Xtandi 40 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xtandi 40 mg měkké tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna měkká tobolka obsahuje enzalutamidum 40 mg.
Pomocná látka se známým účinkem:
Jedna měkká tobolka obsahuje 52,4 mg sorbitolu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Měkká tobolka.
Bílé až krémově bílé podlouhlé měkké tobolky (přibližně 20 mm x 9 mm) s potiskem „ENZ“ černým inkoustem na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Xtandi je indikován k:
• léčbě dospělých mužů s metastatickým kastračně rezistentním karcinomem prostaty, kteří jsou asymptomatičtí nebo mírně symptomatičtí po selhání androgen deprivační terapie a u nichž dosud nebyla chemoterapie klinicky indikována (viz bod 5.1)
• léčbě dospělých mužů s metastatickým kastračně rezistentním karcinomem prostaty, u jejichž onemocnění došlo k progresi při nebo po léčbě docetaxelem.
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je 160 mg enzalutamidu (čtyři 40 mg tobolky) jako jedna denní perorální dávka.
U pacientů, kteří nepodstoupili chirurgickou kastraci, je zapotřebí během léčby pokračovat ve farmakologické kastraci pomocí analogu LHRH.
Pokud pacient zapomene užít Xtandi v obvyklou dobu, má užít předepsanou dávku co nejblíže k obvyklé době. Jestliže pacient vynechá dávku celý den, má být léčba opět zahájena následující den obvyklou dávkou.
Pokud se u pacienta vyskytne toxicita > stupni 3 nebo netolerovatelný nežádoucí účinek má být přípravek vysazen po dobu jednoho týdne, nebo dokud se nezlepší příznaky na stupeň 2 nebo nižší, pak lze pokračovat se stejnou nebo pokud je třeba nižší dávkou (120 mg nebo 80 mg).
Současné užívání se silnými inhibitory CYP2C8
Pokud je to možné, je třeba se vyhnout současnému užívání silných inhibitorů CYP2C8. Jestliže pacientům musí být současně podáván silný inhibitor CYP2C8, dávka enzalutamidu má být snížena na 80 mg jednou denně. Jestliže dojde k přerušení současného podávání silného inhibitoru CYP2C8, měla by se dávka enzalutamidu vrátit na tu, která byla podávána před zahájením užívání silného inhibitoru CYP2C8 (viz bod 4.5).
Starší pacienti
U starších lidí není nutná žádná úprava dávky (viz bod 5.1 a 5.2).
Porucha funkce jater
U pacientů s mírnou, středně těžkou ani závažnou poruchou funkce jater (Child-Pugh třídy A, B resp. C) není nutná žádná úprava dávky. Nicméně u pacientů se závažnou poruchou funkce jater bylo zjištěno zvýšení poločasu léku (viz body 4.4 a 5.2).
Porucha funkce ledvin
U pacientů s mírnou až středně těžkou poruchou ledvin (viz bod 5.2) není nutná žádná úprava dávky. Opatrnost se doporučuje u pacientů s těžkou poruchou ledvin nebo s onemocněním ledvin v konečném stadiu (viz bod 4.4).
Pediatrická populace
Neexistuje žádné relevantní použití enzalutamidu u pediatrické populace při indikaci, kterou je léčba dospělých mužů s metastatickým kastračně rezistentním karcinomem prostaty.
Způsob podání
Xtandi se užívá perorálně. Tobolky se polykají celé s vodou a lze je užívat s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Ženy, které jsou těhotné nebo mohou otěhotnět (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití Riziko epileptického záchvatu
Opatrnosti je třeba při podávání přípravku Xtandi pacientům s anamnézou epileptických záchvatů nebo jiných predispozičních faktorů, včetně, mimo jiné, poranění mozku, mrtvice, primárních nádorů mozku nebo mozkových metastáz nebo alkoholismu. Riziko epileptického záchvatu navíc může být zvýšeno u pacientů, kteří současně užívají léčivé přípravky, jež snižují práh vzniku epileptických záchvatů.
Syndrom posteriorní reverzibilní encefalopatie
Vzácně došlo k hlášení syndromu reverzibilní posteriorní encefalopatie (PRES) u pacientů užívajících Xtandi (viz bod 4.8). PRES je vzácná, reverzibilní, neurologická porucha, která se může projevit rychle se vyvíjecími symptomy, včetně epileptického záchvatu, bolesti hlavy, zmatenosti, slepoty a dalšími vizuálními a neurologickými poruchami, doprovázenými hypertenzí nebo bez hypertenze. Diagnostika PRES vyžaduje potvrzení pomocí zobrazení mozku, přednostně zobrazení magnetickou rezonancí (MRI). U pacientů, u kterých se projevil PRES, je doporučeno přerušení léčby přípravkem Xtandi.
Současné užívání s jinými léčivými přípravky
Enzalutamid je silný induktor enzymů a to může vést ke ztrátě účinnosti mnoha běžně užívaných léčivých přípravků (viz příklady v bodu 4.5). Přehled současně podávaných léčivých přípravků má být stanoven při zahájení léčby enzalutamidem. Současnému podávání enzalutamidu s léčivými přípravky, které jsou citlivými substráty mnoha metabolizujících enzymů nebo transportérů (viz bod 4.5) se má vyhnout, pokud má léčebný účinek pro pacienta velký význam a jestliže nelze snadno provádět úpravy dávky na základě sledování účinnosti nebo plazmatické koncentrace.
Je třeba se vyhnout současnému podávání warfarinu a antikoagulancií kumarinového typu. Pokud je Xtandi podáván současně s antikoagulanciem, které je metabolizováno pomocí CYP2C9 (jako např. warfarin nebo acenokumarol), má se provádět dodatečné monitorování hodnot International Normalised Ratio (INR) (viz bod 4.5).
Porucha funkce ledvin
Opatrnosti je zapotřebí u pacientů s těžkou poruchou funkce ledvin, neboť u této populace pacientů nebyly provedeny žádné studie enzalutamidu.
Závažná porucha funkce jater
U pacientů se závažnou poruchou funkce jater bylo zjištěno zvýšení poločasu léku, pravděpodobně v souvislosti se zvýšenou distribucí v tkáních. Klinický význam tohoto zjištění zůstává neznámý. Nicméně očekává se prodloužení doby do dosažení rovnovážného stavu koncentrací a doba k dosažení maximálního farmakologického účinku, stejně jako doba pro nástup a pokles indukce enzymů (viz bod 4.5) může být zvýšena.
Nedávné kardiovaskulární onemocnění
Ze studií 3. fáze byli vyloučeni pacienti s nedávným infarktem myokardu (v posledních 6 měsících) nebo nestabilní anginou pectoris (v posledních 3 měsících), srdeční selhání třídy III nebo IV podle Newyorské kardiologické asociace (NYHA), s výjimkou případů, kdy levá ventrikulární ejekční frakce (LVEF) > 45%, nebo jde o bradykardii nebo neléčenou hypertenzi. To je třeba vzít v úvahu, je-li přípravek Xtandi předepisován těmto pacientům.
Androgen-deprivační léčba může prodlužovat QT interval.
Před zahájením léčby přípravkem Xtandi by měl lékař zvážit poměr přínosů a rizik, včetně rizika torsade de pointes, u pacientů s rizikovými faktory pro prodloužení QT intervalu v anamnéze a u pacientů souběžně užívajících léčivé přípravky, které mohou prodlužovat QT interval (viz bod 4.5).
Použití s chemoterapií
Bezpečnost a účinnost současného užívání přípravku Xtandi s cytotoxickou chemoterapií nebyly stanoveny. Současné podávání enzalutamidu nemělo žádný klinicky významný účinek na farmakokinetiku intravenózně podávaného docetaxelu (viz bod 4.5), zvýšení výskytu neutropenie vyvolané docetaxelem však nelze vyloučit.
Pomocné látky
Xtandi obsahuje sorbitol (E420). Pacienti se vzácnými vrozenými problémy s intolerancí fruktózy by neměli tento léčivý přípravek užívat.
Reakce hypersenzitivity
Reakce hypersenzitivity zde uvedené, aniž by byl jejich výčet úplný, jako jsou edém jazyka, edém rtů a edém faryngu, byly pozorovány v souvislosti s enzalutamidem (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Možné účinky jiných léčivých přípravků na expozice enzalutamidu
Inhibitory CYP2C8
CYP2C8 hraje důležitou úlohu při eliminaci enzalutamidu a při tvorbě jeho aktivního metabolitu.
Po perorálním podání silného inhibitoru CYP2C8 gemfibrozilu (600 mg dvakrát denně) zdravým mužským subjektům došlo ke zvýšení hodnoty AUC u enzalutamidu o 326 %, zatímco hodnota Cmax enzalutamidu se snížila o 18 %. Hodnota AUC pro volný enzalutamid včetně volného aktivního metabolitu se zvýšila o 77 %, zatímco hodnota Cmax se snížila o 19%. Během léčby enzalutamidem je třeba se vyhnout silným inhibitorům (např. gemfibrozil) CYP2C8, nebo je používat s opatrností. Pokud pacientům musí být současně podáván silný inhibitor CYP2C8, má být dávka enzalutamidu snížena na 80 mg jednou denně (viz bod 4.2).
Inhibitory CYP3A4
CYP3A4 hraje nevýznamnou úlohu při metabolizmu enzalutamidu. Po perorálním podávání silného inhibitoru CYP3A4 itrakonazolu (200 mg jednou denně) zdravým mužským subjektům došlo ke zvýšení hodnoty AUC enzalutamidu o 41%, zatímco hodnota Cmax zůstala beze změny. Hodnota AUC pro volný enzalutamid včetně volného aktivního metabolitu se zvýšila o 27 %, zatímco hodnota Cmax zůstala opět beze změny. Při podávání Xtandi současně s inhibitory CYP3A4 není nutná žádná úprava dávky.
Induktory CYP2C8 a CYP3A4
Po perorálním podání středně silného induktoru CYP2C8 a silného induktoru CYP3A4 rifampicinu (600 mg jednou denně) zdravým mužským subjektům, se hodnota AUC pro enzalutamid včetně aktivního metabolitu snížila o 37 %, zatímco hodnota Cmax zůstala beze změny. Při podávání Xtandi současně s induktory CYP2C8 a CYP3A4 není nutná žádná úprava dávky.
Možné účinky enzalutamidu na expozici jiných léčivých přípravků
Indukce enzymů
Enzalutamid je silný induktor enzymů a zvyšuje syntézu mnoha enzymů a transportérů; proto je očekávaná interakce s mnoha běžně užívanými léčivými přípravky, které jsou substráty těchto enzymů nebo transportérů. Pokles plazmatické koncentrace může být značný a vede ke ztrátě nebo snížení klinické účinnosti. Je zde také riziko zvýšené tvorby aktivních metabolitů. Mezi enzymy, které mohou být indukované, patří CYP3A v játrech a střevech CYP2B6, CYP2C9, CYP2C19 a uridin 5'-difosfo-glukuronosyltransferáza (UGT - glukuronid-konjugující enzymy). Může být indukován také transportní protein P-gp a pravděpodobně i jiné transportéry, např. multirezistentní protein 2 (MRP2), protein rezistence karcinomu prsu (BCRP) a polypeptidový transportér organických aniontů 1B1 (OATP1B1).
Studie in vivo ukázaly, že enzalutamid je silný induktor CYP3A4 a středně silný induktor CYP2C9 a CYP2C19. Současné podávání enzalutamidu (160 mg jednou denně) s jednou perorální dávkou citlivých substrátů CYP u pacientů s karcinomem prostaty mělo za následek 86% pokles hodnoty AUC midazolamu (substrát CYP3A4), 56% pokles hodnoty AUC S-warfarinu (substrát CYP2C9) a 70% pokles hodnoty AUC omeprazolu (substrát CYP2C19). UGT1A1 mohla být indukována také.
V klinické studii u pacientů s metastatickým CRPC neměl přípravek Xtandi (160 mg jednou denně) žádný klinicky významný účinek na farmakokinetiku intravenózně podávaného docetaxelu (75 mg/m2 infuzí každé 3 týdny). AUC docetaxelu poklesl o 12 % [Poměr geometrického průměru (GMR) =
0,882 (90% CI: 0,767; 1,02)], zatímco Cmax poklesla o 4 % [GMR = 0,963 (90% CI: 0,834; 1,11)].
Jsou očekávané interakce s určitými léčivými přípravky, které jsou vylučovány metabolismem nebo aktivním transportem. Pokud má léčebný účinek pro pacienta velký význam a nelze snadno provádět úpravy dávky na základě sledování účinnosti nebo plazmatické koncentrace, je třeba se těmto léčivým přípravkům vyhnout nebo je používat s opatrností. Existuje podezření vyššího rizika poškození jater po podání paracetamolu u pacientů, kteří jsou současně léčeni induktory enzymů.
Skupina léčivých přípravků, které mohou být ovlivněny, zahrnuje mimo jiné:
• analgetika (např. fentanyl, tramadol)
• antibiotika (např. klarithromycin, doxycyklin)
• cytostatika (např. kabazitaxel)
• antikoagulancia (např. acenokumarol, warfarin)
• antiepileptika (např. karbamazepin, klonazepam, fenytoin, primidon, kyselina valproová)
• antipsychotika (např. haloperidol)
• betablokátory (např. bisoprolol, propranolol)
• blokátory kalciových kanálů (např. diltiazem, felodipin, nikardipin, nifedipin, verapamil)
• srdeční glykosidy (např. digoxin)
• kortikosteroidy (např. dexamethason, prednisolon)
• antivirotika proti viru HIV (např. indinavir, ritonavir)
• hypnotika (např. diazepam, midazolam, zolpidem)
• statiny metabolizované CYP3A4 (např. atorvastatin, simvastatin)
• léky na onemocnění štítné žlázy (např. levothyroxin)
Plná indukční schopnost enzalutamidu se může projevit přibližně až za 1 měsíc po zahájení léčby, kdy bude dosaženo ustáleného stavu plazmatické koncentrace enzalutamidu, ačkoli některé účinky indukce mohou být patrné dříve. U pacientů užívajících léčivé přípravky, které jsou substráty CYP2B6, CYP3A4, CYP2C9, CYP2C19, nebo UGT1A1 by měla být hodnocena případná ztráta farmakologických účinků (nebo zvýšení účinků v případech, kdy dochází k tvorbě aktivních metabolitů) během prvního měsíce léčby enzalutamidem, a měla by se zvážit vhodná úprava dávky.
S ohledem na dlouhý poločas rozpadu enzalutamidu (5,8 dne, viz bod 5.2) mohou účinky na enzymy přetrvávat po dobu jednoho měsíce nebo déle po ukončení užívání enzalutamidu. Při ukončení léčby enzalutamidem může být nezbytné postupné snižování dávky současně podávaných léčivých přípravků.
Substráty CYP1A2 a CYP2C8
Enzalutamid (160 mg jednou denně) nezpůsoboval klinicky významné změny v AUC nebo Cmax kofeinu (substrát CYP1A2) nebo pioglitazonu (substrát CYP2C8). Hodnota AUC pioglitazonu se zvýšila o 20 %, zatímco hodnota Cmax se snížila o 18 %. Hodnota AUC kofeinu se snížila o 11 % a Cmax o 4 %. Při současném podávání substrátu CYP1A2 nebo CYP2C8 s Xtandi není indikována žádná úprava dávky.
Substráty P-gp
In vitro údaje naznačují, že enzalutamid může být inhibitorem efluxního transportéru P-gp. Účinek enzalutamidu na substráty P-gp nebyl hodnocen in vivo; avšak za podmínek klinického použití může být enzalutamid induktorem P-gp prostřednictvím aktivace nuklearního pregnanového receptoru (PXR). Léčivé přípravky s úzkým terapeutickým rozpětím, které jsou substráty pro P-gp (např. kolchicin, dabigatran etexilát, digoxin) by při současném podávání Xtandi měly být používány s opatrností a mohou vyžadovat úpravu dávky k udržení optimální koncentrace v plazmě.
Substráty BCRP, MRP2, OAT3 a OCT1
Na základě údajů in vitro nelze vyloučit inhibici proteinu BCRP a MRP2 (ve střevě), stejně jako inhibici organického aniontového transportéru 3 (OAT3) a organického kationového transportéru 1 (OCT1) (systémovou inhibici). Teoreticky je možná indukce i těchto transportérů, celkový efekt není v současné době znám.
Léčivé přípravky, které prodlužují QT interval
Kvůli souvislosti androgen-deprivační léčby a prodloužení QT intervalu by měla být pečlivě zvážena souběžná léčba přípravkem Xtandi s léčivými přípravky, o kterých je známo, že prodlužují QT interval a léčba přípravky, které mohou vyvolat torsade de pointes, jako jsou antiarytmika třídy I A (např. chinidin, disopyramid), třídy III (např. amiodaron, sotalol, dofetilid, ibutilid), methadon, moxifloxacin, antipsychotika a další (viz bod 4.4.).
Vliv jídla na expozice enzalutamidu
Jídlo nemá žádný klinicky významný účinek na rozsah expozice enzalutamidu. V klinických studiích byl přípravek Xtandi podáván bez ohledu na jídlo.
4.6 Fertilita, těhotenství a kojení
Ženy, které mohou otěhotnět
Nejsou k dispozici žádné údaje týkající se použití přípravku Xtandi v těhotenství u lidí a tento léčivý přípravek není určen k použití u žen, které mohou otěhotnět.
Antikoncepce u mužů a žen
Není známo, zda enzalutamid nebo jeho metabolity jsou přítomny ve spermatu. V průběhu léčby enzalutamidem a 3 měsíce po léčbě pacient musí používat kondom, pokud je sexuálně aktivní s těhotnou ženou. Pokud má pacient pohlavní styk s ženou v plodném věku, musí používat kondom a další formu antikoncepce v průběhu léčby a 3 měsíce po jejím skončení. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Enzalutamid není určen k použití u žen. Enzalutamid je kontraindikován u žen, které jsou nebo mohou být těhotné (viz body 4.3 a 5.3).
Kojení
Enzalutamid není určen k použití u žen.
Fertilita
Studie na zvířatech ukázaly, že enzalutamid měl vliv na reprodukční systém u samců potkanů a psů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Enzalutamid může mít středně závažný vliv na schopnost řídit nebo obsluhovat stroje, protože byly hlášeny psychiatrické a neurologické příhody včetně epileptického záchvatu (viz bod 4.8). Pacienti, u kterých se vyskytly epileptické záchvaty nebo jiné predisponující faktory (viz bod 4.4), by měli být poučeni o riziku při řízení nebo obsluhování strojů. Nebyly ale provedeny žádné studie, které by stanovily účinky enzalutamidu na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastějšími nežádoucími účinky jsou astenie/únava, návaly horka, bolest hlavy a hypertenze. Ostatními důležitými nežádoucími účinky j sou pády, nepatologické zlomeniny, kognitivní poruchy a neutropenie.
K epileptickému záchvatu došlo u 0,5 % pacientů léčených enzalutamidem, 0,1 % u pacientů léčených placebem a 0,3% u pacientů léčených bikalutamidem. Vzácně došlo k hlášení syndromu reverzibilní posteriorní encefalopatie u pacientů léčených enzalutamidem (viz bod 4.4).
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky pozorované v klinických studiích jsou uvedeny níže podle kategorie frekvence výskytu. Kategorie frekvence výskytu jsou definovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000); není známo (z dostupných údajů nelze určit). V každé skupině frekvencí jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky identifikované v kontrolovaných klinických studiích a po uvedení na trh
|
Třídy orgánových systémů podle databáze MedDRA |
F rekvence |
|
Poruchy krve a lymfatického systému |
méně časté: leukopenie, neutropenie není známo*: trombocytopenie |
|
Poruchy imunitního systému |
není známo*: edém jazyka, edém rtů, edém faryngu |
|
Celkové poruchy a reakce v místě aplikace |
velmi časté: astenie/únava |
|
Psychiatrické poruchy |
časté: úzkost méně časté: zrakové halucinace |
|
Poruchy nervového systému |
velmi časté: bolest hlavy časté: poruchy paměti, amnézie, porucha pozornosti, syndrom neklidných nohou méně časté: kognitivní poruchy, epileptický záchvat není známo*: syndrom posteriorní reverzibilní encefalopatie |
|
Srdeční poruchy |
není známo*: prodloužení QT intervalu (viz bod 4.4 a 4.5) |
|
Poruchy reprodukčního systému a prsu |
časté: gynekomastie |
|
Cévní poruchy |
velmi časté: návaly horka, hypertenze |
|
Gastrointestinální poruchy | |
|
Poruchy kůže a podkožní tkáně | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
časté: zlomeniny** není známo*: myalgie, svalové spasmy, svalová slabost, bolest zad |
|
Poranění, otravy a procedurální komplikace |
časté: pády |
* Spontánní hlášení ze zkušeností po uvedení na trh
** Zahrnují všechny zlomeniny s výjimkou patologických zlomenin
Popis vybraných nežádoucích účinků
Epileptický záchvat
V kontrolovaných klinických studiích se epileptické záchvaty objevily u 10 pacientů (0,5 %) z 2051 pacientů léčených denní dávkou 160 mg enzalutamidu, zatímco epileptický záchvat byl zaznamenán u jednoho pacienta (<0,1 %) užívajícího placebo a u jednoho pacienta (0,3%) užívajícího bikalutamid. Zdá se, že dávka je důležitým predikčním faktorem ohledně rizika epileptického záchvatu, jak o tom svědčí preklinické údaje a údaje ze studie s eskalací dávky. V obou kontrolovaných klinických studiích byli vyřazeni pacienti s předchozím epileptickým záchvatem nebo rizikovými faktory pro vznik epileptického záchvatu.
V klinické studii AFFIRM došlo při léčbě enzalutamidem v denní dávce 160 mg k epileptickému záchvatu u šesti (0,8 %) z 800 chemoterapeuticky v minulosti již léčených pacientů, zatímco u pacientů dostávajících placebo nedošlo k žádnému epileptickému záchvatu. U několika z těchto pacientů byly přítomny potenciálně přispívající faktory, které mohly samostatně zvýšit riziko epileptického záchvatu. V klinické studii PREVAIL u pacientů, kteří dosud nepodstoupili chemoterapii, došlo k epileptickému záchvatu u jednoho pacienta (0,1 %) z 871 pacientů, léčených denní dávkou 160 mg enzalutamidu, a u jednoho pacienta (0,1 %) užívajícího placebo. V kontrolované studii s bikalutamidem došlo k epileptickému záchvatu u 3 pacientů (0,8 %) z 380 pacientů léčených enzalutamidem, kteří dosud nepodstoupili chemoterapii a u 1pacienta (0,3 %) z 387 pacientů užívajících bikalutamid.
Mechanizmus, kterým enzalutamid může snižovat práh vzniku epileptických záchvatů, není znám, ale mohl by souviset s údaji ze studií in vitro, které prokázaly, že enzalutamid a jeho aktivní metabolity se váží na chloridový kanál s GABA a mohou inhibovat jeho aktivitu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Proti enzalutamidu neexistuje žádná protilátka. V případě předávkování má být léčba enzalutamidem přerušena a mají být zahájena obecná podpůrná opatření beroucí v úvahu poločas rozpadu 5,8 dní. U pacientů po předávkování se může vyskytnout zvýšené riziko epileptických záchvatů.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: dosud nepřidělena, ATC kód: dosud nepřidělen Mechanismus účinku
O karcinomu prostaty je známo, že je citlivý na androgeny a reaguje na inhibici signálních drah androgenních receptorů. I přes nízké nebo dokonce nedetekovatelné hladiny androgenu v séru signální dráhy androgenních receptorů i nadále podporují progresi onemocnění. Stimulace růstu nádorových buněk prostřednictvím androgenního receptoru vyžaduje jeho translokaci do jádra buňky a vazbu na DNA. Enzalutamid je silný inhibitor signalizace androgenních receptorů, který blokuje několik kroků androgenní signální dráhy. Enzalutamid kompetitivně inhibuje vazbu androgenů na androgenní receptory, inhibuje translokaci aktivovaných receptorů do jádra a inhibuje spojení aktivovaného androgenního receptoru s DNA dokonce i v případě nadměrné exprese androgenních receptorů a v buňkách karcinomu prostaty rezistentních na antiandrogeny. Léčba enzalutamidem snižuje růst buněk karcinomu prostaty a umí vyvolat zánik rakovinných buněk a regresi nádoru. V preklinických studiích enzalutamidu nebyla prokázána aktivita agonisty androgenního receptoru.
Farmakodynamické účinky
Ve fázi 3 klinické studie pacientů, u nichž došlo k progresi po předchozí chemoterapii docetaxelem, vykázalo 54 % pacientů léčených enzalutamidem nejméně 50% pokles hladiny PSA oproti výchozímu stavu oproti 1,5 % pacientů, kteří dostávali placebo.
Klinická účinnost a bezpečnost
Účinnost enzalutamidu byla zjišťována ve dvou randomizovaných placebem kontrolovaných multicentrických klinických studiích 3. fáze_[CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] u pacientů s progredujícím metastatickým karcinomem prostaty, u nichž došlo k selhání androgen deprivační terapie [analog hormonu uvolňujícího luteinizační hormon (LHRH) nebo po bilaterální orchiektomii]. Do studie PREVAIL byli zařazeni pacienti, kteří dosud nepodstoupili terapii docetaxelem, zatímco pacienti zařazení do studie AFFIRM již předtím byli docetaxelem léčeni.
Všichni pacienti pokračovali užíváním analogu LHRH nebo podstoupili bilaterální orchiektomii.V ramenu s účinnou léčbou byl perorálně podáván přípravek Xtandi v dávce 160 mg denně. V obou klinických studiích pacienti dostávali placebo v kontrolním ramenu a pacienti měli povoleno, ale nevyžadovalo se, aby užívali prednison (maximální povolená denní dávka byla 10 mg prednisonu nebo ekvivalent).
Změny v sérových koncentracích PSA nezávisle nepredikují vždy klinický přínos. Proto bylo v obou studiích doporučeno, aby pacienti pokračovali ve studijní léčbě, dokud nebudou splněna kritéria pro vysazení, jak je dále pro každou studii uvedeno.
Studie MDV3100-03 (PREVAIL) (chemoterapeuticky naivní pacienti)
Bylo celkem randomizováno v poměru 1:1 1717 asymptomatických nebo mírně symptomatických pacientů, kteří neabsolvovali chemoterapii, k podávání buď enzalutamidu perorálně v dávce 160 mg jednou denně (N = 872) nebo placeba perorálně jednou denně (N = 845). Zařazení pacientů s viscerálním_onemocněním, s anamnézou mírného až středně závažného srdečního selhání (NYHA třída 1 nebo 2), nebo užívajících léky spojené se snížením prahu epileptického záchvatu bylo povoleno. Byli vyloučeni pacienti s předchozí anamnézou epileptického záchvatu nebo onemocněním, které by mohlo k epileptickému záchvatu predisponovat a pacienti se střední nebo silnou bolestí způsobenou karcinomem prostaty. Hodnocená léčba pokračovala do progrese onemocnění (průkazu radiografické progrese, výskytu skeletální příhody nebo klinické progrese) a iniciace podávání buď cytotoxické chemoterapie, nebo jiného hodnoceného přípravku, nebo do nepřijatelné toxicity.
Demografické parametry a charakteristiky onemocnění pacientů při vstupu do studie byly mezi léčebnými rameny vyváženy. Střední věk byl 71 let (rozsah 42 až 93) a rasové rozdělení bylo 77 % bělochů, 10 % Asiatů, 2 % černochů a 11 % jiných nebo neznámých ras. Šedesát osm procent (68 %) pacientů mělo výkonnostní stav dle ECOG 0 a 32 % pacientů mělo ECOG 1. Výchozí hodnocení bolesti bylo 0-1 (asymptomatický) u 67 % pacientů a 2-3 (mírně symptomatický) u 32 % pacientů, jak je definováno dle krátkého formuláře hodnocení bolesti (Brief Pain Inventory Short Form) (nejhorší bolest za posledních 24 hodin na stupnici od 0 do 10). Přibližně 45 % pacientů mělo při vstupu do studie měřitelné onemocnění v měkkých tkáních a 12 % pacientů mělo viscerální metastázy (plíce a/nebo játra).
Společné primární cílové parametry účinnosti zahrnovaly celkové přežití a přežití bez radiografické progrese (radiographic progression-free survival, rPFS). Kromě společných primárních cílových parametrů se rovněž hodnotil přínos léčby prostřednictvím doby do zahájení cytotoxické chemoterapie, nejlepší celkové léčebné odpovědi v měkkých tkáních, doby do první kostní příhody, PSA odpovědi (>50 % pokles vůči výchozímu stavu), doby do progrese PSA a doby do zhoršení celkového skóre dle FACT-P.
Radiografická progrese byla hodnocena pomocí sekvenčního zobrazovacího sledování, jak je definují kritéria pracovní skupiny Prostate Cancer Clinical Trials Working Group 2 (PCWG2) (pro kostní léze) a/nebo kritéria hodnocení odpovědi na léčbu u solidních tumorů (RECIST v 1.1) (pro léze v měkkých tkáních). Při hodnocení rPFS bylo využito centrálního hodnocení.
V předem specifikované průběžné analýze celkového přežití, kde bylo zaznamenáno 540 úmrtí, bylo prokázáno při léčbě enzalutamidem statisticky významné zlepšení celkového přežití v porovnání s léčbou placebem se snížením rizika úmrtí o 29,4 % [HR = 0,706, (95% CI: 0,596; 0,837), p < 0,0001]. Aktualizovaná analýza přežití byla provedena, když bylo zaznamenáno 784 úmrtí. Výsledky této analýzy byly shodné s výsledky průběžné analýzy (tabulka 2, obrázek 1). 52 % pacientů léčených enzalutamidem a 81 % pacientů léčených placebem dostávalo následnou terapii pro metastatický CRPC , která může prodloužit celkové přežití.
|
Enzalutamid (N = 872) |
Placebo (N = 845) | |
|
Plánovaná interim analýza | ||
|
Počet úmrtí (%) |
241 (27,6%) |
299 (35,4%) |
|
Medián přežití, měsíce (95% CI) |
32,4 (30,1; NR) |
30.2 (28.0; NR) |
|
P-hodnotaa |
< 0,0001 | |
|
Poměr rizika (95% CI) b |
0,71 (0,60; 0,84) | |
|
Aktualizovaná analýza přežití | ||
|
Počet úmrtí (%) |
368 (42,2%) |
416 (49,2%) |
|
Medián přežití, měsíce (95% CI) |
35,3 (32,2; NR) |
31,3 (28,8; 34,2) |
|
P hodnota3 |
0,0002 | |
|
Poměr rizika (95% CI) b |
0,77 (0,67; 0,88) | |
a P hodnota odvozena z nestratifíkovaného log-rank testu
b Poměr rizika je odvozen z nestratifíkovaného proporcionálního modelu rizik. Poměr rizika < 1 favorizuje enzalutamid. NR - nedosaženo.
Obrázek 1: Kaplanovy-Meierovy křivky celkového přežití na základě aktualizované analýzy přežití ve studii PREVAIL (analýza intent-to-treat)
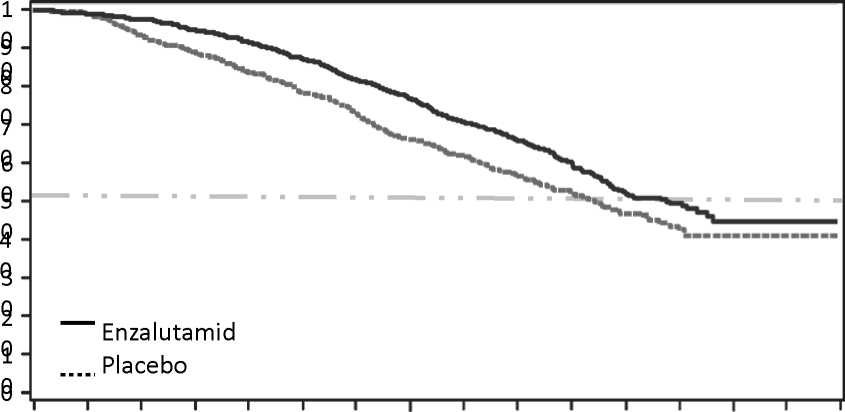
P
ř e ž it
(
% )
|
0 |
3 |
6 |
9 |
1 |
1 |
12 2 |
2 |
33 |
3 |
3 |
4 |
4 |
|
Pacienti v riziku |
2 |
5 |
8 Měsíce 4 |
7 |
0 3 |
6 |
9 |
2 |
5 | |||
|
Enzaluta 8 |
863 |
850 |
824 |
798 |
758 |
710 665 597 |
441 |
289 174 |
86 |
21 |
2 |
0 |
|
maí 8 ebo 5 |
835 |
782 |
745 |
702 |
657 |
612 551 504 |
365 |
254 153 |
72 |
16 |
2 |
0 |
Obrázek 2: Aktualizovaná analýza celkového přežití v jednotlivých podskupinách: poměr rizika a 95% interval spolehlivosti (CI - konfidence interval) ve studii PREVAIL (analýza intent-to-treat)
Podskupina Všichni pacienti
Stav výkonnosti dle ECOG = 0 Stav výkonnosti dle ECOG= 1 Věk <75 let Věk >75 let
Geografickýregion-Severni Amerika Geografickýregion- Eviopa Geografickýregion- Zbytek světa Viscerální onemocněni (plicní a/nebo ledvinové)- Ano Viscerální onemocněni (plicní afnebo ledvinové)- Ne
Počet pacientů Enzalutamidí Placebo
872/845
584/585
288/260
555/553
317/292
218/208
465/446
189/191
98/106
774/739
Poměr rizika pro úmrtí
(95% Cl)
0.77(0.67, 0.88) 0.80(0.67,0.96) 0.68(0.54,0.86) 0.87(0.72,1.04) 0.62(0.50,0.78) 0.88(0.66,1.17) 0.74(0.61,0.90) 0.71 (0.52,0.97) 0.69(0.48,1.01) 0.78(0.67,0.91)
0 < 0. 1._J,
Upřednostňuje 0 Upřednostňuje Enzalutamid Placebo
V předem specifikované analýze rPFS byl prokázán statisticky významný rozdíl mezi léčebnými skupinami se snížením rizika radiografické progrese nebo úmrtí o 81,4 % [HR = 0,186 (95% CI:
0,149; 0,231), p<0,0001]. Jedno sto osmnáct (14 %) pacientů léčených enzalutamidem a 321 (40 %) pacientů léčených placebem mělo příhodu. Mediánu rPFS nebylo dosaženo (95% CI: 13,8; nedosaženo) ve skupině léčené enzalutamidem, ve skupině léčené placebem (obrázek 3) byl dosažen medián rPFS 3,9 měsíců (95% CI: 3,7; 5,4). Konzistentní benefit pro rPFS při léčbě enzalutamidem byl prokázán v rámci všech předem specifikovaných podskupin pacientů (např. dle věku, výchozího výkonnostního stavu dle ECOG , výchozího PSA a LDH, Gleasonova skóre při diagnóze a přítomnosti viscerálního onemocnění při screeningu). Předem specifikovaná analýza rPFS založená na hodnocení radiografické progrese zkoušejícím lékařem prokázala statisticky významné zlepšení mezi léčebnými skupinami s 69,3% snížením rizika radiografické progrese či úmrtí [HR = 0,307 (95% CI: 0,267; 0,353), p<0,0001]. Medián rPFS byl 19,7 měsíců ve skupině s enzalutamidem a 5,4 měsíců ve skupině s placebem.
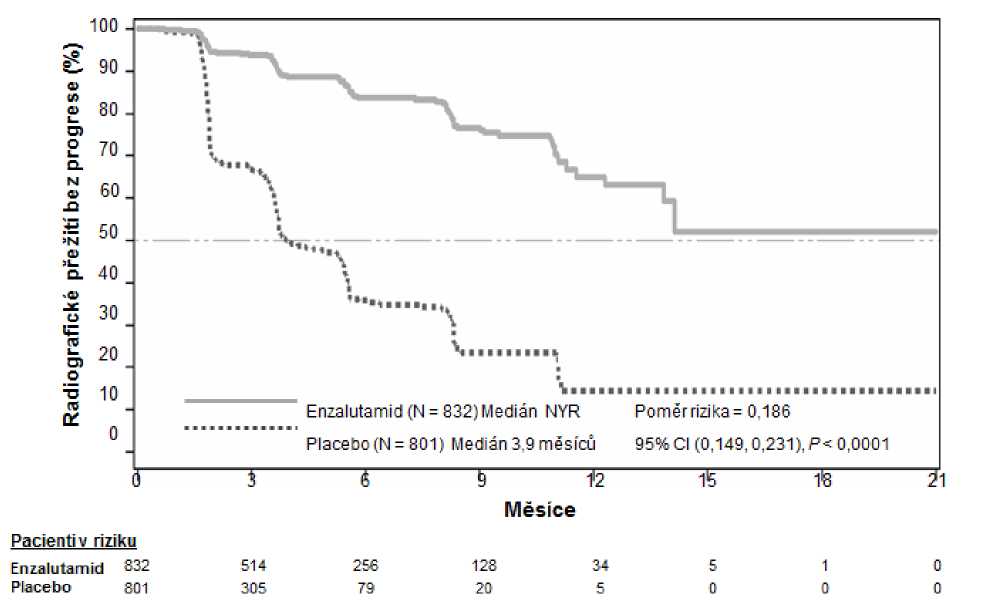
V době primární analýzy bylo randomizováno 1633 pacientů.
Kromě společných primárních cílových parametrů účinnosti byla rovněž prokázána statisticky významná zlepšení v následujících prospektivně definovaných cílových parametrech.
Střední doba do zahájení cytotoxické chemoterapie byla 28,0 měsíců u pacientů užívajících enzalutamid a 10,8 měsíců u pacientů dostávající placebo (HR=0,350, 95% CI: [0,303; 0,403],
p < 0,0001).
Podíl pacientů léčených enzalutamidem s měřitelným onemocněním při vstupu do studie, kteří měli objektivní léčebnou odpověď v měkkých tkáních, byl 58,8 % (95% CI. 53,8; 63,7) v porovnání s 5,0 % (95% CI: 3,0; 7,7) pacientů užívajících placebo. Absolutní rozdíl v objektivní odpovědi v měkkých tkáních mezi rameny s enzalutamidem a placebem byl 53,9 % (95% CI: 48,5 %, 59,1 %, p < 0,0001). Kompletní léčebné odpovědi byly hlášeny u 19,7 % pacientů léčených enzalutamidem v porovnání s 1,0 % pacientů léčených placebem a částečné odpovědi na léčbu byly hlášeny u 39,1 % pacientů léčených enzalutamidem versus 3,9 % pacientů léčených placebem.
Enzalutamid významně snížil riziko první skeletální příhody o 28 % [HR = 0,718 (95% CI: 0,610; 0,844) hodnota p<0,0001]. Skeletální příhoda byla definována jako radiační terapie nebo chirurgický výkon na kostech pro karcinom prostaty, patologická fraktura, komprese míchy nebo změna antineoplastické terapie pro léčbu kostní bolesti. Analýza zahrnovala 587 skeletálních příhod, z nichž 389 příhod (66,3 %) představovalo radiační léčbu, 79 příhod (13,5 %) kompresi míchy, 70 příhod (11,9 %) patologickou frakturu, 45 příhod (7,6 %) změnu antineoplastické terapie pro léčbu kostní bolesti a 22 příhod (3,7 %) chirurgický zákrok na kostech.
U pacientů léčených enzalutamidem byla prokázána významně vyšší celková míra odpovědi PSA (definovaná jako > 50% snížení od výchozí hodnoty) v porovnání s pacienty užívajícími placebo, a to 78,0 % versus 3,5 % (rozdíl = 74,5 %, p<0,0001).
Střední doba do progrese PSA podle PCWG2 kritérií byla 11,2 měsíců pro pacienty léčené enzalutamidem a 2,8 měsíců pro pacienty, kteří užívali placebo [HR=0,169, (95% CI: 0,147; 0,195),
p < 0,0001].
Při léčbě enzalutamidem došlo ke snížení rizika zhoršení celkového skóre FACT-P o 37,5 % v porovnání s placebem (p < 0,001). Střední doba do zhoršení FACT-P skóre byla 11,3 měsíců ve skupině s enzalutamidem a 5,6 měsíců ve skupině s placebem.
Studie 9785-CL-0222 (TERRAIN) chemoterapeuticky naivní pacienti
Do studie TERRAIN bylo celkem zařazeno 375 chemo-naivních pacientů a pacientů antiandrogeny neléčených, kteří byli randomizováni buď k užívání enzalutamidu v dávce 160 mg jednou denně (N = 184), nebo bikalutamidu v dávce 50 mg jednou denně (N = 191). Střední doba PFS bylo 15,7 měsíců pro pacienty léčené enzalutamidem oproti 5,8 měsíců u pacientů na bikalutamidu [HR = 0.44 (95% CI: 0,34; 0,57), p < 0,0001]. Přežití bez progrese bylo definováno jako potvrzená radiografická progrese onemocnění podle nezávislého centrálního vyhodnocení, skeletální příhoda, zahájení nové antineoplastické léčby nebo smrt z jakékoliv příčiny, podle toho co nastane dříve.
Shodný PFS benefit byl pozorován napříč všemi předem specifikovanými podskupinami pacientů. Studie CRPC2 (AFFIRM) (pacienti, kteří byli chemoterapeuticky již léčeni)
Účinnost a bezpečnost enzalutamidu u pacientů s metastatickým kastračně rezistentním karcinomem prostaty, kteří dostávali docetaxel, užívali analog LHRH nebo podstoupili orchiektomii, byla hodnocena v randomizované, placebem kontrolované, multicentrické klinické studii fáze 3. Celkem 1199 pacientů bylo randomizováno v poměru 2:1 k perorálnímu užívání buď enzalutamidu v dávce 160 mg jednou denně (N = 800), nebo placeba jednou denně (N = 399). Pacientům bylo povoleno, ale nebylo vyžadováno, aby užívali prednison (povolená maximální denní dávka činila 10 mg prednisonu nebo ekvivalent). Pacienti randomizovaní do jedné z těchto skupin pokračovali v léčbě až do progrese onemocnění (definované jako potvrzená radiografická progrese nebo výskyt nepříznivé skeletální příhody) a zahájení nové systémové antineoplastické léčby, nepřijatelné toxicity, nebo odstoupení ze studie.
Následující demografické údaje pacientů a základní charakteristiky onemocnění byly mezi léčenými skupinami vyrovnané. Střední věk byl 69 let (rozmezí 41 - 92) a rasové rozdělení bylo 93 % bělochů, 4 % černochů, 1 % Asiatů a 2 % ostatních. Hodnocení účinnosti podle ECOG skóre činilo 0 - 1 u 91,5 % pacientů a 2 u 8,5 % pacientů; 28 % mělo střední skóre bolesti pomocí stručných dotazníků bolesti (Brief Pain Inventory) >4 (pacientem hlášená střední nejhorší bolest v průběhu předchozích 24 hodin počítaná po dobu sedm dní před randomizací). Většina (91 %) pacientů měla metastázy do kostí a 23 % mělo viscerální metastázy do plic nebo do jater. Při vstupu do studie mělo 41 % randomizovaných pacientů pouze PSA progresi, zatímco 59 % pacientů mělo radiografickou progresi. Padesát jedno procento (51 %) pacientů bylo ve výchozím stavu na bisfosfonátech.
Ze studie AFFIRM byli vyloučeni pacienti se zdravotními potížemi, které je mohly predisponovat k epileptickým záchvatům (viz bod 4.8), a pacienti užívající léčivé přípravky, o nichž je známo, že snižují práh pro vznik epileptických záchvatů, stejně jako pacienti s klinicky významným kardiovaskulárním onemocněním, jako je nekontrolovaná hypertenze, nedávno prodělaný infarkt myokardu nebo nestabilní angina pectoris, selhání srdce funkční třídy III nebo IV podle Newyorské kardiologické asociace (pokud ejekční frakce nebyla > 45 %), klinicky významné komorové arytmie nebo AV blok (bez permanentního kardiostimulátoru).
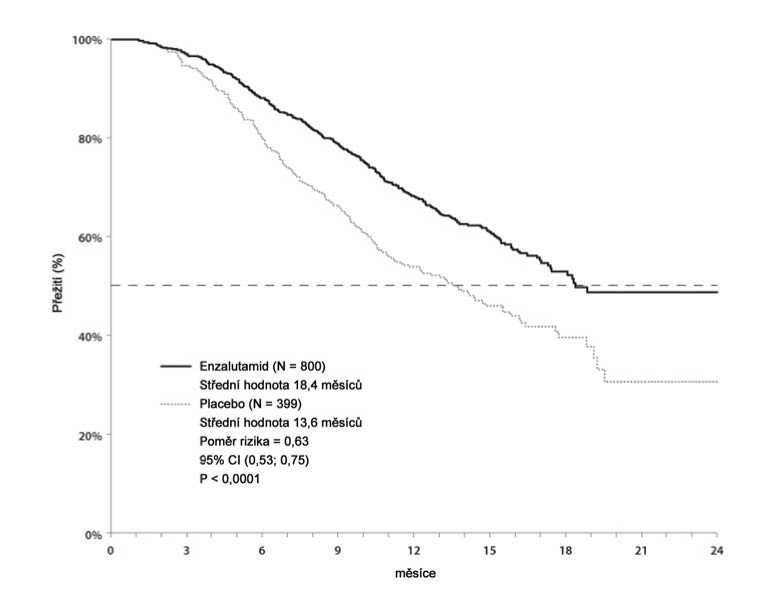
Protokolem předem specifikovaná předběžná analýza po 520 úmrtích ukázala statisticky významnou převahu v celkovém přežití u pacientů léčených enzalutamidem ve srovnání s placebem (tabulka 3 a obr. 4 a 5).
Tabulka 3: Celkové přežití pacientůlLéčených buď enzalutamidem, nebo placebem ve studii AFFIRM (analýza intent-to-treat)
|
Enzalutamid (N = 800) |
Placebo (N = 399) | |
|
Úmrtí (%) |
308 (38,5 %) |
212 (53,1 %) |
|
Střední přežití (měsíce) (95% CI) |
18,4 (17,3; NR) |
13,6 (11,3; 15,8) |
|
P hodnota3 |
< 0,0001 | |
|
Poměr rizika (95% CI)b |
0,631 (0,529; 0,752) | |
a) P hodnota je odvozena od log-rank testu stratifikovaného podle ECOG skóre výkonnosti (0 -a středního skóre bolesti (< 4 vs. > 4)
b) Poměr rizika je odvozen od stratifikovaného proporcionálního modelu rizika. Poměr rizika < podporuje enzalutamid
NR, nedosaženo.
1 vs. 2) 1
Obr. 4: Křivky celkového přežití podle Kaplan-Meiera ve studii AFFIRM (analýza ITT -
intent-to-treat)
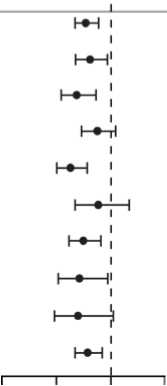
Obr. 5: Celkové přežití podle podskupin ve studii AFFIRM - poměr rizika a 95 % interval Spolehlivosti (CI - confidence interval)
Celková střední hodnota přežiti (més) enzalutamidřplacebo
Počet pacientů Poměr rizika úmrtí
Podskupina enzalutamid / placebo [95% Cl)
|
Všichni pacienti |
80C/399 |
0,63(0,53-0,75) | |
|
Věk | |||
|
<65 |
2327130 |
t ♦ \ |
0,63(0,46-0,87) |
|
265 |
568/269 |
h#-H |
0,63 (0,51-0,78) |
|
Výchozí ECOG skóre účinnosti | |||
|
0-1 |
730/367 |
»-l |
0,62 (0,52-0,75) |
|
2 |
70732 |
I * t |
0,65 (0,39-1,07) |
|
Výchozí střední skóre bolesti na BPt-SF (otázka č. 3) | |||
|
<4 |
574/284 |
l-e—l |
0,59(0,47-0,74) |
|
24 |
226/115 |
-1 |
0,71 (0,54-0,94) |
|
Počet předchozích chemoterapeutických režimů | |||
|
1 |
579/296 |
rt—\ |
0,59(0,48-0,73) |
|
>2 |
2217103 |
i—•-1 |
0,74 (0,54-1,03) |
|
Typ progrese při vstupu do studie | |||
|
Pouze PSA progrese |
3267164 |
\ t i |
0,62(0,46-0,83) |
|
Rentgenologická progrese + PSA progrese |
470/234 |
hf—i |
0,64 (0,52-0.80) |
|
Výchozí hodnota PSA í střední hodnota (111,2 pg/l) |
4127188 |
0,67(0,50-0,89) | |
|
> střední hodnota (111,2 pg/l) |
368/211 |
i—•—i |
0,62 (0,50-0,78) |
|
Výchozí hodnota LDH | |||
|
s střední hodnota (211IU/I) |
411/192 |
i—«—i |
0,63(0,46-0,86) |
|
> středni hodnota (211 IU/I) |
389/205 |
h-#H |
0,61 (0,50-0,76) |
|
Celkové Gleason skóre v době stanoveni diagnózy <7 |
3807175 |
1 • 1 |
0,67(0,51-0,88) |
|
>8 |
3667193 |
h-#—I |
0,60 (0,47-0,76) |
|
Viscerálni onemocněni plic nebo jater při screeningu Ano |
196/82 |
, , 1, |
0,78(0,56-1,09) |
|
Ne |
604/317 |
h#H |
0,56 (0,46-0,69) |
18,4/13,6
~/12,4
18,4/13,9 -/14,2
10,5/7,2
—/16.2 12,4/9,1
-/14,2
15,9/12,3 -/19,5
17,3/13,0
-/19,2
15,3/10,3 —719,2
12,4/85 18,4/14,8
18,2/11,3
13,4/9,5
—714,2
0,0 05 to t5 O0
Zvýhodňuje enzalutamid Zvýhodňuje placebo
ECOG: Eastem Cooperative Oncology Group; BPI-SF: Brief Pain Inventroy-Short Form PSA: Prostatě Specific Antigen
Kromě pozorovaného zlepšení celkového přežití, upřednostnily klíčové sekundární cílové parametry (progrese PSA, přežití bez rentgenologické progrese a doba do první skeletální příhody) enzalutamid a byly statisticky významné po úpravě pro vícenásobné testování.
Přežití bez progrese podle rentgenologického hodnocení ze strany zkoušejícího pomocí RECIST v1.1 pro měkké tkáně a výskytu 2 nebo více kostních lézí na skenu kostí činilo 8,3 měsíců u pacientů léčených enzalutamidem a 2,9 měsíce u pacientů, kteří dostávali placebo (poměr rizika, HR = 0,404; 95% CI: [0,350; 0,466]; p < 0,0001). Analýza zahrnovala 216 úmrtí bez zdokumentované progrese a 645 zdokumentovaných případů progrese, z nichž 303 (47 %) bylo v důsledku progrese do měkké tkáně, 268 (42 %) bylo v důsledku progrese kostních lézí a 74 (11 %) bylo v důsledku progrese do měkké tkáně i kostních lézí.
Potvrzený pokles PSA o 50 % nebo o 90 % byl 54,0 % a 24,8 % u pacientů léčených enzalutamidem a 1,5 % a 0,9 % u pacientů, kteří dostávali placebo (p < 0,0001). Střední doba do progrese PSA činila 8,3 měsíce u pacientů léčených enzalutamidem a 3,0 měsíce u pacientů, kteří dostávali placebo (HR = 0,248; 95% CI: [0,204; 0,303]; p < 0,0001).
Střední doba do první nepříznivé skeletální příhody činila 16,7 měsíců u pacientů léčených enzalutamidem a 13,3 měsíce u pacientů, kteří dostávali placebo (HR = 0,688; 95% CI: [0,566; 0,835]; p < 0,0001). Nepříznivá skeletální příhoda byla definována jako radiační terapie nebo operace kostí, patologická fraktura kostí, komprese míchy nebo změna protinádorové terapie k léčbě bolesti kostí. Analýza zahrnovala 448 nepříznivých skeletálních příhod, z nichž 277 příhod (62 %) bylo ozařování kostí, 95 příhod (21 %) byly komprese míchy, 47 příhod (10 %) byly patologické zlomeniny kostí,
36 příhod (8 %) byly změny v protinádorové terapii k léčbě bolesti kostí a 7 příhod (2 %) byly operace kostí.
Účinnost enzalutamidu u pacientů, kteří dříve užívali abirateron-acetát, nebyla studována.
Starší pacienti
Ze 1671 pacientů ve studiích fáze 3, kteří dostávali enzalutamid, bylo 1261 pacientů (75 %) ve věku 65 let a vyšším a 516 pacientů (31 %) bylo ve věku 75 let a vyšším. U těchto starších pacientů nebyly pozorovány žádné rozdíly v bezpečnosti nebo účinnosti ve srovnání s mladšími pacienty.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s enzalutamidem u všech podskupin pediatrické populace u karcinomu prostaty (viz bod 4.2 -informace pro pediatrické použití).
5.2 Farmakokinetické vlastnosti
Enzalutamid je špatně rozpustný ve vodě. U tohoto přípravku se rozpustnost zvyšuje emulgátorem/detergentem glyceromakrogol-oktanodekanoátem. Vpreklinických studiích byla absorpce enzalutamidu zvyšována rozpuštěním v glyceromakrogol-oktanodekanoátu.
Farmakokinetika enzalutamidu byla hodnocena u pacientů s karcinomem prostaty a u zdravých mužských subjektů. Průměrný konečný poločas rozpadu (t1/2) u enzalutamidu u pacientů po jedné perorální dávce je 5,8 dne (v rozmezí od 2,8 do 10,2 dnů) a ustáleného stavu je dosaženo přibližně za jeden měsíc. Při každodenním perorálním podání se enzalutamid akumuluje přibližně 8,3násobně v porovnání s jednou dávkou. Denní kolísání plazmatických koncentrací jsou nízká (poměr minimálních a maximálních hodnot - peak-to-trough, 1,25). Ke clearance enzalutamidu dochází hlavně prostřednictvím metabolismu v játrech, přičemž vzniká aktivní metabolit, který je srovnatelně aktivní jako enzaluamid a cirkuluje přibližně ve stejných plazmatických koncentracích jako enzalutamid.
Absorpce
Maximální plazmatické koncentrace (Cmax) enzalutamidu u pacientů jsou pozorovány 1 až 2 hodiny po podání. Na základě balanční studie celkového příjmu a výdeje u lidí se perorální absorpce enzalutamidu odhaduje nejméně na 84,2 %. Enzalutamid není substrátem efluxních transportérů P-gp nebo BCRP. V ustáleném stavu jsou průměrné hodnoty Cmax pro enzalutamid a jeho aktivní metabolity 16,6 pg/ml (23 % variační koeficient [CV]) a 12,7 pg/ml (30 % CV).
Jídlo nemá klinicky významný účinek na míru absorpce. V klinických studiích byl přípravek Xtandi podáván bez ohledu na jídlo.
Distribuce
Průměrný zdánlivý distribuční objem (V/F) enzalutamidu u pacientů po jedné perorální dávce je 110 l (29 % CV). Distribuční objem enzalutamidu je větší než objem celkové tělesné tekutiny, což svědčí o rozsáhlé extravaskulární distribuci. Studie u hlodavců naznačují, že enzalutamid a jeho aktivní metabolit mohou překonat hematoencefalickou bariéru.
Enzalutamid se z 97 % až 98 % váže na plazmatické proteiny, především na albumin. Aktivní metabolit je z 95 % vázán na plazmatické proteiny. Nedošlo k vytěsňování proteinové vazby mezi enzalutamidem a dalšími vysoce vázanými léčivými přípravky (warfarin, ibuprofen a kyselina salicylová) in vitro.
Biotransformace
Enzalutamid je rozsáhle metabolizován. V lidské plazmě existují dva hlavní metabolity: N-desmethyl enzalutamid (aktivní) a derivát kyseliny karboxylové (neaktivní). Enzalutamid je metabolizován cytochromem CYP2C8 a v menším rozsahu CYP3A4/5 (viz bod 4.5), které oba hrají roli v tvorbě aktivního metabolitu. In vitro se N-desmethyl enzalutamid metabolizuje na metabolit karboxylové kyseliny pomocí karboxylesterázy 1, která rovněž hraje menší úlohu v metabolismu enzalutamidu na metabolit karboxylové kyseliny. N-desmethyl enzalutamid nebyl metabolizován CYP in vitro.
Za podmínek klinického použití je enzalutamid silný induktor cytochromu CYP3A4, středně silný induktor CYP2C9 a CYP2C19, a nemá žádný klinicky významný účinek na CYP2C8 (viz bod 4.5).
Eliminace
Průměrná zdánlivá clearance (CL/F) enzalutamidu u pacientů se pohybuje mezi 0,520 a 0,564 l/h.
Po perorálním podání 14C-enzalutamidu je 84,6 % radioaktivity vyloučeno do 77 dní po dávce: 71,0 % je vyloučeno močí (převážně v podobě neaktivního metabolitu se stopovým množstvím enzalutamidu a aktivního metabolitu), a 13,6 % je vyloučeno stolicí (0,39 % dávky jako nezměněný enzalutamid).
Údaje in vitro ukazují, že enzalutamid není substrátem pro OATP1B1, OATP1B3, nebo OCT1 a N-desmethyl enzalutamid není substrátem pro P-gp nebo BCRP.
Údaje in vitro ukazují, že enzalutamid a jeho hlavní metabolity neinhibují následující transportéry v klinicky relevantních koncentracích: OATP1B1, OATP1B3, OCT2 nebo OAT1.
Linearita/nelinearita
Při dávkách v rozmezí od 40 do 160 mg nebyly pozorovány žádné závažné odchylky od proporcionality dávky. V ustáleném stavu Cmin hodnoty enzalutamidu a aktivního metabolitu zůstaly konstantní u jednotlivých pacientů po dobu více než jednoho roku chronické terapie, což prokazuje časově lineární farmakokinetiku, jakmile je jednou dosaženo ustáleného stavu.
Porucha funkce ledvin
Nebyla provedena žádná formální studie enzalutamidu při poškození ledvin. Pacienti s hladinou sérového kreatininu > 177 ^mol/l (2 mg/dl) byli z klinických studií vyloučeni. Na základě populační farmakokinetické analýzy není nutná úprava dávky u pacientů s vypočítanými hodnotami clearance kreatininu (CrCL) > 30 ml/min (odhadnutými podle vzorce Cockcrofta a Gaulta). Enzalutamid nebyl hodnocen u pacientů s těžkou poruchou funkce ledvin (CrCL < 30 ml/min) nebo v konečném stadiu onemocnění ledvin, a při léčbě těchto pacientů se doporučuje opatrnost. Je nepravděpodobné, že enzalutamid bude významně eliminován při intermitentní hemodialýze nebo kontinuální ambulantní peritoneální dialýze.
Porucha funkce jater
Porucha funkce jater nemá výrazný vliv na celkovou expozici enzalutamidu nebo jeho aktivních metabolitů. Nicméně poločas rozpadu byl u pacientů s těžkou poruchou funkce jater ve srovnání se zdravými kontrolními pacienty zdvojnásoben (10,4 dnů v porovnání k 4,7 dnům), pravděpodobně v souvislosti se zvýšenou distribucí v tkáních.
Farmakokinetika enzalutamidu byla zkoumána u subjektů s výchozí mírnou (N = 6), středně těžkou (N = 8) nebo závažnou (N=8) poruchou funkce jater (Child-Pugh třída A, B, resp. C) a u 22 odpovídajících kontrolních subjektů s normální funkcí jater. V porovnání se zdravými kontrolními pacienty se po jednorázové perorální dávce 160 mg enzalutamidu u pacientů s mírnou poruchou jater hodnota AUC enzalutamidu zvýšila o 5 % a Cmax o 24 %, u pacientů se středně těžkou poruchou jater se AUC enzalutamidu zvýšila o 29 %, resp. Cmax klesla o 11 % a u pacientů se závažnou poruchou funkce jater se AUC enzalutamidu zvýšila o 5 % a Cmax klesla o 41 %. V součtu nevázaného enzalutamidu a nevázaného aktivního metabolitu se u pacientů s mírnou poruchou jater zvýšila AUC o 14% a Cmax o 19%, u pacientů se středně těžkou poruchou jater se AUC zvýšila o 14%, resp. Cmax klesla o 17% a u pacientů se závažnou poruchou funkce jater se AUC enzalutamidu zvýšila o 34 % a Cmax klesla o 27 % v porovnání se zdravými kontrolními pacienty.
Rasa
Většina pacientů v klinických studiích (> 84 %) byli běloši. Na základě farmakokinetických údajů ze studie u japonských pacientů s karcinomem prostaty nebyly zjištěny žádné klinicky významné rozdíly v expozici mezi Japonci a bělochy. K vyhodnocení potenciálních rozdílů ve farmakokinetice enzalutamidu u jiných ras není dostatek údajů.
Starší pacienti
V populační farmakokinetické analýze nebyl shledán žádný klinicky relevantní vliv věku na farmakokinetiku enzalutamidu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Léčba enzalutamidem u březích myší vedla ke zvýšené incidenci úmrtí embryí a plodů a k externím a skeletálním změnám. Studie reprodukční toxicity enzalutamidu nebyly provedeny, ale při studiích u potkanů (4 a 26 týdnů) a psů (4, 13 a 39 týdnů) byly zjištěny atrofie, aspermie/hypospermie a hypertrofie/hyperplazie reprodukčních orgánů, v souladu s farmakologickou aktivitou enzalutamidu. Při studiích u myší (4 týdny), potkanů (4 a 26 týdnů) a psů (4, 13 a 39 týdnů), byly ve spojení s enzalutamidem pozorovány změny u reprodukčních orgánů, a to pokles hmotnosti orgánů s atrofií prostaty a nadvarlete. Hypertrofile Leydingových buněk a/nebo hyperplazie byly pozorovány u myší (4 týdny) a psů (39 týdnů). Další změny reprodukčních tkání zahrnovaly hypertrofii/hyperplazii hypofýzy a atrofii semenných váčků u potkanů a testikulární hypospermii a degeneraci semenných tubulů u psů. Rozdíly mezi pohlavími byly zaznamenány u prsních žláz potkanů (atrofie u samců a lobulámí hyperplazie u samic). Změny u reprodukčních orgánů u obou druhů byly konzistentní s farmakologickou aktivitou enzalutamidu a k jejich zvratu nebo částečnému vyřešení došlo po 8 týdenním období zotavení. Ani u jednoho z těchto druhů zvířat nebyly žádné další významné změny v klinické patologii nebo histopatologii v jakémkoliv dalším orgánovén systému, včetně jater.
Enzalutamid nevyvolával mutace v testu mikrobiální mutageneze (Amesův test) a nebyl klastogenní v žádném testu in vitro s lymfomovými buňkami myší, ani v mikronukleovém testu in vivo na myších. Dlouhodobé studie na zvířatech k vyhodnocení karcinogenního potenciálu enzalutamidu nebyly provedeny. Enzalutamid nebyl fototoxický in vitro.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
glyceromakrogol-oktanodekanoát butylhydroxyanisol (E320) butylhydroxytoluen (E321)
Tobolka
želatina
dehydratovaný sorbitol glycerol
oxid titaničitý (E171) čištěná voda
Černý inkoust
černý oxid železitý (E172)
polyvinyl-acetát-ftalát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Kartonové pouzdro obsahující PVC/PCTFE/Al blistry obsahující 28 měkkých tobolek. Jedna krabička obsahuje 4 pouzdra (112 měkkých tobolek).
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Astellas Pharma Europe B.V.
Sylviusweg 62 2333 BE Leiden Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/846/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. června 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ /VÝROB CI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Astellas Pharma Europe B.V.
Sylviusweg 62 2333 BE Leiden Nizozemsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 8 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xtandi 40 mg měkké tobolky enzalutamidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje enzalutamidum 40 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sorbitol (E420).
Další informace viz Příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
112 měkkých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Astellas Pharma Europe B.V. Sylviusweg 62 2333 BE Leiden Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/846/001 112 měkkých tobolek
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xtandi 40 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xtandi 40 mg měkké tobolky enzalutamidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje enzalutamidum 40 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sorbitol (E420).
Další informace viz Příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 měkkých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Perorální podání.
pondělí
úterý
středa
čtvrtek
pátek
sobota
neděle
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Astellas Pharma Europe B.V. Sylviusweg 62 2333 BE Leiden Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xtandi 40 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Xtandi 40 mg
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI_
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE_
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Xtandi 40 mg měkké tobolky
enzalutamidum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Xtandi a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Xtandi užívat
3. Jak se Xtandi užívá
4. Možné nežádoucí účinky
5. Jak Xtandi uchovávat
6. Obsah balení a další informace
1. Co je Xtandi a k čemu se používá
Xtandi obsahuje léčivou látku enzalutamid. Xtandi se používá k léčbě dospělých mužů s rakovinou prostaty, která se rozšířila do dalších částí těla.
Jak Xtandi účinkuje
Xtandi je lék, který způsobuje zablokování aktivity hormonů nazývaných androgeny (např. testosteron). Blokováním androgenů enzalutamid zastavuje růst a dělení rakovinných buněk.
2. Čemu musíte věnovat pozornost, než začnete Xtandi užívat Neužívejte Xtandi:
- Jestliže jste alergický(á) (přecitlivělý(á)) na enzalutamid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže jste těhotná nebo můžete otěhotnět (viz „Těhotenství, kojení a fertilita“)
Upozornění a opatření
Epileptický záchvat
Epileptické záchvaty byly hlášeny asi u 5 z 1000 lidí užívajících Xtandi a u méně než jednoho z 1000 lidí užívajících placebo (viz také „Další léčivé přípravky a Xtandi“ v tomto bodu a v bodu 4, „Možné nežádoucí účinky“).
Mezi některé situace, při nichž můžete mít vyšší riziko epileptického záchvatu, patří:
- Pokud jste měl dřívější epizody epileptického záchvatu
- Pokud jste prodělal vážné poranění hlavy nebo máte poranění hlavy v anamnéze
- Pokud jste prodělal určité druhy cévní mozkové příhody
- Pokud jste měl nádor na mozku nebo rakovinu, která se rozšířila do mozku
- Pokud pijete velké množství alkoholu buď pravidelně, nebo čas od času
- Pokud užíváte léky, které mohou vyvolat epileptické záchvaty nebo které mohou zvýšit náchylnost k epileptickým záchvatům (viz „Další léčivé přípravky a Xtandi“ níže)
Pokud máte epileptický záchvat během léčby:
Přestaňte užívat Xtandi a už neužívejte žádné další tobolky. Co nejdříve navštivte svého lékaře. Syndrom posteriorní reverzibilní encefalopatie (PRES)
Vzácně došlo u pacientů léčených přípravkem Xtandi k hlášení PRES, což je vzácný vratný stav postihující mozek. Jestliže máte epileptický záchvat, zhoršující se bolesti hlavy, zmatenost, oslepnutí nebo jiné poruchy zraku, kontaktujte co nejdříve svého lékaře. (Viz také bod 4 „Možné nežádoucí účinky“).
Před užitím Xtandi se poraďte se svým lékařem
- Pokud užíváte nějaké léky k prevenci vzniku krevních sraženin (např. warfarin, acenokumarol)
- Pokud máte potíže s játry
- Pokud máte potíže s ledvinami
Informujte prosím svého lékaře, pokud trpíte něčím z následujícího:
Jakékoliv srdeční či cévní onemocnění, včetně poruch srdečního rytmu (arytmie), nebo léčba těchto onemocnění. Při užívání přípravku Xtandi může být zvýšeno riziko poruch srdečního rytmu.
Před užitím přípravku se poraďte se svým lékařem, pokud se Vás týká některý z výše uvedených bodů, nebo si nejste jist.
Děti a dospívající
Tento léčivý přípravek není určen pro podávání dětem a dospívajícím.
Další léčivé přípravky a Xtandi
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval nebo které možná budete užívat. Je potřeba, abyste znal(a) názvy léků, které užíváte. Mějte u sebe jejich seznam a ukažte jej svému lékaři, pokud dostanete nový lék. Nezačínejte nebo nepřestávejte užívat žádný lék dříve, než se poradíte s lékařem, který Vám předepsal Xtandi.
Informujte svého lékaře, pokud užíváte některý z následujících léků. Při současném užívání s přípravkem Xtandi mohou tyto léky zvyšovat riziko epileptického záchvatu:
- Některé léky používané k léčbě astmatu nebo dalších onemocnění dýchacích cest (např. aminofylin, theofylin)
- Léky používané k léčbě některých duševních poruch, jako jsou deprese a schizofrenie (např. klozapin, olanzapin, risperidon, ziprasidon, bupropion, lithium, chlorpromazin, mesoridazin, thioridazin, amitriptylin, desipramin, doxepin, imipramin, maprotilin, mirtazapin)
- Některé léky k léčbě bolesti (např. pethidin)
Informujte svého lékaře, pokud užíváte některý z následujících léků. Tyto léky mohou ovlivnit účinek přípravku Xtandi, nebo Xtandi může ovlivnit účinek těchto léků:
Jsou to některé z léků, které se používají:
- Ke snižování hladiny cholesterolu (např. gemfibrozil, atorvastatin, simvastatin)
- K léčbě bolesti (např. fentanyl, tramadol)
- K léčbě rakoviny (např. kabazitaxel)
- K léčbě epilepsie (např. karbamazepin, klonazepam, fenytoin, primidon, kyselina valproová)
- K léčbě určitých psychiatrických poruch, jako jsou těžké stavy úzkosti nebo schizofrenie (např. diazepam, midazolam, haloperidol)
- K léčbě poruch spánku (např. zolpidem)
- K léčbě srdečních onemocnění nebo k snížení krevního tlaku (např. bisoprolol, digoxin, diltiazem, felodipin, nikardipin, nifedipin, propranolol, verapamil)
- K léčbě závažných onemocnění souvisejících se zánětem (např. dexamethason, prednisolon)
- K léčbě nákazy HIV (např. indinavir, ritonavir)
- K léčbě bakteriálních infekcí (např. klarithromycin, doxycyklin)
- K léčbě poruch štítné žlázy (např. levothyroxin)
- K léčbě dny (např. kolchicin)
- K prevenci srdečních onemocnění nebo mrtvice (dabigatran-etexilát)
Přípravek Xtandi a některé souběžně užívané přípravky k léčbě poruch srdečního rytmu (např. chinidin, prokainamid, amiodaron a sotalol) se mohou navzájem ovlivňovat. Přípravek Xtandi může zvyšovat riziko poruch srdečního rytmu, pokud je užíván s některými dalšími přípravky (např. methadon (užívaný k úlevě od bolesti nebo jako část odvykací terapie), moxifloxacin (antibiotikum), antipsychotika užívaná k léčbě závažných duševních poruch).
Informujte svého lékaře, pokud užíváte některý z výše uvedených léků. Možná bude nutné změnit dávku přípravku Xtandi nebo jiných léků, které užíváte.
Těhotenství, kojení a plodnost
- Přípravek Xtandi není určen k použití u žen. Tento přípravek může poškodit nenarozené dítě, pokud jej užívají těhotné ženy. Nesmí jej užívat ženy, které jsou těhotné, mohou otěhotnět, nebo kojí.
- Tento přípravek by mohl mít vliv na mužskou plodnost.
- Máte-li pohlavní styk se ženou, která může otěhotnět, používejte kondom a další účinnou antikoncepční metodu v průběhu léčby tímto přípravkem a 3 měsíce po jejím ukončení. Pokud máte pohlavní styk s těhotnou ženou, používejte kondom k ochraně nenarozeného dítěte.
Řízení dopravních prostředků a obsluha strojů
Tento léčivý přípravek může mít středně závažný vliv na schopnost řídit, používat nástroje nebo obsluhovat stroje, protože nežádoucí účinky přípravku Xtandi zahrnují epileptické záchvaty. Pokud je u Vás zvýšené riziko epileptických záchvatů (viz bod 2), poraďte se s lékařem.
Xtandi obsahuje sorbitol
Tento lék obsahuje sorbitol (typ cukru). Pokud Vám lékař řekl, že trpíte nesnášenlivostí některých cukrů, před užitím tohoto přípravku se poraďte se svým lékařem.
3. Jak se přípravek Xtandi užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý, poraďte se se svým lékařem.
Obvyklá dávka přípravku je 160 mg (čtyři tobolky), užívaná najednou jednou denně.
Užívání přípravku Xtandi
- Tobolky polykejte celé a zapíjejte je vodou.
- Před polykáním tobolky nežvýkejte, nerozpouštějte ani neotvírejte.
- Přípravek Xtandi může být užíván s jídlem nebo bez jídla.
Během užívání přípravku Xtandi Vám může lékař také předepsat jiné léky.
Jestliže jste užil více přípravku Xtandi, než jste měl
Jestliže jste užil více tobolek, než je předepsáno, přestaňte užívat přípravek Xtandi a kontaktujte svého lékaře. Můžete mít zvýšené riziko epileptického záchvatu nebo jiných nežádoucích účinků.
Jestliže jste zapomněl užít přípravek Xtandi
- Jestliže jste zapomněl užít přípravek Xtandi v obvyklou dobu, vezměte si obvyklou dávku, jakmile si vzpomenete.
- Jestliže jste zapomněl užít přípravek Xtandi celý den, vezměte si obvyklou dávku následující den.
- Jestliže j ste zapomněl užít přípravek Xtandi déle než jeden den, ihned to sdělte svému lékaři.
- Nezdvojnásobujte následující dávku, abyste nahradil vynechanou dávku.
Jestliže jste přestal užívat přípravek Xtandi
Nepřestávejte užívat tento přípravek, dokud Vám to lékař neřekne.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Epileptické záchvaty
Epileptické záchvaty byly hlášeny u 5 z 1000 lidí užívajících Xtandi a u méně než jednoho z 1000 lidí užívajících placebo.
Epileptické záchvaty jsou pravděpodobnější, pokud užijete větší než doporučenou dávku tohoto přípravku, pokud užíváte určité jiné léky, nebo je-li u Vás vyšší než obvyklé riziko záchvatů (viz bod 2).
Pokud máte epileptický záchvat, co nejdříve navštivte svého lékaře. Neužívejte už žádnou další dávku přípravku Xtandi.
Syndrom posteriorní reverzibilní encefalopatie (PRES)
Vzácně došlo u pacientů léčených přípravkem Xtandi k hlášení PRES (může postihnout až 1 z 1000 osob), což je vzácný vratný stav postihující mozek. Jestliže máte epileptický záchvat, zhoršující se bolesti hlavy, zmatenost, oslepnutí nebo jiné poruchy zraku, kontaktujte prosím co nejdříve svého lékaře.
Další možné nežádoucí účinky zahrnují:
Velmi časté (mohou postihnout více než 1 z 10 osob)
Únava, bolest hlavy, návaly horka, vysoký krevní tlak
Časté (mohou postihnout až 1 z 10 osob)
Pády, zlomeniny kostí, pocity úzkosti, suchá kůže, svědění, potíže se zapamatováním, zvětšení prsů u mužů (gynekomastie), příznaky syndromu neklidných nohou (nekontrolovatelné nutkání pohybovat částí těla, obvykle nohou), snížená koncentrace, zapomětlivost
Méně časté (mohou postihnout až 1 ze 100 osob)
Halucinace, potíže s jasným uvažováním, nízkých počet bílých krvinek
Není známo (z dostupných údajů nelze určit)
Bolest svalů, svalové křeče, svalová slabost, bolest zad, EKG změny (prodloužení QT intervalu), podrážděný žaludek s pocitem na zvracení (nevolnost), vyrážka, zvracení, otok rtů, jazyka a/nebo hrdla, snížení počtu krevních destiček (což zvyšuje riziko krvácení nebo tvorby podlitin), průjem
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na kartonovém pouzdru a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Neužívejte žádnou tobolku, která prosakuje, je poškozená nebo vykazuje známky falšování.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní
prostředí.
6. Obsah balení a další informace
Co Xtandi obsahuje
- Léčivou látkou je enzalutamidum. Jedna tobolka obsahuje enzalutamidum 40 mg.
- Dalšími složkami tobolky jsou glyceromakrogol-oktanodekanoát, butylhydroxyanisol (E320) a butylhydroxytoluen (E321).
- Složkami obalu tobolky jsou želatina, dehydratovaný sorbitol (viz bod 2), glycerol, oxid titaničitý (E171) a čištěná voda.
- Složkami barvy jsou: černý oxid železitý (E172) a polyvinyl-acetát-ftalát.
Jak Xtandi vypadá a co obsahuje toto balení
- Tobolky Xtandi jsou bílé až krémově bílé podlouhlé měkké tobolky (přibližně 20 mm x 9 mm) s vytištěným „ENZ“ na jedné straně.
- Jedna krabička obsahuje 112 tobolek ve 4 blistrových pouzdrech po 28 tobolkách.
Držitel rozhodnutí o registraci a výrobce
Astellas Pharma Europe B.V.
Sylviusweg 62 2333 BE Leiden Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Astellas Pharma B.V. Branch Astellas Pharma a/s
Tél/Tel: + 32 (0)2 5580710
Danija
Tel: +45 4343 0355
Ten.: + 359 2 862 53 72
Tél/Tel: + 32 (0)2 5580710
|
Danmark Astellas Pharma a/s Tlf: + 45 43 430355 |
Malta E.J. Busuttil Ltd. Tel: + 356 21 447184 |
|
Deutschland Astellas Pharma GmbH Tel: + 49 (0)89 454401 |
Nederland Astellas Pharma B.V. Tel: + 31 (0)71 5455745 |
|
Eesti Astellas Pharma a/s Taani Tel: +45 4343 0355 |
Norge Astellas Pharma Tlf: + 47 66 76 46 00 |
|
EXXáSa Astellas Pharmaceuticals AEBE Tr|^: + 30 210 8189900 |
Osterreich Astellas Pharma Ges.m.b.H. Tel: + 43 (0)1 8772668 |
|
Espaňa Astellas Pharma S.A. Tel: + 34 91 4952700 |
Polska Astellas Pharma Sp.z.o.o. Tel.: + 48 225451 111 |
|
France Astellas Pharma S.A.S. Tél: + 33 (0)1 55917500 |
Portugal Astellas Farma, Lda. Tel: + 351 21 4401320 |
|
Hrvatska Astellas d.o.o. Tel: + 385 1 670 01 02 |
Románia S.C.Astellas Pharma SRL Tel: + 40 (0)21 361 04 95 /96 /92 |
|
Ireland Astellas Pharma Co. Ltd. Tel: + 353 (0)1 4671555 |
Slovenija Astellas Pharma d.o.o. Tel: + 386 14011 400 |
|
Ísland Vistor hf Sími: + 354 535 7000 |
Slovenská republika Astellas Pharma s.r.o., Tel: + 421 2 4444 2157 |
|
Italia Astellas Pharma S.p.A. Tel: + 39 02 921381 |
Suomi/Finland Astellas Pharma Puh/Tel: + 358 (0)9 85606000 |
|
Kúnpoq Astellas Pharmaceuticals AEBE EAAáSa Tr|^: + 30 210 8189900 |
Sverige Astellas Pharma AB Tel: + 46 (0)40-650 15 00 |
|
Latvija Astellas Pharma a/s Danija Tel: +45 4343 0355 |
United Kingdom Astellas Pharma Ltd. Tel: + 44 (0)203 379 8700 |
Tato příbalová informace byla naposledy revidována MM/RRRR.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
37