Xydalba 500 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xydalba 500 mg prášek pro koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje dalbavancini hydrochloridum v množství odpovídajícímu dalbavancinum 500 mg.
Po rekonstituci 1 ml obsahuje dalbavancinum 20 mg.
Naředěný infuzní roztok musí obsahovat dalbavancinum v koncentraci 1 až 5 mg/ml (viz bod 6.6). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok (prášek pro koncentrát). Bílý, téměř bílý až světle žlutý prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Xydalba je indikován k léčbě akutních bakteriálních infekcí kůže a kožních struktur (ABSSSI) u dospělých (viz body 4.4 a 5.1).
Je třeba dbát oficiálních doporučení pro správné používání antibakteriálních látek.
4.2 Dávkování a způsob podání
Dávkování
Doporučené dávkování a délka léčby u dospělých
Doporučená dávka dalbavancinu u dospělých pacientů trpících ABSSSI je 1 500 mg podávaných buď jako jediná infuze 1 500 mg, nebo jako 1 000 mg v prvním týdnu a 500 mg následující týden (viz body 5.1 a 5.2).
Starší pacienti
Dávkování není třeba žádným způsobem upravovat (viz bod 5.2).
Porucha funkce ledvin
U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin (clearance kreatininu > 30 až 79 ml/min) není třeba dávkování upravovat. U pacientů podstupujících pravidelnou hemodialýzu (třikrát týdně) není třeba dávkování upravovat a dalbavancin lze podávat bez ohledu na harmonogram hemodialýzy.
U pacientů s chronickou poruchou funkce ledvin a s clearance kreatininu < 30 ml/min, kteří nepodstupují hemodialýzu pravidelně, je doporučená dávka snížena buď na 1 000 mg podávaných jako jediná infuze, nebo na 750 mg první týden a 375 mg následující týden (viz bod 5.2).
Porucha funkce jater
U pacientů s lehkou poruchou funkce jater není třeba dávkování dalbavancinu upravovat (Child-Pugh skóre A). Při předepisování dalbavancinu pacientů se středně těžkou až těžkou poruchou funkce jater (Child-Pugh skóre B a C) je třeba dbát opatrnosti, protože nejsou dostupné žádné údaje pro stanovení správného dávkování (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost dalbavancinu u dětí ve věku od narození do 18 let dosud nebyla prokázána. Údaje, které jsou v současnosti dostupné, jsou popsané v bodě 5.2, avšak nelze stanovit žádné doporučení ohledně dávkování.
Způsob podání
Intravenózní podání
Přípravek Xydalba je třeba nejméně 30 minut před podáním intravenózní infuze rekonstituovat a poté naředit. Pokyny pro rekonstituci a ředění přípravku před podáváním viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivní reakce
Při podávání přípravku Xydalba pacientům, u kterých se objevila hypersenzitivita na další glykopeptidy, je třeba dbát opatrnosti, protože se může objevit zkřížená hypersenzitivita. Pokud dojde k alergické reakci na přípravek Xydalba, je třeba podávání přípravku přerušit a nasadit vhodnou léčbu alergické reakce.
Průjmová onemocnění související s infekcí Clostridium difficile
V souvislosti s použitím téměř všech antibiotik byla hlášena kolitida související s antibakteriálními látkami a pseudomembranózní kolitida, které se mohou vyskytnout v mírné až život ohrožující formě. Proto je nutné tuto diagnózu zvážit u pacientů, u kterých se během nebo po podání dalbavancinu vyskytne průjem (viz bod 4.8). Za takových okolností je třeba zvážit přerušení léčby dalbavancinem a zavedení podpůrných opatření spolu s podáním specifické léčby Clostridium difficile. Tito pacienti nesmějí být léčeni přípravky, které potlačují peristaltiku.
Reakce související s infuzí
Přípravek Xydalba se podává intravenózní infuzí trvající 30 minut, aby se minimalizovalo riziko reakcí souvisejících s podáním infuze. Rychlé intravenózní infuze glykopeptidových antibakteriálních látek mohou způsobit reakce podobné „syndromu červeného muže“, včetně zrudnutí horní poloviny těla, kopřivky, svědění kůže anebo vyrážky. Tyto reakce mohou vymizet po zastavení nebo zpomalení podávání infuze.
Porucha funkce ledvin
Informace o účinku a bezpečnosti dalbavancinu u pacientů s clearance kreatininu < 30 ml/min jsou omezené. Na základě simulací je nutné upravit dávkování u pacientů s chronickou poruchou funkce ledvin s clearance kreatininu < 30 ml/min, kteří nepodstupují pravidelně hemodialýzu (viz body 4.2 a 5.2).
Smíšené infekce
Při smíšených infekcích s podezřením na gramnegativní bakterie je třeba pacientům podávat i příslušné antibakteriální látky proti gramnegativním bakteriím (viz bod 5.1).
Rezistentní organismy
Použití antibiotik může vést k přemnožení rezistentních mikroorganismů. Pokud dojde během léčby k superinfekci, je třeba přijmout náležitá opatření.
Omezené klinické údaje
Existují omezené klinické údaje týkající se bezpečnosti a účinnosti dalbavancinu v případě podání více než dvou dávek (s časovým odstupem jednoho týdne). V rámci hlavních hodnocení bezpečnosti a účinnosti v případě ABSSSI se typy léčených infekcí omezovaly pouze na celulitidu/erysipel, abscesy a infekce ran. Nejsou žádné zkušenosti s použitím dalbavancinu při léčbě pacientů se závažnou poruchou imunity.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Z výsledků in vitro screeningové studie receptorů nevyplývá případná interakce s jinými terapeutickými cíli nebo potenciál pro klinicky relevantní farmakodynamické interakce (viz bod 5.1).
Klinické studie lékových interakcí nebyly v případě dalbavancinu provedeny.
Potenciál jiných léčivých přípravků ovlivnit farmakokinetiku dalbavancinu.
Dalbavancin není in vitro metabolizován enzymy CYP, proto není pravděpodobné, že by současné podávání induktorů nebo inhibitorů CYP ovlivnilo farmakokinetiku dalbavancinu.
Není známo, zda je dalbavancin substrát pro jaterní vychytávání a efluxní transportéry. Podávání spolu s inhibitory těchto transportérů může způsobit zvýšení expozice dalbavancinu. Příklady inhibitorů transportérů jsou posílené inhibitory proteázy, verapamil, chinidin, itrakonazol, klarithromycin a cyklosporin.
Potenciál dalbavancinu ovlivnit farmakokinetiku jiných léčivých přípravků.
Předpokládá se, že potenciál interakce dalbavancinu s léčivými přípravky odbourávanými enzymy CYP je nízký, protože tato látka není ani inhibitorem ani induktorem enzymů CYP in vitro. Neexistují žádná data ukazující na to, že by dalbavancin byl inhibitorem CYP2C8.
Není známo, zda je dalbavancin inhibitorem transportérů. Při kombinaci s dalbavancinem nelze vyloučit zvýšenou expozici substrátům transportérů citlivých na inhibovanou aktivitu transportéru, jako jsou statiny a digoxin.
4.6 Fertilita, těhotenství a kojení
0 podávání dalbavancinu v těhotenství nejsou k dispozici žádné údaje. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Kromě nejnutnějších případů se nedoporučuje podávat přípravek Xydalba v těhotenství.
Kojení
Není známo, zda je dalbavancin vylučován do mateřského mléka. Dalbavancin je však vylučován do mléka laktujících potkanů a je pravděpodobné, že bude při kojení vylučován i do mateřského mléka. Dalbavancin se špatně vstřebává perorálně, avšak nelze vyloučit dopad na gastrointestinální flóru nebo ústní flóru kojence. Je třeba rozhodnout, zda pokračovat v kojení nebo zda ukončit léčbu Xydalbou při zvážení přínosů kojení pro dítě a přínosů léčby pro ženu.
Fertilita
Studie na zvířatech ukázaly sníženou fertilitu (viz bod 5.3). Potenciální riziko pro lidi není známo.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xydalba má mírný vliv na schopnost řídit nebo obsluhovat stroje, protože u malého počtu pacientů se vyskytly závratě (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Ve fázi II a III klinických studií byl dalbavancin podáván 2 473 pacientům buď jako jediná dávka
1 500 mg, nebo 1 000 mg s následným podáním 500 mg o týden později. Nejčastějšími nežádoucími účinky u > 1 % pacientů léčených dalbavancinem byla nauzea (2,4 %), průjem (1,9 %) a bolest hlavy (1,3 %) a tyto příznaky byly většinou mírné nebo středně závažné.
Tabulkový seznam nežádoucích účinků (Tabulka 1)
Ve fázi II a III klinických studií dalbavancinu byly zjištěny následující nežádoucí účinky. Nežádoucí účinky jsou rozděleny podle systémové třídy orgánů a četnosti. Skupiny frekvencí jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000).
Tabulka 1
|
Třída orgánového systému |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
vulvovaginální mykotická infekce, infekce močových cest, mykotická infekce, kolitida způsobená Clostridium difficile, orální kandidóza | ||
|
Poruchy krevního oběhu a lymfatického systému |
anémie, trombocytóza, eosinofilie, leukopenie, neutropenie | ||
|
Poruchy imunitního systému |
anafylaktoidní reakce | ||
|
Poruchy metabolismu a výživy |
snížená chuť k jídlu | ||
|
Psychiatrické poruchy |
insomnie | ||
|
Poruchy nervové soustavy |
dysguezie, závratě | ||
|
Cévní poruchy |
zarudnutí, flebitida |
|
Třída orgánového systému |
Časté |
Méně časté |
Vzácné |
|
Respirační, hrudní a mediastinální poruchy |
bronchospasmus | ||
|
Gastrointestinální poruchy |
zácpa, bolest břicha, dyspepsie, břišní diskomfort, zvracení | ||
|
Poruchy kůže a podkožní tkáně | |||
|
Poruchy reprodukčního systému a prsu |
vulvovaginální pruritus | ||
|
Celkové poruchy a reakce v místě aplikace |
reakce související s infuzí | ||
|
Vyšetření |
zvýšená krevní hladina laktátdehydrogenázy, zvýšená hladina alaninaminotransferázy, zvýšená hladina aspartátaminotransferázy, zvýšené hodnoty kyseliny močové v krvi, abnormální hodnoty jaterních testů, zvýšené transaminázy, zvýšená krevní hladina alkalické fosfatázy, zvýšený počet trombocytů, zvýšená teplota, zvýšené jaterní enzymy, zvýšená hladina gamma-glutamyltransferázy |
Popis vybraných nežádoucích účinků
Skupinové nežádoucí účinky
S používáním glykopeptidů (vankomycinu a teikoplaninu) je spojována ototoxicita, u pacientů s konkomitantní léčbou s ototoxickou látkou, jako je např. aminoglykosid, může existovat zvýšené riziko.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Žádné konkrétní informace o léčbě předávkování dalbavancinem nejsou známy, protože při klinických studiích nebyla pozorována toxicita limitující dávkování. Ve studiích fáze I byly zdravým dobrovolníkům podávány jednotlivé dávky až 1 500 mg akumulativní dávky ve výši až 4 500 mg po dobu až osmi týdnů bez známek toxicity nebo klinicky významných laboratorních výsledků.
Ve studiích fáze III byly pacientům podávány jednotlivé dávky až 1 500 mg.
Léčba předávkování dalbavancinem má spočívat v pozorování a obecných podpůrných opatřeních. Přestože nejsou k dispozici informace týkající se konkrétně použití hemodialýzy k léčbě předávkování, je třeba poznamenat, že ve studii fáze I prováděné s pacienty s poruchou funkce ledvin bylo po třech hodinách hemodialýzy odstraněno méně než 6 % doporučené dávky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antibakteriální léčiva pro systémovou aplikaci, glykopeptidová antibiotika, AtC kód: J01XA04.
Mechanismus účinku
Dalbavancin je baktericidní lipoglykopeptid.
Jeho mechanismus účinku u citlivých kmenů grampozitivních bakterií spočívá v přerušení syntézy buněčné stěny navázáním se na terminální D-alanyl-D-alanin kmenového peptidu v peptidoglykanu stěny vznikající buňky, čímž se zamezí vzniku křížové vazby (transpeptidaci a transglykosylaci) podjednotek disacharidů a dojde k odumření bakteriální buňky.
Mechanismus rezistence
Všechny gramnegativní bakterie jsou vůči dalbavancinu přirozeně rezistentní.
Rezistence na dalbavancin v případě bakterií Staphylococcus spp. a Enterococcus spp. je umožněna rezistencí VanA, genotypem, u kterého dochází k modifikaci cílového peptidu ve stěně vznikající buňky. Na základě studií in vitro není aktivita dalbavancinu ovlivněna jinými třídami genů rezistence na vankomycin.
MIC dalbavancinu jsou vyšší u stafylokoků intermediárně rezistentních na vankomycin (VISA) než u kmenů plně citlivých na vankomycin. Vzhledem k tomu, že izoláty s vyššími MIC dalbavancinu představují stabilní fenotypy a současně vykazují rezistenci na ostatní glykopeptidy, spočívá pravděpodobný mechanismus ve zvýšení počtu glykopeptidových cílů ve vznikajícím peptidoglykanu.
Zkřížená rezistence mezi dalbavancinem a jinými třídami antibiotik nebyla ve studiích in vitro prokázána. Rezistence na methicilin nemá na působení dalbavancinu žádný vliv.
Interakce s jinými antibakteriálními látkami
Při studiích in vitro, které byly provedeny na 12 druzích gramnegativních patogenů (viz bod 4.5), nebyl zjištěn antagonismus mezi dalbavancinem a jinými běžně používanými antibiotiky (tj. cefepimem, ceftazidimem, ceftriaxonem, imipenemem, meropenemem, amikacinem, aztreonamem, ciprofloxacinem, piperacilinem/tazobaktamem a trimethoprimem/sulfamethoxazolem).
Hraniční hodnoty testování citlivosti
Níže jsou uvedeny hraniční hodnoty minimální inhibiční koncentrace (MIC) stanovené Evropským výborem pro testování antimikrobiální citlivosti (EUCAST):
• Staphylococcus spp.: citlivý < 0,125 mg/l; rezistentní > 0,125 mg/l,
• Beta-hemolytické streptokoky skupin A, B, C, G: citlivý < 0,125 mg/l; rezistentní > 0,125 mg/l,
• Viridující streptokoky (pouze skupina Streptococcus anginosus): citlivý < 0,125 mg/l; rezistentní > 0,125 mg/l.
Vztah PK/PD
Baktericidní aktivita vůči stafylokokům v podmínkách in vitro je závislá na čase při koncentracích dalbavancinu v séru podobných těm, jichž se dosahuje při podávání doporučených dávek u lidí.
V podmínkách in vivo byl zkoumán vztah PK/PD dalbavancinu u bakterií S. aureus pomocí neutropenického modelu zvířecí infekce. Tento model prokázal, že nejvyššího čistého snížení počtu (logio) kolonie tvořících jednotek (CFU) bylo dosaženo, když byly podávány větší dávky v méně častých intervalech.
Klinická účinnost vůči konkrétním patogenům
Při klinických studiích byla prokázána účinnost na patogeny způsobující onemocnění ABSSSI, které byly v podmínkách in vitro citlivé na dalbavancin:
• Staphylococcus aureus,
• Streptococcus pyogenes,
• Streptococcus agalactiae,
• Streptococcus dysgalactiae,
• skupina bakterií Streptococcus anginosus (zahrnuje S. anginosus, S. intermedius a S. constellatus),
Antibakteriální aktivita vůči jiným relevantním patogenům
Vůči níže uvedeným patogenům nebyla stanovována klinická účinnost, studie in vitro však naznačují, že v případě nepřítomnosti získaných mechanismů rezistence by patogeny byly na dalbavancin citlivé:
• streptokoky skupiny G,
• Clostridium perfringens,
• Peptostreptococcus spp.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xydalba u jedné nebo více podskupin pediatrické populace trpící ABSSSI (více informací o použití u dětí viz body 4.2 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti dalbavancinu byly stanoveny při použití u zdravých jedinců, pacientů i u zvláštních populací. Systémová expozice vůči dalbavancinu je závislá na dávce po podání jednotlivých dávek v rozmezí od 140 do 1 120 mg, přičemž vykazovaná farmakokinetika dalbavancinu je lineární. Po několikanásobném podávání intravenózních infuzí jednou týdně po dobu až osmi týdnů (1 000 mg v den 1, poté až sedm týdenních dávek po 500 mg) u zdravých dospělých jedinců nebyla zjištěna žádná akumulace dalbavancinu.
Průměrný terminální eliminační poločas (tJ/2) činil 372 (rozmezí od 333 do 405) hodin. Farmakokinetické vlastnosti dalbavancinu lze nejlépe popsat pomocí modelu se třemi kompartmenty (distribuční fáze a a P následované terminální eliminační fází). Distribuční poločas (ti/2p), který představuje zásadní součást klinicky relevantní závislosti koncentrace na času, se tak pohyboval mezi 5 až 7 dny a odpovídá dávkování jednou týdně.
Odhadované farmakokinetické parametry dalbavacinu po dvoudávkovém režimu, respektive jednodávkovém režimu jsou vedeny v Tabulce 2 níže.
Tabulka 2:
Průměrné (SD) farmakokinetické parametry dalbavancinu s použitím populační FK analýzy1
|
Parametr |
Dvoudávkový režim2 |
Jednodávkový režim3 |
|
Cmax (mg/l) |
Den 1: 281 (52) |
Den 1: 411 (86) |
|
Den 8: 141 (26) | ||
|
AUC0-den14 (mg^h/l) |
18 100 (4 600) |
20 300 (5 300) |
|
CL (l/h) |
0,048 (0,0086) |
0,049 (0,0096) |
|
1 Zdroj: DAL-MS-01. 2 1 000 mg v den 1 + 500 mg v den 8; Studie DUR001-303 pacienti s hodnotitelným FK vzorkem. 3 1 500 mg; Studie DUR001-303 pacienti s hodnotitelným FK vzorkem. | ||
Plazmatická koncentrace dalbavancinu v čase po dvoudávkovém, respektive jednodávkovém režimu je zobrazena na Obrázku 1.
Obrázek 1. Koncentrace dalbavancinu v plazmě versus čas u typického pacienta s ABSSSI (simulace pomocí populačně farmakokinetického modelu) jak u jednodávkových, tak u dvoudávkových režimů.
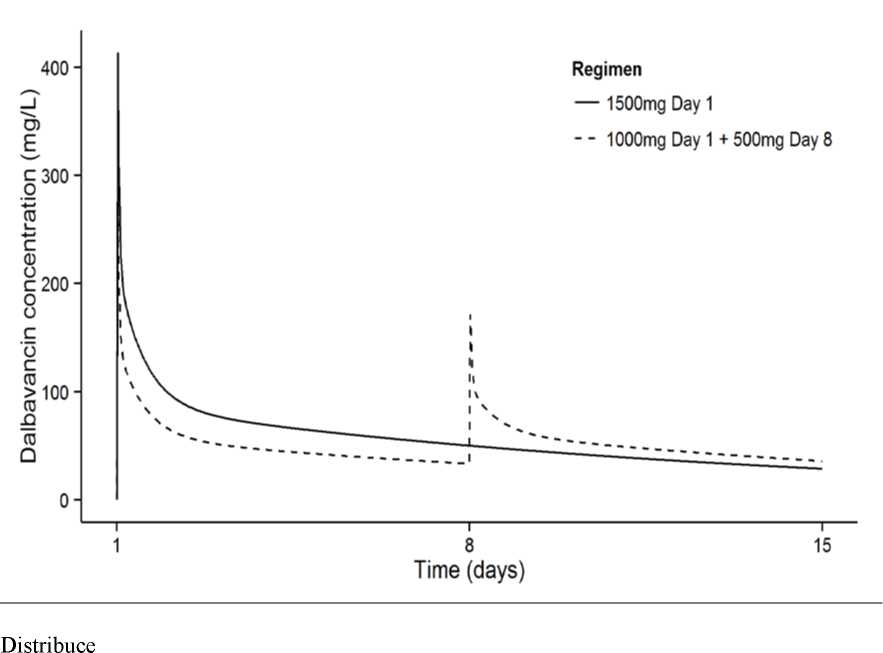
Clearance a distribuční objem v ustáleném stavu jsou u zdravých jedinců a pacientů zasažených infekcí srovnatelné. Distribuční objem v ustáleném stavu byl podobný objemu extracelulární tekutiny. Dalbavancin se reverzibilně váže na proteiny lidské plazmy, zejména na albumin. Vázání dalbavancinu na proteiny plazmy dosahuje 93 % a nemění se v závislosti na koncentraci léčivého přípravku ani v případě ledvinové či jaterní nedostatečnosti. Po podání jednotlivé intravenózní dávky 1 000 mg u zdravých dobrovolníků dosahovala hodnota AUC v tekutině puchýře na kůži (navázaného i nenavázaného dalbavancinu) přibližně 60 % celkové plazmatické AUC v den 7 po podání dávky.
V lidské plazmě nebylo zjištěno významné množství metabolitů. V moči byla zachycena přítomnost metabolitů hydroxy-dalbavancinu a mannosyl aglykonu (< 25 % podané dávky). Metabolické dráhy, které jsou za vznik těchto metabolitů odpovědné, nebyly určeny. Vzhledem k tomu, že metabolismus k celkové eliminaci dalbavancinu přispívá poměrně malou měrou, se však nepředpokládá, že by docházelo k lékovým interakcím prostřednictvím inhibice nebo indukce metabolismu dalbavancinu. Hydroxy-dalbavancin a mannosyl aglykon vykazují v porovnání s dalbavancinem výrazně nižší antibakteriální aktivitu.
Eliminace
Po podání jednotlivé dávky 1 000 mg u zdravých subjektů bylo v průměru 19 % až 33 % podaného dalbavancinu vyloučeno močí jako dalbavancin a 8 % až 12 % jako metabolit hydroxy-dalbavancin. Přibližně 20 % podané dávky bylo vyloučeno stolicí.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetické vlastnosti dalbavancinu byly hodnoceny na 28 subjektech s různě těžkou poruchou funkce ledvin a na 15 srovnatelných subjektech s normální funkcí ledvin. Po podání jednotlivé dávky 500 mg nebo 1 000 mg dalbavancinu se průměrná plazmatická clearance (CLT) u subjektů s lehkou (CLcr 50-79 ml/min), středně těžkou (CLcr 30-49 ml/min) a těžkou (CLcr < 30 ml/min) poruchou funkce ledvin snížila v porovnání se subjekty s normální funkcí ledvin o 11 %, resp. 35 % a 47 %. Průměrná hodnota AUC u subjektů s clearance kreatininu < 30 ml/min byla přibližně dvakrát vyšší. Klinická významnost snížení průměrné plazmatické CLT a s tím souvisejícího zvýšení hodnoty AUC0 -<» zaznamenaného v těchto farmakokinetických studiích dalbavancinu u subjektů s těžkou poruchou funkce ledvin nebyla potvrzena. Farmakokinetické vlastnosti dalbavancinu u pacientů s onemocněním ledvin v konečném stadiu, kteří pravidelně docházejí na dialýzu ledvin (3x týdně) byly podobné jako vlastnosti zjištěné u subjektů trpících lehkou nebo středně těžkou poruchou funkce ledvin a tři hodiny po hemodialýze bylo odstraněno méně než 6 % podané dávky. Pokyny k dávkování u subjektů s onemocněním ledvin jsou uvedeny v bodě 4.2.
Porucha funkce jater
Farmakokinetické vlastnosti dalbavancinu byly hodnoceny na 17 subjektech s lehkou, středně těžkou nebo těžkou poruchou funkce jater a porovnány s devíti zdravými subjekty s normální jaterní funkcí.
U subjektů s lehkou poruchou funkce jater se v porovnání se subjekty s normální jaterní funkcí průměrná hodnota AUC nezměnila. U subjektů se středně těžkou a těžkou poruchou funkce jater se však průměrná hodnota AUC snížila o 28 %, resp. 31 %. Příčina a klinická významnost snížené expozice u subjektů se středně těžkou a těžkou poruchou funkce jater nejsou známy. Pokyny k dávkování u subjektů s onemocněním jater jsou uvedeny v bodě 4.2.
Pohlaví
U zdravých subjektů ani u pacientů trpících infekcemi nebyly zjištěny žádné klinicky významné rozdíly ve farmakokinetických vlastnostech dalbavancinu související s pohlavím subjektů. Nedoporučuje se upravovat dávkování v závislosti na pohlaví.
Starší pacienti
Farmakokinetické vlastnosti dalbavancinu se s věkem významně neměnily, dávkování tedy není třeba upravovat v závislosti na věku (viz bod 4.2). Zkušenosti s podáváním dalbavancinu u seniorů jsou omezené: Do druhé a třetí fáze klinických studií bylo zahrnuto 276 pacientů ve věku > 75 let, přičemž 173 z nich byl podáván dalbavancin. Do klinických studií byli zařazeni pacienti ve věku do 93 let.
Bezpečnost a účinnost přípravku Xydalba u dětí ve věku od narození do 18 let dosud nebyla potvrzena. Celkem deseti dětským pacientům ve věku od 12 do 16 let s ustupujícími infekcemi byla podána jednotlivá dávka dalbavancinu 1 000 mg (tělesná hmotnost > 60 kg) nebo dalbavancinu 15 mg/kg (tělesná hmotnost < 60 kg).
Průměrná plazmatická expozice vůči dalbavancinu na základě hodnoty AUCinf (17,495 pg^h/ml a 16,248 pg •h/ml) a Cmax (212 pg/ml a 191 pg/ml) byla podobná, když byl přípravek podán s obsahem 1 000 mg u dětí (12 - 16 let) s tělesnou hmotností > 60 kg (61,9-105,2 kg) nebo s obsahem 15 mg/kg u dětských pacientů s tělesnou hmotností < 60 kg (47,9-58,9 kg). Zdánlivý terminální poločas t/ byl u dávek dalbavancinu s obsahem 1 000 mg a 15 mg/kg podobný, průměrné hodnoty dosahovaly 227, resp. 202 hodin. Bezpečnostní profil dalbavancinu u subjektů ve věku od 12 do 16 let zahrnutých do této studie se shodoval s bezpečnostním profilem zjištěným u dospělých léčených dalbavancinem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxicita dalbavancinu byla hodnocena po každodenním intravenózním podávání přípravku po dobu až tří měsíců u potkanů a psů. Toxicita podle dávky byla stanovena na základě chemických rozborů séra a histologických nálezů poškození ledvin a jater, snížených parametrů červených krvinek a podráždění v místě vpichu. Reakce na infuze související s dávkou podávaného přípravku byly zjištěny pouze u psů a vyznačovaly se otokem, příp. zarudnutím kůže (nesouvisejícím s místem vpichu), zblednutím sliznic, sliněním, dávením, zklidněním a mírným poklesem krevního tlaku a zvýšením srdečního tepu.
Popsané reakce na infuze byly přechodné povahy (pominuly během jedné hodiny po podání dávky) a byly přičítány uvolňování histaminu. Profil toxicity dalbavancinu u potkaních mláďat se shodoval s profilem dříve zjištěným u dospělých potkanů při podání stejných dávek (mg/kg/den).
Studie zabývající se reprodukční toxicitou u potkanů a králíků nijak neprokázaly teratogenní účinky.
U potkanů, jejichž expozice byla zhruba třikrát vyšší než klinická expozice, byla zjištěna snížená plodnost a zvýšená úmrtnost embryí, nižší hmotnost plodu, snížená osifikace kostí a vyšší úmrtnost narozených mláďat. U samic králíka, které byly v období březosti vystaveny expozici na úrovni pod léčebným rozmezím pro lidské pacienty, docházelo v důsledku toxicity k potratům.
Dlouhodobé studie karcinogenity nebyly provedeny. Na základě baterie testů genotoxicity provedených in vitro a in vivo nebyly u dalbavancinu potvrzeny mutagenní ani klastogenní účinky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mannitol (E421)
Monohydrát laktózy
Kyselina chlorovodíková (pro úpravu pH)
Hydroxid sodný (pro úpravu pH)
6.2 Inkompatibility
Roztoky obsahující chlorid sodný mohou způsobit vysrážení přípravku a nesmí se používat pro rekonstituci koncentrátu a ředění (viz bod 6.6).
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky ani intravenózními roztoky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Suchý prášek: 3 roky
Chemická a fyzikální stabilita po otevření či rekonstituci (in-use stabilita) přípravku Xydalba byla prokázána jak u rekonstituovaného koncentrátu, tak u ředěného roztoku po dobu 48 hodin při teplotě 25 °C nebo nižší. Celková in-use stabilita od rekonstituce do podání přípravku nemá přesáhnout 48 hodin.
Z mikrobiologického hlediska má být infuzní roztok použit okamžitě. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by tato doba neměla být delší než 24 hodin při teplotě 2-8 °C, pokud rekonstituce/ředění neproběhly za kontrolovaných a validovaných aseptických podmínek.
Chraňte před mrazem.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci nebo naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Skleněná lahvička třídy I pro jedno použití o objemu 48 ml uzavřená elastomerovou zátkou a zeleným odtrhovacím víčkem.
Každé balení obsahuje jednu lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Pro podání formou infuze je nutné přípravek Xydalba rekonstituovat sterilní vodou na injekci a následně naředit roztokem glukózy o koncentraci 50 mg/ml (5 %).
Lahvičky s přípravkem Xydalba jsou určeny pouze pro jednorázové použití.
Pokyny pro rekonstituci a ředění
Rekonstituci a ředění přípravku Xydalba je třeba provádět za aseptických podmínek.
1. Obsah lahvičky je třeba rekonstituovat pomalým přidáváním 25 ml vody pro injekce.
2. Neprotřepávat. Abyste předešli pěnění, lahvičkou střídavě otáčejte jemným krouživým pohybem a převracejte dnem vzhůru, dokud nebude obsah zcela rozpuštěn. Rekonstituce koncentrátu může trvat až 5 minut.
3. Rekonstituovaný koncentrát v lahvičce obsahuje 20 mg dalbavancinu v 1 ml přípravku.
4. Rekonstituovaný koncentrát musí být čirý, bezbarvý až nažloutlý bez viditelných částic.
5. Pro podání formou infuze je třeba rekonstituovaný koncentrát dále naředit roztokem glukózy o koncentraci 50 mg/ml (5 %).
6. Pro naředění rekonstituovaného koncentrátu přeneste odpovídající objem koncentrátu pro ředění 20 mg/ml z injekční lahvičky do infuzního vaku nebo lahve obsahující infuzní roztok glukózy o koncentraci 50 mg/ml (5 %). Například: 25 ml koncentrátu obsahuje 500 mg dalbavancinu.
7. Po naředění musí mít infuzní roztok koncentraci dalbavancinu 1 až 5 mg/ml.
8. Infuzní roztok musí být čirý, bezbarvý až nažloutlý bez viditelných částic.
9. V případě výskytu částic nebo nesprávného zabarvení je třeba roztok zlikvidovat.
Přípravek Xydalba se nesmí mísit s dalšími léčivými přípravky nebo infuzními roztoky. Roztoky obsahující chlorid sodný mohou způsobit vysrážení přípravku a NESMÍ se používat pro rekonstituci koncentrátu a ředění. Kompatibilita rekonstituovaného koncentrátu přípravku Xydalba byla ověřena pouze s infuzním roztokem glukózy o koncentraci 50 mg/ml (5 %).
Veškerý nepoužitý rekonstituovaný přípravek zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Durata Therapeutics International B.V.
Spaces Zuidas II,
Barbara Strozzilaan 101,
1083HN Amsterdam Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/986/001
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce/výrobců odpovědných za propouštění šarží
Almac Pharma Services Limited
Seagoe Industrial Estate
Craigavon
Co Armagh
BT63 5UA
Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do šesti měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c, odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskukteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP j e třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k význaným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Krabička
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xydalba 500 mg prášek pro koncentrát pro infuzní roztok Dalbavancinum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jedna lahvička obsahuje dalbavancini hydrochloridum v množství odpovídajícím dalbavancinum 500 mg.
Po rekonstituci obsahuje 1 ml přípravku dalbavancinum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol (E421), monohydrát laktózy,
hydroxid sodný a/nebo kyselina chlorovodíková (pro úpravu pH.)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok
1 lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podávání po rekonstituci a naředění. Pouze pro jednorázové použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Durata Therapeutics International B.V.
Spaces Zuidas II
Barbara Strozzilaan 101
1083HN Amsterdam
Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/986/001
13. ČÍSLO ŠARŽE
Č. šarže
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU Štítek na lahvičce
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Xydalba 500 mg prášek pro koncentrát dalbavancinum
intravenózní podání po rekonstituci a naředění
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Č. šarže
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Durata Therapeutics International B.V.
B. PŘÍBALOVÁ INFORMACE
Xydalba 500 mg prášek pro koncentrát pro infuzní roztok
dalbavancinum
VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si tuto příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Xydalba a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Xydalba používat
3. Jak Vám bude přípravek Xydalba podáván
4. Možné nežádoucí účinky
5. Jak přípravek Xydalba uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xydalba a k čemu se používá
Přípravek Xydalba obsahuje léčivou látku dalbavancin, což je antibiotikum ze skupiny glykopeptidů.
Přípravek Xydalba se používá k léčbě dospělých s infekcí kůže a podkožních tkání.
Přípravek Xydalba působí tak, že zahubí určité bakterie, které mohou způsobovat závažné infekce. Tyto bakterie hubí tak, že jim brání v tvorbě buněčných stěn.
Pokud je Vaše infekce způsobena i jinými typy bakterií, může se Váš lékař rozhodnout léčit Vás kromě přípravku Xydalba ještě dalšími antibiotiky.
2. Co musíte vědět, než začnete přípravek Xydalba používat
Nepoužívejte přípravek Xydalba, jestliže jste alergický(á) na dalbavancin nebo kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před podáním přípravku Xydalba se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou:
• Pokud máte nebo jste měli problémy s ledvinami. V závislosti na stavu Vašich ledvin může lékař přistoupit ke snížení podávané dávky.
• Pokud se u Vás objevily příznaky průjmu, nebo pokud jste průjmem trpěli při dřívější léčbě antibiotiky.
• Pokud trpíte alergií na jiná antibiotika, jako jsou vankomycin nebo teikoplanin.
Pokud se u Vás v průběhu léčby nebo po ní vyskytne průjem, informujte o tom neprodleně svého lékaře. Bez konzultace s lékařem neberte žádné léky na léčbu průjmu.
Reakce související s infuzí
Při podávání formou nitrožilní infuze mohou tyto typy antibiotik způsobit zarudnutí horní části těla, výskyt kopřivky, svědění a/nebo vyrážky. Pokud se u Vás tyto typy reakcí objeví, Váš lékař může rozhodnout o ukončení podávání infuze, nebo podávání zpomalit.
Další infekce
Podávání antibiotik někdy umožní rozvoj nové, odlišné infekce. Pokud k rozvoji takové infekce dojde, informujte o tom lékaře, a ten rozhodne o dalším postupu.
Děti a dospívající
Nepodávejte tento léčivý přípravek dětem mladším 18 let. Použití přípravku Xydalba u dětí mladších 18 let nebylo doposud studováno.
Další léčivé přípravky a Xydalba
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Kromě nejnutnějších případů se nedoporučuje podávat přípravek Xydalba v těhotenství. Je tomu tak proto, že není známo, jaké účinky by přípravek mohl mít na nenarozené dítě. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. O podání přípravku Xydalba rozhodujete Vy a lékař.
Není známo, zda se přípravek Xydalba vylučuje do mateřského mléka. Před kojením se poraďte se svým lékařem. O podání přípravku Xydalba rozhodujete Vy a lékař. Při používání přípravku Xydalba nemáte kojit.
Řízení dopravních prostředků a obsluha strojů
Přípravek Xydalba může vyvolávat závratě. Po použití přípravku buďte opatrní při řízení a obsluze strojů.
Přípravek Xydalba obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě „bez sodíku“.
3. Jak Vám bude přípravek Xydalba podáván
Přípravek Xydalba Vám bude podávat lékař nebo zdravotní sestra.
Přípravek Xydalba se podává v jedné dávce 1 500 mg nebo ve dvou dávkách s týdenním odstupem: 1 000 mg přípravku v den 1 a 500 mg přípravku v den 8.
Přípravek Xydalba Vám bude podáván prostřednictvím kapací infuze přímo do Vašeho krevního oběhu (intravenózně) po dobu 30 minut.
Pacienti s chronickým onemocněním ledvin
Pokud trpíte chronickým onemocněním ledvin, může lékař rozhodnout o snížení podávané dávky.
Jestliže Vám bylo podáno více přípravku Xydalba, než byste měl(a) dostat
Jestliže se domníváte, že Vám možná byla podána příliš vysoká dávka přípravku Xydalba, informujte o tom okamžitě svého lékaře nebo zdravotní sestru.
Jestliže Vám kvůli opomenutí nebyla podána dávka přípravku Xydalba
Pokud se domníváte, že Vám nebyla podána druhá dávka přípravku, informujte o tom okamžitě svého lékaře nebo zdravotní sestru.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejete se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Závažné nežádoucí účinky
Pokud se u Vás vyskytne kterýkoli z následujících příznaků, neprodleně to sdělte svému lékaři -
možná budete potřebovat neodkladnou lékařskou pomoc:
• Náhlý otok rtů, obličeje, krku nebo jazyka, závažná vyrážka, svědění, stažení hrdla, pokles krevního tlaku, obtíže při polykání a/nebo dýchání. Může se jednat o příznaky život ohrožující reakce přecitlivělosti. Takto závažná reakce se jakožto nežádoucí účinek vyskytuje jen vzácně. Objevuje se nanejvýše u jednoho z tisíce pacientů.
• Bolesti břicha (bolest žaludku), a/nebo vodnatý průjem. Závažnost těchto příznaků se může zvyšovat nebo příznaky nemusí odeznít a stolice může obsahovat krev nebo hlen. Může se jednat o příznaky infekce střev. V takovém případě neberte žádné léky na zastavení nebo zpomalení střevního pohybu. Infekce střev je uváděna jako méně častý nežádoucí účinek. Objevuje se nanejvýše u jednoho ze sta pacientů.
• Poruchy sluchu. Tento nežádoucí účinek se vyskytl u pacientů používajících podobný lék. Četnost výskytu tohoto nežádoucího účinku není známa. Z dostupných údajů nelze provést odhad četnosti výskytu.
Další nahlášené nežádoucí účinky přípravku Xydalba jsou uvedeny níže.
Pokud se u Vás vyskytne kterýkoli z níže uvedených nežádoucích účinků, sdělte to svému lékaři,
lékárníkovi nebo zdravotní sestře:
Časté - vyskytuji se až u 1 z 10 pacientů:
• bolesti hlavy
• pocit na zvracení
• průjem
Méně časté - vyskytují se až u 1 ze 100 pacientů:
• poševní infekce, plísňové infekce, moučnivka
• infekce močového ústrojí
• anémie (snížený počet červených krvinek), zvýšený počet krevních destiček (trombocytóza), zvýšený počet bílých krvinek zvaných eosinofily (eosinofilie), nízký počet jiných typů bílých krvinek (leukopenie, neutropenie)
• změny ve výsledcích krevních testů
• snížená chuť k j ídlu
• potíže se spánkem
• závratě
• změny ve vnímání chuti
• zánět a otok povrchových žil, zarudnutí
• kašel
• bolest břicha a nepříjemné pocity v břiše, poruchy trávení, zácpa
• abnormální výsledky jaterních testů
• zvýšená hladina alkalické fosfatázy (enzymu přítomného v těle)
• svědění, kopřivka
• svědění pohlavních orgánů (ženy)
• bolest, zarudnutí či otok v místě aplikace infuze
• pocity horka
• zvýšená krevní hladina gama-glutamyltransferázy (enzym vytvářený v játrech a dalších tělních tkáních)
• vyrážka
• zvracení
Vzácné - vyskytující se až u 1 z 1 000 pacientů:
• potíže s dýcháním (bronchospasmus)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Xydalba uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání, pokud je uchováván v neotevřeném původním obalu.
Připravený infuzní roztok přípravku Xydalba se nesmí použít, pokud jsou v roztoku přítomny jakékoli částice nebo pokud je roztok zakalený.
Přípravek Xydalba je určen pouze pro jednorázové použití.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Co přípravek Xydalba obsahuje
• Léčivou látkou přípravku je dalbavancinum. Jedna lahvička s práškem obsahuje dalbavancini hydrochloridum v množství odpovídajícím dalbavancinum 500 mg.
• Dalšími pomocnými látkami jsou mannitol (E421), monohydrát laktózy, kyselina chlorovodíková a/nebo hydroxid sodný (pouze pro úpravu pH).
Jak přípravek Xydalba vypadá a co obsahuje toto balení
Xydalba prášek pro koncentrát pro infuzní roztok se dodává ve 48ml skleněných lahvičkách opatřených zeleným odtrhovacím víčkem. Lahvička obsahuje bílý až světle žlutý prášek.
Dodává se v baleních obsahujících jednu lahvičku.
Držitel rozhodnutí o registraci
Durata Therapeutics International B.V.
Spaces Zuidas II, Barbara Strozzilaan 101, 1083HN Amsterdam, Nizozemsko
Výrobce
Almac Pharma Services Ltd
Seagoe Industrial Estate, Craigavon, Country Armagh BT63 5UA Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgium/ Belgie/Belgique/Belgien
Cardiome UK Limited Tél/Tel: +32 (0)28 08 86 20
Lithuania/ Lietuva
UAB MRA Totorip str. 20-9 LT-01121 Vilnius Tel: + 370 5264 9010
Bulgaria/ Etarapnu
AHg^eauHH OapMa Etarapua EOOfl 6ya. AceH HopgaHOB 10 BG-Co$ua 1592 Tea.: + 359 2 975 1395 office@angelini.bg
Česká republika
Angelini Pharma Česká republika s.r.o.
Páteřní 1216/7
CZ-635 00 Brno
Tel: + 420 546 123 111
info@angelini .cz
Denmark/ Danmark
Cardiome UK Limited Tlf: +45 8082 6022
Germany/ Deutschland
Cardiome UK Limited Tel: +49 (0)69 33 29 62 76
Hungary/ Magyarország
Angelini Pharma Magyarország Kft Dayka Gábor u. 3., 214-215. számú iroda H-1118 Budapest Tel: + 36 1 336 1614 office@angelini.hu
Malta
Cardiome UK Limited Tel: +41 848 00 79 70
Netherlands/ Nederland
Cardiome UK Limited Tel: +31 (0)20 808 32 06
|
Estonia/ Eesti Lorenzo Pharma OU Koidu str. 20-19 EE-10136 Tallinn Tel: + 372 604 1669 |
Norway/ Norge Cardiome UK Limited Tlf: +41 848 00 79 70 |
|
Greece/ EXláSa ANGELINI PHARMA HELLAS ABEE nAPAmrai & EMnopiAi oapmakqn A%aíag 4 & TpoiZnvía? GR-14564 Néa Kn^ioiá Tqk + 30 210 626 9200 info@angelinipharma.gr |
Austria/ Osterreich Angelini Pharma Osterreich GmbH GewerbestraBe 18-20 A-2102 Bisamberg Tel: + 43 2262 6060 office@angelini.at |
|
Spain/ Espana Angelini Farmaceutica S.A. C. Osi, 7 E-08034 Barcelona Tel: + 34 93 253 4500 |
Poland/ Polska Angelini Pharma Polska Sp. z o.o. ul. Podlesna 83 PL-05-552 Eazy Tel.: + 48 22 70 28 200 |
|
France CORREVIO Tél: +33 (0)1 77 68 89 17 |
Portugal Angelini Farmaceutica, Lda Rua Joao Chagas, 53, Piso 3 P-1499-040 Cruz Quebrada- Dafundo Tel: + 351 21 414 8300 apoio.utente @angelini.pt |
|
Croatia/ Hrvatska Angelini Pharma Osterreich GmbH, Podružnica, za promidžbu Zagreb Hektoroviceva 2/5 HR-10000 Zagreb Tel: + 385 1 644 8232 |
Romania/ Románia Angelini Pharmaceuticals Romania SRL Str. Drumea Rádulescu, Nr. 52, Sector 4 RO-Bucuresti 040336 Tel: + 40 21 331 6767 office@angelini.ro |
|
Ireland Cardiome UK Limited Tel: +41 848 00 79 70 |
Slovenia/ Slovenija Angelini Pharma d.o.o. Koprska ulica 108 A SI-1000 Ljubljana Tel: +386 1 544 65 79 info@angelini.si |
|
Iceland/ Ísland Island Cardiome UK Limited Sími: +41 848 00 79 70 |
Slovak Republic/ Slovenská republika Angelini Pharma Slovenská republika s.r.o Júnová 33 SK-831 01 Bratislava Tel: + 421 2 59 207 320 office@angelini.sk |
|
Italy/ Italia Angelini S.p.A Viale Amelia 70 I-00181 Roma Tel: + 39 06 78 0531 |
Suomi/Finland Cardiome UK Limited Puh/Tel: +41 848 00 79 70 |
|
Cyprus/ Kúnpoq |
Sweden/ Sverige |
ANGELINI PHARMA HELLAS ABEE
Cardiome UK Limited Tel: +46 (0)8 408 38440
United Kingdom
Cardiome UK Limited Tel: +44 (0)203 002 8114
nAPArQEH! & EMnOPIA! OAPMAKQN
A%aíag 4 & Tpo@nvíag
GR-14564 Néa Kn^rará
TpA.: + 30 210 626 9200
Latvia/ Latvija
SIA Livorno Pharma Vilandes str. 17-1 LV-1010 Riga Tel: + 371 6721 1124
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Následující informace jsou určeny pouze zdravotnické pracovníky:
Důležité: Před předepsáním tohoto léčivého přípravku se seznamte s informacemi obsaženými v souhrnu údajů o přípravku.
Pro podání formou infuze je nutné přípravek Xydalba rekonstituovat sterilní vodou na injekci a následně naředit roztokem glukózy o koncentraci 50 mg/ml (5 %).
Lahvičky s přípravkem Xydalba jsou určeny pouze pro jednorázové použití.
Pokyny pro rekonstituci a ředění
Rekonstituci a ředění přípravku Xydalba je třeba provádět za aseptických podmínek.
1. Obsah lahvičky je třeba rekonstituovat pomalým přidáváním 25 ml vody na injekci.
2. Neprotřepávat. Abyste předešli pěnění, lahvičkou střídavě otáčejte jemným krouživým pohybem a převracejte dnem vzhůru, dokud nebude obsah zcela rozpuštěn. Rekonstituce koncentrátu může trvat až 5 minut.
3. Rekonstituovaný koncentrát v injekční lahvičce obsahuje 20 mg dalbavancinu v 1 ml přípravku.
4. Rekonstituovaný koncentrát musí být čirý, bezbarvý až nažloutlý bez viditelných částic.
5. Pro podání formou infuze je třeba rekonstituovaný koncentrát dále naředit roztokem glukózy o koncentraci 50 mg/ml (5 %).
6. Pro naředění rekonstituovaného koncentrátu přeneste odpovídající objem koncentrátu pro ředění 20 mg/ml z injekční lahvičky do infuzního vaku nebo lahve obsahující infuzní roztok glukózy o koncentraci 50 mg/ml (5 %). Například: 25 ml koncentrátu obsahuje 500 mg dalbavancinu.
7. Po naředění musí mít infuzní roztok koncentraci dalbavancinu 1 až 5 mg/ml
8. Infuzní roztok musí být čirý, bezbarvý až nažloutlý bez viditelných částic.
9. V případě výskytu částic nebo nesprávného zabarvení je třeba roztok zlikvidovat.
Přípravek Xydalba se nesmí mísit s dalšími léčivými přípravky nebo infuzními roztoky. Roztoky obsahující chlorid sodný mohou způsobit vysrážení přípravku a NESMÍ se používat pro rekonstituci koncentrátu a ředění. Kompatibilita rekonstituovaného koncentrátu přípravku Xydalba byla ověřena pouze s infuzním roztokem glukózy o koncentraci 50 mg/ml (5 %).
Likvidace
Veškerý nepoužitý rekonstituovaný přípravek zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
29