Xultophy 100 Jednotek/Ml + 3,6 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xultophy 100 jednotek/ml + 3,6 mg/ml injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
1 ml roztoku obsahuje 100 jednotek insulinum degludecum* a 3,6 mg liraglutidum*.
*Je vyráběn rekombinantní DNA technologií v Saccharomyces cerevisiae.
Jedno předplněné pero obsahuje 3 ml odpovídající 300 jednotkám insulinu degludek a 10,8 mg liraglutidu.
Jedna dávkovací jednotka obsahuje 1 jednotku insulinu degludek a 0,036 mg liraglutidu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý, bezbarvý, izotonický roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Xultophy je určen k léčbě dospělých s diabetes mellitus 2. typu a slouží ke zlepšení kontroly hladiny glukózy v krvi v kombinaci s perorálními přípravky snižujícími hladinu glukózy, pokud tyto přípravky samotné nebo v kombinaci s agonisty receptoru GLP-1 či s bazálním inzulinem neposkytují dostatečnou kompenzaci hladiny glukózy (dostupné údaje o různých kombinacích viz bod 4.4 a 5.1).
4.2 Dávkování a způsob podání
Dávkování
Přípravek Xultophy se podává jednou denně subkutánním podáním. Lze jej podat kdykoli v průběhu dne, přednostně ve stejnou denní dobu.
Přípravek Xultophy je třeba dávkovat dle individuálních potřeb pacienta. Doporučuje se optimalizovat kompenzaci hladiny glukózy úpravou dávky na základě plazmatické hladiny glukózy nalačno.
Úprava dávky může být nutná v případě, že je pacient vystaven zvýšené fyzické námaze, mění svou obvyklou dietu, nebo po dobu souběžně probíhajícího onemocnění.
Pacienti, kteří dávku vynechají, mají být poučeni, aby si tuto dávku aplikovali ihned po zjištění této skutečnosti a dále pokračovali v obvyklém dávkování jednou denně. Mezi injekcemi musí být vždy zajištěna alespoň 8hodinová prodleva. To se rovněž týká situací, kdy podání ve stejnou denní dobu není možné.
Přípravek Xultophy se podává v dávkovačích jednotkách. Jedna dávkovači jednotka obsahuje 1 jednotku inzulínu degludek a 0,036 mg liraglutidu. Předplněné pero umožňuje podat přípravek v dávce o velikosti 1 až 50 dávkovačích jednotek v jedné injekci v přírůstcích po jedné dávkovači jednotce. Maximální denní dávka přípravku Xultophy je 50 dávkovacích jednotek (50 jednotek inzulinu degludek a 1,8 mg liraglutidu). Počítadlo dávky na peru ukazuje počet dávkovacích jednotek.
Přídatná léčba k_perorálním léčivým _přípravkům snižujícím hladinu glukózy v krvi Doporučená počáteční dávka přípravku Xultophy je 10 dávkovacích jednotek (10 jednotek inzulinu degludek a 0,36 mg liraglutidu).
Přípravek Xultophy lze přidat ke stávající léčbě perorálními antidiabetiky. V případě, že je přípravek Xultophy přidán k terapii deriváty sulfonylurey, je třeba zvážit snížení dávky derivátů sulfonylurey (viz bod 4.4).
Převedení z agonistů receptoru GLP-1
Před zahájením léčby přípravkem Xultophy má být ukončena léčba agonisty receptoru GLP-1. Při převedení z agonistů receptoru GLP-1 je počáteční doporučovaná dávka přípravku Xultophy 16 dávkovacích jednotek (16 jednotek inzulinu degludek a 0,6 mg liraglutidu) (viz bod 5.1). Doporučená počáteční dávka nesmí být překročena. Při převedení z agonistů receptoru GLP-1 s dlouhodobým účinkem (například podávaných jednou týdně) je nutno zvážit prodloužený účinek. Léčbu přípravkem Xultophy je nutno zahájit v okamžiku, kdy má být podána další dávka dlouhodobě působícího agonisty receptoru GLP-1. Během převedení a v následujících týdnech je doporučeno pečlivé monitorování hladiny glukózy.
Převedení z bazálního inzulinu
Před zahájením léčby přípravkem Xultophy je třeba ukončit léčbu bazálním inzulinem. Při přechodu z léčby bazálním inzulinem se doporučuje počáteční dávka přípravku Xultophy o velikosti 16 dávkovacích jednotek (16 jednotek inzulinu degludek a 0,6 mg liraglutidu) (viz bod 4.4 a 5.1). Doporučená počáteční dávka nesmí být překročena. Během převedení a v následujících týdnech je doporučeno pečlivé monitorování hladiny glukózy.
Zvláštní skupiny _pacientů
Starší pacienti (věk > 65 let)
Přípravek Xultophy může být podáván starším pacientům. Sledování hladiny glukózy má být intenzivnější a dávka má být upravena individuálně. Zkušenosti s léčbou pacientů ve věku > 75 let jsou omezené.
Porucha funkce ledvin
Pokud je přípravek Xultophy používán u pacientů s mírnou nebo středně závažnou poruchou funkce ledvin, sledování hladiny glukózy má být intenzivnější a dávka má být upravena individuálně. Přípravek Xultophy není doporučen pro použití u pacientů se závažnou poruchou funkce ledvin, včetně pacientů v konečném stadiu renálního onemocnění (viz bod 5.2).
Porucha funkce jater
Zkušenosti s léčbou přípravkem Xultophy u pacientů s poruchou funkce jater jsou v současnosti příliš omezené na to, aby mohlo u těchto pacientů být jeho použití doporučeno (viz bod 5.2).
Pediatrická populace
Neexistuje žádné relevantní použití přípravku Xultophy u pediatrické populace.
Způsob podání
Přípravek Xultophy je určen pouze pro subkutánní podání. Přípravek Xultophy nesmí být podán intravenózně ani intramuskulárně.
Přípravek Xultophy se aplikuje subkutánní injekcí do stehna, horní části paže nebo břicha. Místa aplikace mají být v rámci jedné oblasti vždy obměňována, aby se snížilo riziko lipodystrofie. Další pokyny týkající se podávání viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na jednu nebo obě léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek Xultophy se nesmí podávat pacientům s diabetes mellitus 1. typu nebo k léčbě diabetické ketoacidózy.
Hypoglykemie
Pokud je dávka přípravku Xultophy větší, než je potřeba, může dojít k hypoglykemii. Vynechání jídla nebo neplánovaná namáhavá fyzická zátěž mohou vést k hypoglykemii. Při léčbě v kombinaci s deriváty sulfonylurey se může riziko hypoglykemie zmenšit snížením dávky derivátů sulfonylurey. Přidružené onemocnění ledvin, jater či onemocnění postihující nadledviny, podvěsek mozkový nebo štítnou žlázu může vyžadovat změny v dávce přípravku Xultophy. Pacienti, kteří mají výrazně zlepšenou kompenzaci hladiny glukózy v krvi (například při intenzifikované terapii), mohou zaznamenat změnu obvyklých varovných symptomů hypoglykemie a musí být patřičně poučeni.
U pacientů s dlouholetým diabetem mohou běžné varovné symptomy hypoglykemie (viz bod 4.8) vymizet. Prodloužený účinek přípravku Xultophy může opozdit zotavení se z hypoglykemie.
Hyperglykemie
Nedostatečné dávkování a/nebo přerušení léčby antidiabetiky může vést k hyperglykemii a potenciálně k hyperosmolárnímu kómatu. V případě přerušení léčby přípravkem Xultophy zajistěte, že budou dodrženy instrukce pro zahájení alternativní antidiabetické léčby. Dále mohou souběžná onemocnění, hlavně infekce, vést k hyperglykemii, a tím vyvolat vyšší potřebu léčby antidiabetiky. První symptomy hyperglykemie se obvykle v průběhu hodin nebo dní stupňují. Patří mezi ně žízeň, zvýšená frekvence močení, nauzea, zvracení, ospalost, zarudlá suchá kůže, sucho v ústech a ztráta chuti k jídlu či acetonový zápach dechu.
V případech závažné hyperglykemie je třeba zvážit podání inzulinu s rychlým účinkem. Neléčené hyperglykemické stavy v konečném důsledku vedou až k hyperosmolárnímu kómatu/diabetické ketoacidóze, což je potenciálně letální.
Kombinace pioglitazonu a inzulínových léčivých přípravků
Pokud byl pioglitazon užíván v kombinaci s inzulinovými léčivými přípravky, byly hlášeny případy srdečního selhání, a to zvláště u pacientů s rizikovými faktory pro vznik srdečního selhání. Tuto skutečnost je nutno vzít v úvahu, pokud je zvažována léčba pioglitazonem v kombinaci s přípravkem Xultophy. Pokud je tato kombinace použita, měli by být pacienti sledováni s ohledem na známky a příznaky srdečního selhání, zvýšení hmotnosti a edém. Pioglitazon musí být vysazen, pokud se objeví jakékoliv zhoršení srdečních příznaků.
Poruchy oka
Intenzifikovaná léčba inzulinem, složkou přípravku Xultophy, s prudkým zlepšením kompenzace hladiny glukózy v krvi může být spojena s přechodným zhoršením diabetické retinopatie, zatímco dlouhodobé zlepšení kompenzace hladiny glukózy v krvi snižuje riziko progrese diabetické retinopatie.
Tvorba protilátek
Podávání přípravku Xultophy může vést k tvorbě protilátek proti inzulínu degludek a/nebo liraglutidu. Ve vzácných případech může přítomnost těchto protilátek vyžadovat úpravu dávky přípravku Xultophy, aby se usměrnila tendence k hyperglykemii či hypoglykemii. U několika málo pacientů se po léčbě přípravkem Xultophy vytvořily specifické protilátky proti inzulinu degludek, protilátky se zkříženou reaktivitou proti humánnímu inzulinu nebo protilátky proti liraglutidu. Tvorba protilátek nebyla spojena se sníženou účinností přípravku Xultophy.
Akutní pankreatitida
Použití agonistů receptoru GLP-1, včetně liraglutidu - složky přípravku Xultophy, bylo spojeno s rizikem rozvoje akutní pankreatitidy. Bylo hlášeno několik případů akutní pankreatitidy. Pacienty je třeba informovat o charakteristických příznacích akutní pankreatitidy. Je-li podezření na pankreatitidu, je třeba podávání přípravku Xultophy přerušit. Pokud je akutní pankreatitida potvrzena, nesmí se léčba přípravkem Xultophy již obnovit. U pacientů s pankreatitidou v anamnéze je třeba dbát zvláštní opatrnosti.
Nežádoucí účinky na štítnou žlázu
V klinických studiích s agonisty receptoru GLP-1, včetně liraglutidu - složky přípravku Xultophy, byly hlášeny nežádoucí účinky na štítnou žlázu, včetně zvýšené hladiny kalcitoninu v krvi, zvětšení štítné žlázy a neoplazie štítné žlázy, a to zvláště u pacientů s již dříve existujícím onemocněním štítné žlázy. Přípravek Xultophy proto musí být u těchto pacientů používán s opatrností.
Zánětlivé střevní onemocnění a diabetická gastroparéza
Nejsou žádné zkušenosti s podáváním přípravku Xultophy pacientům se zánětlivým onemocněním střev a diabetickou gastroparézou. Použití přípravku Xultophy se proto u těchto pacientů nedoporučuje.
Dehydratace
V klinických studiích s agonisty receptoru GLP-1, včetně liraglutidu - složky přípravku Xultophy, byly hlášeny známky a příznaky dehydratace, včetně poruchy funkce ledvin a akutního selhání ledvin. Pacienti léčení přípravkem Xultophy musí být v souvislosti s gastrointestinálními nežádoucími účinky upozorněni na potenciální riziko dehydratace a musí být seznámeni s bezpečnostními opatřeními, která musí učinit, aby zabránili úbytku tekutin.
Zamezení chybám v medikaci
Pacienty je zapotřebí poučit, aby před každou aplikací zkontrolovali štítek pera, aby nedošlo k náhodné záměně mezi přípravkem Xultophy a jinými injekčními léčivými přípravky k léčbě diabetu.
Nestudované populace
Přechod na přípravek Xultophy z dávek bazálního inzulinu < 20 a > 50 jednotek nebyl studován.
Přípravek Xultophy nebyl studován v kombinaci s inhibitory dipeptidylpeptidázy 4 (DPP-4), glinidy ani prandiálním inzulinem.
S podáváním pacientům s městnavým srdečním selháním třídy I-II podle New York Heart Association (NYHA) jsou omezené zkušenosti. Přípravek Xultophy proto musí být u těchto pacientů používán s opatrností. S podáváním přípravku pacientům s městnavým srdečním selháním třídy III-IV podle NYHA nejsou žádné zkušenosti. Použití přípravku Xultophy se proto u těchto pacientů nedoporučuje.
Pomocné látky
Přípravek Xultophy obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě je „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakodynamické interakce
Nebyly provedeny studie interakcí s přípravkem Xultophy.
Rada látek ovlivňuje metabolismus glukózy a může vyžadovat úpravu dávky přípravku Xultophy. Následující látky mohou snížit potřebu_přípravku Xultophy:
Antidiabetika, inhibitory monoaminooxidázy (IMAO), beta-blokátory, inhibitory angiotensin konvertujícího enzymu (ACE), salicyláty, anabolické steroidy a sulfonamidy.
Následující látky mohou zvýšit potřebu_přípravku Xultophy:
Perorální antikoncepce, thiazidy, glukokortikoidy, hormony štítné žlázy, sympatomimetika, růstové hormony a danazol.
Beta-blokátory mohou překrývat příznaky hypoglykemie.
Oktreotid/lanreotid může jak zvýšit, tak snížit potřebu přípravku Xultophy.
Alkohol může zesílit nebo zeslabit hypoglykemický účinek přípravku Xultophy.
Farmakokinetické interakce
Údaje in vitro naznačují, že potenciální farmakokinetické lékové interakce související s interakcí s cytochromem CYP a s vazbou na proteiny jsou u liraglutidu i inzulinu degludek nízké.
Malé zpoždění ve vyprazdňování žaludku při používání liraglutidu může ovlivnit absorpci současně podávaných perorálních léčivých přípravků. Studie interakcí neprokázaly žádné klinicky významné zpoždění absorpce.
Warfarin a další deriváty kumarinu
Nebyla provedena žádná studie interakcí. Klinicky významné interakce s léčivými látkami se špatnou rozpustností nebo s úzkým terapeutickým indexem, jako je warfarin, nemohou být vyloučeny. Po zahájení léčby přípravkem Xultophy se u pacientů užívajících warfarin nebo další deriváty kumarinu doporučuje častější monitorování INR (International Normalised Ratio).
Paracetamol
Liraglutid neměnil celkovou expozici paracetamolem po podání jednorázové dávky 1 000 mg paracetamolu. Hodnota Cmax paracetamolu byla snížena o 31 % a střední hodnota tmax byla zpožděna až na 15 min. Při souběžném užívání paracetamolu není nutná žádná úprava dávkování.
Atorvastatin
Liraglutid neměnil celkovou expozici atorvastatinem v klinicky významné míře po podání jednorázové dávky 40 mg atorvastatinu. Při podávání s liraglutidem proto není nutná žádná úprava dávky atorvastatinu. Při podávání s liraglutidem se hodnota Cmax atorvastatinu snížila o 38 % a střední hodnota tmax se zpozdila z 1 hod na 3 hod.
Griseofulvin
Liraglutid neměnil celkovou expozici griseofulvinem po podání jednorázové dávky 500 mg griseofulvinu. Hodnota Cmax griseofulvinu byla zvýšena o 37 %, zatímco ke změně střední hodnoty tmax nedošlo. Úprava dávky griseofulvinu a jiných látek s nízkou rozpustností a vysokou permeabilitou není nutná.
Digoxin
Podání jednorázové dávky 1 mg digoxinu spolu s liraglutidem vedlo ke snížení AUC digoxinu o 16 %, hodnota Cmax byla snížena o 31 %. Střední hodnota doby do dosažení maximální koncentrace (tmax) digoxinu byla zpožděna z 1 h na 1,5 h. Na základě těchto výsledků není nutná úprava dávky digoxinu.
Lisinopril
Podání jednorázové dávky 20 mg lisinoprilu spolu s liraglutidem vedlo ke snížení AUC lisinoprilu o 15 %, hodnota Cmax byla snížena o 27 %. Střední hodnota tnax lisinoprilu byla při podávání liraglutidu zpožděna z 6 hod na 8 hod. Na základě těchto výsledků není nutná úprava dávky lisinoprilu.
Perorální antikoncepce
Po podání jednorázové dávky perorálního antikoncepčního přípravku snižoval liraglutid hodnotu Cmax ethinylestradiolu o 12 % a levonorgestrelu o 13 %. Hodnota tmax byla u obou látek při podání liraglutidu zpožděna o 1,5 hod. Nedošlo k žádnému klinicky významnému účinku na celkovou expozici ethinylestradiolem ani levonorgestrelem. Předpokládá se proto, že antikoncepční účinek není při současném podávání liraglutidu ovlivněn.
4.6 Fertilita, těhotenství a kojení
S použitím přípravku Xultophy, inzulinu degludek nebo liraglutidu u těhotných žen neexistují žádné klinické zkušenosti. Pokud si pacientka přeje otěhotnět nebo otěhotní, musí být léčba přípravkem Xultophy přerušena.
Co se týče embryotoxicity a teratogenity, reprodukční studie s inzulinem degludek na zvířatech neprokázaly žádné rozdíly mezi inzulinem degludek a humánním inzulinem. Studie s liraglutidem na zvířatech prokázaly reprodukční toxicitu, viz bod 5.3. Potenciální riziko pro člověka není známé.
Kojení
S používáním přípravku Xultophy během kojení neexistují žádné klinické zkušenosti. Není známo, zda dochází k exkreci inzulinu degludek nebo liraglutidu do lidského mateřského mléka. Vzhledem k chybějícím zkušenostem se přípravek Xultophy během kojení nemá podávat.
U potkanů se inzulin degludek vylučoval do mléka. Koncentrace v mléku byla nižší než v plazmě. Studie na zvířatech prokázaly, že přenos liraglutidu a strukturně blízkých metabolitů do mléka byl nízký. Neklinické studie s liraglutidem prokázaly snížení neonatálního růstu u kojených potkaních mláďat spojené s léčbou (viz bod 5.3).
Fertilita
S vlivem přípravku Xultophy s ohledem na fertilitu neexistují žádné klinické zkušenosti.
Reprodukční studie na zvířatech s inzulinem degludek neodhalily žádné nežádoucí účinky na fertilitu. S výjimkou lehkého snížení počtu živých nidovaných vajíček neprokázaly studie s liraglutidem na zvířatech škodlivé účinky v souvislosti s fertilitou.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacientova schopnost koncentrace a reakce se může v důsledku hypoglykemie snížit. Uvedená skutečnost může znamenat riziko v situacích, kdy jsou tyto schopnosti zvláště důležité (například při řízení motorového vozidla nebo obsluhování strojů).
Pacienti musí být poučeni o opatřeních, jak se vyvarovat vzniku hypoglykemie při řízení. To je zvláště důležité především u pacientů, kteří mají nevýrazné nebo žádné varovné příznaky hypoglykemie nebo kteří mají časté epizody hypoglykemie. V těchto případech je třeba vhodnost řízení zvážit.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Program klinického vývoje přípravku Xultophy zahrnoval přibližně 1 900 pacientů léčených tímto přípravkem.
Nejčastěji hlášenými nežádoucími účinky během léčby přípravkem Xultophy byla hypoglykemie a gastrointestinální nežádoucí účinky (viz bod „Popis vybraných nežádoucích účinků“ níže).
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky spojené s přípravkem Xultophy jsou uvedeny níže, jsou řazeny podle třídy orgánových systémů a frekvence výskytu. Kategorie frekvence výskytu jsou definovány takto: Velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 1 Nežádoucí účinky hlášené ve fázi ^ 3 kontrolovaných studií
|
Třída orgánových systémů podle databáze MedDRA |
Frekvence |
Nežádoucí účinek |
|
Poruchy imunitního systému |
Méně časté |
Kopřivka |
|
Méně časté |
Přecitlivělost | |
|
Není známo |
Anafylaktická reakce | |
|
Poruchy metabolismu a výživy |
Velmi časté |
Hypoglykemie |
|
Časté |
Snížená chuť k jídlu | |
|
Méně časté |
Dehydratace | |
|
Gastrointestinální poruchy |
Časté |
Nauzea, průjem, zvracení, zácpa, dyspepsie, gastritida, bolest břicha, gastroezofageální refluxní choroba, břišní distenze |
|
Méně časté |
Říhání, flatulence | |
|
Není známo |
Pankreatitida (včetně nekrotizující pankreatitidy) | |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Méně časté |
Získaná lipodystrofie | |
|
Celkové poruchy a reakce v místě aplikace |
Časté |
Reakce v místě vpichu |
|
Není známo |
Periferní edém | |
|
Vyšetření |
Méně časté |
Zvýšená tepová frekvence |
Popis vybraných nežádoucích účinků
Hypoglykemie
Pokud je dávka přípravku Xultophy větší, než je potřeba, může dojít k hypoglykemii. Těžká hypoglykemie může vést k bezvědomí a/nebo křečím a může vyústit v přechodné nebo trvalé poškození mozkové funkce, či dokonce v úmrtí. Symptomy hypoglykemie se obvykle objevují náhle. Mohou zahrnovat studený pot, chladnou bledou pokožku, únavu, nervozitu nebo třes, úzkost, neobvyklou únavu nebo slabost, zmatenost, problémy s koncentrací, ospalost, přílišný hlad, změny vidění, bolest hlavy, nauzeu a palpitaci. Četnost hypoglykemií viz bod 5.1.
Alergické reakce
U přípravku Xultophy byly hlášeny alergické reakce projevující se známkami a příznaky jako je kopřivka (0,3 % pacientů léčených přípravkem Xultophy), vyrážka (0,7 %), pruritus (0,5 %) a/nebo otok obličeje (0,2 %). Po uvedení liraglutidu na trh bylo hlášeno několik případů anafylaktických reakcí s dalšími příznaky jako hypotenzí, palpitacemi, dušností a edémem. Anafylaktické reakce mohou být potenciálně život ohrožující.
Gastrointestinální nežádoucí účinky
Gastrointestinální nežádoucí účinky se mohou častěji objevovat na začátku léčby přípravkem Xultophy a obvykle vymizí do několika dnů nebo týdnů pokračující léčby. Nevolnost byla hlášena u 7,8 % pacientů a u většiny z nich byla přechodného charakteru. Podíl pacientů uvádějících nevolnost byl v kterémkoli čase léčby nižší než 4 % za týden. Průjem byl hlášen u 7,5 % pacientů, zvracení u 3,9 % pacientů. Četnost nevolnosti a průjmu byla u přípravku Xultophy „Častá“ a u liraglutidu „Velmi častá“. Kromě toho byla až u 3,6 % pacientů léčených přípravkem Xultophy hlášena zácpa, dyspepsie, gastritida, bolest břicha, gastroezofageální refluxní choroba, břišní distenze, říhání, flatulence a snížená chuť k jídlu.
Reakce v místě vpichu
U 2,6 % pacientů léčených přípravkem Xultophy byly hlášeny reakce v místě vpichu (včetně hematomu v místě vpichu, bolesti, krvácení, erytému, uzlíků, otoku, změny zabarvení kůže, pruritu, pocitu tepla v místě vpichu a zduření v místě vpichu). Tyto reakce byly obvykle mírné a přechodné a obvykle v průběhu léčby vymizely.
Lipodystrofie
V místě vpichu může vzniknout lipodystrofie (včetně lipohypertrofie, lipoatrofie). Průběžná cyklická změna místa vpichu v dané oblasti může pomoci snížit riziko vzniku těchto reakcí.
Zvýšená tepová _ frekvence
V klinických studiích s přípravkem Xultophy bylo pozorováno průměrné zvýšení tepové frekvence
o 2 až 3 tepy za minutu oproti výchozí hodnotě. Dlouhodobé klinické účinky zvýšení tepové frekvence nebyly stanoveny.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Jsou k dispozici omezené údaje týkající se předávkování přípravkem Xultophy.
Pokud je pacientovi podána větší dávka přípravku Xultophy, než je zapotřebí, může se u něj rozvinout hypoglykemie.
• Mírné hypoglykemické epizody lze léčit perorálním podáním glukózy nebo jiných potravin obsahujících cukr. Pacientům se proto doporučuje, aby s sebou vždy nosili potraviny s obsahem cukru.
• Závažné hypoglykemické příhody, kdy si pacient není sám schopen zajistit léčbu, lze léčit intramuskulárním nebo subkutánním podáním glukagonu (0,5 až 1 mg) zaškolenou osobou nebo intravenózní aplikací glukózy zdravotnickým pracovníkem. Glukózu je též nutné podat intravenózně, jestliže pacient na glukagon nereaguje do 10 až 15 minut. Jakmile se pacient probere k vědomí, doporučuje se podat mu perorálně sacharidy jako prevenci relapsu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: léčiva k terapii diabetu. Inzuliny a analogy dlouze působící, k injekční aplikaci. ATC kód: A10AE56.
Mechanismus účinku
Přípravek Xultophy je kombinovaný přípravek obsahující inzulin degludek a liraglutid, jež mají komplementární mechanismus účinku zlepšující kompenzaci hladiny glukózy v krvi.
Inzulin degludek je bazální inzulin tvořící po subkutánní injekci rozpustné multihexamery vedoucí ke vzniku depotního úložiště, ze kterého se inzulin degludek postupně a pomalu absorbuje do krevního oběhu, což vede k rovnoměrnému a stabilnímu účinku inzulinu degludek na snížení hladiny glukózy s nízkou mezidenní variabilitou účinku inzulinu.
Inzulin degludek se specificky váže na humánní inzulinový receptor a má stejné farmakologické účinky jako humánní inzulin.
Účinek inzulinu degludek na snížení hladiny glukózy v krvi je způsoben usnadněným vychytáváním glukózy, které následuje po navázání inzulinu na receptory svalových a tukových buněk, a současnou inhibicí výdeje glukózy z jater.
Liraglutid je analog GLP-1 (Glucagon-Like Peptide-1) s 97 % sekvenční homologií s lidským GLP-1, který se váže na receptor GLP-1 (GLP-1R) a aktivuje jej. Po subkutánním podání je protrahovaný profil účinku založen na třech mechanismech: na shlukování, které má za následek pomalou absorpci, na vazbě na albumin a na vyšší enzymatické stabilitě vůči enzymům dipeptidylpeptidáze IV (DPP-IV) a neutrální endopeptidáze (NEP), což vede k delšímu plazmatickému poločasu.
Účinek liraglutidu je zprostředkován přes specifickou interakci s receptory GLP-1 a zlepšuje kompenzaci hladiny glukózy v krvi snížením hladiny glukózy v krvi nalačno i postprandiálně. Liraglutid stimuluje sekreci inzulinu a snižuje nepřiměřeně vysokou sekreci glukagonu v závislosti na koncentraci glukózy. Když je tedy koncentrace glukózy v krvi vysoká, je stimulována sekrece inzulinu a sekrece glukagonu je inhibována. A naopak, při hypoglykemii liraglutid snižuje sekreci inzulinu a nesnižuje sekreci glukagonu. Mechanismus snižování koncentrace glukózy v krvi zahrnuje rovněž mírné zpomalení vyprazdňování žaludku.
Liraglutid snižuje tělesnou hmotnost a množství tělesného tuku mechanismem, který zahrnuje snížení hladu a snížení příjmu energie.
GLP-1 je fyziologický regulátor chuti k jídlu a příjmu potravy, ale přesný mechanismus účinku není zcela jasný. Ve studiích na zvířatech vedla periferní aplikace liraglutidu k vychytávání ve specifických oblastech mozku zapojených do regulace chuti k jídlu, kde liraglutid prostřednictvím specifické aktivace GLP-1R zvyšoval klíčové signály sytosti a snižoval klíčové signály hladu, čímž docházelo ke snižování tělesné hmotnosti.
Farmakodynamické účinky
Přípravek Xultophy má stabilní farmakodynamický profil s trváním účinku odrážejícím kombinaci jednotlivých profilů účinku inzulinu degludek a liraglutidu, což umožňuje podávání přípravku Xultophy jednou denně v kteroukoli denní dobu s jídlem či bez něj. Přípravek Xultophy zlepšuje kompenzaci hladiny glukózy prostřednictvím trvalého snižování plazmatických hladin glukózy nalačno a postprandiálních hladin glukózy po všech jídlech.
Redukce postprandiální glukózy byla potvrzena v dílčí studii 4hodinovým testem se standardizovaným jídlem u pacientů, u nichž užívání metforminu samotného nebo v kombinaci s pioglitazonem nevedlo k dostatečné kompenzaci hladiny glukózy v krvi. Přípravek Xultophy snižoval výkyvy hladiny postprandiální plazmatické glukózy (v průměru přes 4 hodiny) významněji než inzulin degludek. Výsledky přípravku Xultophy a liraglutidu byly podobné.
Klinická účinnost a bezpečnost
Přídatná léčba kperorálním přípravkům snižujícím hladinu glukózy Přídatná léčba k metforminu v monoterapii nebo v kombinaci s pioglitazonem
Účinnost a bezpečnost přípravku Xultophy v porovnání s inzulínem degludek a liraglutidem (všechny podávány jednou denně) byly studovány v 26týdenní randomizované, kontrolované, otevřené studii k cílovým hodnotám (treat-to-target) u pacientů s diabetes mellitus 2. typu s 26týdenním prodloužením. Počáteční dávka přípravku Xultophy byla 10 dávkovacích jednotek (10 jednotek inzulinu degludek a 0,36 mg liraglutidu) a počáteční dávka inzulinu degludek byla 10 jednotek. Tato dávka byla titrována dvakrát týdně podle níže uvedené tabulky 2.
Pacienti ve větvi s liraglutidem dodržovali fixní navyšování dávky s počáteční dávkou 0,6 mg a přírůstkem dávky o 0,6 mg týdně, dokud nebylo dosaženo udržovací dávky 1,8 mg. Maximální dávka přípravku Xultophy byla 50 dávkovacích jednotek, zatímco ve větvi s inzulinem degludek nebyla dána žádná maximální dávka.
Tabulka 2 Titrace přípravku Xultophy a bazálního inzulinu
|
Hladina glukózy v plazmě před snídaní* |
Úprava dávky | ||
|
Xultophy |
Bazální inzulin | ||
|
mmol/l |
mg/dl |
(dávkovací j ednotky) |
(jednotky) |
|
< 4,0 |
< 72 |
-2 |
-2 |
|
4,0-5,0 |
72-90 |
0 |
0 |
|
> 5,0 |
> 90 |
+2 |
+2 |
* Hladiny glukózy v plazmě změřené pacientem
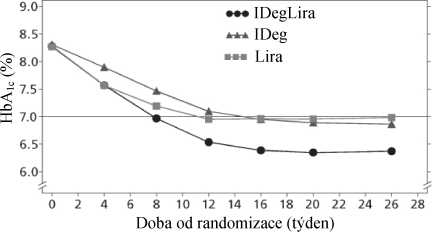
Nejdůležitější výsledky studie jsou uvedeny na obrázku 1 a v tabulce 3.
Po 26 týdnech léčby dosáhlo 60,4 % pacientů léčených přípravkem Xultophy cílové hodnoty HbA1c < 7 % bez potvrzených hypoglykemických epizod. Tento poměr byl výrazně vyšší, než jaký byl pozorován u inzulinu degludek (40,9 %; poměr pravděpodobnosti 2,28; p<0,0001), a podobný jako u liraglutidu (57,7 %; poměr pravděpodobnosti 1,13; p=0,3184).
Četnosti potvrzených hypoglykemií byly nižší u přípravku Xultophy než u inzulinu degludek bez ohledu na kompenzaci hladiny glukózy, viz obrázek 1.
9.0
8.5
8.0
7.5
7.0
6.5
6.0
6.5
• IDegLira poz. čet.
----IDegLira
- IDeg
A IDeg poz. čet.
5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0
HbAic (%) na konci léčby
Křivky jsou průměrné četnosti hypoglykemií z negativního binominálního modelu s unikátními trajektoriemi léčby a symboly jsou pozorované četnosti hypoglykemie versus průměrné hodnoty HbÁ1c podle kvartilů.
6.0
5.5
5.0 1 4.5
4.0 . 3.5
3.0
2.5
2.0 i 1.5 . 1.0 ' 0.5
0.0
IDegLira = Xultophy, IDeg = inzulin degludek, Lira = liraglutid, poz.čet. = pozorovaná četnost, REP = na jednoho pacienta a rok léčby
Obrázek 1 Průměrný HbAic (%) v závislosti na týdnech léčby (vlevo) a četnost potvrzených hypoglykemií na jednoho pacienta a rok léčby versus průměrný HbAic (%) (vpravo) u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných metforminem v monoterapii nebo v kombinaci s pioglitazonem
Četnost závažných hypoglykemií, definovaných jako epizoda vyžadující pomoc další osoby, na jednoho pacienta a rok léčby (vyjádřeno v procentech pacientů) byla 0,01 (2 pacienti z 825) u přípravku Xultophy, 0,01 (2 pacienti z 412) u inzulinu degludek a 0,00 (0 pacientů z 412) u liraglutidu. Četnost nočních hypoglykemických příhod byla u léčby přípravkem Xultophy a inzulinem degludek podobná.
Pacienti léčení přípravkem Xultophy měli celkově méně gastrointestinálních nežádoucích účinků než pacienti léčení liraglutidem. Důvodem může být pomalejší zvyšování dávky složky liraglutidu během zahájení léčby přípravkem Xultophy v porovnání s použitím liraglutidu v monoterapii.
Dlouhodobé údaje (52 týdnů) u pacientů nedostatečně kompenzovaných metforminem v monoterapii nebo v kombinaci s pioglitazonem
Účinnost a bezpečnost přípravku Xultophy byly sledovány až po dobu 52 týdnů léčby. Pokles HbA1c z výchozí hodnoty za 52 týdnů byl 1,84 % u přípravku Xultophy s odhadovaným léčebným rozdílem - 0,65 % v porovnání s liraglutidem (p<0,0001) a -0,46 % v porovnání s inzulinem degludek (p<0,0001). Tělesná hmotnost se snížila o 0,4 kg s odhadovaným léčebným rozdílem mezi přípravkem Xultophy a inzulinem degludek -2,80 kg (p<0,0001) a četnost potvrzených hypoglykemií zůstala na hodnotě 1,8 příhody na jednoho pacienta a rok léčby se zachováním významného snížení celkového rizika potvrzených hypoglykemií v porovnání s inzulinem degludek.
Přídatná léčba k derivátům sulfonylurey v monoterapii nebo v kombinaci s metforminem Účinnost a bezpečnost přípravku Xultophy jako přídatné léčby k derivátům sulfonylurey v monoterapii nebo v kombinaci s metforminem byly studovány v 26týdenní randomizované, placebem kontrolované, dvojitě zaslepené studii k cílovým hodnotám (treat-to-target) u 435 pacientů s diabetes mellitus 2. typu, z nichž bylo 289 léčeno přípravkem Xultophy. Počáteční dávka přípravku Xultophy byla 10 dávkovacích jednotek (10 jednotek inzulinu degludek a 0,36 mg liraglutidu) a dávka byla titrována dvakrát týdně. Titrace byla prováděna podle tabulky 2 s cílovou titrací 4 - 6 mmol/l.
Nej důležitější výsledky studie jsou uvedeny na obrázku 2 a v tabulce 3.

0 2 4 6 8 10 12 14 16 18 20 22 24 26 28
Doba od randomizace (týden)
IDegLira = Xultophy
9.0
8.5
8.0
7.5
7.0
6.5
6.0
Obrázek 2 Průměrný HbA1c (%) v závislosti na týdnech léčby u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných deriváty sulfonylurey v monoterapii nebo v kombinaci s metforminem
Četnost závažných hypoglykemií na jednoho pacienta a rok léčby (vyjádřeno v procentech pacientů) byla 0,02 (2 pacienti z 288) u přípravku Xultophy a 0,00 (0 pacientů ze 146) u placeba.
|
Předchozí léčba metforminem ± pioglitazonem |
Předchozí léčba deriváty sulfonylurey ± metforminem | ||||
|
Xultophy |
Inzulin degludek |
Liraglutid |
Xultophy |
Placebo | |
|
N |
833 |
413 |
414 |
289 |
146 |
|
HbAjc (%) Výchozí hodnota^konec studie Průměrná změna Odhadovaný rozdíl |
8,3^6,4 -1,91 |
8,3^6,9 -1,44 -0,47ab[-0,58; -0,36] |
8,3^7,0 -1,28 -0,64ab[-0,75; -0,53] |
7,9^6,4 -1,45 |
7,9^7,4 -0,46 -1,02ab[-1, 18; -0,87] |
Tabulka 3 Výsledky z 26týdenních studií s přípravkem Xultophy u pacientů s diabetes mellitus
2. typu buď nedostatečně kompenzovaných metforminem v monoterapii nebo v kombinaci s pioglitazonem (vlevo), nebo nedostatečně kompenzovaných deriváty sulfonylurey v monoterapii nebo v kombinaci s metforminem (vpravo)__
|
Pacienti (%) dosahující HbA1c < 7 % Všichni pacienti Odhadovaný poměr pravděpodobnosti |
80,6 |
65,1 2,38b [1,78; 3,18] |
60,4 3,26b [2,45; 4,33] |
79,2 |
28,8 11,95b [7,22; 19,77] |
|
Pacienti (%) dosahující HbAic < 6,5 % Všichni pacienti Odhadovaný poměr pravděpodobnosti |
69,7 |
47,5 2,82b [2,17; 3,67] |
41,1 3,98b [3,05; 5,18] |
64,0 |
12,3 16,36b [9,05; 29,56] |
|
Četnost potvrzených hypoglykemií* na jednoho pacienta a rok léčby (procento pacientů) Odhadovaný poměr |
1,80 (31,9 %) |
2,57 (38,6 %) 0,68ac[0,53; 0,87] |
0,22 (6,8 %) 7,61b [5,17; 11,21] |
3,52 (41,7 %) |
1,35 (17,1 %) 3, 74b [2,28; 6,13] |
|
Tělesná hmotnost (kg) Výchozí hodnota—konec studie Průměrná změna Odhadovaný rozdíl |
87,2—86,7 -0,5 |
87,4—89,0 1,6 -2,22ab [-2,64; -1,80] |
87,4—84,4 -3,0 2,44b [2,02; 2,86] |
87,2—87,7 0,5 |
89,3—88,3 -1,0 1,48b [0,90; 2,06] |
|
FPG (mmol/l) Výchozí hodnota—konec studie Průměrná změna Odhadovaný rozdíl |
9,2—5,6 -3,62 |
9,4—5,8 -3,61 -0,17[-0,41; 0,07] |
9,0—7,3 -1,75 -1,76b [-2,0; -1,53] |
9,1—»6,5 -2,60 |
9,1—8,8 -0,31 -2,30b [-2,72; -1,89] |
|
Dávka na konci studie Inzulin degludek (jednotky) Liraglutid (mg) Odhadovaný rozdíl, dávka inzulinu degludek |
CO ^ |
53 -14,90ab [-17,14; -12,66] |
1,8 |
28 1,0 |
- |
Výchozí hodnota, hodnota na konci studie a změna hodnot jsou pozorovaným přeneseným posledním pozorováním. 95 % interval spolehlivosti je uveden v „[]“.
*Potvrzené hypoglykemie definované jako závažná hypoglykemie (epizoda vyžadující pomoc další osoby) a/nebo lehká hypoglykemie (hladina glukózy v plazmě < 3,1 mmol/l bez ohledu na příznaky).
A Cílové parametry s potvrzenou superioritou přípravku Xultophy versus komparátor.
B p<0,0001
C p<0,05
Převedení z terapie agonisty receptoru GLP-1
Účinnost a bezpečnost přípravku Xultophy (podávaného jedenkrát denně) byly studovány v porovnání s nezměněnou léčbou agonisty receptoru GLP-1 (podávaných dle schválené dokumentace) ve 26týdenní randomizované otevřené studii k cílovým hodnotám (treat-to target) u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných léčbou samotnými agonisty receptoru GLP-1 a metforminem (74,2 %) či v kombinaci s pioglitazonem (2,5 %), sulfonylureou (21,2 %) nebo oběma (2,1 %).
Počáteční dávka přípravku Xultophy byla 16 dávkovacích jednotek (16 jednotek inzulinu degludek a 0,6 mg liraglutidu) a dávka byla titrována dvakrát týdně dle tabulky 2. Pacienti ve větvi s agonisty receptoru GLP-1 měli pokračovat v předchozí léčbě agonisty receptoru GLP-1.
Nejdůležitější výsledky studie jsou uvedeny v tabulce 4 a na obrázku 3.
|
Předchozí léčba agonisty receptoru GLP-1 | ||
|
Xultophy |
Agonisté receptoru GLP-1 | |
|
N |
292 |
146 |
|
HbAic (%) Výchozí hodnota—konec studie |
7,8—6,4 |
7,7—7,4 |
|
Průměrná změna |
-1,3 |
-0,3 -0,94ab[-1,11; -0,78] |
Tabulka 4 Výsledky 26týdenní studie s přípravkem Xultophy u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných ^ agonisty receptoru GLP-1_
3 7.5
<
S 70
6.5
|
- |
*** IDegLira GLP-1 RA nezměněn | |
|
" -—m—- ■- |
-m | |
|
; |
----•---- |
-* - |
9.0
8.5
8.0
7.5
7.0
6.5
6.0
4 6 8 10 12 14 16 18 20 22 24 26 28
Doba od randomizace (týden)
|
Odhadovaný rozdíl | ||
|
Pacienti (%) dosahující HbAic < 7 % Všichni pacienti Odhadovaný poměr pravděpodobnosti |
75,3 |
35,6 6,84B [4,28; 10,94] |
|
Pacienti (%) dosahující HbAic < 6,5 % Všichni pacienti Odhadovaný poměr pravděpodobnosti |
63,0 |
22,6 7,53b [4,58; 12,38] |
|
Četnost potvrzených hypoglykemií* na jednoho pacienta a rok léčby (procento pacientů) Odhadovaný poměr |
2,82 (32,0%) |
0,12 (2,8%) 25,36b [10,63; 60,51] |
|
Tělesná hmotnost (kg) Výchozí hodnota^konec studie Průměrná změna Odhadovaný rozdíl |
95,6^-97,5 2,0 |
95,5^-94,7 -0,8 2,89b [2,17; 3,62] |
|
FPG (mmol/l) Výchozí hodnota^konec studie Průměrná změna Odhadovaný rozdíl |
9,0^6,0 -2,98 |
9,4^8,8 -0,60 -2,64b [-3,03; -2,25] |
|
Dávka na konci studie Inzulin degludek (jednotky) Liraglutid (mg) Odhadovaný rozdíl, dávka inzulinu degludek |
43 1,6 |
Dávka agonistů receptoru GLP-1 měla pokračovat beze změny oproti výchozí hodnotě |
Výchozí hodnota, hodnota na konci studie a změna hodnot jsou pozorovaným přeneseným posledním pozorováním. 95 % interval spolehlivosti je uveden v „[]“.
*Potvrzené hypoglykemie definované jako závažná hypoglykemie (epizoda vyžadující pomoc další osoby) a/nebo lehká hypoglykemie (hladina glukózy v plazmě < 3,1 mmol/l bez ohledu na příznaky).
A Cílové parametry s potvrzenou superioritou přípravku Xultophy versus komparátor.
B p<0,001
IDegLira=Xultophy, GLP-1 RA=agonisté receptoruGLP-1
Obrázek 3 Průměrný HbA1c (%) v závislosti na týdnech léčby u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných agonisty receptoru GLP-1
Četnost na jednoho pacienta a rok léčby (vyjádřeno v procentech pacientů) u závažných hypoglykemií byla 0,01 (1 pacient z 291) u přípravku Xultophy a 0,00 (0 pacientů ze 199) u agonistů receptoru GLP-1.
Převod z terapie bazálním inzulinem
Účinnost a bezpečnost přípravku Xultophy v porovnání s inzulinem glargin (oba podávány jednou denně) byly studovány v 26týdenní randomizované, otevřené studii k cílovým hodnotám (treat-to-target) u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných inzulinem glargin (20 -50 jednotek) a metforminem. Počáteční dávka přípravku Xultophy byla 16 dávkovacích jednotek a počáteční dávka inzulinu glargin byla totožná s denní dávkou před započetím studie. Dávka byla v obou větvích titrována dvakrát týdně podle tabulky 2. Maximální povolená dávka byla 50 dávkovacích jednotek přípravku Xultophy, zatímco pro inzulin glargin maximální dávka neexistovala.
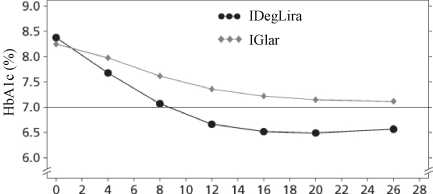
Nejdůležitější výsledky studie jsou uvedeny v tabulce 5 a na obrázku 4.
54,3 % pacientů léčených přípravkem Xultophy dosáhlo cílové hodnoty HbA1c <7 % bez potvrzených
hypoglykemických epizod oproti 29,4 % pacientů léčených inzulínem glargin (poměr pravděpodobnosti 3,24; p<0,001).
Doba od randomizace (týden)
IDegLira = Xultophy, IGlar = inzulin glargin
Obrázek 4 Průměrný HbA1c (%) v závislosti na týdnech léčby u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných inzulínem glargin
Četnost závažných hypoglykemií na jednoho pacienta a rok léčby (vyjádřeno v procentech pacientů) byla 0,00 (0 pacientů z 278) u přípravku Xultophy a 0,01 (1 pacient z 279) u inzulinu glargin. Četnost nočních hypoglykemických příhod byla významně nižší u léčby přípravkem Xultophy v porovnání s léčbou inzulinem glargin (odhadovaný léčebný poměr 0,17; p<0,001).
Účinnost a bezpečnost přípravku Xultophy v porovnání s inzulinem degludek (oba podávány jednou denně) byly studovány v 26týdenní randomizované, dvojitě zaslepené studii k cílovým hodnotám (treat-to-target) u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných bazálním inzulinem (20 - 40 jednotek) a metforminem v monoterapii nebo v kombinaci s deriváty sulfonylurey/glinidy. Podávání bazálního inzulinu a derivátů sulfonylurey/glinidů bylo při randomizaci přerušeno.
Počáteční dávka přípravku Xultophy byla 16 dávkovacích jednotek (16 jednotek inzulinu degludek a 0,6 mg liraglutidu) a počáteční dávka inzulinu degludek byla 16 jednotek. Dávka byla titrována dvakrát týdně podle tabulky 2. Maximální povolená dávka byla 50 dávkovacích jednotek přípravku Xultophy a 50 jednotek inzulinu degludek.
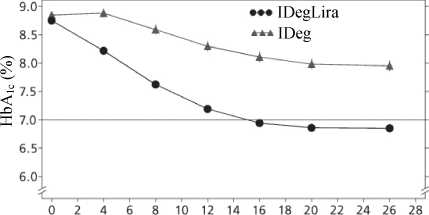
Nejdůležitější výsledky studie jsou uvedeny v tabulce 5 a na obrázku 5.
48,7 % pacientů dosáhlo cílové hodnoty HbAic < 7 % bez potvrzených hypoglykemických epizod, což byl významně vyšší poměr, než jaký byl pozorován u inzulinu degludek (15,6 %; poměr pravděpodobnosti 5,57; p<0,0001).
9.0
8.5
8.0
7.5
7.0
6.5
6.0
Doba od randomizace (týden)
IDegLira = Xultophy, IDeg = inzulin degludek
Obrázek 5 Průměrný HbA1c (%) v závislosti na týdnech léčby u pacientů s diabetes mellitus 2. typu nedostatečně kompenzovaných bazálním inzulinem
Četnost na jednoho pacienta a rok léčby (vyjádřeno v procentech pacientů) u závažných hypoglykemií byla 0,01 (1 pacient ze 199) u přípravku Xultophy a 0,00 (0 pacientů ze 199) u inzulinu degludek.
Četnost nočních hypoglykemických příhod byla u léčby přípravkem Xultophy a inzulínem degludek podobná.
|
Předchozí léčba inzulinem glargin |
Předchozí léčba bazálním inzulinem (NPH, inzulin detemir, inzulin glargin) | ||||
|
Xultophy |
Inzulin glargin, žádné omezení dávky |
Xultophy |
Inzulin degludek, maximální dovolená dávka 50 jednotek | ||
|
N |
278 |
279 |
199 |
199 | |
|
HbA1c (%) Výchozí hodnota—konec studie Průměrná změna Odhadovaný rozdíl |
8,4—6,6 -1,81 |
8,2—7,1 -1,13 -0,59AB[-0,74; -0,45] |
8,7—6,9 -1,90 |
8,8—8,0 -0,89 -1,05ab[-1,25; -0,84] | |
|
Pacienti (%) dosahující HbA1C <7 % Všichni pacienti Odhadovaný poměr pravděpodobnosti |
71,6 |
47,0 3,45b [2,36;5,05] |
60,3 |
23,1 5,44b [3,42; 8,66] | |
|
Pacienti (%) dosahující HbAic <6,5 % Všichni pacienti Odhadovaný poměr pravděpodobnosti |
55,4 |
30,8 3,29b [2,27; 4,75] |
45,2 |
13,1 5,66b [3,37; 9,51] | |
|
Četnost potvrzených hypoglykemií* na jednoho pacienta a rok léčby (procento pacientů) Odhadovaný poměr |
2,23 (28,4%) |
5,05 (49,1%) 0,43ab [0,30;0,61] |
1,53 (24,1%) |
2,63 (24,6%) 0,66 [0,39; 1,13] | |
|
Tělesná hmotnost (kg) Výchozí hodnota—konec studie Průměrná změna Odhadovaný rozdíl |
88,3—86,9 -1,4 |
87,3—89,1 1,8 -3,20ab [-3,77; -2,64] |
95,4—92,7 -2,7 |
93,5—93,5 0,0 -2,51b [-3,21; -1,82] | |
|
FPG (mmol/l) Výchozí hodnota—konec studie Průměrná změna Odhadovaný rozdíl |
8,9—>6,1 -2,83 |
8,9—6,1 -2,77 -0,01 [-0,35; 0,33] |
9,7—6,2 -3,46 |
9,6—7,0 -2,58 -0,73c[-1,19; -0,27] | |
|
Dávka na konci studie Inzulin (jednotky) Liraglutid (mg) Odhadovaný rozdíl, dávka bazálního inzulinu |
41 1,5 |
66d -25,47b [-28,90;-22,05] |
45 1,7 |
45 -0,02 [-1,88; 1,84] | |
Tabulka 5 Výsledky 26týdenní studie s přípravkem Xultophy u pacientů s diabetes mellitus 2. typu buď nedostatečně kompenzovaných inzulínem glargin (vlevo) nebo bazálním inzulínem (vpravo)__^_
Výchozí hodnota, hodnoty na konci studie a změna hodnot jsou pozorovaným přeneseným posledním pozorováním. 95 % interval spolehlivosti je uveden v „[]“.
*Potvrzené hypoglykemie definované jako závažná hypoglykemie (epizoda vyžadující pomoc další osoby) a/nebo lehká hypoglykemie (hladina glukózy v plazmě < 3,1 mmol/l bez ohledu na příznaky).
A Cílové parametry s potvrzenou superioritou přípravku Xultophy versus komparátor.
B p<0,0001
C p<0,05
D Průměrná dávka inzulinu glargin před započetím studie byla 32 jednotek
Další klinické údaje
Sekrece inzulinu/funkce beta buněk
Přípravek Xultophy v porovnání s inzulinem degludek zlepšuje funkci beta buněk (měřeno pomocí homeostatického modelového stanovení funkce beta buněk (HOMA-P)). Po 52 týdnech léčby byla u 260 pacientů s diabetes mellitus 2. typu prokázána zlepšená sekrece inzulinu ve srovnání s inzulinem degludek v odpovědi na test se standardizovaným jídlem. Pro léčbu přesahující 52 týdnů léčby nejsou dostupné žádné údaje.
Krevní tlak
U pacientů nedostatečně kompenzovaných samotným metforminem nebo v kombinaci s pioglitazonem snížil přípravek Xultophy průměrný systolický krevní tlak o 1,8 mmHg v porovnání s inzulínem degludek (snížení o 0,7 mmHg) a s liraglutidem (snížení o 2,7 mmHg). U pacientů, kteří byli nedostatečně kompenzováni samotnou sulfonylureou nebo v kombinaci s metforminem bylo snížení u přípravku Xultophy 3,5 mmHg a u placeba 3,2 mmHg. Rozdíly nebyly statisticky významné. Ve dvou studiích s pacienty nedostatečně kompenzovanými léčbou bazálním inzulinem bylo snížení systolického krevního tlaku u přípravku Xultophy 5,4 mmHg a 1,7 mmHg u inzulinu degludek, s odhadovaným statisticky významným léčebným rozdílem -3,71 mmHg (p=0,0028) a 3,7 mmHg u přípravku Xultophy oproti 0,2 mmHg u inzulinu glargin s odhadovaným statisticky významným léčebným rozdílem -3,57 mmHg (p<0,001).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xultophy u všech podskupin pediatrické populace při léčbě diabetes mellitus 2. typu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika inzulinu degludek a liraglutidu při podávání v podobě přípravku Xultophy nebyla v porovnání se samostatnými injekcemi inzulinu degludek a liraglutidu významným způsobem celkově klinicky ovlivněna.
Následující informace odráží farmakokinetické vlastnosti přípravku Xultophy, pokud není uvedeno, že předkládané údaje byly získány ze samostatného podávání inzulinu degludek nebo liraglutidu.
Absorpce
Celková expozice inzulinem degludek po podávání přípravku Xultophy byla ekvivalentní v porovnání s inzulinem degludek v monoterapii, zatímco hodnota Cmax byla vyšší o 12 %. Celková expozice liraglutidem po podávání přípravku Xultophy byla ekvivalentní v porovnání s liraglutidem v monoterapii, zatímco hodnota Cmax byla nižší o 23 %. Tyto rozdíly nemají žádný klinický význam, neboť podávání přípravku Xultophy se zahajuje a titruje podle cílových hodnot glukózy v krvi u jednotlivých pacientů.
Na základě populační farmakokinetické analýzy narůstá expozice inzulinem degludek a liraglutidem proporcionálně s dávkou přípravku Xultophy v celém dávkovacím rozmezí.
Farmakokinetický profil přípravku Xultophy je konzistentní při dávkování jednou denně a ustáleného stavu koncentrace inzulinu degludek a liraglutidu je dosaženo za 2-3 dny každodenní aplikace.
Distribuce
Inzulin degludek a liraglutid se ve velké míře vážou na bílkoviny v plazmě (> 99 % respektive > 98 %).
Biotransformace
Inzulin degludek
Odbourávání inzulinu degludek je podobné jako u humánního inzulinu; všechny vzniklé metabolity jsou inaktivní.
Liraglutid
Během 24 hodin po podání jednorázové dávky radioaktivně značeného liraglutidu s [3H] zdravým subjektům byla hlavní složka v plazmě intaktní liraglutid. V plazmě byly zjištěny dva méně významné metabolity (< 9 % a < 5 % celkové expozice plazmatické radioaktivity). Liraglutid je metabolizován podobným způsobem jako velké bílkoviny, přičemž nebyl zjištěn určitý orgán jako hlavní cesta eliminace.
Poločas inzulínu degludek je přibližně 25 hodin a poločas liraglutidu je přibližně 13 hodin.
Zvláštní skupiny pacientů
Starší _ pacienti
Podle výsledků populační farmakokinetické analýzy u dospělých pacientů až do věku 83 let léčených přípravkem Xultophy nemá věk žádný klinicky významný vliv na farmakokinetiku přípravku Xultophy.
Pohlaví
Podle výsledků populační farmakokinetické analýzy nemá pohlaví žádný klinicky významný vliv na farmakokinetiku přípravku Xultophy.
Etnický původ
Podle výsledků populační farmakokinetické analýzy, která zahrnovala pacienty z bělošských, černošských, indických, asijských a hispánských skupin, nemá etnický původ žádný klinicky významný vliv na farmakokinetiku přípravku Xultophy.
Porucha _ funkce ledvin Inzulin degludek
Mezi zdravými subjekty a pacienty s poruchou funkce ledvin nebyly zjištěny rozdíly ve farmakokinetice inzulinu degludek.
Liraglutid
U pacientů s poruchou funkce ledvin byla expozice liraglutidem ve srovnání s jedinci s normální funkcí ledvin snížena. U pacientů s mírnou (clearance kreatininu Clkr 50 - 80 ml/min) poruchou funkce ledvin byla expozice liraglutidem snížena o 33 %, u pacientů se středně závažnou (Clkr 30 -50 ml/min) poruchou funkce ledvin o 14 %, se závažnou (Clkr < 30 ml/min) poruchou funkce ledvin o 27 % a u terminálního selhání ledvin vyžadujícího dialýzu byla expozice liraglutidem snížena o 28 %. Podobně ve 26týdenní klinické studii s pacienty s onemocněním diabetes mellitus 2. typu a se středně závažnou poruchou funkce ledvin (Clkr 30 - 59 ml/min) byla expozice liraglutidu o 26 % nižší v porovnání se samostatnou klinickou studií, v níž byli zahrnuti pacienti s onemocněním diabetes mellitus 2. typu s normální funkcí ledvin či s mírnou poruchou funkce ledvin.
Porucha _ funkce _ jater Inzulin degludek
Mezi zdravými subjekty a pacienty s poruchou funkce jater nebyly zjištěny rozdíly ve farmakokinetice inzulinu degludek.
Liraglutid
Farmakokinetika liraglutidu byla hodnocena u subjektů s různým stupněm poruchy funkce jater ve studii s jednorázovou dávkou. U subjektů s lehkou až středně závažnou poruchou funkce jater byla expozice liraglutidem snížená ve srovnání se zdravými subjekty o 13 - 23 %. Expozice byla významně nižší (44 %) u subjektů se závažnou poruchou funkce jater (skóre Child Pugh > 9).
Pediatrická _ populace
U dětí a dospívajících mladších 18 let nebyly provedeny žádné studie s přípravkem Xultophy.
5.3 Předklinické údaje vztahující se k bezpečnosti
Program neklinického vývoje inzulinu degludek/liraglutidu zahrnoval pivotní studie kombinační toxicity trvající až 90 dnů u jediného relevantního druhu (potkani Wistar) k podpoře programu klinického vývoje. Lokální tolerance byla hodnocena u králíků a prasat.
Neklinické údaje vztahující se k bezpečnosti získané na základě studií toxicity po opakovaném podávání neodhalily žádné bezpečnostní riziko pro člověka.
Lokální reakce tkáně ve dvou studiích u králíků a prasat byly omezeny na mírné zánětlivé reakce.
Nebyly provedeny žádné studie s kombinací inzulin degludek/liraglutid ke zhodnocení kancerogeneze, mutageneze či snížení fertility. Následující údaje jsou založeny na studiích s inzulinem degludek a liraglutidem použitých jednotlivě.
Inzulin degludek
Neklinické údaje získané na základě farmakologických studií bezpečnosti, toxicity po opakovaném podávání, hodnocení kancerogenního potenciálu a reprodukční toxicity neodhalily žádné bezpečnostní riziko pro člověka.
Poměr mitogenního k metabolickému potenciálu inzulinu degludek je nezměněný ve srovnání s tímto poměrem pro humánní inzulin.
Liraglutid
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání nebo genotoxicity neodhalily žádné zvláštní riziko pro člověka. Ve dvouletých studiích kancerogenity na potkanech a myších byly pozorovány tumory z C-buněk štítné žlázy, které nebyly letální. U potkanů nebyl pozorován NOAEL (No observed adverse effect level). Tyto tumory nebyly pozorovány u opic léčených po dobu 20 měsíců. Tyto nálezy u hlodavců jsou způsobeny negenotoxickým a receptory zprostředkovaným mechanismem specifickým pro GLP-1, na který jsou hlodavci zvláště citliví. Význam pro člověka je pravděpodobně nízký, ale nemůže být zcela vyloučen. Žádné jiné tumory spojené s léčbou nebyly zjištěny.
Studie na zvířatech neprokázaly přímý škodlivý vliv týkající se fertility, prokázaly však lehce zvýšenou míru časných úmrtí embryí po nejvyšší dávce. Podávání liraglutidu ve střední fázi těhotenství vedlo ke snížení tělesné hmotnosti matky a zpomalení růstu plodu s neprůkazným vlivem na žebra u potkanů a kosterní změny u králíků. Růst novorozenců byl u potkanů vystavených působení liraglutidu snížen a tento efekt přetrvával u skupiny s vysokou dávkou i po odstavení. Není známo, zda je redukovaný růst mláďat způsoben sníženým příjmem mléka v důsledku přímého vlivu GLP-1, nebo sníženou tvorbou mateřského mléka způsobenou snížením kalorického příjmu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Glycerol
Fenol
Zinkum-acetát
Kyselina chlorovodíková (pro úpravu pH)
Hydroxid sodný (pro úpravu pH)
Voda na injekci
6.2 Inkompatibility
Látky přidané k přípravku Xultophy mohou způsobit degradaci léčivých látek. Přípravek Xultophy se nesmí přidávat do infuzních roztoků.
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Po prvním otevření lze přípravek uchovávat po dobu 21 dnů při maximální teplotě 30 °C. Přípravek je nutné 21 dnů po prvním otevření zlikvidovat.
6.4 Zvláštní opatření pro uchovávání
Před prvním otevřením: uchovávejte v chladničce (2 °C - 8 °C). Neuchovávejte v blízkosti mrazicího zařízení. Chraňte před mrazem. Uchovávejte předplněné pero s nasazeným uzávěrem, aby byl přípravek chráněn před světlem.
Po prvním otevření: uchovávejte při maximální teplotě do 30 °C nebo uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte předplněné pero s nasazeným uzávěrem, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
3 ml roztoku v zásobní vložce (sklo typu 1), s pístem (halobutyl) a zátkou (halobutyl/polyisopren) vložené do jednorázového předplněného pera pro vícenásobné dávkování zhotoveného z polypropylenu, polykarbonátu a akrylonitrilbutadienstyrenu.
Velikosti balení: po 1, 3, 5 předplněných perech a skupinové balení obsahující 10 předplněných per (2 balení po 5).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Předplněné pero je určené k použití s injekčními jehlami NovoTwist nebo NovoFine o délce do 8 mm a tloušťce do 32 G.
Předplněné pero je určeno k použití pouze jednou osobou.
Přípravek Xultophy nesmí být použit v případě, že roztok není čirý a bezbarvý.
Přípravek Xultophy, který byl zmražen, nesmí být použit.
Pacient musí po každé aplikaci zlikvidovat použitou jehlu.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Podrobné pokyny k použití naleznete v příbalové informaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/14/947/001
EU/1/14/947/002
EU/1/14/947/003
EU/1/14/947/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18. září 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCI BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologických léčivých látek
Novo Nordisk A/S
Hallas Alle, Kalundborg, 4400, Dánsko
Novo Nordisk A/S
Novo Allé, Bagsv^rd, 2880, Dánsko
Název a adresa výrobce odpovědného za propouštění šarží
Novo Nordisk A/S
Novo Allé, Bagsv^rd, 2880, Dánsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci poskytne před uvedením přípravku na trh edukační materiály určené všem lékařům a zdravotním sestrám, u nichž se předpokládá, že budou zapojeni do léčby a péče o
diabetické pacienty, a rovněž všem lékárníkům, u nichž se předpokládá, že budou přípravek Xultophy vydávat.
Držitel rozhodnutí o registraci musí před distribucí edukačních materiálů v příslušném členském státě nechat odsouhlasit konečný obsah a způsob distribuce edukačních materiálů společně s komunikačním plánem příslušnou národní agenturou členského státu.
Účelem edukačních materiálů je zvýšit povědomí o skutečnosti, že přípravek Xultophy obsahuje fixní kombinaci inzulinu degludek a liraglutidu (založeného na bázi GLP-1), aby se tak minimalizovalo riziko chyb v medikaci při používání přípravku Xultophy.
Držitel rozhodnutí o registraci musí zajistit, aby zdravotničtí pracovníci byli informováni o tom, že všichni pacienti, kterým byl předepsán přípravek Xultophy, musí být před předepsáním či vydáním přípravku Xultophy proškoleni ve správném používání předplněného pera.
Edukační materiály musí obsahovat:
- Souhrn údajů o přípravku a příbalovou informaci
- Informační brožurku pro zdravotnické pracovníky, která musí obsahovat následující klíčové údaje:
• tento přípravek obsahuje fixní kombinaci inzulinu degludek
s liraglutidem (založeného na bázi GLP-1), což představuje nový způsob pohledu na léčbu pacientů s diabetes mellitus 2. typu. V tomto kontextu je nutno zdůraznit příslušná opatření, jak jsou uvedena v souhrnu údajů o přípravku.
• jasné vysvětlení dávkování přípravku a objasnění významu pojmu „dávkovací jednotka“ ve vztahu k dávce každé jednotlivé složky v této dávkovací jednotce
• připomenutí nutnosti hlásit všechny chyby v medikaci bez ohledu na to, zda měly za následek nežádoucí účinek či nikoliv.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xultophy 100 jednotek/ml + 3,6 mg/ml injekční roztok insulinům degludecum + liraglutidum
2. OBSAH LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje 300 jednotek insulinum degludecum a 10,8 mg liraglutidum ve 3 ml roztoku
1 ml roztoku obsahuje 100 jednotek insulinum degludecum a 3,6 mg liraglutidum
Jedna dávkovači jednotka obsahuje 1 jednotku insulinum degludecum a 0,036 mg liraglutidum
3. SEZNAM POMOCNÝCH LÁTEK
Glycerol, fenol, zinkum-acetát, kyselinu chlorovodíkovou a hydroxid sodný (pro úpravu pH) a vodu na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1x3 ml 3x3 ml 5x3 ml
5. ZPŮSOB A CESTA PODÁNÍ
Doporučeno k použití s jednorázovými jehlami NovoTwist nebo NovoFine
Jehly nejsou součástí balení
Před použitím si přečtěte příbalovou informaci
Subkutánní podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čirý bezbarvý roztok Určeno k použití pouze jednou osobou
8. POUŽITELNOST
Použitelné do
Po prvním otevření: spotřebujte do 21 dnů
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce Chraňte před mrazem
Po prvním otevření: uchovávejte při maximální teplotě do 30 °C nebo uchovávejte v chladničce Uchovávejte pero s nasazeným uzávěrem, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Po každé aplikaci jehlu zlikvidujte
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLA
EU/1/14/947/001 1 předplněné pero EU/1/14/947/002 3 předplněná pera EU/1/14/947/003 5 předplněných per
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xultophy
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Xultophy 100 jednotek/ml + 3,6 mg/ml injekční roztok insulinům degludecum + liraglutidum
2. ZPŮSOB PODÁNÍ
Subkutánní podání
3. POUŽITELNOST
Použitelné do
4. ČÍSLO ŠARŽE
č.S.
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
Novo Nordisk A/S
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xultophy 100 jednotek/ml + 3,6 mg/ml injekční roztok insulinům degludecum + liraglutidum
2. OBSAH LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje 300 jednotek insulinum degludecum a 10,8 mg liraglutidum ve 3 ml roztoku
1 ml roztoku obsahuje 100 jednotek insulinum degludecum a 3,6 mg liraglutidum
Jedna dávkovači jednotka obsahuje 1 jednotku insulinum degludecum a 0,036 mg liraglutidum
3. SEZNAM POMOCNÝCH LÁTEK
Glycerol, fenol, zinkum-acetát, kyselinu chlorovodíkovou a hydroxid sodný (pro úpravu pH) a vodu na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Skupinové balení: 10 předplněných per o obsahu 3 ml (2 balení po 5 perech)
5. ZPŮSOB A CESTA PODÁNÍ
Doporučeno k použití s jednorázovými jehlami NovoTwist nebo NovoFine
Jehly nejsou součástí balení
Před použitím si přečtěte příbalovou informaci
Subkutánní podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čirý bezbarvý roztok Určeno k použití pouze jednou osobou
8. POUŽITELNOST
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce Chraňte před mrazem
Po prvním otevření: uchovávejte při maximální teplotě do 30 °C nebo uchovávejte v chladničce Uchovávejte pero s nasazeným uzávěrem, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Po každé aplikaci jehlu zlikvidujte
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLA
EU/1/14/947/004 10 (2 x 5) předplněných per
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xultophy
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU KRABIČKA SKUPINOVÉHO BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xultophy 100 jednotek/ml + 3,6 mg/ml injekční roztok insulinům degludecum + liraglutidum
2. OBSAH LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje 300 jednotek insulinum degludecum a 10,8 mg liraglutidum ve 3 ml roztoku
1 ml roztoku obsahuje 100 jednotek insulinum degludecum a 3,6 mg liraglutidum
Jedna dávkovači jednotka obsahuje 1 jednotku insulinum degludecum a 0,036 mg liraglutidum
3. SEZNAM POMOCNÝCH LÁTEK
Glycerol, fenol, zinkum-acetát, kyselinu chlorovodíkovou a hydroxid sodný (pro úpravu pH) a vodu na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
5 předplněných per o obsahu 3 ml. Součást skupinového balení, nelze prodávat odděleně
5. ZPŮSOB A CESTA PODÁNÍ
Doporučeno k použití s jednorázovými jehlami NovoTwist nebo NovoFine
Jehly nejsou součástí balení
Před použitím si přečtěte příbalovou informaci
Subkutánní podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čirý bezbarvý roztok Určeno k použití pouze jednou osobou
8. POUŽITELNOST
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce Chraňte před mrazem
Po prvním otevření: uchovávejte při maximální teplotě do 30 °C nebo uchovávejte v chladničce Uchovávejte pero s nasazeným uzávěrem, aby byl přípravek chráněn před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Po každé aplikaci jehlu zlikvidujte
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLA
EU/1/14/947/004 10 (2 x 5) předplněných per
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xultophy
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Xultophy 100 jednotek/ml + 3,6 mg/ml injekční roztok insulinům degludecum + liraglutidum
VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Xultophy a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Xultophy používat
3. Jak se přípravek Xultophy používá
4. Možné nežádoucí účinky
5. Jak přípravek Xultophy uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xultophy a k čemu se používá K čemu se přípravek Xultophy používá
Přípravek Xultophy se používá ke zlepšení hladin glukózy (cukru) v krvi u dospělých pacientů s diabetes mellitus (cukrovkou) 2. typu. Diabetes se u člověka rozvine tehdy, když jeho tělo:
• nevytváří dostatek inzulinu ke kontrole hladiny cukru v krvi, nebo
• není schopno inzulin řádně využít.
Jak přípravek Xultophy působí
Přípravek Xultophy obsahuje dvě léčivé látky pomáhající tělu kontrolovat hladinu cukru v krvi:
• inzulin degludek - dlouhodobě působící bazální inzulin, který snižuje hladiny cukru v krvi.
• liraglutid - „analog GLP-1“, který pomáhá tělu vytvářet větší množství inzulinu během jídel a snižuje množství cukru, který tělo vytváří.
Přípravek Xultophy a perorální přípravky k léčbě diabetu
Přípravek Xultophy se používá s perorálními antidiabetiky (například metforminem, pioglitazonem a deriváty sulfonylurey). Předepisuje se v případě, že tyto léky (používané samostatně nebo s léčbou GLP-1 či s bazálním inzulinem) dostatečně nekontrolují hladiny cukru v krvi.
Pokud se léčíte přípravky GLP-1
Před zahájením léčby přípravkem Xultophy musíte ukončit léčbu přípravky GLP-1.
Pokud užíváte bazální inzulin
Při zahájení léčby přípravkem Xultophy musíte ukončit léčbu bazálním inzulinem.
2. Čemu musíte věnovat pozornost, než začnete přípravek Xultophy používat
Nepoužívejte přípravek Xultophy:
• jestliže j ste alergický(á) na inzulin degludek nebo liraglutid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Xultophy se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
• Pokud také užíváte deriváty sulfonylurey (například glimepirid nebo glibenklamid), může vám lékař v závislosti na hladinách cukru v krvi doporučit snížení dávky derivátů sulfonylurey.
• Přípravek Xultophy nepoužívejte, pokud máte diabetes mellitus 1. typu nebo máte-li „ketoacidózu“ (stav, kdy dochází k nahromadění kyseliny v krvi).
• Použití přípravku Xultophy se nedoporučuje pacientům se zánětlivým onemocněním střev nebo opožděným vyprazdňováním žaludku (diabetická gastroparéza).
Při používání přípravku Xultophy věnujte zvláštní pozornost následujícím okolnostem:
• nízká hladina cukru v krvi (hypoglykemie) - máte-li nízkou hladinu cukru v krvi, postupujte dle instrukcí v bodě 4 „Nízká hladina cukru v krvi (hypoglykemie)“.
• vysoká hladina cukru v krvi (hyperglykemie) - máte-li vysokou hladinu cukru v krvi, postupujte dle instrukcí v bodě 4 „Vysoká hladina cukru v krvi (hyperglykemie)“.
Důležité okolnosti, o kterých musíte vědět, než začnete tento přípravek používat:
Sdělte svému lékaři, pokud:
• máte problémy s očima. Rychlé zlepšení kontroly hladiny cukru v krvi může na krátkou dobu zhoršit problémy s očima způsobené diabetem. Dlouhodobé zlepšení kontroly hladiny cukru v krvi může problémy s očima zmírnit.
• máte nebo jste měl(a) onemocnění štítné žlázy.
Důležité okolnosti, o kterých musíte vědět, když tento přípravek používáte:
• pokud máte silné bolesti břicha, které neustupují, sdělte to svému lékaři. Mohla by to být známka zánětu slinivky (akutní pankreatitidy).
• dehydratace (ztráta tekutin). Může k ní dojít, pokud pociťujete nevolnost nebo zvracíte nebo máte průjem. Je důležité pít velké množství tekutin, aby se dehydratace zastavila.
Děti a dospívající
Nepodávejte tento léčivý přípravek dětem ani dospívajícím. S podáváním přípravku Xultophy dětem a dospívajícím mladším 18 let nejsou žádné zkušenosti.
Další léčivé přípravky a přípravek Xultophy
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Některé léky ovlivňují hladinu cukru v krvi, a proto může být zapotřebí změnit dávku přípravku Xultophy.
Seznam nejběžnějších léčivých přípravků, které mohou ovlivnit vaši léčbu přípravkem Xultophy, je uveden níže.
Vaše hladina cukru v krvi se může snížit, pokud užíváte:
• jiné přípravky k léčbě diabetu (tablety nebo injekce)
• sulfonamidy - k léčbě infekcí
• anabolické steroidy - jako například testosteron
• beta-blokátory - k léčbě vysokého krevního tlaku. Mohou ztížit rozeznání varovných příznaků nízké hladiny cukru v krvi (viz bod 4 „Varovné příznaky nízké hladiny cukru v krvi - mohou se objevit náhle“)
• kyselinu acetylsalicylovou (a léčivé přípravky zvané salicyláty) - k úlevě od bolesti nebo ke snížení mírné horečky
• inhibitory monoaminooxidázy (MAO) - k léčbě deprese
• inhibitory angiotenzin konvertujícího enzymu (ACE) - k léčbě některých potíží se srdcem nebo vysokého krevního tlaku.
Vaše hladina cukru v krvi se může zvýšit, pokud užíváte:
• danazol - léčivý přípravek ovlivňující ovulaci
• perorální antikoncepci - pilulky ke kontrole početí
• hormony štítné žlázy - k léčbě onemocnění štítné žlázy
• růstový hormon - při nízkých hladinách růstového hormonu
• léčivé přípravky zvané „glukokortikoidy“ jako například kortizon - k léčbě zánětů
• léčivé přípravky zvané „sympatomimetika“ jako například epinefrin (adrenalin), salbutamol nebo terbutalin - k léčbě astmatu
• tablety na odvodnění zvané „thiazidy“ - k léčbě vysokého krevního tlaku nebo pokud tělo zadržuje příliš velké množství vody (retence vody).
Oktreotid a lanreotid - používané k léčbě akromegalie (vzácné onemocnění spočívající v příliš vysoké hladině růstového hormonu). Tyto léky mohou jak zvyšovat, tak snižovat hladinu cukru v krvi.
Pioglitazon - tablety používané k léčbě diabetes mellitus 2. typu. U některých pacientů s dlouhodobým onemocněním diabetem 2. typu a srdečním onemocněním či prodělanou cévní mozkovou příhodou, kteří byli léčeni pioglitazonem a inzulinem, došlo k rozvoji srdečního selhání. Ihned informujte ošetřujícího lékaře, pokud pocítíte příznaky srdečního selhání, jako jsou neobvyklá dušnost nebo rychlý nárůst hmotnosti či lokalizovaný otok (edém).
Warfarin nebo jiné látky k ředění krve - léčivé přípravky používané k prevenci tvorby krevních sraženin. Sdělte svému lékaři, pokud užíváte warfarin nebo jiné látky k ředění krve, neboť u vás může být zapotřebí častěji provádět krevní testy sloužící ke změření srážlivosti krve (parametr zvaný „International Normalised Ratio - Mezinárodní normalizovaný poměr” neboli test INR).
Přípravek Xultophy s alkoholem
Pokud pijete alkohol, může se vaše potřeba přípravku Xultophy změnit. Vaše hladina cukru v krvi se může jak zvýšit, tak snížit. Měl(a) byste si proto hladinu cukru v krvi kontrolovat častěji, než obvykle.
Těhotenství a kojení
Pokud jste těhotná nebo plánujete otěhotnět, přípravek Xultophy nepoužívejte. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, sdělte to svému lékaři. Není známo, zda přípravek Xultophy ovlivňuje dítě.
Pokud kojíte, přípravek Xultophy nepoužívejte. Není známo, zda přípravek Xultophy přechází do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Vaše schopnost řídit dopravní prostředky nebo používat jakékoli nástroje či stroje může být ovlivněna, pokud máte hladinu cukru v krvi nízkou nebo vysokou. Pokud je hladina cukru v krvi nízká nebo vysoká, může být ovlivněna vaše schopnost soustředit se nebo reagovat. To může být nebezpečné pro vás nebo ostatní. Zeptejte se svého lékaře, jestli můžete řídit dopravní prostředky, pokud:
• máte často nízkou hladinu cukru v krvi,
• máte potíže rozpoznat stav s nízkou hladinou cukru v krvi
Důležitá informace o některých složkách přípravku Xultophy
Přípravek Xultophy obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě je „bez sodíku“.
3. Jak se přípravek Xultophy používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Váš lékař vám sdělí:
• kolik přípravku Xultophy budete každý den potřebovat,
• kdy si máte zkontrolovat hladinu cukru v krvi,
• jak si nastavit dávku.
Dávka přípravku Xultophy se podává po „dávkovacích jednotkách“. Počítadlo dávky na peru ukazuje počet dávkovacích jednotek.
Čas podání dávky
• Přípravek Xultophy si aplikujte jednou denně, pokud možno každý den ve stejnou dobu.
Vyberte si denní dobu, která vám bude nejlépe vyhovovat.
• Pokud si nemůžete přípravek Xultophy aplikovat každý den ve stejnou dobu, lze ho podat během dne v jiném čase. Ujistěte se, že mezi dávkami uplynulo minimálně 8 hodin.
• Přípravek Xultophy si nemusíte aplikovat s jídlem.
• Při nastavení dávky a její úpravě postupujte vždy dle pokynů svého lékaře.
• Pokud chcete svou obvyklou stravu změnit, domluvte se nejprve se svým lékařem, lékárníkem nebo zdravotní sestrou, jelikož změna ve stravování může změnit vaši potřebu přípravku Xultophy.
Jak zacházet s přípravkem Xultophy
Přípravek Xultophy je předplněné dávkovací pero.
• Přípravek Xultophy se podává po „dávkovacích jednotkách“. Počítadlo dávky na peru ukazuje počet dávkovacích jednotek.
• Jedna dávkovací jednotka obsahuje 1 jednotku inzulinu degludek a 0,036 mg liraglutidu.
• Maximální denní dávka přípravku Xultophy je 50 dávkovacích jednotek (50 jednotek inzulinu degludek a 1,8 mg liraglutidu).
Důkladně si pročtěte „Pokyny k použití“ na druhé straně této příbalové informace a používejte pero podle návodu.
Před aplikací přípravku vždy zkontrolujte štítek pera, abyste se ujistil(a), že používáte správné pero. Jak si aplikovat injekci
Před prvním použitím přípravku Xultophy vám váš lékař nebo zdravotní sestra ukážou, jak si injekci aplikovat.
• Přípravek Xultophy se podává jako injekce pod kůži (subkutánně). Neaplikujte jej do žíly nebo svalu.
• Nejlepší místa k aplikaci jsou přední strana stehen, horní části paží nebo přední část pasu (břicho).
• Místo v oblasti, kam injekci podáváte, denně měňte, aby se snížilo riziko vzniku bouliček nebo důlků v kůži (viz bod 4).
Podrobné pokyny k použití jsou uvedeny na druhé straně této příbalové informace.
Nepoužívejte přípravek Xultophy:
• Pokud je pero poškozeno nebo nebylo uchováváno správně (viz bod 5).
• Pokud roztok v okénku pera není čirý a bezbarvý.
Použití u starších pacientů (65 let nebo starších)
Přípravek Xultophy lze používat u starších pacientů. Pokud jste však vyššího věku, může být zapotřebí kontrolovat vaši hladinu cukru v krvi častěji. Promluvte si se svým lékařem o změnách v dávkování.
Pokud máte potíže s ledvinami nebo játry
Pokud máte potíže s ledvinami nebo játry, může být zapotřebí hladinu cukru v krvi kontrolovat častěji. Promluvte si se svým lékařem o změnách v dávkování.
Jestliže jste užil(a) více přípravku Xultophy, než jste měl(a)
Jestliže jste užil(a) více přípravku Xultophy, než jste měl(a), může vaše hladina cukru v krvi poklesnout na nízkou hodnotu (hypoglykemie) nebo vám může být nevolno nebo můžete zvracet. Máte-li nízkou hladinu cukru v krvi, postupujte dle pokynů v bodě 4 „Nízká hladina cukru v krvi (hypoglykemie)“.
Jestliže jste zapomněl(a) použít přípravek Xultophy
Pokud si zapomenete vzít dávku tohoto přípravku, aplikujte vynechanou dávku ihned po zjištění této skutečnosti. Zajistěte minimální odstup 8 hodin mezi jednotlivými dávkami. Pokud zjistíte, že jste si zapomněl(a) vzít předchozí dávku ve chvíli, kdy je čas si vzít další pravidelnou dávku, dávku nezdvojnásobujte.
Jestliže jste přestal(a) používat přípravek Xultophy
Nepřestávejte přípravek Xultophy používat, aniž byste si o tom promluvil(a) se svým lékařem. Pokud přestanete přípravek Xultophy používat, mohlo by to vést k velmi vysoké hladině cukru v krvi, viz informace v bodě 4 „Vysoká hladina cukru v krvi (hyperglykemie)“.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. U tohoto přípravku mohou nastat následující závažné nežádoucí účinky:
• Nízká hladina cukru v krvi (velmi časté: může se objevit u více než 1 pacienta z 10).
Pokud vám hladina cukru v krvi poklesne, můžete omdlít (upadnout do bezvědomí). Těžká hypoglykemie může způsobit poškození mozku a může být život ohrožující. Pocítíte-li příznaky nízké hladiny cukru v krvi, učiňte okamžitě opatření k jejímu zvýšení. Postupujte dle pokynů „Nízká hladina cukru v krvi (hypoglykemie)“ uvedených v tomto bodě níže.
• Závažná alergická reakce (anafylaktická reakce) (není známo: četnost nelze z dostupných údajů určit).
Pokud u vás dojde k závažné alergické reakci na kteroukoli ze složek přípravku Xultophy, přestaňte jej používat a ihned vyhledejte svého lékaře. Mezi příznaky závažné alergické reakce patří:
• místní reakce rozšířené na další části těla
• náhle se budete cítit špatně a začnete se potit
• budete mít potíže s dýcháním
• budete mít rychlou tepovou frekvenci nebo závrať.
Další nežádoucí účinky jsou:
Časté (mohou se projevit až u 1 pacienta z 10)
• Snížená chuť k jídlu, pocit na zvracení, zvracení, průjem, zácpa, zažívací potíže (dyspepsie), zánět sliznice žaludku (gastritida), bolest břicha, pálení žáhy nebo nadýmání - tyto potíže obvykle po několika dnech nebo týdnech odezní.
• Reakce v místě vpichu. Tyto známky mohou zahrnovat podlitiny, krvácení, bolest, zarudnutí, kopřivku, otok nebo svědění - tyto projevy obvykle po několika dnech odezní. Pokud do několika týdnů nevymizí, navštivte svého lékaře. Pokud začnou být reakce závažné, přestaňte přípravek Xultophy používat a ihned vyhledejte svého lékaře.
Méně časté (mohou se projevit až u 1 pacienta ze 100)
• Kopřivka (červené hrbolky na kůži, které mohou někdy svědit)
• Alergické reakce (hypersenzitivita) jako vyrážka, svědění a otok obličeje
• Dehydratace (ztráta tekutin) - je důležité pít velké množství tekutin, aby se dehydratace zastavila
• Říhání a větry (flatulence)
• Vyrážka
• Svědění
• Změny na kůži v místě vpichu („lipodystrofie”) - tuková tkáň pod kůží se může ztenčit („lipoatrofie“) nebo zesílit („lipohypertrofie“). Obměňování míst vpichu při každé injekci snižuje riziko takových změn kůže. Pokud zaznamenáte takovéto změny kůže, informujte svého lékaře nebo zdravotní sestru. Pokud budete dál injekce podávat do stejného místa, tyto reakce se mohou stávat závažnějšími a ovlivňovat množství léku aplikovaného perem.
• Zvýšená tepová frekvence.
Není známo (četnost nelze z dostupných údajů určit)
• Zánět slinivky břišní (pankreatitida)
• Otékání paží nebo dolních končetin (periferní edém) - když začnete lék používat, vaše tělo může zadržovat více vody, než by mělo. To vede k otoku kotníků a dalších kloubů. Tento problém většinou trvá pouze krátce.
Obecné důsledky léčby diabetů
► Nízká hladina cukru v krvi (hypoglykemie)
K nízké hladině cukru v krvi může dojít, pokud:
• pijete alkohol
• vaše fyzická aktivita je větší než obvykle
• jíte příliš málo nebo vynecháte jídlo
• použijete příliš mnoho přípravku Xultophy.
Varovné příznaky nízké hladiny cukru v krvi - mohou se objevit náhle
Bolest hlavy, nesrozumitelná artikulace, rychlá tepová frekvence, studený pot, chladná bledá pokožka, pocit na zvracení (nevolnost), pocit velkého hladu, třes, pocit nervozity či obavy, neobvyklá únava, slabost a ospalost nebo zmatenost, potíže s koncentrací, krátkodobé změny vidění.
Co dělat, pokud máte nízkou hladinu cukru v krvi:
• Vezměte si glukózové tablety nebo jinou přesnídávku s vysokým obsahem cukrů - například sladkosti, sušenky nebo ovocnou šťávu (vždy s sebou pro případ potřeby noste glukózové tablety nebo přesnídávku s vysokým obsahem cukrů).
• Pokud je to možné, změřte si hladinu cukru v krvi a odpočívejte. Může být nutné si změřit hladinu cukru v krvi vícekrát. To proto, že ke zlepšení hladiny cukru v krvi nemusí dojít hned.
• Počkejte, dokud příznaky nízké hladiny cukru v krvi neustoupí nebo dokud se hladina cukru v krvi neupraví. Poté pokračujte v léčbě obvyklým způsobem.
Co musí udělat ostatní, když ztratíte vědomí:
Řekněte lidem ve svém okolí, že máte diabetes. Informujte je o tom, k čemu může dojít, pokud vaše hladina cukru v krvi poklesne, včetně rizika ztráty vědomí.
Informujte je, že pokud ztratíte vědomí, musí postupovat následovně:
• otočit vás na bok,
• ihned vyhledat lékařskou pomoc,
• nedávat vám žádné jídlo ani nápoj, jelikož byste se mohl(a) udusit.
Z bezvědomí se můžete zotavit rychleji, pokud vám bude injekčně podán glukagon. Ten může podat pouze osoba, která ví, jak jej použít.
• Pokud dostanete glukagon, budete ihned poté, co nabudete vědomí, potřebovat cukr nebo sladkou přesnídávku.
• Pokud nebudete reagovat na podání glukagonu, budete muset být léčen(a) v nemocnici.
• Pokud není déletrvající závažná nízká hladina cukru v krvi léčena, může způsobit poškození mozku. To může být krátkodobé, ale i dlouhodobé. Může dokonce způsobit úmrtí.
Promluvte si se svým lékařem, pokud:
• vaše hladina cukru v krvi poklesla do takové míry, že jste ztratil(a) vědomí
• byl vám injekčně aplikován glukagon
• jste již několikrát v poslední době měl(a) nízkou hladinu cukru v krvi,
protože dávka přípravku Xultophy, jídla nebo fyzická aktivita může v těchto případech vyžadovat úpravu.
Vysoká hladina cukru může nastat, pokud:
• pijete alkohol
• vaše fyzická aktivita je menší než obvykle
• jíte více než obvykle
• máte infekci nebo horečku
• nepoužil(a) jste dostatek přípravku Xultophy, trvale používáte méně přípravku Xultophy, než potřebujete, zapomněl(a) jste přípravek Xultophy použít nebo jste jej přestal(a) používat bez přechozí domluvy se svým lékařem.
Varovné známky vysoké hladiny cukru v krvi - obvykle se objevují postupně
Zrudlá suchá pokožka, pocit ospalosti nebo únavy, sucho v ústech, ovocný (acetonový) zápach dechu, častější močení, pocit žízně, ztráta chuti k jídlu, pocit na zvracení nebo zvracení.
Mohou to být příznaky velmi závažného stavu nazývaného „ketoacidóza“. Jedná se o nahromadění kyseliny v krvi, jelikož tělo odbourává tuk místo cukru. Pokud tento stav není léčen, může vést až k diabetickému kómatu a v konečném důsledku k úmrtí.
Co dělat, pokud máte vysokou hladinu cukru v krvi:
• Otestujte si hladinu cukru v krvi.
• Otestujte krev nebo moč na přítomnost ketonů.
• Ihned vyhledejte lékařskou pomoc.
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Xultophy uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku pera a na krabičce za „Použitelné do“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Před otevřením
Uchovávejte v chladničce (2° C až 8 °C). Neuchovávejte v blízkosti mrazicího zařízení. Chraňte před mrazem.
Během používání
Chraňte před mrazem. Přípravek Xultophy můžete nosit s sebou a uchovávat jej při pokojové teplotě (ne vyšší než 30 °C) nebo v chladničce (2 °C až 8 °C) nejdéle po dobu 21 dnů. Přípravek je nutné 21 dnů po prvním otevření zlikvidovat.
Na předplněném peru mějte nasazený uzávěr vždy, když jej nepoužíváte, aby byl přípravek chráněn před světlem.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Xultophy obsahuje
• Léčivými látkami jsou insulinům degludecum a liraglutidum. Jeden ml roztoku obsahuje 100 jednotek inzulínu degludek a 3,6 mg liraglutidu. Jedno nepoužité předplněné pero (3 ml) obsahuje 300 jednotek inzulinu degludek a 10,8 mg liraglutidu.
• Dalšími složkami jsou glycerol, fenol, zinkum-acetát, kyselina chlorovodíková a hydroxid sodný (k úpravě pH) a voda na injekci.
Jak přípravek Xultophy vypadá a co obsahuje toto balení
Přípravek Xultophy je čirý a bezbarvý roztok.
Velikosti balení: 1, 3, 5 předplněných per a skupinové balení obsahující 10 předplněných per (2 balení po 5) o obsahu 3 ml. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Novo Nordisk A/S Novo Allé
DK-2880 Bagsv^rd, Dánsko
Nyní otočte na druhou stranu a přejděte k informacím, jak předplněné pero používat.
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Před použitím předplněného pera Xultophy si pečlivě přečtěte tyto pokyny. Nepoužívejte pero bez odpovídajícího proškolení lékařem nebo zdravotní sestrou.
Začněte kontrolou svého pera tím, že se ujistíte, že obsahuje přípravek Xultophy 100 jednotek/ml + 3,6 mg/ml. Poté si prostudujte následující obrázky a seznamte se se všemi částmi pera a jehly.
Pokud jste slepý(á) či slabozraký(á) a nejste schopen(schopna) přečíst údaje na počítadle dávky svého pera, nepoužívejte toto pero bez pomoci.
Požádejte o pomoc druhou osobu, která má dobrý zrak a je proškolena v používání předplněného pera Xultophy.
Přípravek Xultophy je léčivý přípravek, který obsahuje inzulin degludek a liraglutid. Přípravek Xultophy se podává po „dávkovacích jednotkách“.
Jedna dávkovací jednotka obsahuje 1 jednotku inzulinu degludek + 0,036 mg liraglutidu.
Vaše pero je předplněné dávkovací pero. Obsahuje 3 ml roztoku Xultophy. Slouží k aplikaci dávek od:
- 1 dávkovací jednotky
- až po maximálně 50 dávkovacích jednotek (50 jednotek inzulinu degludek + 1,8 mg liraglutidu)
Pero umožňuje podávání dávky v přírůstcích po 1 dávkovací jednotce. Neprovádějte žádný přepočet své dávky. Nastavený počet dávkovacích jednotek se rovná číslu zobrazenému na počítadle dávky.
Vaše pero je určeno k použití s jednorázovými jehlami NovoTwist nebo NovoFine o délce do 8 mm a tloušťce do 32 G. Jehly nejsou součástí balení.
A Důležité upozornění
Věnujte těmto poznámkám zvláštní pozornost, neboť jsou důležité pro bezpečné používání pera.
Předplněné pero Xultophy a jehla (příklad)

Uzávěr
pera

Vnější kryt jehly

Vnitřní kryt jehly

Jehla

Stupnice
pera
Okénko
pera
Štítek
pera

Papírový
kryt

-Počítadlo dávky
-Ukazatel dávky
Volič dávky
Dávkovací
tlačítko
Dávkovací
tlačítko
hladkým
ovrchem
1. Připravte si pero a novou jehlu
• Zkontrolujte název a barevný štítek pera a ujistěte se, že obsahuje přípravek Xultophy.
To je obzvlášť důležité, pokud užíváte více než jeden typ léku aplikovaného injekčně. Užití špatného léku vám může poškodit zdraví.
• Sejměte uzávěr pera.
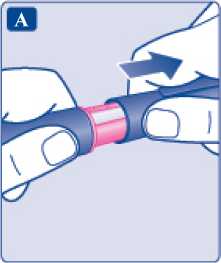
|
• Zkontrolujte, zda je roztok v peru čirý a bezbarvý. Podívejte se přes okénko pera. Pokud je roztok zakalený, pero nepoužívejte. |
O ,_ |
|
• Vezměte si novou jehlu a odtrhněte papírový kryt. | |
|
• Nasaďte jehlu rovně na pero. Našroubujte ji na doraz. | |
|
• Sejměte vnější kryt jehly a ponechte si jej na později. Budete jej potřebovat po podání injekce, abyste mohl(a) jehlu bezpečně sejmout z pera. |
o |
|
• Sejměte vnitřní kryt jehly a vyhoďte jej. Pokud byste se pokusil(a) jej opět nasadit, mohl(a) byste se o jehlu nechtěně píchnout. Na hrotu jehly se může objevit kapka roztoku. To je zcela normální, i přesto však musíte zkontrolovat průtok. Nenasazujte na pero novou jehlu, dokud nejste připraven(a) si injekci aplikovat. Jk Pro každou aplikaci vždy použijte novou jehlu. Tím se může předejít ucpání jehel, kontaminaci, infekci a nepřesnému dávkování. A Nikdy nepoužívejte ohnuté či poškozené jehly. |
I w. |
2. Zkontrolujte průtok
• Otočením voliče dávky zvolte 2 dávkovači jednotky. Ujistěte se, že počítadlo dávky ukazuje hodnotu 2.
• Počítadlo dávky a ukazatel dávky ukazují, kolik dávkovacích jednotek přípravku Xultophy jste zvolil(a).
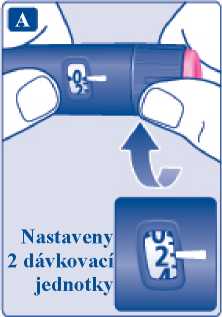
Pero držte s jehlou otočenou směrem nahoru.
Jemně několikrát klepněte na horní část pera, aby se nahoře nahromadily vzduchové bubliny.
Stiskněte a podržte dávkovací tlačítko, dokud se počítadlo dávky nevrátí na 0.
0 musí být proti ukazateli dávky.
Na hrotu jehly se musí objevit kapka roztoku.
Na špičce jehly může zůstat malá kapka, kterou si však neaplikujete. Pokud se neobjeví žádná kapka, zopakujte kroky 2A až 2C, a to až 6krát. Pokud se stále žádná kapka neobjevuje, vyměňte jehlu a zopakujte kroky 2A až 2C ještě jednou.
Pokud se kapka roztoku ani poté neobjeví, pero zlikvidujte a použijte nové.
A Vždy před podáním injekce zkontrolujte, zda se na špičce jehly objeví kapka. Tak se ujistíte, že roztok protéká.
Pokud se neobjeví žádná kapka, neaplikoval(a) byste si žádný lék, i když by se počítadlo dávky pohybovalo. V takovém případě je možné, že došlo k ucpání nebo poškození jehly.
A Před aplikací injekce je důležité vždy zkontrolovat průtok. Pokud tak neučiníte, můžete si aplikovat příliš málo léku, nebo dokonce vůbec žádný. To může vést k vysoké hladině cukru v krvi.
3. Nastavení dávky
• Otočením voliče dávky zvolte potřebnou dávku.
Počítadlo dávky ukazuje dávku v dávkovačích jednotkách.
Pokud zvolíte špatnou dávku, můžete ji opravit otočením voliče dávky směrem dopředu nebo dozadu.
Na peru lze nastavit maximálně 50 dávkovacích jednotek.
Volič dávky mění počet dávkovacích jednotek.
Pouze počítadlo dávky a ukazatel dávky ukazují, kolik dávkovacích jednotek na dávku jste zvolil(a).
Můžete zvolit až 50 dávkovacích jednotek na dávku. Pokud vaše pero obsahuje méně než 50 dávkovacích jednotek, počítadlo dávky se zastaví na počtu zbývajících dávkovacích jednotek.
Volič dávky cvaká jiným způsobem při otáčení dopředu, zpět nebo přes počet zbývajících dávkovacích jednotek. Cvakání pera nepočítejte.

Před aplikací léku vždy pomocí počítadla dávky a ukazatele dávky zkontrolujte, kolik dávkovacích jednotek jste zvolil(a).
Cvakání pera nepočítejte. Pokud si nastavíte a aplikujete špatnou dávku, hladina cukru v krvi vám může stoupnout nebo klesnout.
Nepoužívejte stupnici pera - ukazuje pouze přibližné zbývající množství roztoku v peru.

Přesné množství zbývajícího roztoku lze zjistit pomocí počítadla dávky:
Otáčejte voličem dávky, dokud se počítadlo dávky nezastaví.
Pokud se zobrazí hodnota 50, zbývá v peru ještě alespoň 50 dávkovacích jednotek. Pokud se zobrazí hodnota nižší než 50, uvedený počet představuje počet dávkovacích jednotek zbývajících ve vašem peru.
Pokud potřebujete více léku, než kolik ve vašem peru zbývá, můžete dávku rozdělit mezi dvě pera.
Při dělení dávky dávejte velký pozor na správný výpočet.
Pokud máte pochybnosti, aplikujte si celou dávku z nového pera. Pokud dávku rozdělíte špatně, aplikujete si příliš málo nebo příliš mnoho léku. To může vaši hladinu cukru v krvi zvýšit nebo snížit.
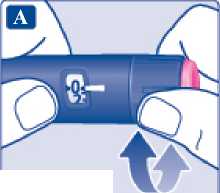
Příklady
Nastaveno 5 dávkovacích jednotek

Nastaveno 24 dávkovacích jednotek

Přibližné
množství
zbývajícího
roztoku

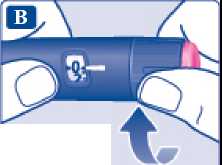
Příklad
Počítadlo dávky se zastavilo: zbývá 42 dávkovacích jednotek

A
4. Aplikace dávky
• Zaveďte jehlu pod kůži, jak vám ukázal váš lékař nebo zdravotní sestra.
• Ujistěte se, že vidíte na počítadlo dávky. Nezakrývejte je prsty. Mohlo by to vést k přerušení aplikace.
Stiskněte a podržte dávkovací tlačítko, dokud počítadlo dávky neukazuje 0.
0 musí být zarovnána s ukazatelem dávky. Můžete uslyšet nebo pocítit cvaknutí.
Po návratu počítadla dávky na 0 přidržte jehlu v kůži a pomalu počítejte do 6.
Pokud jehlu vytáhnete dřív, můžete vidět proud roztoku vytékající z hrotu jehly. Pokud se tak stane, nebyla aplikována celá dávka a bude nutné, abyste svou hladinu cukru v krvi kontroloval(a) častěji.
Vytáhněte jehlu z kůže.
Pokud se v místě vpichu objeví krev, jemně na něj zatlačte. Oblast netřete.
Po podání injekce se na konci jehly může objevit kapka roztoku. To je zcela normální a neovlivňuje to vaši dávku.
Vždy sledujte počítadlo dávky, abyste věděl(a), kolik dávkovacích jednotek podáváte. Podržte stisknuté dávkovací tlačítko, dokud počítadlo dávky neukazuje 0. Pokud se počítadlo dávky na 0 nevrátí, nebyla podána celá dávka, což může vést k vysoké hladině cukru v krvi.
Jak lze zjistit, že je jehla ucpaná nebo poškozená?
• Pokud se při nepřerušovaném stisknutí dávkovacího tlačítka na počítadle dávky neobjeví 0, je možné, že jste použil(a) ucpanou nebo poškozenou jehlu.
• V takovém případě jste si neaplikoval(a) žádný lék, ačkoli se počítadlo dávky posunulo z původní dávky, kterou jste nastavil(a).
Jak nakládat s ucpanou jehlou?
Vyměňte jehlu podle popisu v části 5 a opakujte všechny kroky počínaje od části 1: Připravte si pero a novou jehlu. Ujistěte se, že jste zvolil(a) celou potřebnou dávku.
Nikdy se při aplikaci nedotýkejte počítadla dávky. Může to vést k přerušení aplikace.
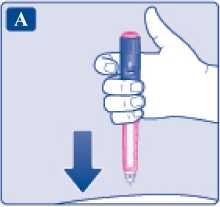
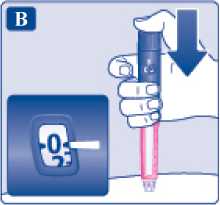
Pomalu počítejte
|
5. Po aplikaci • Na hladkém povrchu zaveďte špičku jehly do vnějšího krytu jehly. Nedotýkejte se při tom jehly ani vnějšího krytu. |
° 4- |
|
• Po zakrytí jehly opatrně vnější kryt jehly zcela dotlačte. • Odšroubujte jehlu a opatrně ji zlikvidujte dle pokynů svého lékaře nebo zdravotní sestry. |
0\^ |
|
• Uzávěr pera nasaďte na pero po každém použití, aby byl roztok chráněn před světlem. Po každé aplikaci vždy jehlu zlikvidujte. Zajistíte tím použití ostré jehly a zabráníte ucpání jehel. Pokud je jehla ucpaná, nelze aplikovat žádný lék. Prázdné pero bez nasazené jehly vyhoďte dle pokynů svého lékaře, zdravotní sestry, lékárníka nebo místních úřadů. A Nikdy se nepokoušejte nasadit vnitřní kryt jehly zpět na jehlu. Mohl(a) byste se o jehlu píchnout. A Po každé aplikaci vždy jehlu z pera sejměte. Tím se může předejít ucpání jehel, kontaminaci, infekci, úniku roztoku a nepřesnému dávkování. |
Q |
|
Další důležité informace • Vždy s sebou mějte další pero a nové jehly pro případ ztráty či poškození. • Pero a jehly vždy uchovávejte mimo dohled a dosah ostatních, zejména pak dětí. • Nikdy pero s nikým nesdílejte. Váš lék by jim mohl poškodit zdraví. • Nikdy jehly s nikým nesdílejte. Mohlo by dojít k přenosu infekce. • Pečující osoby musí být při manipulaci s použitými jehlami velice opatrné, aby nedošlo k poranění jehlou a přenesení infekce. | |
|
Péče o pero • Pero nenechávejte v autě ani na jiném místě, kde může být příliš vysoká nebo příliš nízká teplota. • Neskladujte pero při teplotách nad 30 °C. • Nevystavujte pero prachu, špíně ani tekutinám. • Pero neumývejte, nenamáčejte ani nepromazávejte. V případě potřeby jej očistěte hadříkem navlhčeným ve slabém čisticím prostředku. • Nenechte pero spadnout na tvrdý povrch ani s ním o takový povrch neklepejte. |
Pokud pero upustíte nebo máte podezření, že se poškodilo, našroubujte na něj novou jehlu a před aplikací zkontrolujte průtok roztoku. Nepokoušejte se pero znovu naplnit. Po vypotřebování je nutné jej zlikvidovat.
Nepokoušejte se pero opravit ani ho rozebrat.
50