele, a budete pokračovat ve spánku. Zavřete víčko na druhé nádobce.
Pokud máte pocit, že účinek Xyremu je příliš silný nebo příliš slabý, řekněte to svému lékaři nebo lékárníkovi.
Jestliže jste užil(a) více Xyremu, než jste měl(a)
K příznakům předávkování Xyremu patří neklid, zmatenost, zhoršení pohybů, zhoršení dýchání, rozmazané vidění, nadměrné pocení, bolest hlavy, zvracení, snížené vědomí, které vede až ke kómatu, a záchvaty. Jestliže užijete více Xyremu, než Vám bylo předepsáno, nebo ho užijete omylem, vyhledejte ihned lékařskou pohotovostní službu. Vezměte si s sebou označenou lahev s lékem, i když je prázdná.
Jestliže jste zapomněl(a) užít Xyrem
Jestliže jste zapomněl(a) si vzít první dávku, vezměte si ji ihned, jakmile si vzpomenete, a pak pokračujte v užívání léku jako dříve. Pokud si zapomenete vzít druhou dávku, vynechte tuto dávku a neužívejte Xyrem až do dalšího večera. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat Xyrem
Xyrem užívejte tak dlouho, jak jste byl(a) poučen(a) svým lékařem. Jestliže je léčba Xyremem zastavena, mohou se vrátit Vaše záchvaty kataplexie a může se vyskytnout nespavost, bolest hlavy, úzkost, závratě, problémy se spánkem, spavost, halucinace a abnormální myšlení.
Pokud přerušíte užívání Xyremu na více než 14 po sobě jdoucích dní, měl(a) byste se poradit s lékařem, protože byste měl(a) znovu začít užívat Xyrem ve snížené dávce.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Nežádoucí účinky jsou obvykle mírné až středně závažné. Pokud se některé z nežádoucích účinků vyskytnou, řekněte to ihned svému lékaři.
Velmi časté (mohou postihnout více než 1 pacienta z 10):
Nevolnost, závratě, bolest hlavy.
Časté (mohou postihnout až 1 z 10pacientů):
Poruchy spánku včetně nespavosti, rozmazané vidění, pocit bušení srdce, zvracení, bolest břicha, průjem, nechutenství, snížená chuť k jídlu, snížení tělesné hmotnosti, slabost, abnormální sny, únava, pocit opilosti, spánková obrna, ospalost, třes, zmatenost či dezorientace, noční můry, náměsíčnost, pomočování se v posteli, pocení, deprese, svalové křeče, otoky, pády, bolesti kloubů, bolest zad, nadměrná spavost během dne, poruchy rovnováhy, poruchy pozornosti, poruchy citlivosti zejména na doteky, abnormální pocity doteku, zklidnění, abnormální chuť, úzkost, poruchy usínání v průběhu noci, nervozita, pocit „točení se“ (závrať), neschopnost udržet moč, dušnost, chrápání, překrvení nosní sliznice (ucpání nosu), vyrážka, zánět vedlejších nosních dutin, zánět nosu a hrdla, zvýšený krevní tlak.
Méně časté (mohou postihnout až 1 pacienta ze 100):
Psychóza (duševní porucha, která se může projevit halucinacemi, nesouvislou řečí, zmateným a neklidným chováním), paranoia (nadměrná vztahovačnost), abnormální myšlení, halucinace, neklid, sebevražedný pokus, potíže s usínáním, neklid nohou, zapomnětlivost, myoklonus (mimovolní svalové stahy), mimovolní odchod stolice, přecitlivělost.
Není známo (četnost z dostupných údajů nelze určit):
Křeče, mělké dýchání nebo nižší frekvence dýchání, kopřivka, myšlenky na sebevraždu, krátké přerušení dýchání během spánku, euforie, sucho v ústech, otok obličeje (angioedém), dehydratace, prudký záchvat paniky, mánie (nadměrná veselost a zvýšená aktivita) / bipolární porucha (střídání nadměrné veselosti a depresivní nálady) , blud, bruxismus (skřípání zubů a svírání čelistí), časté močení/nutkání k močení (zvýšená potřeba močení), ušní šelest (šum v uších, jako je zvonění nebo bzučení), poruchy příjmu potravy spojené se spánkem (noční jedlictví) a ztráta vědomí.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xyrem uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvi (EXP). Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Přípravek zředěný v dávkovacích nádobkách by měl být spotřebován během 24 hodin.
Po prvním otevření lahve Xyremu musí být po 40 dnech všechen nespotřebovaný roztok zlikvidován.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Xyrem obsahuje
- Léčivou látkou je natrii oxybutyras. Jeden ml roztoku obsahuje natrii oxybutyras 500 mg.
- Pomocnými látkami jsou čištěná voda, kyselina jablečná a hydroxid sodný.
Jak Xyrem vypadá a co obsahuje toto balení
Xyrem se dodává jako perorální roztok (roztok k vnitřnímu užití) v plastové lahvi jantarově hnědé barvy o objemu 240 ml, která obsahuje 180 ml roztoku a je uzavřena dětským bezpečnostním uzávěrem. Při dodání je pod uzávěrem na hrdle lahve plastová fólie. Balení obsahuje jednu lahev, zatlačovací adaptér lahve, plastovou odměrnou stříkačku a dvě dávkovací nádobky s dětským bezpečnostním uzávěrem.
Xyrem je čirý až lehce opalizující roztok.
Držitel rozhodnutí o registraci a výrobce
UCB Pharma Ltd, 208 Bath Road, Slough, Berkshire, SL1 3WE, Velká Británie
Od svého lékaře byste měl(a) obdržet informační balíček o Xyremu, který obsahuje následující: Příručku pacienta s informacemi, jak používat tento přípravek, Často kladené otázky, které mohou mít pacienti ohledně bezpečnosti přípravku a Kartičku pro pacienta.
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien Lietuva
UCB Pharma SA/NV UCB Pharma Oy Finland
Tel/Tél: +32 / (0)2 559 92 00 Tel: + 358 10 234 6800 (Suomija)
|
BtnrapHH ro CH EH EtnrapHH EOOfl Ten.: + 359 (0) 2 962 30 49 |
Luxembourg/Luxemburg UCB Pharma SA/NV Tél/Tel: +32 / (0)2 559 92 00 |
|
Česká republika UCB s.r.o. Tel: + 420 221 773 411 |
Magyarország UCB Magyarország Kft. Tel.: + 36-(1) 391 0060 |
|
Danmark UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
Malta Pharmasud Ltd. Tel: +356 / 21 37 64 36 |
|
Deutschland UCB Pharma GmbH Tel: + 49 /(0) 2173 48 4848 |
Nederland UCB Pharma B.V. Tel.: +31 / (0)76-573 11 40 |
|
Eesti UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Soome) |
Norge UCB Nordic A/S Tel: +45 / 32 46 24 00 |
|
EXXáSa UCB A.E. Tnk: +30 / 2109974000 |
Osterreich UCB Pharma GmbH Tel: + 43 (1) 291 80 00 |
|
Espaňa UCB Pharma, S.A. Tel: + 34 / 91 570 34 44 |
Polska UCB Pharma Sp. z o.o. Tel.: + 48 22 696 99 20 |
|
France UCB Pharma S.A. Tél: + 33 / (0)1 47 29 44 66 |
Portugal UCB Pharma (Produtos Farmaceuticos) Lda Tel: + 351 / 21 302 5300 |
|
Hrvatska Medis Adria d.o.o. Tel: +385 (0) 1 230 34 46 |
Románia UCB Pharma Romania S.R.L. Tel: +40 21 300 29 04 |
|
Ireland UCB (Pharma) Ireland Ltd. Tel: + 353 / (0)1-46 37 395 |
Slovenija Medis, d.o.o. Tel: + 386 1 589 69 00 |
|
Ísland Vistor hf. Tel: +354 535 7000 |
Slovenská republika UCB s.r.o., organizačná zložka Tel: + 421 (0) 2 5920 2020 |
|
Italia UCB Pharma S.p.A. Tel: + 39 / 02 300 791 |
Suomi/Finland UCB Pharma Oy Finland Puh/ Tel: + 358 10 234 6800 |
|
Kúrcpog Lifepharma (Z.A.M.) Ltd Tnk: + 357 22 34 74 40 |
Sverige UCB Nordic A/S Tel: + 46 / (0) 40 29 49 00 |
Latvija
United Kingdom
UCB Pharma Ltd.
Tel : +44 / (0)1753 534 655
UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Somija)
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
31