Xolair 150 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Xolair 75 mg prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje omalizumabum* 75 mg.
Po rozpuštění obsahuje jedna injekční lahvička omalizumabum 125 mg/ml (75 mg v 0,6 ml).
*Omalizumab je humanizovaná monoklonální protilátka vyrobená technologií rekombinantní DNA v linii savčích buněk vaječníků čínských křečků (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: bílý až téměř bílý lyofilizát Rozpouštědlo: čirý a bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Xolair je indikován k léčbě dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Léčbu Xolairem je možno použít pouze u pacientů s astmatem vyvolaným prokazatelně IgE (imunoglobulinem E) (viz bod 4.2).
Dospělí a dospívající (12 let a starší)
Xolair se doporučuje jako doplňková léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a kteří mají sníženou funkci plic (FEV1 <80 %), stejně jako časté symptomy během dne nebo probouzení v noci, a kteří mají dokumentované těžké exacerbace astmatu navzdory vysokým denním dávkám inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
Děti (6 až <12 let)
Xolair se doporučuje jako přídatná léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a časté denní nebo noční příznaky buzení a kteří mají prokázané četné vážné exacerbace přesto, že užívají vysoké denní dávky inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
4.2 Dávkování a způsob podání
Léčba Xolairem by měla být zahájena lékařem zkušeným v diagnostice a léčbě těžkého perzistujícího astmatu.
Dávkování
Vhodná dávka a frekvence podávání Xolairu se určí podle výchozích hodnot IgE (IU/ml), které se stanoví před zahájením léčby, a dle tělesné hmotnosti (kg). Aby mohla být stanovena dávka, měla by být před podáním počáteční dávky u pacientů stanovena hladina IgE jakýmkoliv komerčním testem pro stanovení celkového IgE v séru. Na základě výsledků může být potřeba podávat 75 až 600 mg Xolairu v 1 až 4 injekcích na každé podání.
U pacientů s IgE nižším než 76 IU/ml je méně pravděpodobné, že se projeví přínos přípravku (viz bod
5.1). Ošetřující lékař se musí ujistit, že dospělí a dospívající pacienti s IgE nižším než 76 IU/ml a děti (6 až <12 let) s IgE nižším než 200 IU/ml mají před začátkem léčby prokázanou in vitro reaktivitu (RAST) na celoroční vzdušný alergen.
Viz Tabulka 1 schéma konverze a Tabulka 2 a 3 schéma stanovení dávky u dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Pacientům, jejichž výchozí hodnoty hladin IgE nebo tělesná hmotnost v kilogramech jsou mimo limity dávkovací tabulky, by Xolair neměl být podáván.
Maximální doporučená dávka je 600 mg omalizumabu každé dva týdny.
Tabulka 1: Konverze dávky na počet injekčních lahviček, počet injekcí a celkový objem injekcí při každé aplikaci
|
Dávka (mg) |
Počet injekčních lahviček 75 mg a 150 mg b |
Počet injekcí |
Celkový objem injekcí (ml) | |
|
75 |
1c |
0 |
1 |
0,6 |
|
150 |
0 |
1 |
1 |
1,2 |
|
225 |
1c |
1 |
2 |
1,8 |
|
300 |
0 |
2 |
2 |
2,4 |
|
375 |
1c |
2 |
3 |
3,0 |
|
450 |
0 |
3 |
3 |
3,6 |
|
525 |
1c |
3 |
4 |
4,2 |
|
600 |
0 |
4 |
4 |
4,8 |
a 0,6 ml = maximální množství obsažené v jedné injekční lahvičce (Xolair 75 mg). b 1,2 ml = maximální množství obsažené v jedné injekční lahvičce (Xolair 150 mg). c nebo užití 0,6 ml z obsahu injekční lahvičky 150 mg.
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE (IU/ml) |
>20- 25 |
>25- 30 |
>30- 40 |
>40- 50 |
>50- 60 |
>60- 70 |
>70- 80 |
>80- 90 |
>90- 125 |
>125- 150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
PODÁVÁNÍ KAŽDÉ 2 TÝDNY | |||||||||
|
>900- 1000 |
VIZ TABULKA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>125- | |
|
(IU/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
>90-125 |
150 |
|
>30-100 |
PODÁVÁNÍ KAŽDÉ 4 TÝDNY | |||||||||
|
>100-200 |
VIZ TABULKA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- 1200 |
300 |
300 |
450 |
525 |
600 |
NEPODÁVAT - pro doporučení dávkování není dostatek údajů | ||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Délka léčby, její sledování a úprava dávky
Xolair je určen pro dlouhodobou léčbu. Klinické studie prokázaly, že Xolair dosahuje účinnosti po minimálně 12-16 týdnech léčby. Po 16 týdnech od zahájení léčby Xolairem by měl lékař před podáváním dalších injekcí zhodnotit účinnost léčby. Rozhodnutí o pokračování léčby Xolairem následně po 16týdenním intervalu, nebo příležitostně později, by mělo být založeno na tom, zdali je patrné výrazné zlepšení z hlediska celkové kontroly astmatu (viz bod 5.1, Celkové hodnocení účinnosti léčby lékařem).
Ukončení léčby Xolairem má obecně za následek návrat ke zvýšeným hladinám volného IgE a s tím spojeným symptomům. Celkové hladiny IgE jsou zvýšené během léčby a zůstávají zvýšené až po dobu jednoho roku po skončení léčby. Proto opětovné stanovení hladin IgE během léčby Xolairem nemůže být použito jako vodítko pro stanovení dávky. Stanovení dávky po přerušení léčby trvající méně než jeden rok by mělo být založeno na výchozích hodnotách hladin sérového IgE získaných při stanovení zahajovací dávky. Pro stanovení dávky mohou být celkové hladiny IgE v séru znovu stanoveny, jestliže léčba Xolairem byla přerušena jeden rok nebo déle.
Dávky by měly být přizpůsobeny významným změnám tělesné hmotnosti (viz Tabulka 2 a 3).
Zvláštní skupiny pacientů
Starší pacienti (65 let a starší)
Pro užití Xolairu u pacientů starších více než 65 let jsou dostupné omezené údaje, ale nebylo zjištěno, že by u starších pacientů bylo nutné podávání jiné dávky než u mladších dospělých pacientů.
Pacienti s poškozením ledvin nebo jater
Nejsou k dispozici žádné studie hodnotící vliv zhoršené funkce ledvin nebo jater na farmakokinetiku Xolairu. Protože clearance omalizumabu je při klinických dávkách ovlivněna hlavně retikuloendotelovým systémem (RES), je nepravděpodobné, že by byla ovlivněna poškozením ledvin nebo jater. Protože není doporučena žádná zvláštní úprava dávkování pro tyto pacienty, Xolair by měl být podáván s opatrností (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost Xolairu u dětí mladších 6 let nebyla dosud stanovena. Nej sou dostupné žádné údaje.
Způsob podání
Pouze pro subkutánní podání. Nepodávejte intravenózně nebo intramuskulárně.
Injekce se aplikují subkutánně do oblasti deltového svalu ramene. Alternativně se může injekce aplikovat do stehna, jestliže je z jakéhokoliv důvodu vyloučena aplikace do oblasti deltového svalu.
Zkušenosti s aplikací injekce Xolairu samotným pacientem jsou omezené. Z toho důvodu má léčbu podávat pouze zdravotnický personál.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6 a také v bodě Informace pro zdravotnické pracovníky v příbalové informaci.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Obecné
Xolair není určen k léčbě exacerbací akutního astmatu, akutního bronchospasmu nebo status asthmaticus.
Xolair nebyl zkoumán u pacientů se syndromem hyperimunoglobulinemie E nebo alergickou bronchopulmonální aspergilózou nebo pro prevenci anafylaktických reakcí včetně takových, které jsou vyvolané alergií na potraviny, atopickou dermatitidou nebo alergickou rinitidou. Xolair není indikován pro léčbu těchto stavů.
Léčba Xolairem nebyla zkoumána u pacientů s autoimunitním onemocněním, s onemocněním zprostředkovaným imunokomplexy, nebo s preexistující zhoršenou funkcí ledvin nebo jater (viz bod
4.2). Pokud je Xolair podáván těmto skupinám pacientů, měla by být zachovávána opatrnost.
Náhlé vysazení systémových nebo inhalačních kortikosteroidů po zahájení léčby Xolairem se nedoporučuje. Snižování dávek kortikosteroidů je nutno provádět pod přímým dohledem lékaře a je vhodné jej provádět postupně.
Poruchy imunitního systému Alergické reakce typu I
Lokální nebo systémové alergické reakce typu I včetně anafylaxe a anafylaktického šoku se mohou objevit po podání omalizumabu, přičemž tyto reakce se mohou objevit i po dlouhotrvající léčbě omalizumabem. Většina těchto reakcí se objevila během 2 hodin po první a následné injekci Xolairu, ale některé se objevily až po 2 hodinách a dokonce i po více než 24 hodinách po injekci. Proto by následně po podání Xolairu měly být vždy dostupné k okamžitému užití léčivé přípravky na léčbu anafylaktických reakcí. Pacienti by měli být informováni o možnosti takových reakcí, a pokud se objeví alergická reakce, měli by vyhledat okamžitě lékařskou pomoc. Výskyt anafylaxe nesouvisející s omalizumabem v anamnéze může být rizikový faktor pro anafylaxi následující po podání Xolairu.
U malého počtu pacientů v klinických studiích byly detekovány protilátky proti omalizumabu (viz bod 4.8). Klinický význam protilátek proti Xolairu není dobře prostudován.
Sérová nemoc
Sérová nemoc a reakce podobné sérové nemoci, které j sou opožděnými alergickými reakcemi typu III, byly pozorovány u pacientů léčených humanizovanými monoklonálními protilátkami včetně omalizumabu. Předpokládaný patofyziologický mechanismus zahrnuje tvorbu a ukládání imunokomplexů v důsledku vzniku protilátek proti omalizumabu. Reakce se obvykle objevila 1-5 dní po podání prvních nebo následujících injekcí, i po dlouhodobé léčbě. Příznaky poukazující na sérovou nemoc zahrnují artritidu/artralgie, vyrážku (kopřivku nebo jiné formy), horečku a lymfadenopatii. K léčbě nebo prevenci této nemoci mohou být vhodná antihistaminika a kortikosteroidy. Pacienti by měli být poučeni, aby hlásili jakékoli podezřelé příznaky.
Syndrom Churga-Straussové a hypereozinofilní syndrom
U pacientů s těžkým astmatem se může zřídka vyskytnout systémový hypereozinofilní syndrom nebo alergická eozinofilní granulomatózní vaskulitida (syndrom Churga-Straussové), které se obvykle léčí systémovými kortikoidy.
Ve vzácných případech se mohou u pacientů léčených antiastmatickými léčivými přípravky, včetně omalizumabu, projevit systémová eozinofilie a vaskulitida. Tyto případy jsou často spojeny se snižováním léčby perorálními kortikoidy.
U těchto pacientů by lékaři měli věnovat zvýšenou pozornost možnému rozvoji eozinofilie, vaskulitické vyrážky, zhoršení plicních symptomů, abnormalit paranazálních dutin, srdečních komplikací a/nebo neuropatie.
Ve všech závažných případech výše uvedených poruch imunitního systému by mělo být zváženo vysazení omalizumabu.
Parazitární infekce (helmintóza)
IgE může být spojený s imunologickou odpovědí na některé parazitární infekce. V placebem kontrolované studii u pacientů s chronicky vysokým rizikem helmintóz bylo prokázáno mírné zvýšení podílu infekcí ve skupině s omalizumabem, ačkoliv průběh, závažnost a odpověď na léčbu infekce se nezměnily. Četnost výskytu infekce cizopasnými červy v celkovém klinickém programu, který nebyl navržen tak, aby takové infekce prokázal, byla méně než 1 z 1 000 pacientů. Nicméně měla by být zaručena opatrnost u pacientů s vysokým rizikem infekce cizopasnými červy, zejména pokud cestují do oblastí, kde se infekce cizopasnými červy vyskytují endemicky. Pokud pacienti neodpovídají na doporučenou antiparazitární léčbu, mělo by být zváženo přerušení léčby Xolairem.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Xolair může nepřímo snížit účinnost léčivých přípravků používaných k léčbě helmintóz nebo jiných parazitárních infekcí, protože v imunologické odpovědi na některé helmintózy může být zapojen IgE. (viz bod 4.4).
Enzymy cytochromu P450, pumpy zajišťující efflux a mechanismy vazby na proteiny se nepodílejí na clearance omalizumabu; takže je zde malá možnost lékových interakcí. Nebyly provedeny žádné studie interakce Xolairu s léčivými přípravky ani vakcínami. Není žádný farmakologický důvod očekávat, že by se běžně předepisované léčivé přípravky užívané v léčbě astmatu vzájemně ovlivňovaly s omalizumabem.
V klinických studiích byl Xolair běžně užíván v kombinaci s inhalačními a perorálními kortikosteroidy, inhalačními krátkodobě a dlouhodobě působícími beta agonisty, modifikátory leukotrienů, theofyliny a perorálními antihistaminiky. Nevyskytly se žádné údaje, že bezpečnost Xolairu byla změněna těmito dalšími běžně užívanými antiastmatickými léčivými přípravky. Jsou dostupné omezené údaje o užívání Xolairu v kombinaci se specifickou imunoterapií (hyposenzitizační terapie). V klinické studii, kde byl Xolair podáván společně s imunoterapií, nebyl zjištěn žádný rozdíl v bezpečnosti a účinnosti Xolairu v kombinaci se specifickou imunoterapií v porovnání s podáváním samotného Xolairu.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání omalizumabu těhotným ženám jsou omezené. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky (viz bod 5.3). Omalizumab přestupuje přes placentu, ale možnost poškození plodu není známa. U primátů byl omalizumab spojován se snížením na věku závislém počtu krevních destiček, s relativní větší citlivostí u nedospělých zvířat (viz bod
5.3). Xolair by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda se omalizumab vylučuje do lidského mateřského mléka. Dostupné údaje u samic ostatních primátů prokázaly vylučování omalizumabu do mléka (viz bod 5.3). Riziko pro kojené novorozence/děti nelze vyloučit. Omalizumab by neměl být podáván během kojení.
Fertilita
Pro omalizumab nejsou k dispozici žádné údaje týkající se lidské fertility. Ve specificky navržených neklinických studiích fertility u primátů (s výjimkou člověka), včetně studií páření, nebylo pozorováno poškození samčí ani samičí fertility následně po opakovaném podávání omalizumabu v dávkách až 75 mg/kg. Dále nebyly pozorovány žádné genotoxické účinky v samostatných neklinických studiích genotoxicity (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xolair nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Během klinických studií u dospělých a dospívajících pacientů starších 12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy a reakce v místě aplikace injekce, včetně bolestivosti, zduření, zarudnutí a svědění. V klinických studiích u dětí od 6 do <12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy, pyrexie a bolest v nadbřišku. Ve většině případů byly reakce mírné nebo středně závažné.
Tabulkový seznam nežádoucích účinků
Tabulka 4 zaznamenává nežádoucí účinky hlášené v klinických studiích celkové bezpečnosti u populace léčené Xolairem podle MedDRA orgánové klasifikace a četnosti výskytu. V každé skupině četností jsou nežádoucí reakce seřazeny podle klesající závažnosti. Kategorie četností jsou definovány jako: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000) a velmi vzácné (<1/10 000). Reakce hlášené z postmarketingového sledování jsou uvedeny s frekvencí není známo (z dostupných údajů nelze určit).
Tabulka 4: Nežádoucí účinky
|
Infekce a infestace | |
|
Méně časté |
Faryngitida |
|
Vzácné |
Parazitární infekce |
|
Poruchy krve a lymfatického systému | |
|
Není známo |
Idiopatická trombocytopenie, včetně těžkých případů |
|
Poruchy imunitního systému | |
|
Vzácné |
Anafylaktická reakce, jiné závažné alergické stavy, vývoj |
|
protilátek proti omalizumabu | |
|
Není známo |
Sérová nemoc, která může zahrnovat horečku a lymfadenopatii |
|
Poruchy nervového systému | |
|
Časté |
Bolest hlavy* |
|
Méně časté |
Synkopa, parestezie, somnolence, závrať |
|
Cévní poruchy | |
|
Méně časté |
Posturální hypotenze, zčervenání |
|
Respirační, hrudní a mediastinální poruchy | |
|
Méně časté |
Alergický bronchospasmus, kašel |
|
Vzácné |
Otok laryngu |
|
Není známo |
Alergická granulomatózní vaskulitida (tzv. syndrom Churga- |
|
Straussové) | |
|
Gastrointestinální poruchy | |
|
Časté |
Bolest v nadbřišku** |
|
Méně časté | |
|
Poruchy kůže a podkožní tkáně | |
|
Méně časté | |
|
Vzácné |
Angioedém |
|
Není známo |
Alopecie |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Vzácné |
Systémový lupus erythematodes (SLE) |
|
Není známo |
Artralgie, myalgie, otoky kloubů |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Pyrexie** |
|
Časté |
Reakce v místě aplikace jako zduření, zarudnutí, bolest, svědění |
|
Méně časté |
Onemocnění podobné chřipce, otoky paží, zvýšení hmotnosti, |
*: Velmi časté u dětí od 6 do <12 let **: U dětí od 6 do <12 let
Popis vybraných nežádoucích účinků Poruchy imunitního systému Další informace viz bod 4.4.
Anafylaxe
Anafylaktické reakce byly v klinických studiích vzácné. Nicméně v datech po uvedení přípravku na trh se následně po kumulativním vyhledávání v bezpečnostní databázi našlo celkem 898 případů anafylaxe. Na základě očekávané expozice 566 923 pacientoroků to mělo za následek míru hlášení přibližně 0,20 %.
Arteriální tromboembolické příhody (ATE)
V kontrolovaných klinických studiích a během interim analýz observační studie byla pozorována numerická nerovnováha v počtu ATE. Definice složeného endpointu ATE zahrnovala cévní mozkovou příhodu, transitorní ischemickou ataku, infarkt myokardu, nestabilní anginu pectoris a kardiovaskulární úmrtí (včetně úmrtí z neznámé příčiny). V konečné analýze observační studie byl poměr ATE na 1 000 pacientoroků 7,52 (115/15 286 pacientoroků) pro pacienty léčené Xolairem a 5,12 (51/9 963 pacientoroků) pro kontrolní skupinu. V multivariační analýze kontrolující základní dostupné kardiovaskulární rizikové faktory byl poměr rizik 1,32 (95 % interval spolehlivosti 0,91-1,91). V samostatné analýze poolovaných klinických studií, která zahrnovala všechny randomizované, dvojitě zaslepené, placebem kontrolované klinické studie trvající 8 nebo více týdnů, byl poměr ATE na 1 000 pacientoroků 2,69 (5/1 856 pacientoroků) pro pacienty léčené Xolairem a 2,38 (4/1 680 pacientoroků) pro pacienty užívající placebo (relativní riziko 1,13, 95 % interval spolehlivosti 0,24-5,71).
Krevní destičky
V klinických studiích mělo několik pacientů počet krevních destiček pod spodní hranicí normálního laboratorního rozmezí. Žádná z těchto změn nebyla spojena s epizodami krvácení nebo se snížením hemoglobinu. U lidí (pacientů nad 6 let) nebylo hlášeno trvalé snížení počtu krevních destiček, které bylo pozorováno u ostatních primátů (viz bod 5.3), přestože z postmarketingového sledování byly ojediněle hlášeny případy idiopatické trombocytopenie, včetně těžkých případů.
Parazitární infekce
Placebem kontrolovaná studie u pacientů s chronicky vysokým rizikem helmintóz ukázala mírně zvýšenou četnost výskytu infekcí u pacientů užívajících omalizumab ve srovnání s kontrolní skupinou. Toto zvýšení nebylo statisticky signifikantní. Průběh, závažnost a odpověď na léčbu infekcí se nezměnily (viz bod 4.4).
Systémový lupus erythematodes
U pacientů se středně těžkým až těžkým astmatem a chronickou spontánní urtikarií (CSU) byly hlášeny případy systémového lupus erythematodes (SLE) z klinických studií a postmarketingových hlášení. Patogeneze SLE není dobře známa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Maximální tolerovaná dávka Xolairu nebyla stanovena. Pacientům byly podány jednorázové intravenózní dávky až do 4 000 mg, aniž by se prokázala dávkou vymezená toxicita. Nejvyšší úhrnná dávka podaná pacientům byla 44 000 mg po dobu 20 týdnů a tato dávka neměla za následek žádné akutní nežádoucí účinky.
Pokud je podezření z předávkování, pacient by měl být sledován pro výskyt jakýchkoli neobvyklých známek nebo příznaků. Měla by být vyhledána lékařská pomoc a zahájena vhodná opatření.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva pro onemocnění spojená s obstrukcí dýchacích cest, jiná systémová léčiva pro onemocnění spojená s obstrukcí dýchacích cest, ATC kód: R03DX05
Omalizumab je z rekombinantní DNA odvozená humanizovaná monoklonální protilátka, která se selektivně váže na lidský imunoglobulin E (IgE). Protilátka je typu IgG1 kappa, která sestává ze skeletu lidské protilátky a z části odvozené z myší monoklonální protilátky, která váže IgE.
Mechanismus účinku
Omalizumab se váže na IgE a předchází vazbě IgE k FcsRI (receptorům s vysokou afinitou k IgE), čímž se redukuje množství volného IgE, který je využitelný ke spuštění alergické kaskády. Léčba Xolairem u atopických pacientů měla za následek nápadný pokles FcsRI receptorů na bazofilech.
Farmakodynamické účinky
Uvolnění histaminu in vitro z bazofilů izolovaných od jedinců léčených Xolairem bylo snížené přibližně o 90 % po stimulaci alergenem oproti hodnotám před léčbou.
V klinických studiích byly sérové hladiny volného IgE sníženy v závislosti na dávce během 1 hodiny od první dávky a zůstaly snížené mezi dávkami. Po jednom roce od ukončení léčby Xolairem se hladiny IgE vrátily na úroveň před léčbou, aniž by byl pozorován rebound fenomén v hladinách IgE po vysazení léčivého přípravku.
Klinická účinnost a bezpečnost
Dospělí a dospívající >12 let
Účinnost a bezpečnost Xolairu byla prokázána ve 28týdenní dvojitě zaslepené placebem kontrolované studii (studie 1) zahrnující 419 těžkých alergických astmatiků ve věku 12-79 let, kteří měli sníženou plicní funkci (FEVi 40-80 % predikční hodnoty) a nedostatečnou kontrolu astmatických symptomů, přestože dostávali vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta2-agonistů. Pacienti přicházející v úvahu prodělali četné exacerbace astmatu vyžadující systémovou léčbu kortikosteroidy nebo byli hospitalizováni nebo navštívili lékařskou pohotovostní službu kvůli těžké exacerbaci astmatu v posledním roce navzdory průběžné léčbě vysokými dávkami inhalačních kortikosteroidů a dlouhodobě působícími beta2-agonisty. Subkutánně podávaný Xolair nebo placebo byly podávány jako doplňková léčba k >1 000 mikrogramům beklometason-dipropionátu (nebo jeho ekvivalentu) plus beta2-agonisty s dlouhodobým účinkem. Perorální kortikosteroid, theofylin a modifikátory leukotrienů byly dovoleny jako udržovací terapie (22 %, 27 %, resp. 35 % pacientů).
Primárním endpointem bylo sledování výskytu exacerbací astmatu vyžadujících léčbu vysokými dávkami systémových kortikosteroidů. Omalizumab redukoval četnost výskytu exacerbací astmatu o 19 % (p = 0,153). Další hodnocení, která ukázala statistický význam (p<0,05) ve prospěch Xolairu, zahrnovala snížení závažných exacerbací (kde funkce plic u pacientů byla redukována až pod 60 % nejlepší osobní hodnoty a bylo zapotřebí systémových kortikosteroidů) a neodkladných návštěv lékaře souvisejících s astmatem (zahrnovaly hospitalizaci, pohotovostní lékařskou službu a neplánované návštěvy u lékaře) a zlepšení astmatických symptomů a funkce plic podle celkového zhodnocení účinnosti léčby lékařem, Asthma-related Quality of Life (AQL).
Podle analýzy podskupin klinicky významný prospěch léčby Xolairem měli spíše pacienti s celkovou hladinou IgE >76 IU/ml před léčbou. U těchto pacientů ve studii 1 Xolair redukoval četnost výskytu exacerbací astmatu o 40 % (p = 0,002). Kromě toho více pacientů mělo klinicky významnou odpověď v populaci s celkovým IgE >76 IU/ml v programu Xolair při těžkém astmatu. Tabulka 5 zahrnuje výsledky u celkové populace ve studii 1.
Tabulka 5: Výsledky studie 1
Populace studie 1 celkem Xolair Placebo
N=209 N=210
Exacerbace astmatu
Četnost výskytu za 28týdenní 0,74 0,92
období
% redukce, p-hodnota pro poměr 19,4 %, p = 0,153
četnosti
|
Exacerbace závažného astmatu | ||
|
Četnost výskytu za 28týdenní období |
0,24 |
0,48 |
|
% redukce, p-hodnota pro poměr četnosti |
50,1 %, p = |
0,002 |
|
Návštěvy lékaře v naléhavých případech Četnost výskytu za 28týdenní období |
0,24 |
0,43 |
|
% redukce, p-hodnota pro poměr četnosti |
43,9 %, p = |
0,038 |
|
Celkové zhodnocení lékařem | ||
|
% reagujících* |
60,5 % |
42,8 % |
|
Hodnota p ** |
<0,001 | |
|
AQL zlepšení % pacientů se zlepšením >0,5 |
60,8 % |
47,8 % |
|
Hodnota p |
0,008 | |
|
* nápadné zlepšení nebo kompletní kontrola | ||
p-hodnota pro celkové zhodnocení
Studie 2 hodnotila účinnost a bezpečnost Xolairu u skupiny 312 osob s těžkým alergickým astmatem, která odpovídala populaci ve studii 1. Léčba Xolairem v této otevřené studii vedla ke snížení četnosti výskytu klinicky významných exacerbací astmatu o 61 % oproti běžné léčbě astmatu podávané samotné.
Čtyři další velké placebem kontrolované podpůrné studie trvající 28 až 52 týdnů na 1 722 dospělých a dospívajících (studie 3, 4, 5, 6) hodnotily účinnost a bezpečnost Xolairu u pacientů s těžkým perzistujícím astmatem. Kontrola astmatu u většiny pacientů byla nedostatečná, ale užívali méně konkomitantní léčby astmatu než pacienti ve studiích 1 a 2. Studie 3-5 použily exacerbace jako primární endpoint, zatímco studie 6 primárně hodnotila snížení množství inhalačních kortikosteroidů.
Ve studiích 3, 4 a 5 se u pacientů léčených Xolairem snížila četnost výskytu exacerbací astmatu o 37,5 % (p = 0,027), 40,3 % (p<0,001), respektive o 57,6 % (p<0,001) oproti placebu.
Ve studii 6 bylo schopno významně více pacientů s těžkým alergickým astmatem léčených Xolairem redukovat svou dávku flutikasonu až na <500 mikrogramů/den bez zhoršení kontroly astmatu (60,3 %) oproti skupině s placebem (45,8 %, p<0,05).
Skóre kvality života bylo měřeno pomocí dotazníku Juniper Asthma-related Quality of Life Questionnaire. Ve všech šesti studiích došlo ke statisticky významnému zlepšení skóre kvality života oproti výchozím hodnotám u pacientů užívajících Xolair oproti skupině placebo nebo kontrolní skupině.
Celkové hodnocení účinnosti léčby lékařem:
Celkové zhodnocení lékařem bylo provedeno v pěti z výše uvedených studií jako obsáhlé celkové posouzení kontroly astmatu vykonané ošetřujícím lékařem. Lékař vzal do úvahy PEF (peak expiratory flow = vrcholová výdechová rychlost), denní a noční symptomy, užití záchranné medikace, spirometrii a exacerbace. Ve všech pěti studiích se u významně většího podílu pacientů léčených Xolairem vyhodnotilo, že dosáhli buď výrazného zlepšení, nebo kompletní kontroly astmatu oproti skupině pacientů, kteří dostávali placebo.
Děti 6 až <12 let
Primární doklad bezpečnosti a účinnosti Xolairu u skupiny pacientů od 6 do <12 let vycházel z randomizované, dvojitě slepé, placebem kontrolované, multicentrické studie (studie 7).
Studie 7 byla placebem kontrolovaná studie zahrnující podskupinu (N=235) pacientů, u kterých byla určena současná indikace, kteří byli léčeni vysokými dávkami inhalačních kortikosteroidů (ekvivalent >500 pg/den flutikasonu) a dlouhodobě působícími beta-agonisty.
Klinicky významná exacerbace byla definována jako zhoršení příznaků astmatu dle posouzení zkoušejícího lékaře, vyžadující zdvojnásobení výchozí dávky inhalačních kortikosteroidů po dobu minimálně 3 dnů a/nebo vyžadující záchrannou léčbu systémovými (perorálními nebo intravenózními) kortikosteroidy po dobu minimálně 3 dnů.
Ve specifické podskupině pacientů, kteří užívali vysoké dávky inhalačních kortikosteroidů, bylo u skupiny pacientů užívajících omalizumab zaznamenáno statisticky signifikantní snížení výskytu exacerbací ve srovnání se skupinou užívající placebo. Ve 24. týdnu představoval rozdíl mezi léčenými skupinami 34 % (poměr výskytu 0,662, p=0,047) relativní snížení u pacientů léčených omalizumabem oproti skupině pacientů léčených placebem. V druhé, dvojitě slepé, 28týdenní léčebné periodě představoval rozdíl mezi léčenými skupinami 63 % (poměr výskytu 0,37, p<0,001) relativní snížení u pacientů léčených omalizumabem ve srovnání s pacienty léčenými placebem.
Během 52týdenní, dvojitě slepé léčby (zahrnující 24týdenní fázi s fixními dávkami steroidů a 28týdenní fázi s úpravou dávkování steroidů) představoval rozdíl mezi léčebnými skupinami 50 % (poměr výskytu 0,504, p<0,001) relativní snížení počtu exacerbací u pacientů léčených omalizumabem.
Během 52týdenní léčby vykazovala skupina pacientů užívající omalizumab větší pokles použití záchranné medikace beta-agonistů než skupina pacientů užívajících placebo, přestože tento rozdíl mezi léčebnými skupinami nebyl statisticky signifikantní. Z celkového vyhodnocení účinnosti léčby na konci 52týdenní léčby u podskupiny těžkých pacientů užívajících vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta-agonistů vyplývá, že poměr pacientů, kteří prokázali „vynikající“ účinnost, byl u skupiny užívající omalizumab vyšší a poměr pacientů s „průměrnou“ nebo „slabou“ účinností byl nižší ve srovnání s pacienty užívajícími placebo; rozdíl mezi léčebnými skupinami byl statisticky významný (p<0,001), zatímco mezi skupinou s omalizumabem a skupinou s placebem nebyly žádné rozdíly z hlediska subjektivních hodnot kvality života pacienta.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu byla studována u dospělých a dospívajících pacientů s alergickým astmatem.
Absorpce
Průměrná absolutní hodnota biologické dostupnosti omalizumabu je 62 % po subkutánním podání. Po jednorázové subkutánní dávce dospělým a dospívajícím pacientům s astmatem se omalizumab absorboval pomalu a dosáhl maximální koncentrace v séru průměrně po 7-8 dnech. Farmakokinetika omalizumabu je lineární v dávkách vyšších než 0,5 mg/kg. Po opakovaných dávkách omalizumabu byly plochy pod křivkou sérové koncentrace v čase ode dne 0 až po den 14 v rovnovážném stavu až 6x větší oproti těm po první dávce.
Podání Xolairu vyrobeného ve formě lyofilizátu nebo roztoku vedlo k podobným profilům sérové koncentrace omalizumabu v čase.
Distribuce
In vitro omalizumab tvoří s IgE komplexy omezené velikosti. Precipitující komplexy a komplexy s molekulovou hmotností větší než jeden milion daltonů nejsou pozorovány in vitro anebo in vivo. Zdánlivý distribuční objem u pacientů po subkutánním podání byl 78 ± 32 ml/kg.
Eliminace
Clearance omalizumabu zahrnuje průběh clearance IgG stejně tak jako clearance prostřednictvím specifické vazby a tvorby komplexů s cílovým ligandem IgE. Vylučování IgG v játrech zahrnuje odbourávání v retikuloendotelovém systému a endotelových buňkách. Intaktní IgG je také vylučován žlučí. U pacientů s astmatem činil průměrný poločas eliminace omalizumabu ze séra 26 dní, s clearance v průměru 2,4 ± 1,1 ml/kg/den. Kromě toho zdvojnásobení tělesné hmotnosti přibližně zdvojnásobuje danou clearance.
Vlastnosti pacientské populace
Věk,rasa/etnikum, pohlaví, BMI (Body Mass Index)
Populační farmakokinetika Xolairu byla analyzována, aby se zhodnotily účinky demografických charakteristik. Analýzy těchto omezených údajů naznačují, že není nutná žádná úprava dávky pro věk (6-76 let), rasu/etnickou příslušnost, pohlaví nebo BMI (viz bod 4.2).
Zhoršená funkce ledvin a jater
U pacientů se zhoršenou funkcí ledvin nebo jater nejsou žádné farmakokinetické nebo farmakodynamické údaje (viz body 4.2 a 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost omalizumabu byla studována u opic makaka jávského, jelikož omalizumab se váže na jejich i lidský IgE s podobnou afinitou. U některých opic byly zjištěny protilátky proti omalizumabu po opakovaném subkutánním nebo intravenózním podání. Nicméně nebyla pozorována žádná toxicita jako například onemocnění zprostředkované imunokomplexy nebo cytotoxicita podmíněná komplementem. Nevyskytl se žádný důkaz anafylaktické odpovědi následkem degranulace žírných buněk u těchto opic.
Dlouhodobé podávání omalizumabu v dávkách až 250 mg/kg (alespoň 14krát nejvyšší doporučená klinická dávka v mg/kg podle doporučené dávkovači tabulky) nehumánním primátům (dospělým i dospívajícím jedincům) bylo dobře tolerováno s výjimkou snížení počtu krevních destiček, které souviselo s dávkou a bylo závislé na věku, s větší citlivostí u nedospělých zvířat. Hladina koncentrace v séru potřebná k dosažení poklesu trombocytů o 50 % oproti výchozí hodnotě u dospělých opic makaka jávského byla zhruba 4 až 20krát vyšší než očekávané maximální klinické sérové koncentrace. Navíc u makaka jávského bylo pozorováno akutní krvácení a zánět v místě aplikace injekce.
Formální studie kancerogenity nebyly s omalizumabem provedeny.
V reprodukčních studiích u makaka jávského subkutánně podané dávky až do 75 mg/kg týdně (alespoň 8krát nejvyšší doporučená klinická dávka v mg/kg po dobu delší než 4 týdny) nevyvolaly mateřskou toxicitu, embryotoxicitu nebo teratogenní účinky, když byly podávané během organogeneze, a neměly nežádoucí účinky na fetální nebo novorozenecký růst, pokud byly podávané během těhotenství, porodu nebo kojení.
Omalizumab se vylučuje do mateřského mléka makaka jávského. Hladiny omalizumabu v mléce byly 0,15 % koncentrace v séru matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Sacharosa
Histidin
Monohydrát histidin-hydrochloridu Polysorbát 20
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
4 roky.
Po rozpuštění
Chemická a fyzikální stabilita rekonstituovaného léčivého přípravku byla prokázána po dobu 8 hodin při teplotě 2 °C - 8 °C a 4 hodiny při teplotě 30 °C.
Z mikrobiologického hlediska má být léčivý přípravek použit okamžitě po rozpuštění. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 8 hodin při teplotě 2 °C - 8 °C nebo 2 hodiny při teplotě 25 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem: průhledná, bezbarvá injekční lahvička ze skla typu I s pryžovou zátkou a šedou pertlí.
Ampulka s rozpouštědlem: průhledná, bezbarvá ampulka ze skla typu I obsahující 2 ml vody na injekci.
Balení obsahující jednu injekční lahvičku prášku pro přípravu injekčního roztoku a jednu ampulku vody na injekci.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rozpuštění lyofilizovaného léčivého přípravku trvá 15-20 minut, ačkoliv v některých případech to může trvat déle. Zcela rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý a může v něm být několik malých bublinek nebo pěna okolo okraje injekční lahvičky. Vzhledem k viskozitě rekonstituovaného léčivého přípravku se musí věnovat péče tomu, aby se odebral celý obsah léčivého přípravku z injekční lahvičky za účelem získání 0,6 ml před tím, než je vytlačen vzduch nebo přebytečné množství roztoku ze stříkačky.
Pro přípravu Xolairu 75 mg v injekční lahvičce pro subkutánní podání se prosím řiďte následujícími instrukcemi:
1. Odeberte z ampulky 0,9 ml vody na injekci do injekční stříkačky opatřené jehlou o velikosti průměru 18.
2. Za použití standardních aseptických postupů zasuňte jehlu do injekční lahvičky umístěné kolmo na rovný povrch, přidejte vodu na injekci do lahvičky obsahující lyofilizovaný prášek tak, aby voda na injekci směřovala přímo na prášek.
3. Držte lahvičku ve svislé poloze a intenzivně s ní kružte (netřepejte) po dobu přibližně 1 minuty, aby se prášek rovnoměrně namočil.
4. Aby se napomohlo rozpouštění po dokončení kroku 3, jemně kružte lahvičkou po dobu 5-10 sekund přibližně každých 5 minut, aby se rozpustil všechen zbývající prášek.
Pamatujte, že v některých případech to může trvat déle než 20 minut, aby se prášek zcela rozpustil. Jestliže jde o tento případ, opakujte krok 4, dokud jsou v roztoku viditelné částečky podobné gelu.
Když je léčivý přípravek zcela rozpuštěný, neměly by být v roztoku viditelné žádné částečky podobné gelu. Malé bublinky nebo pěna okolo okraje lahvičky jsou obvyklé. Rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý. Nepoužívejte jej, jestliže jsou přítomny pevné částečky.
5. Obraťte injekční lahvičku dnem vzhůru na dobu nejméně 15 sekund, aby roztok natekl až k zátce. Použijte novou 3ml stříkačku opatřenou jehlou o velikosti průměru 18, zasuňte jehlu do obrácené injekční lahvičky. Držte injekční lahvičku v obrácené poloze, jehlu postrčte až na samé dno roztoku v injekční lahvičce, když odebíráte roztok do stříkačky. Před vyjmutím jehly z injekční lahvičky táhněte píst zpátky až na konec těla stříkačky, aby se nasál celý obsah roztoku z obrácené lahvičky.
6. Pro subkutánní injekci nahraďte jehlu o velikosti 18 jehlou velikosti 25.
7. Vytlačte vzduch, velké bubliny a přebytečný roztok tak, aby se získala požadovaná dávka
0,6 ml. Nepatrná vrstva malých bublinek může zůstat v horní části roztoku v injekční stříkačce. Protože je roztok mírně viskózní, může aplikace subkutánně podaného roztoku trvat 5-10 sekund.
Z injekční lahvičky se získá 0,6 ml (75 mg) Xolairu.
8. Injekce se podávájí subkutánně do oblasti deltového svalu ramene nebo do stehna.
Xolair 75 mg prášek a rozpouštědlo pro injekční roztok je dodáván v jednorázové injekční lahvičce.
Z mikrobiologického hlediska by měl být léčivý přípravek použit okamžitě po rozpuštění (viz bod 6.3).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/319/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25. října 2005
Datum posledního prodloužení registrace: 22. června 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje omalizumabum* 150 mg.
Po rozpuštění obsahuje jedna injekční lahvička omalizumabum 125 mg/ml (150 mg v 1,2 ml).
*Omalizumab je humanizovaná monoklonální protilátka vyrobená technologií rekombinantní DNA v linii savčích buněk vaječníků čínských křečků (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: bílý až téměř bílý lyofilizát Rozpouštědlo: čirý a bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Alergické astma
Xolair je indikován k léčbě dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Léčbu Xolairem je možno použít pouze u pacientů s astmatem vyvolaným prokazatelně IgE (imunoglobulinem E) (viz bod 4.2).
Dospělí a dospívající (12 let a starší)
Xolair se doporučuje jako doplňková léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a kteří mají sníženou funkci plic (FEVj <80 %), stejně jako časté symptomy během dne nebo probouzení v noci, a kteří mají dokumentované těžké exacerbace astmatu navzdory vysokým denním dávkám inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
Děti (6 až <12 let)
Xolair se doporučuje jako přídatná léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a časté denní nebo noční příznaky buzení a kteří mají prokázané četné vážné exacerbace přesto, že užívají vysoké denní dávky inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
Chronická spontánní urtikarie (CSU)
Xolair je indikován jako přídatná terapie k léčbě chronické spontánní urtikarie u dospělých a dospívajících pacientů (ve věku 12 let a více) s nedostatečnou odpovědí na léčbu H1 antihistaminiky.
4.2 Dávkování a způsob podání
Léčba Xolairem by měla být zahájena lékařem zkušeným v diagnostice a léčbě těžkého perzistujícího astmatu nebo chronické spontánní urtikarie.
Alergické astma
Dávkování
Vhodná dávka a frekvence podávání Xolairu se určí podle výchozích hodnot IgE (IU/ml), které se stanoví před zahájením léčby, a dle tělesné hmotnosti (kg). Aby mohla být stanovena dávka, měla by být před podáním počáteční dávky u pacientů stanovena hladina IgE jakýmkoliv komerčním testem pro stanovení celkového IgE v séru. Na základě výsledků může být potřeba podávat 75 až 600 mg Xolairu v 1 až 4 injekcích na každé podání.
U pacientů s IgE nižším než 76 IU/ml je méně pravděpodobné, že se projeví přínos přípravku (viz bod
5.1). Ošetřující lékař se musí ujistit, že dospělí a dospívající pacienti s IgE nižším než 76 IU/ml a děti (6 až <12 let) s IgE nižším než 200 IU/ml mají před začátkem léčby prokázanou in vitro reaktivitu (RAST) na celoroční vzdušný alergen.
Viz Tabulka 1 schéma konverze a Tabulka 2 a 3 schéma stanovení dávky u dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Pacientům, jejichž výchozí hodnoty hladin IgE nebo tělesná hmotnost v kilogramech jsou mimo limity dávkovací tabulky, by Xolair neměl být podáván.
Maximální doporučená dávka je 600 mg omalizumabu každé dva týdny.
Tabulka 1: Konverze dávky na počet injekčních lahviček, počet injekcí a celkový objem injekcí při každé aplikaci
|
Dávka (mg) |
Počet injekčních lahviček 75 mg a 150 mg b |
Počet injekcí |
Celkový objem injekcí (ml) | |
|
75 |
1c |
0 |
1 |
0,6 |
|
150 |
0 |
1 |
1 |
1,2 |
|
225 |
1c |
1 |
2 |
1,8 |
|
300 |
0 |
2 |
2 |
2,4 |
|
375 |
1c |
2 |
3 |
3,0 |
|
450 |
0 |
3 |
3 |
3,6 |
|
525 |
1c |
3 |
4 |
4,2 |
|
600 |
0 |
4 |
4 |
4,8 |
a 0,6 ml = maximální množství obsažené v jedné injekční lahvičce (Xolair 75 mg). b 1,2 ml = maximální množství obsažené v jedné injekční lahvičce (Xolair 150 mg). c nebo užití 0,6 ml z obsahu injekční lahvičky 150 mg.
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE (IU/ml) |
>20- 25 |
>25- 30 |
>30- 40 |
>40- 50 |
>50- 60 |
>60- 70 |
>70- 80 |
>80- 90 |
>90- 125 |
>125- 150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
PODÁVÁNÍ KAŽDÉ 2 TÝDNY | |||||||||
|
>900- 1000 |
VIZ TABULKA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(IU/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
PODÁVÁNÍ KAŽDÉ 4 TÝDNY | |||||||||
|
>100-200 |
VIZ TABULKA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- 1200 |
300 |
300 |
450 |
525 |
600 |
NEPODÁVAT - pro doporučení dávkování není dostatek údajů | ||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Délka léčby, její sledování a úprava dávky
Xolair je určen pro dlouhodobou léčbu. Klinické studie prokázaly, že Xolair dosahuje účinnosti po minimálně 12-16 týdnech léčby. Po 16 týdnech od zahájení léčby Xolairem by měl lékař před podáváním dalších injekcí zhodnotit účinnost léčby. Rozhodnutí o pokračování léčby Xolairem následně po 16týdenním intervalu, nebo příležitostně později, by mělo být založeno na tom, zdali je patrné výrazné zlepšení z hlediska celkové kontroly astmatu (viz bod 5.1, Celkové hodnocení účinnosti léčby lékařem).
Ukončení léčby Xolairem má obecně za následek návrat ke zvýšeným hladinám volného IgE a s tím spojeným symptomům. Celkové hladiny IgE jsou zvýšené během léčby a zůstávají zvýšené až po dobu jednoho roku po skončení léčby. Proto opětovné stanovení hladin IgE během léčby Xolairem nemůže být použito jako vodítko pro stanovení dávky. Stanovení dávky po přerušení léčby trvající méně než jeden rok by mělo být založeno na výchozích hodnotách hladin sérového IgE získaných při stanovení zahajovací dávky. Pro stanovení dávky mohou být celkové hladiny IgE v séru znovu stanoveny, jestliže léčba Xolairem byla přerušena jeden rok nebo déle.
Dávky by měly být přizpůsobeny významným změnám tělesné hmotnosti (viz Tabulka 2 a 3).
Chronická spontánní urtikarie (CSU)
Dávkování
Doporučená dávka je 300 mg, podávaných subkutánní injekcí každé čtyři týdny.
Předepisujícím lékařům se doporučuje pravidelně přehodnotit potřebu pokračování léčby.
Zkušenosti z klinického hodnocení dlouhodobé léčby trvající déle než 6 měsíců v této indikaci jsou omezené.
Zvláštní skupiny pacientů
Starší pacienti (65 let a starší)
Pro užití Xolairu u pacientů starších více než 65 let jsou dostupné omezené údaje, ale nebylo zjištěno, že by u starších pacientů bylo nutné podávání jiné dávky než u mladších dospělých pacientů.
Pacienti s poškozením ledvin nebo jater
Nejsou k dispozici žádné studie hodnotící vliv zhoršené funkce ledvin nebo jater na farmakokinetiku omalizumabu. Protože clearance omalizumabu je při klinických dávkách ovlivněna hlavně retikuloendotelovým systémem (RES), je nepravděpodobné, že by byla ovlivněna poškozením ledvin nebo jater. Protože není doporučena žádná zvláštní úprava dávkování pro tyto pacienty, Xolair by měl být podáván s opatrností (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost Xolairu v případě alergického astmatu u pediatrických pacientů mladších 6 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Bezpečnost a účinnost Xolairu v případě chronické spontánní urtikarie u pediatrických pacientů mladších 12 let nebyla dosud stanovena.
Způsob podání
Pouze pro subkutánní podání. Nepodávejte intravenózně nebo intramuskulárně.
Injekce se aplikují subkutánně do oblasti deltového svalu ramene. Alternativně se může injekce aplikovat do stehna, jestliže je z jakéhokoliv důvodu vyloučena aplikace do oblasti deltového svalu.
Zkušenosti s aplikací injekce Xolairu samotným pacientem jsou omezené. Z toho důvodu má léčbu podávat pouze zdravotnický personál.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6 a také v bodě Informace pro zdravotnické pracovníky v příbalové informaci.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Obecné
Xolair není určen k léčbě exacerbací akutního astmatu, akutního bronchospasmu nebo status asthmaticus.
Xolair nebyl zkoumán u pacientů se syndromem hyperimunoglobulinemie E nebo alergickou bronchopulmonální aspergilózou nebo pro prevenci anafylaktických reakcí včetně takových, které jsou vyvolané alergií na potraviny, atopickou dermatitidou nebo alergickou rinitidou. Xolair není indikován pro léčbu těchto stavů.
Léčba Xolairem nebyla zkoumána u pacientů s autoimunitním onemocněním, s onemocněním zprostředkovaným imunokomplexy, nebo s preexistující zhoršenou funkcí ledvin nebo jater (viz bod
4.2). Pokud je Xolair podáván těmto skupinám pacientů, měla by být zachovávána opatrnost.
Náhlé vysazení systémových nebo inhalačních kortikosteroidů po zahájení léčby Xolairem se nedoporučuje. Snižování dávek kortikosteroidů je nutno provádět pod přímým dohledem lékaře a je vhodné jej provádět postupně.
Poruchy imunitního systému Alergické reakce typu I
Lokální nebo systémové alergické reakce typu I včetně anafylaxe a anafylaktického šoku se mohou objevit po podání omalizumabu, přičemž tyto reakce se mohou objevit i po dlouhotrvající léčbě omalizumabem. Většina těchto reakcí se objevila během 2 hodin po první a následné injekci Xolairu, ale některé se objevily až po 2 hodinách a dokonce i po více než 24 hodinách po injekci. Proto by následně po podání Xolairu měly být vždy dostupné k okamžitému užití léčivé přípravky na léčbu anafylaktických reakcí. Pacienti by měli být informováni o možnosti takových reakcí, a pokud se objeví alergická reakce, měli by vyhledat okamžitě lékařskou pomoc. Výskyt anafylaxe nesouvisející s omalizumabem v anamnéze může být rizikový faktor pro anafylaxi následující po podání Xolairu.
U malého počtu pacientů v klinických studiích byly detekovány protilátky proti omalizumabu (viz bod 4.8). Klinický význam protilátek proti Xolairu není dobře prostudován.
Sérová nemoc
Sérová nemoc a reakce podobné sérové nemoci, které jsou opožděnými alergickými reakcemi typu III, byly pozorovány u pacientů léčených humanizovanými monoklonálními protilátkami včetně omalizumabu. Předpokládaný patofyziologický mechanismus zahrnuje tvorbu a ukládání imunokomplexů v důsledku vzniku protilátek proti omalizumabu. Reakce se obvykle objevila 1-5 dní po podání prvních nebo následujících injekcí, i po dlouhodobé léčbě. Příznaky poukazující na sérovou nemoc zahrnují artritidu/artralgie, vyrážku (kopřivku nebo jiné formy), horečku a lymfadenopatii. K léčbě nebo prevenci této nemoci mohou být vhodná antihistaminika a kortikosteroidy. Pacienti by měli být poučeni, aby hlásili jakékoli podezřelé příznaky.
Syndrom Churga-Straussové a hypereozinofilní syndrom
U pacientů s těžkým astmatem se může zřídka vyskytnout systémový hypereozinofilní syndrom nebo alergická eozinofilní granulomatózní vaskulitida (syndrom Churga-Straussové), které se obvykle léčí systémovými kortikoidy.
Ve vzácných případech se mohou u pacientů léčených antiastmatickými léčivými přípravky, včetně omalizumabu, projevit systémová eozinofilie a vaskulitida. Tyto případy jsou často spojeny se snižováním léčby perorálními kortikoidy.
U těchto pacientů by lékaři měli věnovat zvýšenou pozornost možnému rozvoji eozinofilie, vaskulitické vyrážky, zhoršení plicních symptomů, abnormalit paranazálních dutin, srdečních komplikací a/nebo neuropatie.
Ve všech závažných případech výše uvedených poruch imunitního systému by mělo být zváženo vysazení omalizumabu.
Parazitární infekce (helmintóza)
IgE může být spojený s imunologickou odpovědí na některé parazitární infekce. V placebem kontrolované studii s alergickými pacienty bylo u pacientů s chronicky vysokým rizikem helmintóz prokázáno mírné zvýšení podílu infekcí ve skupině s omalizumabem, ačkoliv průběh, závažnost a odpověď na léčbu infekce se nezměnily. Četnost výskytu infekce cizopasnými červy v celkovém klinickém programu, který nebyl navržen tak, aby takové infekce prokázal, byla méně než 1 z 1 000 pacientů. Nicméně měla by být zaručena opatrnost u pacientů s vysokým rizikem infekce cizopasnými červy, zejména pokud cestují do oblastí, kde se infekce cizopasnými červy vyskytují endemicky. Pokud pacienti neodpovídají na doporučenou antiparazitární léčbu, mělo by být zváženo přerušení léčby Xolairem.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Xolair může nepřímo snížit účinnost léčivých přípravků používaných k léčbě helmintóz nebo jiných parazitárních infekcí, protože v imunologické odpovědi na některé helmintózy může být zapojen IgE (viz bod 4.4).
Enzymy cytochromu P450, pumpy zajišťující efflux a mechanismy vazby na proteiny se nepodílejí na clearance omalizumabu; takže je zde malá možnost lékových interakcí. Nebyly provedeny žádné studie interakce Xolairu s léčivými přípravky ani vakcínami. Není žádný farmakologický důvod očekávat, že by se běžně předepisované léčivé přípravky užívané v léčbě astmatu nebo CSU vzájemně ovlivňovaly s omalizumabem.
Alergické astma
V klinických studiích byl Xolair běžně užíván v kombinaci s inhalačními a perorálními kortikosteroidy, inhalačními krátkodobě a dlouhodobě působícími beta agonisty, modifikátory leukotrienů, theofyliny a perorálními antihistaminiky. Nevyskytly se žádné údaje, že bezpečnost Xolairu byla změněna těmito dalšími běžně užívanými antiastmatickými léčivými přípravky. Jsou dostupné omezené údaje o užívání Xolairu v kombinaci se specifickou imunoterapií (hyposenzitizační terapie). V klinické studii, kde byl Xolair podáván společně s imunoterapií, nebyl zjištěn žádný rozdíl v bezpečnosti a účinnosti Xolairu v kombinaci se specifickou imunoterapií v porovnání s podáváním samotného Xolairu.
Chronická spontánní urtikarie (CSU)
V klinických studiích týkajících se CSU byl Xolair podáván v kombinaci s antihistaminiky
(H1 antihistaminiky, H2 antihistaminiky) a antagonisty leukotrienových receptorů (LTRA). Nebylo prokázáno, že by bezpečnost omalizumabu byla pozměněna při podávání s těmito léčivými přípravky, vztaženo k jejich známému bezpečnostnímu profilu u alergického astmatu. Navíc farmakokinetická analýza populace neprokázala žádný relevantní účinek H2 antihistaminik a LTRA na farmakokinetiku omalizumabu (viz bod 5.2).
Pediatrická populace
Klinické studie týkající se CSU zahrnovaly některé pacienty ve věku 12 až 17 let, užívající Xolair v kombinaci s antihistaminiky (H1 antihistaminiky, H2 antihistaminiky) a LTRA. U dětí mladších 12 let nebyly provedeny žádné studie.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání omalizumabu těhotným ženám jsou omezené. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky (viz bod 5.3). Omalizumab přestupuje přes placentu, ale možnost poškození plodu není známa. U primátů byl omalizumab spojován se snížením na věku závislém počtu krevních destiček, s relativní větší citlivostí u nedospělých zvířat (viz bod
5.3). Xolair by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda se omalizumab vylučuje do lidského mateřského mléka. Dostupné údaje u samic ostatních primátů prokázaly vylučování omalizumabu do mléka (viz bod 5.3). Riziko pro kojené novorozence/děti nelze vyloučit. Omalizumab by neměl být podáván během kojení.
Fertilita
Pro omalizumab nejsou k dispozici žádné údaje týkající se lidské fertility. Ve specificky navržených neklinických studiích fertility u primátů (s výjimkou člověka), včetně studií páření, nebylo pozorováno poškození samčí ani samičí fertility následně po opakovaném podávání omalizumabu v dávkách až 75 mg/kg. Dále nebyly pozorovány žádné genotoxické účinky v samostatných neklinických studiích genotoxicity (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xolair nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Alergické astma
Souhrn bezpečnostního profilu
Během klinických studií u dospělých a dospívajících pacientů starších 12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy a reakce v místě aplikace injekce, včetně bolestivosti, zduření, zarudnutí a svědění. V klinických studiích u dětí od 6 do <12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy, pyrexie a bolest v nadbřišku. Ve většině případů byly reakce mírné nebo středně závažné.
Tabulkový seznam nežádoucích účinků
Tabulka 4 zaznamenává nežádoucí účinky hlášené v klinických studiích celkové bezpečnosti u populace léčené Xolairem podle MedDRA orgánové klasifikace a četnosti výskytu. V každé skupině četností jsou nežádoucí reakce seřazeny podle klesající závažnosti. Kategorie četností jsou definovány jako: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000) a velmi vzácné (<1/10 000). Reakce hlášené z postmarketingového sledování jsou uvedeny s frekvencí není známo (z dostupných údajů nelze určit).
Tabulka 4: Nežádoucí účinky u alergického astmatu
|
Infekce a infestace | |
|
Méně časté |
Faryngitida |
|
Vzácné |
Parazitární infekce |
|
Poruchy krve a lymfatického systému | |
|
Není známo |
Idiopatická trombocytopenie, včetně těžkých případů |
|
Poruchy imunitního systému | |
|
Vzácné |
Anafylaktická reakce, jiné závažné alergické stavy, vývoj |
|
protilátek proti omalizumabu | |
|
Není známo |
Sérová nemoc, která může zahrnovat horečku a lymfadenopatii |
|
Poruchy nervového systému | |
|
Časté |
Bolest hlavy* |
|
Méně časté |
Synkopa, parestezie, somnolence, závrať |
|
Cévní poruchy | |
|
Méně časté |
Posturální hypotenze, zčervenání |
|
Respirační, hrudní a mediastinální poruchy | |
|
Méně časté |
Alergický bronchospasmus, kašel |
|
Vzácné |
Otok laryngu |
|
Není známo |
Alergická granulomatózní vaskulitida (tzv. syndrom Churga- |
|
Straussové) | |
|
Gastrointestinální poruchy | |
|
Časté |
Bolest v nadbřišku** |
|
Méně časté | |
|
Poruchy kůže a podkožní tkáně | |
|
Méně časté | |
|
Vzácné |
Angioedém |
|
Není známo |
Alopecie |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Vzácné |
Systémový lupus erythematodes (SLE) |
|
Není známo |
Artralgie, myalgie, otoky kloubů |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Pyrexie** |
|
Časté |
Reakce v místě aplikace jako zduření, zarudnutí, bolest, svědění |
|
Méně časté |
Onemocnění podobné chřipce, otoky paží, zvýšení hmotnosti, |
*: Velmi časté u dětí od 6 do <12 let **: U dětí od 6 do <12 let
Chronická spontánní urtikarie (CSU)
Souhrn bezpečnostního profilu
Bezpečnost a snášenlivost omalizumabu byly studovány podáváním dávek 75 mg, 150 mg a 300 mg každé čtyři týdny u 975 pacientů s CSU, z nichž 242 dostávalo placebo. Celkem 733 pacientů bylo léčeno omalizumabem až 12 týdnů a 490 pacientů až 24 týdnů. Z tohoto množství bylo 412 pacientů léčeno dávkou 300 mg až 12 týdnů a 333 pacientů bylo léčeno dávkou 300 mg až 24 týdnů.
Tabulkový seznam nežádoucích účinků
Samostatná tabulka (Tabulka 5) ukazuje nežádoucí účinky u CSU indikace, vyplývající z rozdílů v dávkování a v léčených populacích (s významně odlišnými rizikovými faktory, komorbiditami, komedikacemi a věkem [např. studie s astmatem zahrnovaly děti od 6-12 let věku]).
Tabulka 5 zaznamenává nežádoucí účinky (příhody vyskytující se u >1 % pacientů v jakékoli léčebné skupině a > 2 % častěji v jakékoli léčebné skupině s omalizumabem než s placebem (po lékařském zhodnocení)) hlášené při dávkách 300 mg ve třech poolovaných studiích fáze III. Uvedené nežádoucí účinky jsou rozděleny do dvou skupin: na účinky identifikované ve 12týdenním a 24týdenním léčebném období.
Nežádoucí účinky jsou uvedeny podle MedDRA orgánové klasifikace. V každé třídě orgánové klasifikace jsou nežádoucí účinky seřazeny podle četnosti, s nejčastějšími reakcemi uvedenými na prvním místě. Odpovídající kategorie četnosti pro každý nežádoucí účinek je stanovena na základě následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 5: Nežádoucí účinky z poolované bezpečnostní databáze s CSU (den 1 až týden 24) při dávce 300 mg omalizumabu
|
Studie s omalizumabem č. 1, 2 a 3, |
Kategorie četnosti | ||
|
Týden 12 |
poolované | ||
|
Placebo N=242 |
300 mg N=412 | ||
|
Infekce a infestace | |||
|
Sinusitida |
5 (2,1 %) |
20 (4,9 %) |
časté |
|
Poruchy nervového systému | |||
|
7 (2,9 %) |
25 (6,1 %) |
časté | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |||
|
Artralgie |
1 (0,4 %) |
12 (2,9 %) |
časté |
|
Celkové poruchy a reakce v místě aplikace | |||
|
Reakce v místě podání injekce* |
2 (0,8 %) |
11 (2,7 %) |
časté |
|
Týden 24 |
Studie s omalizumabem č. 1 a 3, poolované |
Kategorie četnosti | |
|
Placebo N=163 |
300 mg N=333 | ||
|
Infekce a infestace Infekce horních cest dýchacích |
5 (3,1 %) |
19 (5,7 %) |
časté |
* Přestože se neprokázal 2 % rozdíl oproti placebu, reakce v místě podání injekce byly zařazeny, protože u všech případů byla stanovena příčinná souvislost se zkoumanou léčbou.
Popis vybraných nežádoucích účinků relevantních pro indikace alergické astma a CSU Z klinických studií s CSU nebyla získána relevantní data, která by vyžadovala úpravu níže uvedených částí textu.
Poruchy imunitního systému Další informace viz bod 4.4.
Anafylaxe
Anafylaktické reakce byly v klinických studiích vzácné. Nicméně v datech po uvedení přípravku na trh se následně po kumulativním vyhledávání v bezpečnostní databázi našlo celkem 898 případů anafylaxe. Na základě očekávané expozice 566 923 pacientoroků to mělo za následek míru hlášení přibližně 0,20 %.
Arteriální tromboembolické příhody (ATE)
V kontrolovaných klinických studiích a během interim analýz observační studie byla pozorována numerická nerovnováha v počtu ATE. Definice složeného endpointu ATE zahrnovala cévní mozkovou příhodu, transitorní ischemickou ataku, infarkt myokardu, nestabilní anginu pectoris a kardiovaskulární úmrtí (včetně úmrtí z neznámé příčiny). V konečné analýze observační studie byl poměr ATE na 1 000 pacientoroků 7,52 (115/15 286 pacientoroků) pro pacienty léčené Xolairem a 5,12 (51/9 963 pacientoroků) pro kontrolní skupinu. V multivariační analýze kontrolující základní dostupné kardiovaskulární rizikové faktory byl poměr rizik 1,32 (95 % interval spolehlivosti 0,91-1,91). V samostatné analýze poolovaných klinických studií, která zahrnovala všechny randomizované, dvojitě zaslepené, placebem kontrolované klinické studie trvající 8 nebo více týdnů, byl poměr ATE na 1 000 pacientoroků 2,69 (5/1 856 pacientoroků) pro pacienty léčené Xolairem a 2,38 (4/1 680 pacientoroků) pro pacienty užívající placebo (relativní riziko 1,13, 95 % interval spolehlivosti 0,24-5,71).
Krevní destičky
V klinických studiích mělo několik pacientů počet krevních destiček pod spodní hranicí normálního laboratorního rozmezí. Žádná z těchto změn nebyla spojena s epizodami krvácení nebo se snížením hemoglobinu. U lidí (pacientů nad 6 let) nebylo hlášeno trvalé snížení počtu krevních destiček, které bylo pozorováno u ostatních primátů (viz bod 5.3), přestože z postmarketingového sledování byly ojediněle hlášeny případy idiopatické trombocytopenie, včetně těžkých případů.
Parazitární infekce
Placebem kontrolovaná studie u alergických pacientů s chronicky vysokým rizikem helmintóz ukázala mírně zvýšenou četnost výskytu infekcí u pacientů užívajících omalizumab ve srovnání s kontrolní skupinou. Toto zvýšení nebylo statisticky signifikantní. Průběh, závažnost a odpověď na léčbu infekcí se nezměnily (viz bod 4.4).
Systémový lupus erythematodes
U pacientů se středně těžkým až těžkým astmatem a chronickou spontánní urtikarií (CSU) byly hlášeny případy systémového lupus erythematodes (SLE) z klinických studií a postmarketingových hlášení. Patogeneze SLE není dobře známa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Maximální tolerovaná dávka Xolairu nebyla stanovena. Pacientům byly podány jednorázové intravenózní dávky až do 4 000 mg, aniž by se prokázala dávkou vymezená toxicita. Nejvyšší úhrnná dávka podaná pacientům byla 44 000 mg po dobu 20 týdnů a tato dávka neměla za následek žádné akutní nežádoucí účinky.
Pokud je podezření z předávkování, pacient by měl být sledován pro výskyt jakýchkoli neobvyklých známek nebo příznaků. Měla by být vyhledána lékařská pomoc a zahájena vhodná opatření.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva pro onemocnění spojená s obstrukcí dýchacích cest, jiná systémová léčiva pro onemocnění spojená s obstrukcí dýchacích cest, ATC kód: R03DX05
Omalizumab je z rekombinantní DNA odvozená humanizovaná monoklonální protilátka, která se selektivně váže na lidský imunoglobulin E (IgE). Protilátka je typu IgG1 kappa, která sestává ze skeletu lidské protilátky a z části odvozené z myší monoklonální protilátky, která váže IgE.
Alergické astma
Mechanismus účinku
Omalizumab se váže na IgE a předchází vazbě IgE k FcsRI (receptorům s vysokou afinitou k IgE) na bazofilech a mastocytech, čímž se redukuje množství volného IgE, který je využitelný ke spuštění alergické kaskády. Léčba Xolairem u atopických pacientů měla za následek nápadný pokles FcsRI receptorů na bazofilech.
Farmakodynamické účinky
Uvolnění histaminu in vitro z bazofilů izolovaných od jedinců léčených Xolairem bylo snížené přibližně o 90 % po stimulaci alergenem oproti hodnotám před léčbou.
V klinických studiích týkajících se alergických pacientů s astmatem byly sérové hladiny volného IgE sníženy v závislosti na dávce během 1 hodiny od první dávky a zůstaly snížené mezi dávkami. Po jednom roce od ukončení léčby Xolairem se hladiny IgE vrátily na úroveň před léčbou, aniž by byl pozorován rebound fenomén v hladinách IgE po vysazení léčivého přípravku.
Chronická spontánní urtikarie (CSU)
Mechanismus účinku
Omalizumab se váže na IgE a snižuje hladiny volného IgE. Následně klesá počet IgE receptorů (FcsRI) na buňkách. Není úplně jasné, jak tento mechanismus vede ke zlepšení příznaků CSU.
Farmakodynamické účinky
V klinických studiích týkajících se pacientů s CSU byla maximální suprese volného IgE pozorována
3 dny po první subkutánní dávce. Po opakovaných dávkách jednou za 4 týdny zůstaly hladiny volného IgE v séru před podáním dávky stabilní mezi 12 a 24 týdny léčby. Po skončení léčby Xolairem se zvýšily hladiny volného IgE na úroveň před léčbou po 16týdenním období dalšího sledování bez léčby.
Klinická účinnost a bezpečnost u alergického astmatu
Dospělí a dospívající >12 let
Účinnost a bezpečnost Xolairu byla prokázána ve 28týdenní dvojitě zaslepené placebem kontrolované studii (studie 1) zahrnující 419 těžkých alergických astmatiků ve věku 12-79 let, kteří měli sníženou plicní funkci (FEVi 40-80 % predikční hodnoty) a nedostatečnou kontrolu astmatických symptomů, přestože dostávali vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta2-agonistů. Pacienti přicházející v úvahu prodělali četné exacerbace astmatu vyžadující systémovou léčbu kortikosteroidy nebo byli hospitalizováni nebo navštívili lékařskou pohotovostní službu kvůli těžké exacerbaci astmatu v posledním roce navzdory průběžné léčbě vysokými dávkami inhalačních kortikosteroidů a dlouhodobě působícími beta2-agonisty. Subkutánně podávaný Xolair nebo placebo byly podávány jako doplňková léčba k >1 000 mikrogramům beklometason-dipropionátu (nebo jeho ekvivalentu) plus beta2-agonisty s dlouhodobým účinkem. Perorální kortikosteroid, theofylin a modifikátory leukotrienů byly dovoleny jako udržovací terapie (22 %, 27 %, resp. 35 % pacientů).
Primárním endpointem bylo sledování výskytu exacerbací astmatu vyžadujících léčbu vysokými dávkami systémových kortikosteroidů. Omalizumab redukoval četnost výskytu exacerbací astmatu o 19 % (p = 0,153). Další hodnocení, která ukázala statistický význam (p<0,05) ve prospěch Xolairu, zahrnovala snížení závažných exacerbací (kde funkce plic u pacientů byla redukována až pod 60 % nejlepší osobní hodnoty a bylo zapotřebí systémových kortikosteroidů) a neodkladných návštěv lékaře souvisejících s astmatem (zahrnovaly hospitalizaci, pohotovostní lékařskou službu a neplánované návštěvy u lékaře) a zlepšení astmatických symptomů a funkce plic podle celkového zhodnocení účinnosti léčby lékařem, Asthma-related Quality of Life (AQL).
Podle analýzy podskupin klinicky významný prospěch léčby Xolairem měli spíše pacienti s celkovou hladinou IgE >76 IU/ml před léčbou. U těchto pacientů ve studii 1 Xolair redukoval četnost výskytu exacerbací astmatu o 40 % (p = 0,002). Kromě toho více pacientů mělo klinicky významnou odpověď v populaci s celkovým IgE >76 IU/ml v programu Xolair při těžkém astmatu. Tabulka 6 zahrnuje výsledky u celkové populace ve studii 1.
Tabulka 6: Výsledky studie 1
Populace studie 1 celkem Xolair Placebo
_N=209_N=210
Exacerbace astmatu
Četnost výskytu za 28týdenní 0,74 0,92
období
% redukce, p-hodnota pro poměr 19,4 %, p = 0,153
četnosti
Exacerbace závažného astmatu
Četnost výskytu za 28týdenní 0,24 0,48
období
% redukce, p-hodnota pro poměr 50,1 %, p = 0,002
četnosti
|
Návštěvy lékaře v naléhavých případech Četnost výskytu za 28týdenní období % redukce, p-hodnota pro poměr četnosti |
0,24 43,9 %, p = |
0,43 0,038 |
|
Celkové zhodnocení lékařem | ||
|
% reagujících* |
60,5 % |
42,8 % |
|
Hodnota p ** |
<0,001 | |
|
AQL zlepšení | ||
|
% pacientů se zlepšením >0,5 |
60,8 % |
47,8 % |
|
Hodnota p |
0,008 | |
|
* nápadné zlepšení nebo kompletní kontrola | ||
** p-hodnota pro celkové zhodnocení
Studie 2 hodnotila účinnost a bezpečnost Xolairu u skupiny 312 osob s těžkým alergickým astmatem, která odpovídala populaci ve studii 1. Léčba Xolairem v této otevřené studii vedla ke snížení četnosti výskytu klinicky významných exacerbací astmatu o 61 % oproti běžné léčbě astmatu podávané samotné.
Čtyři další velké placebem kontrolované podpůrné studie trvající 28 až 52 týdnů na 1 722 dospělých a dospívajících (studie 3, 4, 5, 6) hodnotily účinnost a bezpečnost Xolairu u pacientů s těžkým perzistujícím astmatem. Kontrola astmatu u většiny pacientů byla nedostatečná, ale užívali méně konkomitantní léčby astmatu než pacienti ve studiích 1 a 2. Studie 3-5 použily exacerbace jako primární endpoint, zatímco studie 6 primárně hodnotila snížení množství inhalačních kortikosteroidů.
Ve studiích 3, 4 a 5 se u pacientů léčených Xolairem snížila četnost výskytu exacerbací astmatu o 37,5 % (p = 0,027), 40,3 % (p<0,001), respektive o 57,6 % (p<0,001) oproti placebu.
Ve studii 6 bylo schopno významně více pacientů s těžkým alergickým astmatem léčených Xolairem redukovat svou dávku flutikasonu až na <500 mikrogramů/den bez zhoršení kontroly astmatu (60,3 %) oproti skupině s placebem (45,8 %, p<0,05).
Skóre kvality života bylo měřeno pomocí dotazníku Juniper Asthma-related Quality of Life Questionnaire. Ve všech šesti studiích došlo ke statisticky významnému zlepšení skóre kvality života oproti výchozím hodnotám u pacientů užívajících Xolair oproti skupině placebo nebo kontrolní skupině.
Celkové hodnocení účinnosti léčby lékařem:
Celkové zhodnocení lékařem bylo provedeno v pěti z výše uvedených studií jako obsáhlé celkové posouzení kontroly astmatu vykonané ošetřujícím lékařem. Lékař vzal do úvahy PEF (peak expiratory flow = vrcholová výdechová rychlost), denní a noční symptomy, užití záchranné medikace, spirometrii a exacerbace. Ve všech pěti studiích se u významně většího podílu pacientů léčených Xolairem vyhodnotilo, že dosáhli buď výrazného zlepšení, nebo kompletní kontroly astmatu oproti skupině pacientů, kteří dostávali placebo.
Děti 6 až <12 let
Primární doklad bezpečnosti a účinnosti Xolairu u skupiny pacientů od 6 do <12 let vycházel z randomizované, dvojitě slepé, placebem kontrolované, multicentrické studie (studie 7).
Studie 7 byla placebem kontrolovaná studie zahrnující podskupinu (N=235) pacientů, u kterých byla určena současná indikace, kteří byli léčeni vysokými dávkami inhalačních kortikosteroidů (ekvivalent >500 pg/den flutikasonu) a dlouhodobě působícími beta-agonisty.
Klinicky významná exacerbace byla definována jako zhoršení příznaků astmatu dle posouzení zkoušejícího lékaře, vyžadující zdvojnásobení výchozí dávky inhalačních kortikosteroidů po dobu minimálně 3 dnů a/nebo vyžadující záchrannou léčbu systémovými (perorálními nebo intravenózními) kortikosteroidy po dobu minimálně 3 dnů.
Ve specifické podskupině pacientů, kteří užívali vysoké dávky inhalačních kortikosteroidů, bylo u skupiny pacientů užívajících omalizumab zaznamenáno statisticky signifikantní snížení výskytu exacerbací ve srovnání se skupinou užívající placebo. Ve 24. týdnu představoval rozdíl mezi léčenými skupinami 34 % (poměr výskytu 0,662, p=0,047) relativní snížení u pacientů léčených omalizumabem oproti skupině pacientů léčených placebem. V druhé, dvojitě slepé, 28týdenní léčebné periodě představoval rozdíl mezi léčenými skupinami 63 % (poměr výskytu 0,37, p<0,001) relativní snížení u pacientů léčených omalizumabem ve srovnání s pacienty léčenými placebem.
Během 52týdenní, dvojitě slepé léčby (zahrnující 24týdenní fázi s fixními dávkami steroidů a 28týdenní fázi s úpravou dávkování steroidů) představoval rozdíl mezi léčebnými skupinami 50 % (poměr výskytu 0,504, p<0,001) relativní snížení počtu exacerbací u pacientů léčených omalizumabem.
Během 52týdenní léčby vykazovala skupina pacientů užívající omalizumab větší pokles použití záchranné medikace beta-agonistů než skupina pacientů užívajících placebo, přestože tento rozdíl mezi léčebnými skupinami nebyl statisticky signifikantní. Z celkového vyhodnocení účinnosti léčby na konci 52týdenní léčby u podskupiny těžkých pacientů užívajících vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta-agonistů vyplývá, že poměr pacientů, kteří prokázali „vynikající“ účinnost, byl u skupiny užívající omalizumab vyšší a poměr pacientů s „průměrnou“ nebo „slabou“ účinností byl nižší ve srovnání s pacienty užívajícími placebo; rozdíl mezi léčebnými skupinami byl statisticky významný (p<0,001), zatímco mezi skupinou s omalizumabem a skupinou s placebem nebyly žádné rozdíly z hlediska subjektivních hodnot kvality života pacienta.
Klinická účinnost a bezpečnost u chronické spontánní urtikarie
Účinnost a bezpečnost Xolairu byla prokázána ve dvou randomizovaných, placebem kontrolovaných studiích fáze III (studie 1 a 2) u pacientů s CSU, kteří zůstali symptomatičtí i přes léčbu H1 antihistaminiky ve schválené dávce. Třetí studie (studie 3) primárně hodnotila bezpečnost Xolairu u pacientů s CSU, kteří zůstali symptomatičtí i přes léčbu H1 antihistaminiky při podání až čtyřnásobku schválené dávky, a při léčbě H2 antihistaminiky a/nebo LTRA. V těchto třech studiích bylo zařazeno 975 pacientů ve věku mezi 12 a 75 lety (průměrný věk 42,3 roků, 39 pacientů ve věku 12-17 let, 54 pacientů >65 let, 259 mužů a 716 žen). U všech pacientů bylo vyžadováno, aby měli nedostačující kontrolu příznaků, což bylo stanoveno pomocí týdenního skóre urtikariální aktivity (UAS7, rozmezí 0-42) > 16, a týdenního skóre závažnosti svědění (což je součást UAS7, rozmezí 0-21) > 8 po dobu 7 dnů před randomizací i přesto, že předtím užívali nějaké antihistaminikum po dobu alespoň 2 týdnů.
Ve studiích 1 a 2 měli pacienti průměrné týdenní skóre závažnosti svědění mezi 13,7 a 14,5 při počátečním vyšetření a průměrné UAS7 skóre 29,5 a 31,7, v uvedeném pořadí. Pacienti v bezpečnostní studii 3 měli průměrné týdenní skóre závažnosti svědění 13,8 a průměrné UAS7 skóre
31,2 při počátečním vyšetření. Napříč všemi třemi studiemi hlásili pacienti před zařazením do studie užívání v průměru 4 až 6 léků (včetně H1 antihistaminik) pro léčbu CSU příznaků. Pacienti dostávali Xolair v dávkách 75 mg, 150 mg nebo 300 mg nebo placebo subkutánní injekcí každé 4 týdny po dobu 24 týdnů a 12 týdnů ve studiích 1 a 2, v tomto pořadí, a 300 mg nebo placebo subkutánní injekcí každé 4 týdny po dobu 24 týdnů ve studii 3. Všechny studie měly 16týdenní období dalšího sledování bez léčby.
Primárním cílem byla změna v týdenním skóre závažnosti svědění od počátečního vyšetření do týdne 12. Omalizumab v dávce 300 mg snižoval týdenní skóre závažnosti svědění o 8,55 na 9,77 (p <0,0001) v porovnání se snížením o 3,63 na 5,14 u placeba (viz Tabulka 7). Statisticky významné výsledky byly dále pozorovány v podílech respondérů pro UAS7 < 6 (v týdnu 12), které byly vyšší u léčebných skupin užívajících 300 mg, v rozmezí od 52-66 % (p<0,0001) v porovnání s 11-19 % u skupiny s placebem, a kompletní odpověď (UAS7=0) byla dosažena u 34-44 % (p<0,0001) pacientů léčených dávkou 300 mg v porovnání s 5-9 % pacientů ve skupinách s placebem. Pacienti v léčebných skupinách s dávkou 300 mg dosáhli nejvyššího průměrného poměru dnů bez angioedému od týdne 4 do týdne 12, (91,0-96,1 %; p<0,001) v porovnání se skupinami s placebem (88,1-89,2 %). Průměrná změna od základní hodnoty do týdne 12 v celkovém DLQI u léčebných skupin s dávkou 300 mg byla vyšší (p<0,001) než u placeba, což ukazuje zlepšení v rozsahu od 9,7-10,3 bodů v porovnání s 5,1-6,1 body pro odpovídající skupiny s placebem.
|
Placebo |
Omalizumab 300 mg | |
|
Studie 1 | ||
|
N |
80 |
81 |
|
Průměr (SD) |
-3,63 (5,22) |
-9,40 (5,73) |
|
Rozdíl v LS průměrech vs. placebo1 |
- |
-5,80 |
|
95 % CI pro rozdíl |
- |
-7,49;-4,10 |
|
p-hodnota vs. placebo2 |
- |
<0,0001 |
|
Studie 2 | ||
|
N |
79 |
79 |
|
Průměr (SD) |
-5,14 (5,58) |
-9,77 (5,95) |
|
Rozdíl v LS průměrech vs. placebo1 |
- |
-4,81 |
|
95 % CI pro rozdíl |
- |
-6,49;-3,13 |
|
p-hodnota vs. placebo2 |
- |
<0,0001 |
|
Studie 3 | ||
|
N |
83 |
252 |
|
Průměr (SD) |
-4,01 (5,87) |
-8,55 (6,01) |
|
Rozdíl v LS průměrech vs. placebo1 |
- |
-4,52 |
|
95 % CI pro rozdíl |
- |
-5,97; -3,08 |
|
p-hodnota vs. placebo2 |
- |
<0,0001 |
*Modifikovaná hodnota „intent-to-treat“ (mITT) populace: zahrnovala všechny pacienty, kteří byli randomizováni a obdrželi alespoň jednu dávku zkoumaného léku.
BOCF (Baseline Observation Carried Forward) bylo použito k připočítání chybějících dat.
1 LS průměr byl odhadován s použitím modelu ANCOVA. Rozvrstvení (strata) byly základní hodnota týdenního skóre závažnosti svědění (<13 vs. >13) a hmotnost na počátku studie (<80 kg vs. >80 kg).
2 p-hodnota je odvozena od ANCOVA t-testu.
Obrázek 1 ukazuje průměrné týdenní skóre závažnosti svědění v průběhu času ve studii 1. Průměrná týdenní skóre závažnosti svědění se významně snížila s maximálním účinkem kolem týdne 12, který se udržel po 24týdenní léčebné období. Ve studii 3 byly výsledky podobné.
Ve všech třech studiích se postupně zvyšovalo průměrné týdenní skóre závažnosti svědění během16týdenního období dalšího sledování bez léčby, v souladu se znovu-objevením se příznaků onemocnění. Průměrné hodnoty na konci období dalšího sledování byly podobné skupině s placebem, ale nižší než příslušné průměrné základní hodnoty.
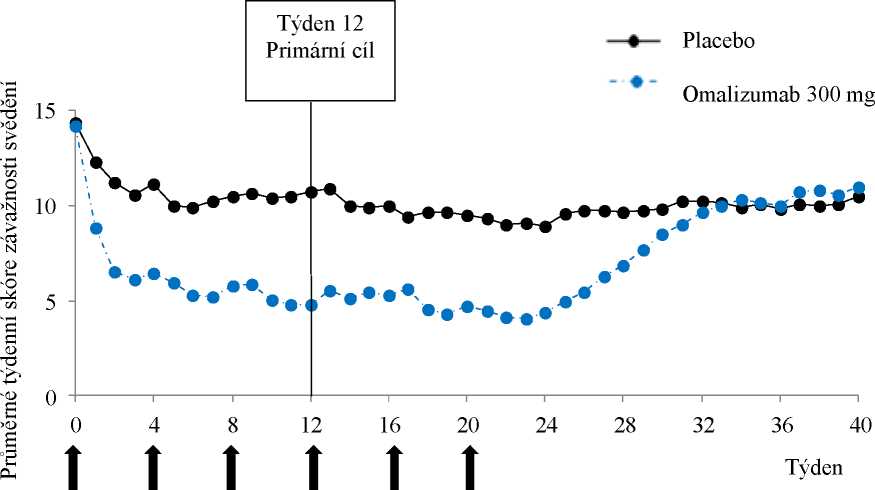
Podávaný omalizumab nebo placebo
BOCF=baseline observation carried forward; mITT=modified intention-to-treat population
Účinnost _po 24 týdnech léčby
Amplituda výsledků účinnosti pozorovaná ve 24. týdnu léčby byla srovnatelná s pozorováním ve 12. týdnu:
Ve studiích 1 a 3 bylo průměrné snížení týdenního skóre závažnosti svědění od počátečního vyšetření u dávky 300 mg 9,8 a 8,6, podíl pacientů s UAS7<6 byl 61,7 % a 55,6 %, a podíl pacientů s kompletní odpovědí (UAS7=0) byl 48,1 % a 42,5 %, v uvedeném pořadí, (všechna p<0,0001, při porovnání s placebem).
S opakovanou léčbou pacientů omalizumabem jsou k dispozici omezené klinické zkušenosti.
Data z klinických studií u dospívajících (12 až 17 let) zahrnovala celkem 39 pacientů, z nichž 11 dostávalo dávku 300 mg. Výsledky pro dávku 300 mg jsou k dispozici u 9 pacientů po týdnu 12 a u 6 pacientů po týdnu 24 a ukazují podobný rozsah odpovědi na léčbu omalizumabem v porovnání s dospělou populací. Průměrná změna od základní hodnoty v týdenním skóre závažnosti svědění ukázala snížení o 8,25 v týdnu 12 a o 8,95 v týdnu 24. Poměry respondérů byly: 33 % v týdnu 12 a 67 % v týdnu 24 pro UAS7=0, a 56 % v týdnu 12 a 67 % v týdnu 24 pro UAS7 <6.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu byla studována u dospělých a dospívajících pacientů s alergickým astmatem, stejně jako u dospělých a dospívajících pacientů s CSU. Obecné farmakokinetické charakteristiky omalizumabu jsou u těchto populací podobné.
Absorpce
Průměrná absolutní hodnota biologické dostupnosti omalizumabu je 62 % po subkutánním podání. Po jednorázové subkutánní dávce dospělým a dospívajícím pacientům s astmatem nebo CSU se omalizumab absorboval pomalu a dosáhl maximální koncentrace v séru průměrně po 6-8 dnech.
U pacientů s astmatem byly po opakovaných dávkách omalizumabu plochy pod křivkou sérové koncentrace v čase ode dne 0 až po den 14 v rovnovážném stavu až 6x větší oproti těm po první dávce.
Farmakokinetika omalizumabu je lineární v dávkách vyšších než 0,5 mg/kg. Po podání dávek 75 mg, 150 mg nebo 300 mg každé 4 týdny u pacientů s CSU se údolní (trough) sérové koncentrace omalizumabu zvýšily úměrně s velikostí dávek.
Podání Xolairu vyrobeného ve formě lyofilizátu nebo roztoku vedlo k podobným profilům sérové koncentrace omalizumabu v čase.
Distribuce
In vitro omalizumab tvoří s IgE komplexy omezené velikosti. Precipitující komplexy a komplexy s molekulovou hmotností větší než jeden milion daltonů nejsou pozorovány in vitro anebo in vivo.
Na základě populační farmakokinetiky byla distribuce omalizumabu u pacientů s alergickým astmatem a pacientů s CSU podobná. Zdánlivý distribuční objem u pacientů s astmatem byl po subkutánním podání 78 ± 32 ml/kg.
Eliminace
Clearance omalizumabu zahrnuje průběh clearance IgG stejně tak jako clearance prostřednictvím specifické vazby a tvorby komplexů s cílovým ligandem IgE. Vylučování IgG v játrech zahrnuje odbourávání v retikuloendotelovém systému a endotelových buňkách. Intaktní IgG je také vylučován žlučí. U pacientů s astmatem činil průměrný poločas eliminace omalizumabu ze séra 26 dní, s clearance v průměru 2,4 ± 1,1 ml/kg/den. Zdvojnásobení tělesné hmotnosti přibližně zdvojnásobuje danou clearance. U pacientů s CSU na základě populačních farmakokinetických simulací dosáhl poločas eliminace omalizumabu ze séra v rovnovážném stavu v průměru 24 dnů a skutečná clearance v rovnovážném stavu u pacienta o hmotnosti 80 kg byla 3,0 ml/kg/den.
Vlastnosti pacientské populace
Pacienti s astmatem: Populační farmakokinetika omalizumabu byla analyzována, aby se zhodnotily účinky demografických charakteristik. Analýzy těchto omezených údajů naznačují, že není nutná žádná úprava dávky u pacientů s astmatem pro věk (6-76 let), rasu/etnickou příslušnost, pohlaví nebo index tělesné hmotnosti (viz bod 4.2).
Pacienti s CSU: Účinky demografických charakteristik a ostatních faktorů na expozici omalizumabu byly hodnoceny na základě populační farmakokinetiky. Kromě toho byly kovarianční účinky hodnoceny analýzou vztahu mezi koncentracemi omalizumabu a klinickými odpověďmi. Tyto analýzy naznačují, že u pacientů s CSU z hlediska věku (12-75 let), rasy/etnika, pohlaví, tělesné hmotnosti, BMI, základní hodnoty IgE, autoprotilátek proti FcsRI nebo z hlediska současného užívání H2 antihistaminik nebo LTRA nejsou nutné žádné úpravy dávkování.
Zhoršená _ funkce ledvin a _jater
U alergických pacientů s astmatem nebo CSU se zhoršenou funkcí ledvin nebo jater nejsou žádné farmakokinetické nebo farmakodynamické údaje (viz body 4.2 a 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost omalizumabu byla studována u opic makaka jávského, jelikož omalizumab se váže na jejich i lidský IgE s podobnou afinitou. U některých opic byly zjištěny protilátky proti omalizumabu po opakovaném subkutánním nebo intravenózním podání. Nicméně nebyla pozorována žádná toxicita jako například onemocnění zprostředkované imunokomplexy nebo cytotoxicita podmíněná komplementem. Nevyskytl se žádný důkaz anafylaktické odpovědi následkem degranulace žírných buněk u těchto opic.
Dlouhodobé podávání omalizumabu v dávkách až 250 mg/kg (alespoň 14krát nejvyšší doporučená klinická dávka v mg/kg podle doporučené dávkovací tabulky) nehumánním primátům (dospělým i dospívajícím jedincům) bylo dobře tolerováno s výjimkou snížení počtu krevních destiček, které souviselo s dávkou a bylo závislé na věku, s větší citlivostí u nedospělých zvířat. Hladina koncentrace v séru potřebná k dosažení poklesu trombocytů o 50 % oproti výchozí hodnotě u dospělých opic makaka jávského byla zhruba 4 až 20krát vyšší než očekávané maximální klinické sérové koncentrace. Navíc u makaka jávského bylo pozorováno akutní krvácení a zánět v místě aplikace injekce.
Formální studie kancerogenity nebyly s omalizumabem provedeny.
V reprodukčních studiích u makaka jávského subkutánně podané dávky až do 75 mg/kg týdně (alespoň 8krát nejvyšší doporučená klinická dávka v mg/kg po dobu delší než 4 týdny) nevyvolaly mateřskou toxicitu, embryotoxicitu nebo teratogenní účinky, když byly podávané během organogeneze, a neměly nežádoucí účinky na fetální nebo novorozenecký růst, pokud byly podávané během těhotenství, porodu nebo kojení.
Omalizumab se vylučuje do mateřského mléka makaka jávského. Hladiny omalizumabu v mléce byly 0,15 % koncentrace v séru matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Sacharosa
Histidin
Monohydrát histidin-hydrochloridu Polysorbát 20
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
4 roky.
Po rozpuštění
Chemická a fyzikální stabilita rekonstituovaného léčivého přípravku byla prokázána po dobu 8 hodin při teplotě 2 °C - 8 °C a 4 hodiny při teplotě 30 °C.
Z mikrobiologického hlediska má být léčivý přípravek použit okamžitě po rozpuštění. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku před použitím j sou v odpovědnosti uživatele a normálně by doba neměla být delší než 8 hodin při teplotě 2 °C - 8 °C nebo 2 hodiny při teplotě 25 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem: průhledná, bezbarvá injekční lahvička ze skla typu I s pryžovou zátkou a modrou pertlí.
Ampulka s rozpouštědlem: průhledná, bezbarvá ampulka ze skla typu I obsahující 2 ml vody na injekci.
Balení, které obsahuje 1 injekční lahvičku prášku a 1 ampulku vody na injekci a vícečetná balení obsahující 4 (4 balení 1+1) nebo 10 (10 balení 1+1) injekčních lahviček prášku a ampulek vody na injekci.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rozpuštění lyofilizovaného léčivého přípravku trvá 15-20 minut, ačkoliv v některých případech to může trvat déle. Zcela rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý a může v něm být několik malých bublinek nebo pěna okolo okraje injekční lahvičky. Vzhledem k viskozitě rekonstituovaného léčivého přípravku se musí věnovat péče tomu, aby se odebral celý obsah léčivého přípravku z injekční lahvičky za účelem získání 1,2 ml před tím, než je vytlačen vzduch nebo přebytečné množství roztoku ze stříkačky.
Pro přípravu Xolairu 150 mg v injekční lahvičce pro subkutánní podání se prosím řiďte následujícími instrukcemi:
1. Odeberte z ampulky 1,4 ml vody na injekci do injekční stříkačky opatřené jehlou o velikosti průměru 18.
2. Za použití standardních aseptických postupů zasuňte jehlu do injekční lahvičky umístěné kolmo na rovný povrch, přidejte vodu na injekci do lahvičky obsahující lyofilizovaný prášek tak, aby voda na injekci směřovala přímo na prášek.
3. Držte lahvičku ve svislé poloze a intenzivně s ní kružte (netřepejte) po dobu přibližně 1 minuty, aby se prášek rovnoměrně namočil.
4. Aby se napomohlo rozpouštění po dokončení kroku 3, jemně kružte lahvičkou po dobu 5-10 sekund přibližně každých 5 minut, aby se rozpustil všechen zbývající prášek.
Pamatujte, že v některých případech to může trvat déle než 20 minut, aby se prášek zcela rozpustil. Jestliže jde o tento případ, opakujte krok 4, dokud jsou v roztoku viditelné částečky podobné gelu.
Když je léčivý přípravek zcela rozpuštěný, neměly by být v roztoku viditelné žádné částečky podobné gelu. Malé bublinky nebo pěna okolo okraje lahvičky jsou obvyklé. Rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý. Nepoužívejte jej, jestliže jsou přítomny pevné částečky.
5. Obraťte injekční lahvičku dnem vzhůru na dobu nejméně 15 sekund, aby roztok natekl až k zátce. Použijte novou 3ml stříkačku opatřenou jehlou o velikosti průměru 18, zasuňte jehlu do obrácené injekční lahvičky. Držte injekční lahvičku v obrácené poloze, jehlu postrčte až na samé dno roztoku v injekční lahvičce, když odebíráte roztok do stříkačky. Před vyjmutím jehly z injekční lahvičky táhněte píst zpátky až na konec těla stříkačky, aby se nasál celý obsah roztoku z obrácené lahvičky.
6. Pro subkutánní injekci nahraďte jehlu o velikosti 18 jehlou velikosti 25.
7. Vytlačte vzduch, velké bubliny a přebytečný roztok tak, aby se získala požadovaná dávka
1,2 ml. Nepatrná vrstva malých bublinek může zůstat v horní části roztoku v injekční stříkačce. Protože je roztok mírně viskózní, může aplikace subkutánně podaného roztoku trvat 5-10 sekund.
Z injekční lahvičky se získá 1,2 ml (150 mg) Xolairu. Pro dávku 75 mg odeberte do injekční stříkačky 0,6 ml a zbylý roztok zlikvidujte.
8. Injekce se podávájí subkutánně do oblasti deltového svalu ramene nebo do stehna.
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok je dodáván v jednorázové injekční lahvičce.
Z mikrobiologického hlediska by měl být léčivý přípravek použit okamžitě po rozpuštění (viz bod 6.3).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/319/002
EU/1/05/319/003
EU/1/05/319/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25. října 2005
Datum posledního prodloužení registrace: 22. června 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Xolair 75 mg injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s roztokem objemu 0,5 ml obsahuje omalizumabum* 75 mg.
*Omalizumab je humanizovaná monoklonální protilátka vyrobená technologií rekombinantní DNA v linii savčích buněk vaječníků čínských křečků (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Xolair je indikován k léčbě dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Léčbu Xolairem je možno použít pouze u pacientů s astmatem vyvolaným prokazatelně IgE (imunoglobulinem E) (viz bod 4.2).
Dospělí a dospívající (12 let a starší)
Xolair se doporučuje jako doplňková léčba ke zlepšení kontroly astmatu pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a kteří mají sníženou funkci plic (FEV1 <80 %), stejně jako časté symptomy během dne nebo probouzení v noci, a kteří mají dokumentované těžké exacerbace astmatu navzdory vysokým denním dávkám inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
Děti (6 až <12 let)
Xolair se doporučuje jako přídatná léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a časté denní nebo noční příznaky buzení a kteří mají prokázané četné vážné exacerbace přesto, že užívají vysoké denní dávky inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
4.2 Dávkování a způsob podání
Léčba Xolairem by měla být zahájena lékařem zkušeným v diagnostice a léčbě těžkého perzistujícího astmatu.
Dávkování
Vhodná dávka a frekvence podávání Xolairu se určí podle výchozích hodnot IgE (IU/ml), které se stanoví před zahájením léčby, a dle tělesné hmotnosti (kg). Aby mohla být stanovena dávka, měla by být před podáním počáteční dávky u pacientů stanovena hladina IgE jakýmkoliv komerčním testem pro stanovení celkového IgE v séru. Na základě výsledků může být potřeba podávat 75 až 600 mg Xolairu v 1 až 4 injekcích na každé podání.
U pacientů s IgE nižším než 76 IU/ml je méně pravděpodobné, že se projeví přínos přípravku (viz bod
5.1). Ošetřující lékař se musí ujistit, že dospělí a dospívající pacienti s IgE nižším než 76 IU/ml a děti (6 až <12 let) s IgE nižším než 200 IU/ml mají před začátkem léčby prokázanou in vitro reaktivitu (RAST) na celoroční vzdušný alergen.
Viz Tabulka 1 schéma konverze a Tabulka 2 a 3 schéma stanovení dávky u dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Pacientům, jejichž výchozí hodnoty hladin IgE nebo tělesná hmotnost v kilogramech jsou mimo limity dávkovací tabulky, by Xolair neměl být podáván.
Maximální doporučená dávka je 600 mg omalizumabu každé dva týdny.
Tabulka 1: Konverze dávky na počet injekčních stříkaček, počet injekcí a celkový objem injekcí při každé aplikaci
|
Dávka (mg) |
Počet injekčních stříkaček 75 mg 150 mg |
Počet injekcí |
Celkový objem injekcí (ml) | |
|
75 |
1 |
0 |
1 |
0,5 |
|
150 |
0 |
1 |
1 |
1,0 |
|
225 |
1 |
1 |
2 |
1,5 |
|
300 |
0 |
2 |
2 |
2,0 |
|
375 |
1 |
2 |
3 |
2,5 |
|
450 |
0 |
3 |
3 |
3,0 |
|
525 |
1 |
3 |
4 |
3,5 |
|
600 |
0 |
4 |
4 |
4,0 |
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE (IU/ml) |
>20- 25 |
>25- 30 |
>30- 40 |
>40- 50 |
>50- 60 |
>60- 70 |
>70- 80 |
>80- 90 |
>90- 125 |
>125- 150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
PODÁVÁNÍ KAŽDÉ 2 TÝDNY | |||||||||
|
>900- 1000 |
VIZ TABULKA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(IU/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
PODÁVÁNÍ KAŽDÉ 4 TÝDNY | |||||||||
|
>100-200 |
VIZ TABULKA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- 1200 |
300 |
300 |
450 |
525 |
600 |
NEPODÁVAT - pro doporučení dávkování není dostatek údajů | ||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Délka léčby, její sledování a úprava dávky
Xolair je určen pro dlouhodobou léčbu. Klinické studie prokázaly, že Xolair dosahuje účinnosti po minimálně 12-16 týdnech léčby. Po 16 týdnech od zahájení léčby Xolairem by měl lékař před podáváním dalších injekcí zhodnotit účinnost léčby. Rozhodnutí o pokračování léčby Xolairem následně po 16týdenním intervalu, nebo příležitostně později, by mělo být založeno na tom, zdali je patrné výrazné zlepšení z hlediska celkové kontroly astmatu (viz bod 5.1, Celkové hodnocení účinnosti léčby lékařem).
Ukončení léčby Xolairem má obecně za následek návrat ke zvýšeným hladinám volného IgE a s tím spojeným symptomům. Celkové hladiny IgE jsou zvýšené během léčby a zůstávají zvýšené až po dobu jednoho roku po skončení léčby. Proto opětovné stanovení hladin IgE během léčby Xolairem nemůže být použito jako vodítko pro stanovení dávky. Stanovení dávky po přerušení léčby trvající méně než jeden rok by mělo být založeno na výchozích hodnotách hladin sérového IgE získaných při stanovení zahajovací dávky. Pro stanovení dávky mohou být celkové hladiny IgE v séru znovu stanoveny, jestliže léčba Xolairem byla přerušena jeden rok nebo déle.
Dávky by měly být přizpůsobeny významným změnám tělesné hmotnosti (viz Tabulka 2 a 3).
Zvláštní skupiny pacientů
Starší pacienti (65 let a starší)
Pro užití Xolairu u pacientů starších více než 65 let jsou dostupné omezené údaje, ale nebylo zjištěno, že by u starších pacientů bylo nutné podávání jiné dávky než u mladších dospělých pacientů.
Pacienti s poškozením ledvin nebo jater
Nejsou k dispozici žádné studie hodnotící vliv zhoršené funkce ledvin nebo jater na farmakokinetiku Xolairu. Protože clearance omalizumabu je při klinických dávkách ovlivněna hlavně retikuloendotelovým systémem (RES), je nepravděpodobné, že by byla ovlivněna poškozením ledvin nebo jater. Protože není doporučena žádná zvláštní úprava dávkování pro tyto pacienty, Xolair by měl být podáván s opatrností (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost Xolairu u dětí mladších 6 let nebyla dosud stanovena. Nej sou dostupné žádné údaje.
Způsob podání
Pouze pro subkutánní podání. Nepodávejte intravenózně nebo intramuskulárně.
Injekce se aplikují subkutánně do oblasti deltového svalu ramene. Alternativně se může injekce aplikovat do stehna, jestliže je z jakéhokoliv důvodu vyloučena aplikace do oblasti deltového svalu.
Zkušenosti s aplikací injekce Xolairu samotným pacientem jsou omezené. Z toho důvodu má léčbu podávat pouze zdravotnický personál (viz bod 6.6 a také bod Informace pro zdravotnické pracovníky v příbalové informaci).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Obecné
Xolair není určen k léčbě exacerbací akutního astmatu, akutního bronchospasmu nebo status asthmaticus.
Xolair nebyl zkoumán u pacientů se syndromem hyperimunoglobulinemie E nebo alergickou bronchopulmonální aspergilózou nebo pro prevenci anafylaktických reakcí včetně takových, které jsou vyvolané alergií na potraviny, atopickou dermatitidou nebo alergickou rinitidou. Xolair není indikován pro léčbu těchto stavů.
Léčba Xolairem nebyla zkoumána u pacientů s autoimunitním onemocněním, s onemocněním zprostředkovaným imunokomplexy, nebo s preexistující zhoršenou funkcí ledvin nebo jater (viz bod
4.2). Pokud je Xolair podáván těmto skupinám pacientů, měla by být zachovávána opatrnost.
Náhlé vysazení systémových nebo inhalačních kortikosteroidů po zahájení léčby Xolairem se nedoporučuje. Snižování dávek kortikosteroidů je nutno provádět pod přímým dohledem lékaře a je vhodné jej provádět postupně.
Poruchy imunitního systému Alergické reakce typu I
Lokální nebo systémové alergické reakce typu I včetně anafylaxe a anafylaktického šoku se mohou objevit po podání omalizumabu, přičemž tyto reakce se mohou objevit i po dlouhotrvající léčbě omalizumabem. Většina těchto reakcí se objevila během 2 hodin po první a následné injekci Xolairu, ale některé se objevily až po 2 hodinách a dokonce i po více než 24 hodinách po injekci. Proto by následně po podání Xolairu měly být vždy dostupné k okamžitému užití léčivé přípravky na léčbu anafylaktických reakcí. Pacienti by měli být informováni o možnosti takových reakcí, a pokud se objeví alergická reakce, měli by vyhledat okamžitě lékařskou pomoc. Výskyt anafylaxe nesouvisející s omalizumabem v anamnéze může být rizikový faktor pro anafylaxi následující po podání Xolairu.
U malého počtu pacientů v klinických studiích byly detekovány protilátky proti omalizumabu (viz bod 4.8). Klinický význam protilátek proti Xolairu není dobře prostudován.
Sérová nemoc
Sérová nemoc a reakce podobné sérové nemoci, které j sou opožděnými alergickými reakcemi typu III, byly pozorovány u pacientů léčených humanizovanými monoklonálními protilátkami včetně omalizumabu. Předpokládaný patofyziologický mechanismus zahrnuje tvorbu a ukládání imunokomplexů v důsledku vzniku protilátek proti omalizumabu. Reakce se obvykle objevila 1-5 dní po podání prvních nebo následujících injekcí, i po dlouhodobé léčbě. Příznaky poukazující na sérovou nemoc zahrnují artritidu/artralgie, vyrážku (kopřivku nebo jiné formy), horečku a lymfadenopatii. K léčbě nebo prevenci této nemoci mohou být vhodná antihistaminika a kortikosteroidy. Pacienti by měli být poučeni, aby hlásili jakékoli podezřelé příznaky.
Syndrom Churga-Straussové a hypereozinofilní syndrom
U pacientů s těžkým astmatem se může zřídka vyskytnout systémový hypereozinofilní syndrom nebo alergická eozinofilní granulomatózní vaskulitida (syndrom Churga-Straussové), které se obvykle léčí systémovými kortikoidy.
Ve vzácných případech se mohou u pacientů léčených antiastmatickými léčivými přípravky, včetně omalizumabu, projevit systémová eozinofilie a vaskulitida. Tyto případy jsou často spojeny se snižováním léčby perorálními kortikoidy.
U těchto pacientů by lékaři měli věnovat zvýšenou pozornost možnému rozvoji eozinofilie, vaskulitické vyrážky, zhoršení plicních symptomů, abnormalit paranazálních dutin, srdečních komplikací a/nebo neuropatie.
Ve všech závažných případech výše uvedených poruch imunitního systému by mělo být zváženo vysazení omalizumabu.
Parazitární infekce (helmintóza)
IgE může být spojený s imunologickou odpovědí na některé parazitární infekce. V placebem kontrolované studii u pacientů s chronicky vysokým rizikem helmintóz bylo prokázáno mírné zvýšení podílu infekcí ve skupině s omalizumabem, ačkoliv průběh, závažnost a odpověď na léčbu infekce se nezměnily. Četnost výskytu infekce cizopasnými červy v celkovém klinickém programu, který nebyl navržen tak, aby takové infekce prokázal, byla méně než 1 z 1 000 pacientů. Nicméně měla by být zaručena opatrnost u pacientů s vysokým rizikem infekce cizopasnými červy, zejména pokud cestují do oblastí, kde se infekce cizopasnými červy vyskytují endemicky. Pokud pacienti neodpovídají na doporučenou antiparazitární léčbu, mělo by být zváženo přerušení léčby Xolairem.
Jedinci senzitivní na latex
Snímatelný kryt jehly této předplněné injekční stříkačky obsahuje derivát přírodního gumového latexu. Ve snímatelném krytu jehly nebyl dosud detekován žádný přírodní gumový latex. Nicméně použití přípravku Xolair injekční roztok v předplněné injekční stříkačce nebylo u jedinců senzitivních na latex studováno, a proto je zde potenciální riziko vzniku hypersenzitivních reakcí, které nemohou být úplně vyloučeny.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Xolair může nepřímo snížit účinnost léčivých přípravků používaných k léčbě helmintóz nebo jiných parazitárních infekcí, protože v imunologické odpovědi na některé helmintózy může být zapojen IgE (viz bod 4.4).
Enzymy cytochromu P450, pumpy zajišťující efflux a mechanismy vazby na proteiny se nepodílejí na clearance omalizumabu; takže je zde malá možnost lékových interakcí. Nebyly provedeny žádné studie interakce Xolairu s léčivými přípravky ani vakcínami. Není žádný farmakologický důvod očekávat, že by se běžně předepisované léčivé přípravky užívané v léčbě astmatu vzájemně ovlivňovaly s omalizumabem.
V klinických studiích byl Xolair běžně užíván v kombinaci s inhalačními a perorálními kortikosteroidy, inhalačními krátkodobě a dlouhodobě působícími beta agonisty, modifikátory leukotrienů, theofyliny a perorálními antihistaminiky. Nevyskytly se žádné údaje, že bezpečnost Xolairu byla změněna těmito dalšími běžně užívanými antiastmatickými léčivými přípravky. Jsou dostupné omezené údaje o užívání Xolairu v kombinaci se specifickou imunoterapií (hyposenzitizační terapie). V klinické studii, kde byl Xolair podáván společně s imunoterapií, nebyl zjištěn žádný rozdíl v bezpečnosti a účinnosti Xolairu v kombinaci se specifickou imunoterapií v porovnání s podáváním samotného Xolairu.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání omalizumabu těhotným ženám jsou omezené. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky (viz bod 5.3). Omalizumab přestupuje přes placentu, ale možnost poškození plodu není známa. U primátů byl omalizumab spojován se snížením na věku závislém počtu krevních destiček, s relativní větší citlivostí u nedospělých zvířat (viz bod
5.3). Xolair by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda se omalizumab vylučuje do lidského mateřského mléka. Dostupné údaje u samic ostatních primátů prokázaly vylučování omalizumabu do mléka (viz bod 5.3). Riziko pro kojené novorozence/děti nelze vyloučit. Omalizumab by neměl být podáván během kojení.
Fertilita
Pro omalizumab nejsou k dispozici žádné údaje týkající se lidské fertility. Ve specificky navržených neklinických studiích fertility u primátů (s výjimkou člověka), včetně studií páření, nebylo pozorováno poškození samčí ani samičí fertility následně po opakovaném podávání omalizumabu v dávkách až 75 mg/kg. Dále nebyly pozorovány žádné genotoxické účinky v samostatných neklinických studiích genotoxicity (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xolair nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Během klinických studií u dospělých a dospívajících pacientů starších 12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy a reakce v místě aplikace injekce, včetně bolestivosti, zduření, zarudnutí a svědění. V klinických studiích u dětí od 6 do <12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy, pyrexie a bolest v nadbřišku. Ve většině případů byly reakce mírné nebo středně závažné.
Tabulkový seznam nežádoucích účinků
Tabulka 4 zaznamenává nežádoucí účinky hlášené v klinických studiích celkové bezpečnosti u populace léčené Xolairem podle MedDRA orgánové klasifikace a četnosti výskytu. V každé skupině četností jsou nežádoucí reakce seřazeny podle klesající závažnosti. Kategorie četností jsou definovány jako: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000) a velmi vzácné (<1/10 000). Reakce hlášené z postmarketingového sledování jsou uvedeny s frekvencí není známo (z dostupných údajů nelze určit).
Tabulka 4: Nežádoucí účinky
|
Infekce a infestace | |
|
Méně časté |
Faryngitida |
|
Vzácné |
Parazitární infekce |
|
Poruchy krve a lymfatického systému | |
|
Není známo |
Idiopatická trombocytopenie, včetně těžkých případů |
|
Poruchy imunitního systému | |
|
Vzácné |
Anafylaktická reakce, jiné závažné alergické stavy, vývoj |
|
protilátek proti omalizumabu | |
|
Není známo |
Sérová nemoc, která může zahrnovat horečku a lymfadenopatii |
|
Poruchy nervového systému | |
|
Časté |
Bolest hlavy* |
|
Méně časté |
Synkopa, parestezie, somnolence, závrať |
|
Cévní poruchy | |
|
Méně časté |
Posturální hypotenze, zčervenání |
|
Respirační, hrudní a mediastinální poruchy | |
|
Méně časté |
Alergický bronchospasmus, kašel |
|
Vzácné |
Otok laryngu |
|
Není známo |
Alergická granulomatózní vaskulitida (tzv. syndrom Churga- |
|
Straussové) | |
|
Gastrointestinální poruchy | |
|
Časté |
Bolest v nadbřišku** |
|
Méně časté | |
|
Poruchy kůže a podkožní tkáně | |
|
Méně časté | |
|
Vzácné |
Angioedém |
|
Není známo |
Alopecie |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Vzácné |
Systémový lupus erythematodes (SLE) |
|
Není známo |
Artralgie, myalgie, otoky kloubů |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Pyrexie** |
|
Časté |
Reakce v místě aplikace jako zduření, zarudnutí, bolest, svědění |
|
Méně časté |
Onemocnění podobné chřipce, otoky paží, zvýšení hmotnosti, |
*: Velmi časté u dětí od 6 do <12 let **: U dětí od 6 do <12 let
Popis vybraných nežádoucích účinků Poruchy imunitního systému Další informace viz bod 4.4.
Anafylaxe
Anafylaktické reakce byly v klinických studiích vzácné. Nicméně v datech po uvedení přípravku na trh se následně po kumulativním vyhledávání v bezpečnostní databázi našlo celkem 898 případů anafylaxe. Na základě očekávané expozice 566 923 pacientoroků to mělo za následek míru hlášení přibližně 0,20 %.
Arteriální tromboembolické příhody (ATE)
V kontrolovaných klinických studiích a během interim analýz observační studie byla pozorována numerická nerovnováha v počtu ATE. Definice složeného endpointu ATE zahrnovala cévní mozkovou příhodu, transitorní ischemickou ataku, infarkt myokardu, nestabilní anginu pectoris a kardiovaskulární úmrtí (včetně úmrtí z neznámé příčiny). V konečné analýze observační studie byl poměr ATE na 1 000 pacientoroků 7,52 (115/15 286 pacientoroků) pro pacienty léčené Xolairem a 5,12 (51/9 963 pacientoroků) pro kontrolní skupinu. V multivariační analýze kontrolující základní dostupné kardiovaskulární rizikové faktory byl poměr rizik 1,32 (95 % interval spolehlivosti 0,91-1,91). V samostatné analýze poolovaných klinických studií, která zahrnovala všechny randomizované, dvojitě zaslepené, placebem kontrolované klinické studie trvající 8 nebo více týdnů, byl poměr ATE na 1 000 pacientoroků 2,69 (5/1 856 pacientoroků) pro pacienty léčené Xolairem a 2,38 (4/1 680 pacientoroků) pro pacienty užívající placebo (relativní riziko 1,13, 95 % interval spolehlivosti 0,24-5,71).
Krevní destičky
V klinických studiích mělo několik pacientů počet krevních destiček pod spodní hranicí normálního laboratorního rozmezí. Žádná z těchto změn nebyla spojena s epizodami krvácení nebo se snížením hemoglobinu. U lidí (pacientů nad 6 let) nebylo hlášeno trvalé snížení počtu krevních destiček, které bylo pozorováno u ostatních primátů (viz bod 5.3), přestože z postmarketingového sledování byly ojediněle hlášeny případy idiopatické trombocytopenie, včetně těžkých případů.
Parazitární infekce
Placebem kontrolovaná studie u pacientů s chronicky vysokým rizikem helmintóz ukázala mírně zvýšenou četnost výskytu infekcí u pacientů užívajících omalizumab ve srovnání s kontrolní skupinou. Toto zvýšení nebylo statisticky signifikantní. Průběh, závažnost a odpověď na léčbu infekcí se nezměnily (viz bod 4.4).
Systémový lupus erythematodes
U pacientů se středně těžkým až těžkým astmatem a chronickou spontánní urtikarií (CSU) byly hlášeny případy systémového lupus erythematodes (SLE) z klinických studií a postmarketingových hlášení. Patogeneze SLE není dobře známa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Maximální tolerovaná dávka Xolairu nebyla stanovena. Pacientům byly podány jednorázové intravenózní dávky až do 4 000 mg, aniž by se prokázala dávkou vymezená toxicita. Nejvyšší úhrnná dávka podaná pacientům byla 44 000 mg po dobu 20 týdnů a tato dávka neměla za následek žádné akutní nežádoucí účinky.
Pokud je podezření z předávkování, pacient by měl být sledován pro výskyt jakýchkoli neobvyklých známek nebo příznaků. Měla by být vyhledána lékařská pomoc a zahájena vhodná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva pro onemocnění spojená s obstrukcí dýchacích cest, jiná systémová léčiva pro onemocnění spojená s obstrukcí dýchacích cest, ATC kód: R03DX05
Mechanismus účinku
Omalizumab je z rekombinantní DNA odvozená humanizovaná monoklonální protilátka, která se selektivně váže na lidský imunoglobulin E (IgE). Protilátka je typu IgG1 kappa, která sestává ze skeletu lidské protilátky a z části odvozené z myší monoklonální protilátky, která váže IgE.
Omalizumab se váže na IgE a předchází vazbě IgE k FcsRI (receptorům s vysokou afinitou k IgE), čímž se redukuje množství volného IgE, který je využitelný ke spuštění alergické kaskády. Léčba Xolairem u atopických pacientů měla za následek nápadný pokles FcsRI receptorů na bazofilech.
Farmakodynamické účinky
Uvolnění histaminu in vitro z bazofilů izolovaných od jedinců léčených Xolairem bylo snížené přibližně o 90 % po stimulaci alergenem oproti hodnotám před léčbou.
V klinických studiích byly sérové hladiny volného IgE sníženy v závislosti na dávce během 1 hodiny od první dávky a zůstaly snížené mezi dávkami. Po jednom roce od ukončení léčby Xolairem se hladiny IgE vrátily na úroveň před léčbou, aniž by byl pozorován rebound fenomén v hladinách IgE po vysazení léčivého přípravku.
Klinická účinnost a bezpečnost
Dospělí a dospívající >12 let
Účinnost a bezpečnost Xolairu byla prokázána ve 28týdenní dvojitě zaslepené placebem kontrolované studii (studie 1) zahrnující 419 těžkých alergických astmatiků ve věku 12-79 let, kteří měli sníženou plicní funkci (FEV1 40-80 % predikční hodnoty) a nedostatečnou kontrolu astmatických symptomů, přestože dostávali vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta2-agonistů. Pacienti přicházející v úvahu prodělali četné exacerbace astmatu vyžadující systémovou léčbu kortikosteroidy nebo byli hospitalizováni nebo navštívili lékařskou pohotovostní službu kvůli těžké exacerbaci astmatu v posledním roce navzdory průběžné léčbě vysokými dávkami inhalačních kortikosteroidů a dlouhodobě působícími beta2-agonisty. Subkutánně podávaný Xolair nebo placebo byly podávány jako doplňková léčba k >1 000 mikrogramům beklometason-dipropionátu (nebo jeho ekvivalentu) plus beta2-agonisty s dlouhodobým účinkem. Perorální kortikosteroid, theofylin a modifikátory leukotrienů byly dovoleny jako udržovací terapie (22 %, 27 %, resp. 35 % pacientů).
Primárním endpointem bylo sledování výskytu exacerbací astmatu vyžadujících léčbu vysokými dávkami systémových kortikosteroidů. Omalizumab redukoval četnost výskytu exacerbací astmatu o 19 % (p = 0,153). Další hodnocení, která ukázala statistický význam (p<0,05) ve prospěch Xolairu, zahrnovala snížení závažných exacerbací (kde funkce plic u pacientů byla redukována až pod 60 % nejlepší osobní hodnoty a bylo zapotřebí systémových kortikosteroidů) a neodkladných návštěv lékaře souvisejících s astmatem (zahrnovaly hospitalizaci, pohotovostní lékařskou službu a neplánované návštěvy u lékaře) a zlepšení astmatických symptomů a funkce plic podle celkového zhodnocení účinnosti léčby lékařem, Asthma-related Quality of Life (AQL).
Podle analýzy podskupin klinicky významný prospěch léčby Xolairem měli spíše pacienti s celkovou hladinou IgE >76 IU/ml před léčbou. U těchto pacientů ve studii 1 Xolair redukoval četnost výskytu exacerbací astmatu o 40 % (p = 0,002). Kromě toho více pacientů mělo klinicky významnou odpověď v populaci s celkovým IgE >76 IU/ml v programu Xolair při těžkém astmatu. Tabulka 5 zahrnuje výsledky u celkové populace ve studii 1.
Tabulka 5: Výsledky studie 1
Populace studie 1 celkem
Xolair
N=209
Placebo
N=210
Exacerbace astmatu
Četnost výskytu za 28týdenní období
0,74
0,92
|
% redukce, p-hodnota pro poměr četnosti |
19,4 %, p = 0,153 |
|
Exacerbace závažného astmatu | |
|
Četnost výskytu za 28týdenní období |
0,24 0,48 |
|
% redukce, p-hodnota pro poměr četnosti |
50,1 %, p = 0,002 |
|
Návštěvy lékaře v naléhavých případech Četnost výskytu za 28týdenní období |
0,24 0,43 |
|
% redukce, p-hodnota pro poměr četnosti |
43,9 %, p = 0,038 |
|
Celkové zhodnocení lékařem % reagujících* Hodnota p ** |
60,5 % <0,001 |
42,8 % |
|
AQL zlepšení | ||
|
% pacientů se zlepšením >0,5 |
60,8 % |
47,8 % |
|
Hodnota p |
0,008 |
* nápadné zlepšení nebo kompletní kontrola
** p-hodnota pro celkové zhodnocení
Studie 2 hodnotila účinnost a bezpečnost Xolairu u skupiny 312 osob s těžkým alergickým astmatem, která odpovídala populaci ve studii 1. Léčba Xolairem v této otevřené studii vedla ke snížení četnosti výskytu klinicky významných exacerbací astmatu o 61% oproti běžné léčbě astmatu podávané samotné.
Čtyři další velké placebem kontrolované podpůrné studie trvající 28 až 52 týdnů na 1 722 dospělých a dospívajících (studie 3, 4, 5, 6) hodnotily účinnost a bezpečnost Xolairu u pacientů s těžkým perzistujícím astmatem. Kontrola astmatu u většiny pacientů byla nedostatečná, ale užívali méně konkomitantní léčby astmatu než pacienti ve studiích 1 a 2. Studie 3-5 použily exacerbace jako primární endpoint, zatímco studie 6 primárně hodnotila snížení množství inhalačních kortikosteroidů.
Ve studiích 3, 4 a 5 se u pacientů léčených Xolairem snížila četnost výskytu exacerbací astmatu o 37,5 % (p = 0,027), 40,3 % (p<0,001), respektive o 57,6 % (p<0,001) oproti placebu.
Ve studii 6 bylo schopno významně více pacientů s těžkým alergickým astmatem léčených Xolairem redukovat svou dávku flutikasonu až na <500 mikrogramů/den bez zhoršení kontroly astmatu (60,3 %) oproti skupině s placebem (45,8 %, p<0,05).
Skóre kvality života bylo měřeno pomocí dotazníku Juniper Asthma-related Quality of Life Questionnaire. Ve všech šesti studiích došlo ke statisticky významnému zlepšení skóre kvality života oproti výchozím hodnotám u pacientů užívajících Xolair oproti skupině placebo nebo kontrolní skupině.
Celkové hodnocení účinnosti léčby lékařem:
Celkové zhodnocení lékařem bylo provedeno v pěti z výše uvedených studií jako obsáhlé celkové posouzení kontroly astmatu vykonané ošetřujícím lékařem. Lékař vzal do úvahy PEF (peak expiratory flow = vrcholová výdechová rychlost), denní a noční symptomy, užití záchranné medikace, spirometrii a exacerbace. Ve všech pěti studiích se u významně většího podílu pacientů léčených Xolairem vyhodnotilo, že dosáhli buď výrazného zlepšení, nebo kompletní kontroly astmatu oproti skupině pacientů, kteří dostávali placebo.
Děti 6 až <12 let
Primární doklad bezpečnosti a účinnosti Xolairu u skupiny pacientů od 6 do <12 let vycházel z randomizované, dvojitě slepé, placebem kontrolované, multicentrické studie (studie 7).
Studie 7 byla placebem kontrolovaná studie zahrnující podskupinu (N=235) pacientů, u kterých byla určena současná indikace, kteří byli léčeni vysokými dávkami inhalačních kortikosteroidů (ekvivalent >500 pg/den flutikasonu) a dlouhodobě působícími beta-agonisty.
Klinicky významná exacerbace byla definována jako zhoršení příznaků astmatu dle posouzení zkoušejícího lékaře, vyžadující zdvojnásobení výchozí dávky inhalačních kortikosteroidů po dobu minimálně 3 dnů a/nebo vyžadující záchrannou léčbu systémovými (perorálními nebo intravenózními) kortikosteroidy po dobu minimálně 3 dnů.
Ve specifické podskupině pacientů, kteří užívali vysoké dávky inhalačních kortikosteroidů, bylo u skupiny pacientů užívajících omalizumab zaznamenáno statisticky signifikantní snížení výskytu exacerbací ve srovnání se skupinou užívající placebo. Ve 24. týdnu představoval rozdíl mezi léčenými skupinami 34 % (poměr výskytu 0,662, p=0,047) relativní snížení u pacientů léčených omalizumabem oproti skupině pacientů léčených placebem. V druhé, dvojitě slepé, 28týdenní léčebné periodě představoval rozdíl mezi léčenými skupinami 63 % (poměr výskytu 0,37, p<0,001) relativní snížení u pacientů léčených omalizumabem ve srovnání s pacienty léčenými placebem.
Během 52týdenní, dvojitě slepé léčby (zahrnující 24týdenní fázi s fixními dávkami steroidů a 28týdenní fázi s úpravou dávkování steroidů) představoval rozdíl mezi léčebnými skupinami 50 % (poměr výskytu 0,504, p<0,001) relativní snížení počtu exacerbací u pacientů léčených omalizumabem.
Během 52týdenní léčby vykazovala skupina pacientů užívající omalizumab větší pokles použití záchranné medikace beta-agonistů než skupina pacientů užívajících placebo, přestože tento rozdíl mezi léčebnými skupinami nebyl statisticky signifikantní. Z celkového vyhodnocení účinnosti léčby na konci 52týdenní léčby u podskupiny těžkých pacientů užívajících vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta-agonistů vyplývá, že poměr pacientů, kteří prokázali „vynikající“ účinnost, byl u skupiny užívající omalizumab vyšší a poměr pacientů s „průměrnou“ nebo „slabou“ účinností byl nižší ve srovnání s pacienty užívajícími placebo; rozdíl mezi léčebnými skupinami byl statisticky významný (p<0,001), zatímco mezi skupinou s omalizumabem a skupinou s placebem nebyly žádné rozdíly z hlediska subjektivních hodnot kvality života pacienta.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu byla studována u dospělých a dospívajících pacientů s alergickým astmatem.
Absorpce
Průměrná absolutní hodnota biologické dostupnosti omalizumabu je 62 % po subkutánním podání. Po jednorázové subkutánní dávce dospělým a dospívajícím pacientům s astmatem se omalizumab absorboval pomalu a dosáhl maximální koncentrace v séru průměrně po 7-8 dnech. Farmakokinetika omalizumabu je lineární v dávkách vyšších než 0,5 mg/kg. Po opakovaných dávkách omalizumabu byly plochy pod křivkou sérové koncentrace v čase ode dne 0 až po den 14 v rovnovážném stavu až 6x větší oproti těm po první dávce.
Podání Xolairu vyrobeného ve formě lyofilizátu nebo roztoku vedlo k podobným profilům sérové koncentrace omalizumabu v čase.
Distribuce
In vitro omalizumab tvoří s IgE komplexy omezené velikosti. Precipitující komplexy a komplexy s molekulovou hmotností větší než jeden milion daltonů nejsou pozorovány in vitro anebo in vivo. Zdánlivý distribuční objem u pacientů po subkutánním podání byl 78 ± 32 ml/kg.
Eliminace
Clearance omalizumabu zahrnuje průběh clearance IgG stejně tak jako clearance prostřednictvím specifické vazby a tvorby komplexů s cílovým ligandem IgE. Vylučování IgG v játrech zahrnuje odbourávání v retikuloendotelovém systému a endotelových buňkách. Intaktní IgG je také vylučován žlučí. U pacientů s astmatem činil průměrný poločas eliminace omalizumabu ze séra 26 dní, s clearance v průměru 2,4 ± 1,1 ml/kg/den. Kromě toho zdvojnásobení tělesné hmotnosti přibližně zdvojnásobuje danou clearance.
Vlastnosti pacientské populace
Věk,rasa/etnikum, pohlaví, BMI (Body Mass Index)
Populační farmakokinetika Xolairu byla analyzována, aby se zhodnotily účinky demografických charakteristik. Analýzy těchto omezených údajů naznačují, že není nutná žádná úprava dávky pro věk (6-76 let), rasu/etnickou příslušnost, pohlaví nebo BMI (viz bod 4.2).
Zhoršená funkce ledvin a jater
U pacientů se zhoršenou funkcí ledvin nebo jater nejsou žádné farmakokinetické nebo farmakodynamické údaje (viz body 4.2 a 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost omalizumabu byla studována u opic makaka jávského, jelikož omalizumab se váže na jejich i lidský IgE s podobnou afinitou. U některých opic byly zjištěny protilátky proti omalizumabu po opakovaném subkutánním nebo intravenózním podání. Nicméně nebyla pozorována žádná toxicita jako například onemocnění zprostředkované imunokomplexy nebo cytotoxicita podmíněná komplementem. Nevyskytl se žádný důkaz anafylaktické odpovědi následkem degranulace žírných buněk u těchto opic.
Dlouhodobé podávání omalizumabu v dávkách až 250 mg/kg (alespoň 14krát nejvyšší doporučená klinická dávka v mg/kg podle doporučené dávkovači tabulky) nehumánním primátům (dospělým i dospívajícím jedincům) bylo dobře tolerováno s výjimkou snížení počtu krevních destiček, které souviselo s dávkou a bylo závislé na věku, s větší citlivostí u nedospělých zvířat. Hladina koncentrace v séru potřebná k dosažení poklesu trombocytů o 50 % oproti výchozí hodnotě u dospělých opic makaka jávského byla zhruba 4 až 20krát vyšší než očekávané maximální klinické sérové koncentrace. Navíc u makaka jávského bylo pozorováno akutní krvácení a zánět v místě aplikace injekce.
Formální studie kancerogenity nebyly s omalizumabem provedeny.
V reprodukčních studiích u makaka jávského subkutánně podané dávky až do 75 mg/kg týdně (alespoň 8krát nejvyšší doporučená klinická dávka v mg/kg po dobu delší než 4 týdny) nevyvolaly mateřskou toxicitu, embryotoxicitu nebo teratogenní účinky, když byly podávané během organogeneze, a neměly nežádoucí účinky na fetální nebo novorozenecký růst, pokud byly podávané během těhotenství, porodu nebo kojení.
Omalizumab se vylučuje do mateřského mléka makaka jávského. Hladiny omalizumabu v mléce byly 0,15 % koncentrace v séru matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Arginin-hydrochlorid Histidin-hydrochlorid Histidin Polysorbát 20 Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti 15 měsíců.
Doba použitelnosti zahrnuje možné odchylky teploty. Přípravek může být ponechán po celkovou dobu 4 hodiny při teplotě 25 °C. Pokud je to nutné, můžete vrátit přípravek do lednice pro pozdější použití, ale tento krok nesmí být proveden více než jednou.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Roztok o objemu 0,5 ml v předplněné injekční stříkačce (ze skla typu I) s vsazenou injekční jehlou (nerezová ocel), pístovou zátkou (typ I) a ochranným krytem jehly.
Balení obsahující 1 předplněnou injekční stříkačku a vícečetná balení obsahující 4 (4 balení po 1) nebo 10 (10 balení po 1) předplněných injekčních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před dokončením kompletace injekce se vyvarujte kontaktu s aktivačními svorkami zařízení, abyste zabránili předčasnému zakrytí jehly krytkou.
Použití injekce
1. Držte injekční stříkačku s jehlou směřující vzhůru, opatrně sejměte kryt z jehly na injekční stříkačce a vyhoďte ho. Nedotýkejte se odkryté jehly. Potom prstem zlehka poklepejte
na injekční stříkačku, dokud stoupá vzduchová bublina k vrcholu injekční stříkačky. Pomalu tlačte píst vzhůru tak, aby byly vzduchové bubliny vytlačeny ze stříkačky bez zbytečné ztráty roztoku.
2. Jemně uchopte kožní řasu v místě vpichu a zasuňte jehlu.
3. Za přidržení obruby prsty, pomalu stlačujte píst injekce, dokud to půjde. Jestliže dochází k úniku roztoku v místě vpichu, zasuňte jehlu do kůže hlouběji
4. Držte píst zcela stlačený a přitom opatrně a přímo vytáhněte jehlu z místa vpichu.
5. Pomalu pusťte píst, tím umožníte automatické zakrytí jehly.
Instrukce pro likvidaci
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/319/005
EU/1/05/319/006
EU/1/05/319/007
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25. října 2005
Datum posledního prodloužení registrace: 22. června 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Xolair 150 mg injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s roztokem objemu 1 ml obsahuje omalizumabum* 150 mg.
*Omalizumab je humanizovaná monoklonální protilátka vyrobená technologií rekombinantní DNA v linii savčích buněk vaječníků čínských křečků (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Alergické astma
Xolair je indikován k léčbě dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Léčbu Xolairem je možno použít pouze u pacientů s astmatem vyvolaným prokazatelně IgE (imunoglobulinem E) (viz bod 4.2).
Dospělí a dospívající (12 let a starší)
Xolair se doporučuje jako doplňková léčba ke zlepšení kontroly astmatu pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a kteří mají sníženou funkci plic (FEVj <80 %), stejně jako časté symptomy během dne nebo probouzení v noci, a kteří mají dokumentované těžké exacerbace astmatu navzdory vysokým denním dávkám inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
Děti (6 až <12 let)
Xolair se doporučuje jako přídatná léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a časté denní nebo noční příznaky buzení a kteří mají prokázané četné vážné exacerbace přesto, že užívají vysoké denní dávky inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů.
Chronická spontánní urtikarie (CSU)
Xolair je indikován jako přídatná terapie k léčbě chronické spontánní urtikarie u dospělých a dospívajících pacientů (ve věku 12 let a více) s nedostatečnou odpovědí na léčbu H1 antihistaminiky.
4.2 Dávkování a způsob podání
Léčba Xolairem by měla být zahájena lékařem zkušeným v diagnostice a léčbě těžkého perzistujícího astmatu nebo chronické spontánní urtikarie.
Alergické astma
Dávkování
Vhodná dávka a frekvence podávání Xolairu se určí podle výchozích hodnot IgE (IU/ml), které se stanoví před zahájením léčby, a dle tělesné hmotnosti (kg). Aby mohla být stanovena dávka, měla by být před podáním počáteční dávky u pacientů stanovena hladina IgE jakýmkoliv komerčním testem pro stanovení celkového IgE v séru. Na základě výsledků může být potřeba podávat 75 až 600 mg Xolairu v 1 až 4 injekcích na každé podání.
U pacientů s IgE nižším než 76 IU/ml je méně pravděpodobné, že se projeví přínos přípravku (viz bod
5.1). Ošetřující lékař se musí ujistit, že dospělí a dospívající pacienti s IgE nižším než 76 IU/ml a děti (6 až <12 let) s IgE nižším než 200 IU/ml mají před začátkem léčby prokázanou in vitro reaktivitu (RAST) na celoroční vzdušný alergen.
Viz Tabulka 1 schéma konverze a Tabulka 2 a 3 schéma stanovení dávky u dospělých, dospívajících a dětí (ve věku 6 až <12 let).
Pacientům, jejichž výchozí hodnoty hladin IgE nebo tělesná hmotnost v kilogramech jsou mimo limity dávkovací tabulky, by Xolair neměl být podáván.
Maximální doporučená dávka je 600 mg omalizumabu každé dva týdny.
Tabulka 1: Konverze dávky na počet injekčních stříkaček, počet injekcí a celkový objem injekcí při každé aplikaci
|
Dávka (mg) |
Počet injekčních stříkaček 75 mg 150 mg |
Počet injekcí |
Celkový objem injekcí (ml) | |
|
75 |
1 |
0 |
1 |
0,5 |
|
150 |
0 |
1 |
1 |
1,0 |
|
225 |
1 |
1 |
2 |
1,5 |
|
300 |
0 |
2 |
2 |
2,0 |
|
375 |
1 |
2 |
3 |
2,5 |
|
450 |
0 |
3 |
3 |
3,0 |
|
525 |
1 |
3 |
4 |
3,5 |
|
600 |
0 |
4 |
4 |
4,0 |
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE (IU/ml) |
>20- 25 |
>25- 30 |
>30- 40 |
>40- 50 |
>50- 60 |
>60- 70 |
>70- 80 |
>80- 90 |
>90- 125 |
>125- 150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
PODÁVÁNÍ KAŽDÉ 2 TÝDNY | |||||||||
|
>900- 1000 |
VIZ TABULKA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Tělesná hmotnost (kg) | ||||||||||
|
Výchozí hodnota IgE |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(IU/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
PODÁVÁNÍ KAŽDÉ 4 TÝDNY | |||||||||
|
>100-200 |
VIZ TABULKA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- 1200 |
300 |
300 |
450 |
525 |
600 |
NEPODÁVAT - pro doporučení dávkování není dostatek údajů | ||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Délka léčby, její sledování a úprava dávky
Xolair je určen pro dlouhodobou léčbu. Klinické studie prokázaly, že Xolair dosahuje účinnosti po minimálně 12-16 týdnech léčby. Po 16 týdnech od zahájení léčby Xolairem by měl lékař před podáváním dalších injekcí zhodnotit účinnost léčby. Rozhodnutí o pokračování léčby Xolairem následně po 16týdenním intervalu, nebo příležitostně později, by mělo být založeno na tom, zdali je patrné výrazné zlepšení z hlediska celkové kontroly astmatu (viz bod 5.1, Celkové hodnocení účinnosti léčby lékařem).
Ukončení léčby Xolairem má obecně za následek návrat ke zvýšeným hladinám volného IgE a s tím spojeným symptomům. Celkové hladiny IgE jsou zvýšené během léčby a zůstávají zvýšené až po dobu jednoho roku po skončení léčby. Proto opětovné stanovení hladin IgE během léčby Xolairem nemůže být použito jako vodítko pro stanovení dávky. Stanovení dávky po přerušení léčby trvající méně než jeden rok by mělo být založeno na výchozích hodnotách hladin sérového IgE získaných při stanovení zahajovací dávky. Pro stanovení dávky mohou být celkové hladiny IgE v séru znovu stanoveny, jestliže léčba Xolairem byla přerušena jeden rok nebo déle.
Dávky by měly být přizpůsobeny významným změnám tělesné hmotnosti (viz Tabulka 2 a 3).
Chronická spontánní urtikarie (CSU)
Dávkování
Doporučená dávka je 300 mg, podávaných subkutánní injekcí každé čtyři týdny.
Předepisujícím lékařům se doporučuje pravidelně přehodnotit potřebu pokračování léčby.
Zkušenosti z klinického hodnocení dlouhodobé léčby trvající déle než 6 měsíců v této indikaci jsou omezené.
Zvláštní skupiny pacientů
Starší pacienti (65 let a starší)
Pro užití Xolairu u pacientů starších více než 65 let jsou dostupné omezené údaje, ale nebylo zjištěno, že by u starších pacientů bylo nutné podávání jiné dávky než u mladších dospělých pacientů.
Pacienti s poškozením ledvin nebo jater
Nejsou k dispozici žádné studie hodnotící vliv zhoršené funkce ledvin nebo jater na farmakokinetiku omalizumabu. Protože clearance omalizumabu je při klinických dávkách ovlivněna hlavně retikuloendotelovým systémem (RES), je nepravděpodobné, že by byla ovlivněna poškozením ledvin nebo jater. Protože není doporučena žádná zvláštní úprava dávkování pro tyto pacienty, Xolair by měl být podáván s opatrností (viz bod 4.4).
Pediatrická populace
Bezpečnost a účinnost Xolairu v případě alergického astmatu u pediatrických pacientů mladších 6 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Bezpečnost a účinnost Xolairu v případě chronické spontánní urtikarie u pediatrických pacientů mladších 12 let nebyla dosud stanovena.
Způsob podání
Pouze pro subkutánní podání. Nepodávejte intravenózně nebo intramuskulárně.
Injekce se aplikují subkutánně do oblasti deltového svalu ramene. Alternativně se může injekce aplikovat do stehna, jestliže je z jakéhokoliv důvodu vyloučena aplikace do oblasti deltového svalu.
Zkušenosti s aplikací injekce Xolairu samotným pacientem jsou omezené. Z toho důvodu má léčbu podávat pouze zdravotnický personál (viz bod 6.6 a také bod Informace pro zdravotnické pracovníky v příbalové informaci).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Obecné
Xolair není určen k léčbě exacerbací akutního astmatu, akutního bronchospasmu nebo status asthmaticus.
Xolair nebyl zkoumán u pacientů se syndromem hyperimunoglobulinemie E nebo alergickou bronchopulmonální aspergilózou nebo pro prevenci anafylaktických reakcí včetně takových, které jsou vyvolané alergií na potraviny, atopickou dermatitidou nebo alergickou rinitidou. Xolair není indikován pro léčbu těchto stavů.
Léčba Xolairem nebyla zkoumána u pacientů s autoimunitním onemocněním, s onemocněním zprostředkovaným imunokomplexy, nebo s preexistující zhoršenou funkcí ledvin nebo jater (viz bod
4.2). Pokud je Xolair podáván těmto skupinám pacientů, měla by být zachovávána opatrnost.
Náhlé vysazení systémových nebo inhalačních kortikosteroidů po zahájení léčby Xolairem se nedoporučuje. Snižování dávek kortikosteroidů je nutno provádět pod přímým dohledem lékaře a je vhodné jej provádět postupně.
Poruchy imunitního systému Alergické reakce typu I
Lokální nebo systémové alergické reakce typu I včetně anafylaxe a anafylaktického šoku se mohou objevit po podání omalizumabu, přičemž tyto reakce se mohou objevit i po dlouhotrvající léčbě omalizumabem. Většina těchto reakcí se objevila během 2 hodin po první a následné injekci Xolairu, ale některé se objevily až po 2 hodinách a dokonce i po více než 24 hodinách po injekci. Proto by následně po podání Xolairu měly být vždy dostupné k okamžitému užití léčivé přípravky na léčbu anafylaktických reakcí. Pacienti by měli být informováni o možnosti takových reakcí, a pokud se objeví alergická reakce, měli by vyhledat okamžitě lékařskou pomoc. Výskyt anafylaxe nesouvisející s omalizumabem v anamnéze může být rizikový faktor pro anafylaxi následující po podání Xolairu.
U malého počtu pacientů v klinických studiích byly detekovány protilátky proti omalizumabu (viz bod 4.8). Klinický význam protilátek proti Xolairu není dobře prostudován.
Sérová nemoc
Sérová nemoc a reakce podobné sérové nemoci, které jsou opožděnými alergickými reakcemi typu III, byly pozorovány u pacientů léčených humanizovanými monoklonálními protilátkami včetně omalizumabu. Předpokládaný patofyziologický mechanismus zahrnuje tvorbu a ukládání imunokomplexů v důsledku vzniku protilátek proti omalizumabu. Reakce se obvykle objevila 1-5 dní po podání prvních nebo následujících injekcí, i po dlouhodobé léčbě. Příznaky poukazující na sérovou nemoc zahrnují artritidu/artralgie, vyrážku (kopřivku nebo jiné formy), horečku a lymfadenopatii. K léčbě nebo prevenci této nemoci mohou být vhodná antihistaminika a kortikosteroidy. Pacienti by měli být poučeni, aby hlásili jakékoli podezřelé příznaky.
Syndrom Churga-Straussové a hypereozinofilní syndrom
U pacientů s těžkým astmatem se může zřídka vyskytnout systémový hypereozinofilní syndrom nebo alergická eozinofilní granulomatózní vaskulitida (syndrom Churga-Straussové), které se obvykle léčí systémovými kortikoidy.
Ve vzácných případech se mohou u pacientů léčených antiastmatickými léčivými přípravky, včetně omalizumabu, projevit systémová eozinofilie a vaskulitida. Tyto případy jsou často spojeny se snižováním léčby perorálními kortikoidy.
U těchto pacientů by lékaři měli věnovat zvýšenou pozornost možnému rozvoji eozinofilie, vaskulitické vyrážky, zhoršení plicních symptomů, abnormalit paranazálních dutin, srdečních komplikací a/nebo neuropatie.
Ve všech závažných případech výše uvedených poruch imunitního systému by mělo být zváženo vysazení omalizumabu.
Parazitární infekce (helmintóza)
IgE může být spojený s imunologickou odpovědí na některé parazitární infekce. V placebem kontrolované studii s alergickými pacienty bylo u pacientů s chronicky vysokým rizikem helmintóz prokázáno mírné zvýšení podílu infekcí ve skupině s omalizumabem, ačkoliv průběh, závažnost a odpověď na léčbu infekce se nezměnily. Četnost výskytu infekce cizopasnými červy v celkovém klinickém programu, který nebyl navržen tak, aby takové infekce prokázal, byla méně než 1 z 1 000 pacientů. Nicméně měla by být zaručena opatrnost u pacientů s vysokým rizikem infekce cizopasnými červy, zejména pokud cestují do oblastí, kde se infekce cizopasnými červy vyskytují endemicky. Pokud pacienti neodpovídají na doporučenou antiparazitární léčbu, mělo by být zváženo přerušení léčby Xolairem.
Jedinci senzitivní na latex
Snímatelný kryt jehly této předplněné injekční stříkačky obsahuje derivát přírodního gumového latexu. Ve snímatelném krytu jehly nebyl dosud detekován žádný přírodní gumový latex. Nicméně použití přípravku Xolair injekční roztok v předplněné injekční stříkačce nebylo u jedinců senzitivních na latex studováno, a proto je zde potenciální riziko vzniku hypersenzitivních reakcí, které nemohou být úplně vyloučeny.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Xolair může nepřímo snížit účinnost léčivých přípravků používaných k léčbě helmintóz nebo jiných parazitárních infekcí, protože v imunologické odpovědi na některé helmintózy může být zapojen IgE (viz bod 4.4).
Enzymy cytochromu P450, pumpy zajišťující efflux a mechanismy vazby na proteiny se nepodílejí na clearance omalizumabu; takže je zde malá možnost lékových interakcí. Nebyly provedeny žádné studie interakce Xolairu s léčivými přípravky ani vakcínami. Není žádný farmakologický důvod očekávat, že by se běžně předepisované léčivé přípravky užívané v léčbě astmatu nebo CSU vzájemně ovlivňovaly s omalizumabem.
Alergické astma
V klinických studiích byl Xolair běžně užíván v kombinaci s inhalačními a perorálními kortikosteroidy, inhalačními krátkodobě a dlouhodobě působícími beta agonisty, modifikátory leukotrienů, theofyliny a perorálními antihistaminiky. Nevyskytly se žádné údaje, že bezpečnost Xolairu byla změněna těmito dalšími běžně užívanými antiastmatickými léčivými přípravky. Jsou dostupné omezené údaje o užívání Xolairu v kombinaci se specifickou imunoterapií (hyposenzitizační terapie). V klinické studii, kde byl Xolair podáván společně s imunoterapií, nebyl zjištěn žádný rozdíl v bezpečnosti a účinnosti Xolairu v kombinaci se specifickou imunoterapií v porovnání s podáváním samotného Xolairu.
Chronická spontánní urtikarie (CSU)
V klinických studiích týkajících se CSU byl Xolair podáván v kombinaci s antihistaminiky
(H1 antihistaminiky, H2 antihistaminiky) a antagonisty leukotrienových receptorů (LTRA). Nebylo prokázáno, že by bezpečnost omalizumabu byla pozměněna při podávání s těmito léčivými přípravky, vztaženo k jejich známému bezpečnostnímu profilu u alergického astmatu. Navíc farmakokinetická analýza populace neprokázala žádný relevantní účinek H2 antihistaminik a LTRA na farmakokinetiku omalizumabu (viz bod 5.2).
Pediatrická populace
Klinické studie týkající se CSU zahrnovaly některé pacienty ve věku 12 až 17 let, užívající Xolair v kombinaci s antihistaminiky (H1 antihistaminiky, H2 antihistaminiky) a LTRA. U dětí mladších 12 let nebyly provedeny žádné studie.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání omalizumabu těhotným ženám jsou omezené. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky (viz bod 5.3). Omalizumab přestupuje přes placentu, ale možnost poškození plodu není známa. U primátů byl omalizumab spojován se snížením na věku závislém počtu krevních destiček, s relativní větší citlivostí u nedospělých zvířat (viz bod
5.3). Xolair by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Není známo, zda se omalizumab vylučuje do lidského mateřského mléka. Dostupné údaje u samic ostatních primátů prokázaly vylučování omalizumabu do mléka (viz bod 5.3). Riziko pro kojené novorozence/děti nelze vyloučit. Omalizumab by neměl být podáván během kojení.
Fertilita
Pro omalizumab nejsou k dispozici žádné údaje týkající se lidské fertility. Ve specificky navržených neklinických studiích fertility u primátů (s výjimkou člověka), včetně studií páření, nebylo pozorováno poškození samčí ani samičí fertility následně po opakovaném podávání omalizumabu v dávkách až 75 mg/kg. Dále nebyly pozorovány žádné genotoxické účinky v samostatných neklinických studiích genotoxicity (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xolair nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Alergické astma
Souhrn bezpečnostního profilu
Během klinických studií u dospělých a dospívajících pacientů starších 12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy a reakce v místě aplikace injekce, včetně bolestivosti, zduření, zarudnutí a svědění. V klinických studiích u dětí od 6 do <12 let byly nejčastěji hlášenými nežádoucími účinky bolest hlavy, pyrexie a bolest v nadbřišku. Ve většině případů byly reakce mírné nebo středně závažné.
Tabulkový seznam nežádoucích účinků
Tabulka 4 zaznamenává nežádoucí účinky hlášené v klinických studiích celkové bezpečnosti u populace léčené Xolairem podle MedDRA orgánové klasifikace a četnosti výskytu. V každé skupině četností jsou nežádoucí reakce seřazeny podle klesající závažnosti. Kategorie četností jsou definovány jako: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000) a velmi vzácné (<1/10 000). Reakce hlášené z postmarketingového sledování jsou uvedeny s frekvencí není známo (z dostupných údajů nelze určit).
Tabulka 4: Nežádoucí účinky u alergického astmatu
|
Infekce a infestace | |
|
Méně časté |
Faryngitida |
|
Vzácné |
Parazitární infekce |
|
Poruchy krve a lymfatického systému | |
|
Není známo |
Idiopatická trombocytopenie, včetně těžkých případů |
|
Poruchy imunitního systému | |
|
Vzácné |
Anafylaktická reakce, jiné závažné alergické stavy, vývoj |
|
protilátek proti omalizumabu | |
|
Není známo |
Sérová nemoc, která může zahrnovat horečku a lymfadenopatii |
|
Poruchy nervového systému | |
|
Časté |
Bolest hlavy* |
|
Méně časté |
Synkopa, parestezie, somnolence, závrať |
|
Cévní poruchy | |
|
Méně časté |
Posturální hypotenze, zčervenání |
|
Respirační, hrudní a mediastinální poruchy | |
|
Méně časté |
Alergický bronchospasmus, kašel |
|
Vzácné |
Otok laryngu |
|
Není známo |
Alergická granulomatózní vaskulitida (tzv. syndrom Churga- |
|
Straussové) | |
|
Gastrointestinální poruchy | |
|
Časté |
Bolest v nadbřišku** |
|
Méně časté | |
|
Poruchy kůže a podkožní tkáně | |
|
Méně časté | |
|
Vzácné |
Angioedém |
|
Není známo |
Alopecie |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Vzácné |
Systémový lupus erythematodes (SLE) |
|
Není známo |
Artralgie, myalgie, otoky kloubů |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Pyrexie** |
|
Časté |
Reakce v místě aplikace jako zduření, zarudnutí, bolest, svědění |
|
Méně časté |
Onemocnění podobné chřipce, otoky paží, zvýšení hmotnosti, |
*: Velmi časté u dětí od 6 do <12 let **: U dětí od 6 do <12 let
Chronická spontánní urtikarie (CSU)
Souhrn bezpečnostního profilu
Bezpečnost a snášenlivost omalizumabu byly studovány podáváním dávek 75 mg, 150 mg a 300 mg každé čtyři týdny u 975 pacientů s CSU, z nichž 242 dostávalo placebo. Celkem 733 pacientů bylo léčeno omalizumabem až 12 týdnů a 490 pacientů až 24 týdnů. Z tohoto množství bylo 412 pacientů léčeno dávkou 300 mg až 12 týdnů a 333 pacientů bylo léčeno dávkou 300 mg až 24 týdnů.
Tabulkový seznam nežádoucích účinků
Samostatná tabulka (Tabulka 5) ukazuje nežádoucí účinky u CSU indikace, vyplývající z rozdílů v dávkování a v léčených populacích (s významně odlišnými rizikovými faktory, komorbiditami, komedikacemi a věkem [např. studie s astmatem zahrnovaly děti od 6-12 let věku]).
Tabulka 5 zaznamenává nežádoucí účinky (příhody vyskytující se u >1 % pacientů v jakékoli léčebné skupině a > 2 % častěji v jakékoli léčebné skupině s omalizumabem než s placebem (po lékařském zhodnocení)) hlášené při dávkách 300 mg ve třech poolovaných studiích fáze III. Uvedené nežádoucí účinky jsou rozděleny do dvou skupin: na účinky identifikované ve 12týdenním a 24týdenním léčebném období.
Nežádoucí účinky jsou uvedeny podle MedDRA orgánové klasifikace. V každé třídě orgánové klasifikace jsou nežádoucí účinky seřazeny podle četnosti, s nejčastějšími reakcemi uvedenými na prvním místě. Odpovídající kategorie četnosti pro každý nežádoucí účinek je stanovena na základě následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 5: Nežádoucí účinky z poolované bezpečnostní databáze s CSU (den 1 až týden 24) při dávce 300 mg omalizumabu
|
Týden 12 |
Studie s omalizumabem č. 1, 2 a 3, poolované |
Kategorie četnosti | |
|
Placebo N=242 |
300 mg N=412 | ||
|
Infekce a infestace Sinusitida 5 (2,1 %) Poruchy nervového systému Bolest hlavy 7 (2,9 %) Poruchy svalové a kosterní soustavy a pojivové tkáně Artralgie 1 (0,4 %) Celkové poruchy a reakce v místě aplikace Reakce v místě podání injekce* 2 (0,8 %) |
20 (4,9 %) 25 (6,1 %) 12 (2,9 %) 11 (2,7 %) |
časté časté časté časté | |
|
Studie s omalizumabem č. 1 a 3, poolované |
Kategorie četnosti | ||
|
Týden 24 |
Placebo N=163 |
300 mg N=333 | |
|
Infekce a infestace Infekce horních cest dýchacích |
5 (3,1 %) |
19 (5,7 %) |
časté |
* Přestože se neprokázal 2 % rozdíl oproti placebu, reakce v místě podání injekce byly zařazeny, protože u všech případů byla stanovena příčinná souvislost se zkoumanou léčbou.
Popis vybraných nežádoucích účinků relevantních _pro indikace alergické astma a CSU Z klinických studií s CSU nebyla získána relevantní data, která by vyžadovala úpravu níže uvedených částí textu.
Poruchy imunitního systému Další informace viz bod 4.4.
Anafylaxe
Anafylaktické reakce byly v klinických studiích vzácné. Nicméně v datech po uvedení přípravku na trh se následně po kumulativním vyhledávání v bezpečnostní databázi našlo celkem 898 případů anafylaxe. Na základě očekávané expozice 566 923 pacientoroků to mělo za následek míru hlášení přibližně 0,20 %.
Arteriální tromboembolické příhody (ATE)
V kontrolovaných klinických studiích a během interim analýz observační studie byla pozorována numerická nerovnováha v počtu ATE. Definice složeného endpointu ATE zahrnovala cévní mozkovou příhodu, transitorní ischemickou ataku, infarkt myokardu, nestabilní anginu pectoris a kardiovaskulární úmrtí (včetně úmrtí z neznámé příčiny). V konečné analýze observační studie byl poměr ATE na 1 000 pacientoroků 7,52 (115/15 286 pacientoroků) pro pacienty léčené Xolairem a 5,12 (51/9 963 pacientoroků) pro kontrolní skupinu. V multivariační analýze kontrolující základní dostupné kardiovaskulární rizikové faktory byl poměr rizik 1,32 (95 % interval spolehlivosti 0,91-1,91). V samostatné analýze poolovaných klinických studií, která zahrnovala všechny randomizované, dvojitě zaslepené, placebem kontrolované klinické studie trvající 8 nebo více týdnů, byl poměr ATE na 1 000 pacientoroků 2,69 (5/1 856 pacientoroků) pro pacienty léčené Xolairem a 2,38 (4/1 680 pacientoroků) pro pacienty užívající placebo (relativní riziko 1,13, 95 % interval spolehlivosti 0,24-5,71).
Krevní destičky
V klinických studiích mělo několik pacientů počet krevních destiček pod spodní hranicí normálního laboratorního rozmezí. Žádná z těchto změn nebyla spojena s epizodami krvácení nebo se snížením hemoglobinu. U lidí (pacientů nad 6 let) nebylo hlášeno trvalé snížení počtu krevních destiček, které bylo pozorováno u ostatních primátů (viz bod 5.3), přestože z postmarketingového sledování byly ojediněle hlášeny případy idiopatické trombocytopenie, včetně těžkých případů.
Parazitární infekce
Placebem kontrolovaná studie u alergických pacientů s chronicky vysokým rizikem helmintóz ukázala mírně zvýšenou četnost výskytu infekcí u pacientů užívajících omalizumab ve srovnání s kontrolní skupinou. Toto zvýšení nebylo statisticky signifikantní. Průběh, závažnost a odpověď na léčbu infekcí se nezměnily (viz bod 4.4).
Systémový lupus erythematodes
U pacientů se středně těžkým až těžkým astmatem a chronickou spontánní urtikarií (CSU) byly hlášeny případy systémového lupus erythematodes (SLE) z klinických studií a postmarketingových hlášení. Patogeneze SLE není dobře známa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Maximální tolerovaná dávka Xolairu nebyla stanovena. Pacientům byly podány jednorázové intravenózní dávky až do 4 000 mg, aniž by se prokázala dávkou vymezená toxicita. Nejvyšší úhrnná dávka podaná pacientům byla 44 000 mg po dobu 20 týdnů a tato dávka neměla za následek žádné akutní nežádoucí účinky.
Pokud je podezření z předávkování, pacient by měl být sledován pro výskyt jakýchkoli neobvyklých známek nebo příznaků. Měla by být vyhledána lékařská pomoc a zahájena vhodná opatření.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva pro onemocnění spojená s obstrukcí dýchacích cest, jiná systémová léčiva pro onemocnění spojená s obstrukcí dýchacích cest, ATC kód: R03DX05
Omalizumab je z rekombinantní DNA odvozená humanizovaná monoklonální protilátka, která se selektivně váže na lidský imunoglobulin E (IgE). Protilátka je typu IgG1 kappa, která sestává ze skeletu lidské protilátky a z části odvozené z myší monoklonální protilátky, která váže IgE.
Alergické astma
Mechanismus účinku
Omalizumab se váže na IgE a předchází vazbě IgE k FcsRI (receptorům s vysokou afinitou k IgE) na bazofilech a mastocytech, čímž se redukuje množství volného IgE, který je využitelný ke spuštění alergické kaskády. Léčba Xolairem u atopických pacientů měla za následek nápadný pokles FcsRI receptorů na bazofilech.
Farmakodynamické účinky
Uvolnění histaminu in vitro z bazofilů izolovaných od jedinců léčených Xolairem bylo snížené přibližně o 90 % po stimulaci alergenem oproti hodnotám před léčbou.
V klinických studiích týkajících se alergických pacientů s astmatem byly sérové hladiny volného IgE sníženy v závislosti na dávce během 1 hodiny od první dávky a zůstaly snížené mezi dávkami. Po jednom roce od ukončení léčby Xolairem se hladiny IgE vrátily na úroveň před léčbou, aniž by byl pozorován rebound fenomén v hladinách IgE po vysazení léčivého přípravku.
Chronická spontánní urtikarie (CSU)
Mechanismus účinku
Omalizumab se váže na IgE a snižuje hladiny volného IgE. Následně klesá počet IgE receptorů (FcsRI) na buňkách. Není úplně jasné, jak tento mechanismus vede ke zlepšení příznaků CSU.
Farmakodynamické účinky
V klinických studiích týkajících se pacientů s CSU byla maximální suprese volného IgE pozorována 3 dny po první subkutánní dávce. Po opakovaných dávkách jednou za 4 týdny zůstaly hladiny volného IgE v séru před podáním dávky stabilní mezi 12 a 24 týdny léčby. Po skončení léčby Xolairem se zvýšily hladiny volného IgE na úroveň před léčbou po 16týdenním období dalšího sledování
bez léčby.
Klinická účinnost a bezpečnost u alergického astmatu
Dospělí a dospívající >12 let
Účinnost a bezpečnost Xolairu byla prokázána ve 28týdenní dvojitě zaslepené placebem kontrolované studii (studie 1) zahrnující 419 těžkých alergických astmatiků ve věku 12-79 let, kteří měli sníženou plicní funkci (FEVi 40-80 % predikční hodnoty) a nedostatečnou kontrolu astmatických symptomů, přestože dostávali vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta2-agonistů. Pacienti přicházející v úvahu prodělali četné exacerbace astmatu vyžadující systémovou léčbu kortikosteroidy nebo byli hospitalizováni nebo navštívili lékařskou pohotovostní službu kvůli těžké exacerbaci astmatu v posledním roce navzdory průběžné léčbě vysokými dávkami inhalačních kortikosteroidů a dlouhodobě působícími beta2-agonisty. Subkutánně podávaný Xolair nebo placebo byly podávány jako doplňková léčba k >1 000 mikrogramům beklometason-dipropionátu (nebo jeho ekvivalentu) plus beta2-agonisty s dlouhodobým účinkem. Perorální kortikosteroid, theofylin a modifikátory leukotrienů byly dovoleny jako udržovací terapie (22 %, 27 %, resp. 35 % pacientů).
Primárním endpointem bylo sledování výskytu exacerbací astmatu vyžadujících léčbu vysokými dávkami systémových kortikosteroidů. Omalizumab redukoval četnost výskytu exacerbací astmatu o 19 % (p = 0,153). Další hodnocení, která ukázala statistický význam (p<0,05) ve prospěch Xolairu, zahrnovala snížení závažných exacerbací (kde funkce plic u pacientů byla redukována až pod 60 % nejlepší osobní hodnoty a bylo zapotřebí systémových kortikosteroidů) a neodkladných návštěv lékaře souvisejících s astmatem (zahrnovaly hospitalizaci, pohotovostní lékařskou službu a neplánované návštěvy u lékaře) a zlepšení astmatických symptomů a funkce plic podle celkového zhodnocení účinnosti léčby lékařem, Asthma-related Quality of Life (AQL).
Podle analýzy podskupin klinicky významný prospěch léčby Xolairem měli spíše pacienti s celkovou hladinou IgE >76 IU/ml před léčbou. U těchto pacientů ve studii 1 Xolair redukoval četnost výskytu exacerbací astmatu o 40 % (p = 0,002). Kromě toho více pacientů mělo klinicky významnou odpověď v populaci s celkovým IgE >76 IU/ml v programu Xolair při těžkém astmatu. Tabulka 6 zahrnuje výsledky u celkové populace ve studii 1.
Tabulka 6: Výsledky studie 1
Populace studie 1 celkem Xolair Placebo
_N=209_N=210
Exacerbace astmatu
Četnost výskytu za 28týdenní 0,74 0,92
období
% redukce, p-hodnota pro poměr 19,4 %, p = 0,153
četnosti
Exacerbace závažného astmatu
Četnost výskytu za 28týdenní 0,24 0,48
období
% redukce, p-hodnota pro poměr 50,1 %, p = 0,002
četnosti
|
Návštěvy lékaře v naléhavých případech Četnost výskytu za 28týdenní období % redukce, p-hodnota pro poměr četnosti |
0,24 43,9 %, p = |
0,43 0,038 |
|
Celkové zhodnocení lékařem | ||
|
% reagujících* |
60,5 % |
42,8 % |
|
Hodnota p ** |
<0,001 | |
|
AQL zlepšení | ||
|
% pacientů se zlepšením >0,5 |
60,8 % |
47,8 % |
|
Hodnota p |
0,008 | |
|
* nápadné zlepšení nebo kompletní kontrola | ||
** p-hodnota pro celkové zhodnocení
Studie 2 hodnotila účinnost a bezpečnost Xolairu u skupiny 312 osob s těžkým alergickým astmatem, která odpovídala populaci ve studii 1. Léčba Xolairem v této otevřené studii vedla ke snížení četnosti výskytu klinicky významných exacerbací astmatu o 61 % oproti běžné léčbě astmatu podávané samotné.
Čtyři další velké placebem kontrolované podpůrné studie trvající 28 až 52 týdnů na 1 722 dospělých a dospívajících (studie 3, 4, 5, 6) hodnotily účinnost a bezpečnost Xolairu u pacientů s těžkým perzistujícím astmatem. Kontrola astmatu u většiny pacientů byla nedostatečná, ale užívali méně konkomitantní léčby astmatu než pacienti ve studiích 1 a 2. Studie 3-5 použily exacerbace jako primární endpoint, zatímco studie 6 primárně hodnotila snížení množství inhalačních kortikosteroidů.
Ve studiích 3, 4 a 5 se u pacientů léčených Xolairem snížila četnost výskytu exacerbací astmatu o 37,5 % (p = 0,027), 40,3 % (p<0,001), respektive o 57,6 % (p<0,001) oproti placebu.
Ve studii 6 bylo schopno významně více pacientů s těžkým alergickým astmatem léčených Xolairem redukovat svou dávku flutikasonu až na <500 mikrogramů/den bez zhoršení kontroly astmatu (60,3 %) oproti skupině s placebem (45,8 %, p<0,05).
Skóre kvality života bylo měřeno pomocí dotazníku Juniper Asthma-related Quality of Life Questionnaire. Ve všech šesti studiích došlo ke statisticky významnému zlepšení skóre kvality života oproti výchozím hodnotám u pacientů užívajících Xolair oproti skupině placebo nebo kontrolní skupině.
Celkové hodnocení účinnosti léčby lékařem:
Celkové zhodnocení lékařem bylo provedeno v pěti z výše uvedených studií jako obsáhlé celkové posouzení kontroly astmatu vykonané ošetřujícím lékařem. Lékař vzal do úvahy PEF (peak expiratory flow = vrcholová výdechová rychlost), denní a noční symptomy, užití záchranné medikace, spirometrii a exacerbace. Ve všech pěti studiích se u významně většího podílu pacientů léčených Xolairem vyhodnotilo, že dosáhli buď výrazného zlepšení, nebo kompletní kontroly astmatu oproti skupině pacientů, kteří dostávali placebo.
Děti 6 až <12 let
Primární doklad bezpečnosti a účinnosti Xolairu u skupiny pacientů od 6 do <12 let vycházel z randomizované, dvojitě slepé, placebem kontrolované, multicentrické studie (studie 7).
Studie 7 byla placebem kontrolovaná studie zahrnující podskupinu (N=235) pacientů, u kterých byla určena současná indikace, kteří byli léčeni vysokými dávkami inhalačních kortikosteroidů (ekvivalent >500 pg/den flutikasonu) a dlouhodobě působícími beta-agonisty.
Klinicky významná exacerbace byla definována jako zhoršení příznaků astmatu dle posouzení zkoušejícího lékaře, vyžadující zdvojnásobení výchozí dávky inhalačních kortikosteroidů po dobu minimálně 3 dnů a/nebo vyžadující záchrannou léčbu systémovými (perorálními nebo intravenózními) kortikosteroidy po dobu minimálně 3 dnů.
Ve specifické podskupině pacientů, kteří užívali vysoké dávky inhalačních kortikosteroidů, bylo u skupiny pacientů užívajících omalizumab zaznamenáno statisticky signifikantní snížení výskytu exacerbací ve srovnání se skupinou užívající placebo. Ve 24. týdnu představoval rozdíl mezi léčenými skupinami 34 % (poměr výskytu 0,662, p=0,047) relativní snížení u pacientů léčených omalizumabem oproti skupině pacientů léčených placebem. V druhé, dvojitě slepé, 28týdenní léčebné periodě představoval rozdíl mezi léčenými skupinami 63 % (poměr výskytu 0,37, p<0,001) relativní snížení u pacientů léčených omalizumabem ve srovnání s pacienty léčenými placebem.
Během 52týdenní, dvojitě slepé léčby (zahrnující 24týdenní fázi s fixními dávkami steroidů a 28týdenní fázi s úpravou dávkování steroidů) představoval rozdíl mezi léčebnými skupinami 50 % (poměr výskytu 0,504, p<0,001) relativní snížení počtu exacerbací u pacientů léčených omalizumabem.
Během 52týdenní léčby vykazovala skupina pacientů užívající omalizumab větší pokles použití záchranné medikace beta-agonistů než skupina pacientů užívajících placebo, přestože tento rozdíl mezi léčebnými skupinami nebyl statisticky signifikantní. Z celkového vyhodnocení účinnosti léčby na konci 52týdenní léčby u podskupiny těžkých pacientů užívajících vysoké dávky inhalačních kortikosteroidů a dlouhodobě působících beta-agonistů vyplývá, že poměr pacientů, kteří prokázali „vynikající“ účinnost, byl u skupiny užívající omalizumab vyšší a poměr pacientů s „průměrnou“ nebo „slabou“ účinností byl nižší ve srovnání s pacienty užívajícími placebo; rozdíl mezi léčebnými skupinami byl statisticky významný (p<0,001), zatímco mezi skupinou s omalizumabem a skupinou s placebem nebyly žádné rozdíly z hlediska subjektivních hodnot kvality života pacienta.
Klinická účinnost a bezpečnost u chronické spontánní urtikarie
Účinnost a bezpečnost Xolairu byla prokázána ve dvou randomizovaných, placebem kontrolovaných studiích fáze III (studie 1 a 2) u pacientů s CSU, kteří zůstali symptomatičtí i přes léčbu H1 antihistaminiky ve schválené dávce. Třetí studie (studie 3) primárně hodnotila bezpečnost Xolairu u pacientů s CSU, kteří zůstali symptomatičtí i přes léčbu H1 antihistaminiky při podání až čtyřnásobku schválené dávky, a při léčbě H2 antihistaminiky a/nebo LTRA. V těchto třech studiích bylo zařazeno 975 pacientů ve věku mezi 12 a 75 lety (průměrný věk 42,3 roků, 39 pacientů ve věku 12-17 let, 54 pacientů >65 let, 259 mužů a 716 žen). U všech pacientů bylo vyžadováno, aby měli nedostačující kontrolu příznaků, což bylo stanoveno pomocí týdenního skóre urtikariální aktivity (UAS7, rozmezí 0-42) > 16, a týdenního skóre závažnosti svědění (což je součást UAS7, rozmezí 0-21) > 8 po dobu 7 dnů před randomizací i přesto, že předtím užívali nějaké antihistaminikum po dobu alespoň 2 týdnů.
Ve studiích 1 a 2 měli pacienti průměrné týdenní skóre závažnosti svědění mezi 13,7 a 14,5 při počátečním vyšetření a průměrné UAS7 skóre 29,5 a 31,7, v uvedeném pořadí. Pacienti v bezpečnostní studii 3 měli průměrné týdenní skóre závažnosti svědění 13,8 a průměrné UAS7 skóre 31,2 při počátečním vyšetření. Napříč všemi třemi studiemi hlásili pacienti před zařazením do studie užívání v průměru 4 až 6 léků (včetně H1 antihistaminik) pro léčbu CSU příznaků. Pacienti dostávali Xolair v dávkách 75 mg, 150 mg nebo 300 mg nebo placebo subkutánní injekcí každé 4 týdny po dobu 24 týdnů a 12 týdnů ve studiích 1 a 2, v tomto pořadí, a 300 mg nebo placebo subkutánní injekcí každé 4 týdny po dobu 24 týdnů ve studii 3. Všechny studie měly 16týdenní období dalšího sledování bez léčby.
Primárním cílem byla změna v týdenním skóre závažnosti svědění od počátečního vyšetření do týdne 12. Omalizumab v dávce 300 mg snižoval týdenní skóre závažnosti svědění o 8,55 na 9,77 (p <0,0001) v porovnání se snížením o 3,63 na 5,14 u placeba (viz Tabulka 7). Statisticky významné výsledky byly dále pozorovány v podílech respondérů pro UAS7 < 6 (v týdnu 12), které byly vyšší u léčebných skupin užívajících 300 mg, v rozmezí od 52-66 % (p<0,0001) v porovnání s 11-19 % u skupiny s placebem, a kompletní odpověď (UAS7=0) byla dosažena u 34-44 % (p<0,0001) pacientů léčených dávkou 300 mg v porovnání s 5-9 % pacientů ve skupinách s placebem. Pacienti v léčebných skupinách s dávkou 300 mg dosáhli nejvyššího průměrného poměru dnů bez angioedému od týdne 4 do týdne 12, (91,0-96,1 %; p<0,001) v porovnání se skupinami s placebem (88,1-89,2 %). Průměrná změna od základní hodnoty do týdne 12 v celkovém DLQI u léčebných skupin s dávkou 300 mg byla vyšší (p<0,001) než u placeba, což ukazuje zlepšení v rozsahu od 9,7-10,3 bodů v porovnání s 5,1-6,1 body pro odpovídající skupiny s placebem.
|
Placebo |
Omalizumab 300 mg | |
|
Studie 1 | ||
|
N |
80 |
81 |
|
Průměr (SD) |
-3,63 (5,22) |
-9,40 (5,73) |
|
Rozdíl v LS průměrech vs. placebo1 |
- |
-5,80 |
|
95 % CI pro rozdíl |
- |
-7,49;-4,10 |
|
p-hodnota vs. placebo2 |
- |
<0,0001 |
|
Studie 2 | ||
|
N |
79 |
79 |
|
Průměr (SD) |
-5,14 (5,58) |
-9,77 (5,95) |
|
Rozdíl v LS průměrech vs. placebo1 |
- |
-4,81 |
|
95 % CI pro rozdíl |
- |
-6,49;-3,13 |
|
p-hodnota vs. placebo2 |
- |
<0,0001 |
|
Studie 3 | ||
|
N |
83 |
252 |
|
Průměr (SD) |
-4,01 (5,87) |
-8,55 (6,01) |
|
Rozdíl v LS průměrech vs. placebo1 |
- |
-4,52 |
|
95 % CI pro rozdíl |
- |
-5,97; -3,08 |
|
p-hodnota vs. placebo2 |
- |
<0,0001 |
*Modifikovaná hodnota „intent-to-treat“ (mITT) populace: zahrnovala všechny pacienty, kteří byli randomizováni a obdrželi alespoň jednu dávku zkoumaného léku.
BOCF (Baseline Observation Carried Forward) bylo použito k připočítání chybějících dat.
1 LS průměr byl odhadován s použitím modelu ANCOVA. Rozvrstvení (strata) byly základní hodnota týdenního skóre závažnosti svědění (<13 vs. >13) a hmotnost na počátku studie (<80 kg vs. >80 kg).
2 p-hodnota je odvozena od ANCOVA t-testu.
Obrázek 1 ukazuje průměrné týdenní skóre závažnosti svědění v průběhu času ve studii 1. Průměrná týdenní skóre závažnosti svědění se významně snížila s maximálním účinkem kolem týdne 12, který se udržel po 24týdenní léčebné období. Ve studii 3 byly výsledky podobné.
Ve všech třech studiích se postupně zvyšovalo průměrné týdenní skóre závažnosti svědění během16týdenního období dalšího sledování bez léčby, v souladu se znovu-objevením se příznaků onemocnění. Průměrné hodnoty na konci období dalšího sledování byly podobné skupině s placebem, ale nižší než příslušné průměrné základní hodnoty.

Podávaný omalizumab nebo placebo
BOCF=baseline observation carried forward; mITT=modified intention-to-treat population
Účinnost _po 24 týdnech léčby
Amplituda výsledků účinnosti pozorovaná ve 24. týdnu léčby byla srovnatelná s pozorováním ve 12. týdnu:
Ve studiích 1 a 3 bylo průměrné snížení týdenního skóre závažnosti svědění od počátečního vyšetření u dávky 300 mg 9,8 a 8,6, podíl pacientů s UAS7<6 byl 61,7 % a 55,6 %, a podíl pacientů s kompletní odpovědí (UAS7=0) byl 48,1 % a 42,5 %, v uvedeném pořadí, (všechna p<0,0001, při porovnání s placebem).
S opakovanou léčbou pacientů omalizumabem jsou k dispozici omezené klinické zkušenosti.
Data z klinických studií u dospívajících (12 až 17 let) zahrnovala celkem 39 pacientů, z nichž 11 dostávalo dávku 300 mg. Výsledky pro dávku 300 mg jsou k dispozici u 9 pacientů po týdnu 12 a u 6 pacientů po týdnu 24 a ukazují podobný rozsah odpovědi na léčbu omalizumabem v porovnání s dospělou populací. Průměrná změna od základní hodnoty v týdenním skóre závažnosti svědění ukázala snížení o 8,25 v týdnu 12 a o 8,95 v týdnu 24. Poměry respondérů byly: 33 % v týdnu 12 a 67 % v týdnu 24 pro UAS7=0, a 56 % v týdnu 12 a 67 % v týdnu 24 pro UAS7 <6.
5.2 Farmakokinetické vlastnosti
Farmakokinetika omalizumabu byla studována u dospělých a dospívajících pacientů s alergickým astmatem, stejně jako u dospělých a dospívajících pacientů s CSU. Obecné farmakokinetické charakteristiky omalizumabu jsou u těchto populací podobné.
Absorpce
Průměrná absolutní hodnota biologické dostupnosti omalizumabu je 62 % po subkutánním podání. Po jednorázové subkutánní dávce dospělým a dospívajícím pacientům s astmatem nebo CSU se omalizumab absorboval pomalu a dosáhl maximální koncentrace v séru průměrně po 6-8 dnech.
U pacientů s astmatem byly po opakovaných dávkách omalizumabu plochy pod křivkou sérové koncentrace v čase ode dne 0 až po den 14 v rovnovážném stavu až 6x větší oproti těm po první dávce.
Farmakokinetika omalizumabu je lineární v dávkách vyšších než 0,5 mg/kg. Po podání dávek 75 mg, 150 mg nebo 300 mg každé 4 týdny u pacientů s CSU se údolní (trough) sérové koncentrace omalizumabu zvýšily úměrně s velikostí dávek.
Podání Xolairu vyrobeného ve formě lyofilizátu nebo roztoku vedlo k podobným profilům sérové koncentrace omalizumabu v čase.
Distribuce
In vitro omalizumab tvoří s IgE komplexy omezené velikosti. Precipitující komplexy a komplexy s molekulovou hmotností větší než jeden milion daltonů nejsou pozorovány in vitro anebo in vivo.
Na základě populační farmakokinetiky byla distribuce omalizumabu u pacientů s alergickým astmatem a pacientů s CSU podobná. Zdánlivý distribuční objem u pacientů s astmatem byl po subkutánním podání 78 ± 32 ml/kg.
Eliminace
Clearance omalizumabu zahrnuje průběh clearance IgG stejně tak jako clearance prostřednictvím specifické vazby a tvorby komplexů s cílovým ligandem IgE. Vylučování IgG v játrech zahrnuje odbourávání v retikuloendotelovém systému a endotelových buňkách. Intaktní IgG je také vylučován žlučí. U pacientů s astmatem činil průměrný poločas eliminace omalizumabu ze séra 26 dní, s clearance v průměru 2,4 ± 1,1 ml/kg/den. Zdvojnásobení tělesné hmotnosti přibližně zdvojnásobuje danou clearance. U pacientů s CSU na základě populačních farmakokinetických simulací dosáhl poločas eliminace omalizumabu ze séra v rovnovážném stavu v průměru 24 dnů a skutečná clearance v rovnovážném stavu u pacienta o hmotnosti 80 kg byla 3,0 ml/kg/den.
Vlastnosti pacientské populace
Pacienti s astmatem: Populační farmakokinetika omalizumabu byla analyzována, aby se zhodnotily účinky demografických charakteristik. Analýzy těchto omezených údajů naznačují, že není nutná žádná úprava dávky u pacientů s astmatem pro věk (6-76 let), rasu/etnickou příslušnost, pohlaví nebo index tělesné hmotnosti (viz bod 4.2).
Pacienti s CSU: Účinky demografických charakteristik a ostatních faktorů na expozici omalizumabu byly hodnoceny na základě populační farmakokinetiky. Kromě toho byly kovarianční účinky hodnoceny analýzou vztahu mezi koncentracemi omalizumabu a klinickými odpověďmi. Tyto analýzy naznačují, že u pacientů s CSU z hlediska věku (12-75 let), rasy/etnika, pohlaví, tělesné hmotnosti, BMI, základní hodnoty IgE, autoprotilátek proti FcsRI nebo z hlediska současného užívání H2 antihistaminik nebo LTRA nejsou nutné žádné úpravy dávkování.
Zhoršená _ funkce ledvin a _jater
U alergických pacientů s astmatem nebo CSU se zhoršenou funkcí ledvin nebo jater nejsou žádné farmakokinetické nebo farmakodynamické údaje (viz body 4.2 a 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost omalizumabu byla studována u opic makaka jávského, jelikož omalizumab se váže na jejich i lidský IgE s podobnou afinitou. U některých opic byly zjištěny protilátky proti omalizumabu po opakovaném subkutánním nebo intravenózním podání. Nicméně nebyla pozorována žádná toxicita jako například onemocnění zprostředkované imunokomplexy nebo cytotoxicita podmíněná komplementem. Nevyskytl se žádný důkaz anafylaktické odpovědi následkem degranulace žírných buněk u těchto opic.
Dlouhodobé podávání omalizumabu v dávkách až 250 mg/kg (alespoň 14krát nejvyšší doporučená klinická dávka v mg/kg podle doporučené dávkovací tabulky) nehumánním primátům (dospělým i dospívajícím jedincům) bylo dobře tolerováno s výjimkou snížení počtu krevních destiček, které souviselo s dávkou a bylo závislé na věku, s větší citlivostí u nedospělých zvířat. Hladina koncentrace v séru potřebná k dosažení poklesu trombocytů o 50 % oproti výchozí hodnotě u dospělých opic makaka jávského byla zhruba 4 až 20krát vyšší než očekávané maximální klinické sérové koncentrace. Navíc u makaka jávského bylo pozorováno akutní krvácení a zánět v místě aplikace injekce.
Formální studie kancerogenity nebyly s omalizumabem provedeny.
V reprodukčních studiích u makaka jávského subkutánně podané dávky až do 75 mg/kg týdně (alespoň 8krát nejvyšší doporučená klinická dávka v mg/kg po dobu delší než 4 týdny) nevyvolaly mateřskou toxicitu, embryotoxicitu nebo teratogenní účinky, když byly podávané během organogeneze, a neměly nežádoucí účinky na fetální nebo novorozenecký růst, pokud byly podávané během těhotenství, porodu nebo kojení.
Omalizumab se vylučuje do mateřského mléka makaka jávského. Hladiny omalizumabu v mléce byly 0,15 % koncentrace v séru matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Arginin-hydrochlorid Histidin-hydrochlorid Histidin Polysorbát 20 Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti 15 měsíců.
Doba použitelnosti zahrnuje možné odchylky teploty. Přípravek může být ponechán po celkovou dobu 4 hodiny při teplotě 25 °C. Pokud je to nutné, můžete vrátit přípravek do lednice pro pozdější použití, ale tento krok nesmí být proveden více než jednou.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Roztok o objemu 1 ml v předplněné injekční stříkačce (ze skla typu I) s vsazenou injekční jehlou (nerezová ocel), pístovou zátkou (typ I) a ochranným krytem jehly.
Balení obsahující 1 předplněnou injekční stříkačku a vícečetná balení obsahující 4 (4 balení po 1) nebo 10 (10 balení po 1) předplněných injekčních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před dokončením kompletace injekce se vyvarujte kontaktu s aktivačními svorkami zařízení, abyste zabránili předčasnému zakrytí jehly krytkou.
Použití injekce
1. Držte injekční stříkačku s jehlou směřující vzhůru, opatrně sejměte kryt z jehly na injekční stříkačce a vyhoďte ho. Nedotýkejte se odkryté jehly. Potom prstem zlehka poklepejte
na injekční stříkačku, dokud stoupá vzduchová bublina k vrcholu injekční stříkačky. Pomalu tlačte píst vzhůru tak, aby byly vzduchové bubliny vytlačeny ze stříkačky bez zbytečné ztráty roztoku.
2. Jemně uchopte kožní řasu v místě vpichu a zasuňte jehlu.
3. Za přidržení obruby prsty, pomalu stlačujte píst injekce, dokud to půjde. Jestliže dochází k úniku roztoku v místě vpichu, zasuňte jehlu do kůže hlouběji
4. Držte píst zcela stlačený a přitom opatrně a přímo vytáhněte jehlu z místa vpichu.
5. Pomalu pusťte píst, tím umožníte automatické zakrytí jehly.
Instrukce pro likvidaci
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/05/319/008
EU/1/05/319/009
EU/1/05/319/010
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25. října 2005
Datum posledního prodloužení registrace: 22. června 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema.europa.eu
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Novartis Pharma S.A.S.
Centre de Biotechnologie 8, rue de l’Industrie F-68330 Huningue Francie
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU SKLÁDACÍ KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 75 mg prášek a rozpouštědlo pro injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje omalizumabum 75 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Prášek: sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 20. Rozpouštědlo: voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 x 75 mg injekční lahvička 1 x 2 ml ampulka s rozpouštědlem
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Použijte okamžitě po rekonstituci (při teplotě 2 °C - 8 °C může být uchováváno až po dobu 8 hodin nebo při teplotě 25 °C po dobu 2 hodin).
Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/319/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 75 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Xolair 75 mg prášek a rozpouštědlo pro injekční roztok
omalizumabum
Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
75 mg
6. JINÉ
Uchovávejte v chladničce (2 °C - 8 °C).
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Rozpouštědlo pro Xolair Voda na injekci
2. ZPŮSOB PODÁNÍ
Použijte 0,9 ml a zbytek zlikvidujte.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2 ml
6. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
SKLÁDACÍ KRABIČKA OBSAHUJÍCÍ 1 INJEKČNÍ LAHVIČKU A 1 AMPULKU (VČETNĚ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje omalizumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Prášek: sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 20. Rozpouštědlo: voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 x 150 mg injekční lahvička 1 x 2 ml ampulka s rozpouštědlem
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Použijte okamžitě po rekonstituci (při teplotě 2 °C - 8 °C může být uchováváno až po dobu 8 hodin nebo při teplotě 25 °C po dobu 2 hodin).
Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/319/002
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 150 mg
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
SKLÁDACÍ KRABIČKA PRO VNITŘNÍ BALENÍ (BEZ BLUE BOXU) U VÍCEČETNÉHO BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje omalizumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Prášek: sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 20. Rozpouštědlo: voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 x 150 mg injekční lahvička 1 x 2 ml ampulka s rozpouštědlem
1 injekční lahvička a 1 ampulka. Součást vícečetného balení. Samostatně neprodejné.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Použijte okamžitě po rekonstituci (při teplotě 2 °C - 8 °C může být uchováváno až po dobu 8 hodin nebo při teplotě 25 °C po dobu 2 hodin).
Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/319/003 Vícečetné balení obsahující 4 balení
EU/1/05/319/004 Vícečetné balení obsahující 10 balení
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 150 mg
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
OZNAČENÍ VÍCEČETNÉHO BALENÍ ZABALENÁ VE FÓLII (VČETNĚ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje omalizumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Prášek: sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 20. Rozpouštědlo: voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
Vícečetné balení: 4 balení, z nichž jedno balení obsahuje 1 injekční lahvičku (150 mg) a 1 ampulku (2 ml).
Vícečetné balení: 10 balení, z nichž jedno balení obsahuje 1 injekční lahvičku (150 mg) a 1 ampulku (2 ml).
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Použijte okamžitě po rekonstituci (při teplotě 2 °C - 8 °C může být uchováváno až po dobu 8 hodin nebo při teplotě 25 °C po dobu 2 hodin).
Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/319/003 Vícečetné balení obsahující 4 balení
EU/1/05/319/004 Vícečetné balení obsahující 10 balení
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok
omalizumabum
Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
150 mg
6. JINÉ
Uchovávejte v chladničce (2 °C - 8 °C).
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Rozpouštědlo pro Xolair Voda na injekci
2. ZPŮSOB PODÁNÍ
Použijte 1,4 ml a zbytek zlikvidujte.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2 ml
6. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU SKLÁDACÍ KRABIČKA PRO JEDNOTLIVÉ BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 75 mg injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka s 0,5 ml roztoku obsahuje omalizumabum 75 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Také obsahuje: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 x 0,5 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Použitou injekční stříkačku ihned odhoďte do k tomuto účelu vyhrazeného kontejneru.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/319/005 75 mg injekční roztok
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 75 mg
SKLÁDACÍ KRABIČKA PRO VÍCEČETNÉ BALENÍ (VČETNĚ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 75 mg injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka s 0,5 ml roztoku obsahuje omalizumabum 75 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Také obsahuje: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Vícečetné balení: 4 balení, každé obsahuje 1 x 0,5 ml. Vícečetné balení: 10 balení, každé obsahuje 1 x 0,5 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/05/319/006 |
75 mg injekční roztok (4) |
|
EU/1/05/319/007 |
75 mg injekční roztok (10) |
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 75 mg
SKLÁDACÍ KRABIČKA PRO VÍCEČETNÉ BALENÍ (BEZ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 75 mg injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka s 0,5 ml roztoku obsahuje omalizumabum 75 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Také obsahuje: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 x 0,5 ml. Součást vícečetného balení.. Samostatně neprodejné.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/05/319/006 |
75 mg injekční roztok (4) |
|
EU/1/05/319/007 |
75 mg injekční roztok (10) |
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 75 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 75 mg injekční roztok omalizumabum Subkutánní podání.
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Xolair 75 mg injekční roztok
omalizumabum
s.c. podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,5 ml
6. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU SKLÁDACÍ KRABIČKA PRO JEDNOTLIVÉ BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka s 1 ml roztoku obsahuje omalizumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Také obsahuje: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 x 1 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Použitou injekční stříkačku ihned odhoďte do k tomuto účelu vyhrazeného kontejneru.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/05/319/008 150 mg injekční roztok
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 150 mg
SKLÁDACÍ KRABIČKA PRO VÍCEČETNÉ BALENÍ (VČETNĚ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka s 1 ml roztoku obsahuje omalizumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Také obsahuje: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Vícečetné balení: 4 balení, každé obsahuje 1 x 1 ml. Vícečetné balení: 10 balení, každé obsahuje 1 x 1 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/05/319/009 |
150 mg injekční roztok (4) |
|
EU/1/05/319/010 |
150 mg injekční roztok (10) |
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 150 mg
SKLÁDACÍ KRABIČKA PRO VÍCEČETNÉ BALENÍ (BEZ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg injekční roztok omalizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka s 1 ml roztoku obsahuje omalizumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Také obsahuje: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20, vodu na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 x 1 ml. Součást vícečetného balení. Samostatně neprodejné.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Užívá se pouze tak, jak stanoví lékař.
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/05/319/009 |
150 mg injekční roztok (4) |
|
EU/1/05/319/010 |
150 mg injekční roztok (10) |
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Xolair 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xolair 150 mg injekční roztok omalizumabum Subkutánní podání.
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Xolair 150 mg injekční roztok
omalizumabum
s.c. podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Xolair 75 mg prášek a rozpouštědlo pro injekční roztok
omalizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Xolair a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán
3. Jak se Xolair podává
4. Možné nežádoucí účinky
5. Jak Xolair uchovávat
6. Obsah balení a další informace
1. Co je Xolair a k čemu se používá
Léčivou látkou v Xolairu je omalizumab. Omalizumab je člověkem vyrobená bílkovina, která se podobá přírodním bílkovinám produkovaným tělem; patří do skupiny léčivých přípravků zvaných monoklonální protilátky. Užívá se k prevenci zhoršení astmatu tím, že kontroluje astmatické příznaky u dospělých a dospívajících (12 let a starší) a dětí (6 až 11 let), kteří již užívají lék proti astmatu, ale u kterých příznaky astmatu nejsou dostatečně kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů.
Xolair způsobuje zablokování látky, která se nazývá imunoglobulin E (IgE) a která se v organismu vytváří. IgE hraje klíčovou roli při vyvolání alergického astmatu.
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán Xolair Vám nesmí být podán
- jestliže jste alergický(á) na omalizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže si myslíte, že můžete být alergický(á) na kteroukoli složku přípravku, řekněte to svému lékaři, protože Xolair by Vám neměl být podán.
Upozornění a opatření
Xolair obsahuje bílkovinu a bílkoviny mohou u některých lidí způsobit závažné alergické reakce. Projevy zahrnují vyrážku, obtíže při dýchání, otok nebo pocit na omdlení. Jestliže se u Vás vyskytnou po užití Xolairu jakékoli alergické reakce, kontaktujte lékaře ihned, jak budete moci.
U pacientů léčených Xolairem byl pozorován určitý druh alergické reakce zvaný sérová nemoc. Projevy sérové nemoci mohou být jeden nebo více z následujících příznaků: bolest kloubu s otokem nebo bez něj nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, svalová bolest. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
U pacientů léčených Xolairem byly pozorovány syndrom Churga-Straussové a hypereozinofilní syndrom. Příznaky mohou zahrnovat jeden nebo více z následujících: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (výrazná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (nosní neprůchodnost), srdeční problémy, bolest, pocit necitlivosti, brnění v rukou a nohou. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
Poraďte se se svým lékařem, než Vám bude podán Xolair:
- jestliže máte problémy s ledvinami nebo játry.
- jestliže máte onemocnění, při kterém Váš imunitní systém napadá části Vašeho těla (autoimunitní onemocnění).
- jestliže žijete v oblasti, kde je častý výskyt parazitárních infekcí, nebo cestujete do takového místa, jelikož Xolair může oslabit Vaši odolnost vůči těmto infekcím.
Xolair neléčí příznaky akutního astmatu, jako je náhlý astmatický záchvat. Proto by Xolair neměl být užíván k léčbě takových příznaků.
Xolair není určen k prevenci nebo léčbě jiných typů alergických stavů, jako jsou náhlé alergické reakce, syndrom hyperimunoglobulinemie E (dědičné imunitní onemocnění), aspergilóza (onemocnění plic vyvolané houbou), alergické jídlo, ekzém nebo senná rýma, protože nebyl u těchto stavů studován.
Děti (mladší 6 let)
Xolair se nemá podávat dětem, které j sou mladší než 6 let. U této věkové skupiny nej sou k dispozici dostatečná data.
Další léčivé přípravky a Xolair
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Toto je obvzlášť důležité, jestliže užíváte:
- léky používané k léčbě parazitární infekce, protože Xolair může oslabit účinek Vaší léčby.
- inhalační kortikosteroidy a jiné léky používané k léčbě astmatu.
Těhotenství a kojení
Xolair by Vám neměl být podáván, jestliže jste těhotná, ledaže by to Váš lékař považoval za nezbytné.
Pokud plánujete otěhotnět, informujte o tom lékaře, než zahájíte léčbu Xolairem. Váš Lékař s Vámi prodiskutuje přínos i potenciální rizika, která mohou nastat při podání tohoto léku během těhotenství.
Pokud jste při léčbě Xolairem otěhotněla, okamžitě to oznamte lékaři.
Xolair by Vám neměl být podáván, jestliže kojíte.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že Xolair bude ovlivňovat Vaši schopnost řídit nebo obsluhovat stroje.
Jak se Xolair podává
3.
Pokyny k užívání Xolairu jsou uvedeny v bodě “Informace pro zdravotnické pracovníky”.
Váš lékař rozhodne, jakou dávku Xolairu potřebujete a jak často Vám bude podána. To záleží na Vaší tělesné hmotnosti a na výsledcích krevních testů stanovujících množství IgE v krvi ještě před tím, než zahájíte léčbu Xolairem.
Xolair je podán lékařem nebo zdravotní sestrou jako injekce rovnou pod kůži (podkožně).
Pečlivě se držte instrukcí svého lékaře nebo zdravotní sestry.
Jaká dávka Vám bude podána
Bude Vám podána 1-4 injekce najednou, každé dva nebo čtyři týdny.
Během léčby Xolairem pokračujte v užívání současných léků na astma. Nepřestávejte užívat žádné léky na astma, aniž byste informoval(a) svého lékaře.
Po zahájení léčby Xolairem nemusíte zpozorovat okamžité zlepšení astmatu. Obvykle to trvá mezi 12 a 16 týdny, než pocítíte plný účinek.
Použití u dětí a dospívajících
Xolair může být podán dětem a dospívajícím ve věku 6 let nebo starším, které již užívají léky na astma, ale u kterých nejsou astmatické příznaky dobře kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů. Váš lékař rozhodne, jakou dávku Xolairu Vaše dítě potřebuje a jak často bude podávána. Bude to záviset na váze Vašeho dítěte a výsledcích krevního vyšetření, provedeného před zahájením léčby kvůli změření množství IgE v jeho/její krvi.
Jestliže jste zmeškal(a) podání dávky přípravku Xolair
Kontaktujte svého lékaře nebo nemocnici ihned, jakmile je to možné, abyste znovu naplánoval(a) návštěvu lékaře.
Jestliže přerušíte léčbu Xolairem
Nepřerušujte léčbu přípravkem Xolair, pokud Vám to nedoporučí Váš lékař. Přerušení nebo ukončení léčby Xolairem může způsobit návrat astmatických symptomů.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Možné nežádoucí účinky
4.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého. Nežádoucí účinky způsobené Xolairem jsou obvykle mírné až středně závažné,
ale příležitostně mohou být závažné.
Závažné nežádoucí účinky jsou:
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• Náhlé těžké alergické reakce: pokud zpozorujete náhlé příznaky alergické reakce nebo kombinace příznaků jako například vyrážku, svědění nebo kopřivku na kůži, otok obličeje, rtů, jazyka, hrtanu, průdušnice nebo jiných částí těla, rychlý srdeční tep, závratě nebo mírné točení hlavy, zkrácení dechu, sípání nebo dýchací potíže, anebo jakékoliv další jiné příznaky, řekněte to okamžitě svému lékaři nebo zdravotní sestře. Pokud máte v anamnéze závažné alergické reakce (anafylaxi) nesouvisející s podáním Xolairu, můžete mít větší riziko vývoje závažné alergické reakce po podání Xolairu.
• Systémový lupus erythematodes (SLE). Příznaky mohou zahrnovat bolest svalů, bolest kloubů a otok, a vyrážku. Mohou se u Vás objevit také další příznaky jako je horečka, úbytek hmotnosti a únava.
Není známo (četnost z dostupných údajů nelze určit)
• Výskyt jednoho nebo více z následujících příznaků: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (významná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (neprůchodnost nosu), srdeční problémy, bolest, znecitlivění, brnění rukou a nohou (příznaky tzv. syndromu „Churga-Straussové nebo hypereozinofilního syndromu“).
• Nízký počet krevních destiček s příznaky jako např. krvácení nebo podlitiny, které vznikají velmi snadno oproti normálnímu stavu.
• Rozvoj jakýchkoli následujících příznaků, obzvláště pokud se vyskytnou v kombinaci: bolest kloubů s otokem nebo bez otoku nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, bolest svalů (známky sérové nemoci).
Jestliže se u Vás cokoli z toho vyskytne, řekněte to ihned svému lékaři nebo zdravotní sestře.
Další možné nežádoucí účinky jsou:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 lidí)
• horečka (u dětí)
Časté nežádoucí účinky (mohou postihovat až 1 z 10 lidí)
• reakce v místě injekce včetně bolesti, zduření, svědění a zčervenání
• bolest v nadbřišku (u dětí)
• bolest hlavy (velmi časté u dětí)
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 lidí)
• pocit závratě, ospalosti nebo únavy
• brnění nebo znecitlivění rukou nebo nohou
• mdloby, nízký krevní tlak při sezení nebo ve stoji (posturální hypotenze), zčervenání
• bolesti v krku, kašel, akutní potíže s dechem
• pocit na zvracení (nausea), průjem, zažívací potíže
• svědění, kopřivka, vyrážka, zvýšená citlivost kůže na slunce
• zvýšení hmotnosti
• symptomy podobné chřipce
• oteklé paže
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• parazitární infekce
Není známo (četnost z dostupných údajů nelze určit)
• bolesti kloubů a svalů, otoky kloubů
• vypadávání vlasů
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xolair uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
6. Obsah balení a další informace Co Xolair obsahuje
- Léčivou látkou je omalizumabum. Jedna injekční lahvička obsahuje omalizumabum 75 mg. Po rozpuštění obsahuje jedna injekční lahvička omalizumabum 125 mg/ml (75 mg v 0,6 ml).
- Pomocnými látkami jsou: sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 20.
Jak Xolair vypadá a co obsahuje toto balení
Xolair 75 mg prášek a rozpouštědlo pro injekční roztok se dodává jako bílý až téměř bílý prášek v malé skleněné injekční lahvičce spolu s ampulkou obsahující 2 ml vody na injekci. Prášek se musí rozpustit ve vodě dříve, než je aplikován lékařem nebo zdravotní sestrou.
Xolair je dostupný v baleních obsahujících jednu injekční lahvičku s práškem pro přípravu injekčního roztoku a jednu ampulku s 2 ml vody na injekci.
Xolair je také dostupný v injekčních lahvičkách se 150 mg omalizumabu.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Btarapna Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EkláSa Novartis (Hellas) A.E.B.E. Tqk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
116 |
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema.europa.eu
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Rozpuštění lyofdizovaného léčivého přípravku trvá 15-20 minut, ačkoliv v některých případech to může trvat déle. Zcela rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý a může v něm být několik malých bublinek nebo pěna okolo okraje injekční lahvičky. Vzhledem k viskozitě rekonstituovaného léčivého přípravku se musí věnovat péče tomu, aby se odebral celý obsah léčivého přípravku z injekční lahvičky za účelem získání 0,6 ml před tím, než je vytlačen vzduch nebo přebytečné množství roztoku ze stříkačky.
Pro přípravu Xolairu 75 mg v injekční lahvičce pro subkutánní podání se prosím řiďte následujícími instrukcemi:
1. Odeberte z ampulky 0,9 ml vody na injekci do injekční stříkačky opatřené jehlou o velikosti průměru 18
2. Za použití standardních aseptických postupů zasuňte jehlu do injekční lahvičky umístěné kolmo na rovný povrch, přidejte vodu na injekci do lahvičky obsahující lyofilizovaný prášek tak, aby voda na injekci směřovala přímo na prášek.
3. Držte lahvičku ve svislé poloze a intenzivně s ní kružte (netřepejte) po dobu přibližně 1 minuty, aby se prášek rovnoměrně namočil.
4. Aby se napomohlo rozpouštění po dokončení kroku 3, jemně kružte lahvičkou po dobu 5-10 sekund přibližně každých 5 minut, aby se rozpustil všechen zbývající prášek.
Pamatujte, že v některých případech to může trvat déle než 20 minut, aby se prášek zcela rozpustil. Jestliže jde o tento případ, opakujte krok 4, dokud jsou v roztoku viditelné částečky podobné gelu.
Když je léčivý přípravek zcela rozpuštěný, neměly by být v roztoku viditelné žádné částečky podobné gelu. Malé bublinky nebo pěna okolo okraje lahvičky jsou obvyklé. Rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý. Nepoužívejte jej, jestliže jsou přítomny pevné částečky.
5. Obraťte injekční lahvičku dnem vzhůru na dobu nejméně 15 sekund, aby roztok natekl až k zátce. Použijte novou 3ml stříkačku opatřenou jehlou o velikosti průměru 18, zasuňte jehlu do obrácené injekční lahvičky. Držte injekční lahvičku v obrácené poloze, jehlu postrčte až na samé dno roztoku v injekční lahvičce, když odebíráte roztok do stříkačky. Před vyjmutím jehly z injekční lahvičky táhněte píst zpátky až na konec těla stříkačky, aby se nasál celý obsah roztoku z obrácené lahvičky.
6. Pro subkutánní injekci nahraďte jehlu o velikosti 18 jehlou velikosti 25.
7. Vytlačte vzduch, velké bubliny a přebytečný roztok tak, aby se získala požadovaná dávka
0,6 ml. Nepatrná vrstva malých bublinek může zůstat v horní části roztoku v injekční stříkačce. Protože je roztok mírně viskózní, může aplikace subkutánně podaného roztoku trvat 5-10 sekund.
Z injekční lahvičky se získá 0,6 ml (75 mg) Xolairu.
8. Injekce se podávájí subkutánně do oblasti deltového svalu ramene nebo do stehna.
Příbalová informace: informace pro uživatele
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok
omalizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Xolair a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán
3. Jak se Xolair podává
4. Možné nežádoucí účinky
5. Jak Xolair uchovávat
6. Obsah balení a další informace
1. Co je Xolair a k čemu se používá
Xolair se používá k léčbě alergického astmatu a chronické spontánní kopřivky (CSU).
Léčivou látkou v Xolairu je omalizumab. Omalizumab je člověkem vyrobená bílkovina, která se podobá přírodním bílkovinám produkovaným tělem; patří do skupiny léčivých přípravků zvaných monoklonální protilátky. Xolair způsobuje zablokování látky, která se nazývá imunoglobulin E (IgE) a která se v organismu vytváří. IgE hraje klíčovou roli při vyvolání alergického astmatu nebo CSU.
Alergické astma
Tento léčivý přípravek se užívá k prevenci zhoršení astmatu tím, že kontroluje astmatické příznaky u dospělých a dospívajících (12 let a starší) a dětí (6 až 11 let), kteří již užívají lék proti astmatu, ale u kterých příznaky astmatu nejsou dostatečně kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů.
Chronická spontánní kopřivka (CSU)
Tento léčivý přípravek se užívá k léčbě chronické spontánní kopřivky u dospělých a dospívajících (ve věku 12 let a starších), kteří již užívají antihistaminika, ale jejichž příznaky CSU nejsou těmito léky dobře kontrolovány.
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán Xolair Vám nesmí být podán
- jestliže jste alergický(á) na omalizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže si myslíte, že můžete být alergický(á) na kteroukoli složku přípravku, řekněte to svému lékaři, protože Xolair by Vám neměl být podán.
Upozornění a opatření
Xolair obsahuje bílkovinu a bílkoviny mohou u některých lidí způsobit závažné alergické reakce. Projevy zahrnují vyrážku, obtíže při dýchání, otok nebo pocit na omdlení. Jestliže se u Vás vyskytnou po užití Xolairu jakékoli alergické reakce, kontaktujte lékaře ihned, jak budete moci.
U pacientů léčených Xolairem byl pozorován určitý druh alergické reakce zvaný sérová nemoc. Projevy sérové nemoci mohou být jeden nebo více z následujících příznaků: bolest kloubu s otokem nebo bez něj nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, svalová bolest. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
U alergických pacientů s astmatem léčených Xolairem byly pozorovány syndrom Churga-Straussové a hypereozinofilní syndrom. Příznaky mohou zahrnovat jeden nebo více z následujících: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (výrazná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (nosní neprůchodnost), srdeční problémy, bolest, pocit necitlivosti, brnění v rukou a nohou. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
Poraďte se se svým lékařem, než Vám bude podán Xolair:
- jestliže máte problémy s ledvinami nebo játry.
- jestliže máte onemocnění, při kterém Váš imunitní systém napadá části Vašeho těla (autoimunitní onemocnění).
- jestliže žijete v oblasti, kde je častý výskyt parazitárních infekcí, nebo cestujete do takového místa, jelikož Xolair může oslabit Vaši odolnost vůči těmto infekcím.
Xolair neléčí příznaky akutního astmatu, jako je náhlý astmatický záchvat. Proto by Xolair neměl být užíván k léčbě takových příznaků.
Xolair není určen k prevenci nebo léčbě jiných typů alergických stavů, jako jsou náhlé alergické reakce, syndrom hyperimunoglobulinemie E (dědičné imunitní onemocnění), aspergilóza (onemocnění plic vyvolané houbou), alergické jídlo, ekzém nebo senná rýma, protože nebyl u těchto stavů studován.
Děti a dospívající
Alergické astma
Xolair se nedoporučuje podávat dětem mladším 6 let.
Chronická spontánní kopřivka (CSU)
Nepodávejte Xolair dětem do 12 let věku. Jeho použití u dětí do 12 let nebylo studováno.
Další léčivé přípravky a Xolair
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Toto je obvzlášť důležité, jestliže užíváte:
- léky používané k léčbě parazitární infekce, protože Xolair může oslabit účinek Vaší léčby.
- inhalační kortikosteroidy a jiné léky používané k léčbě astmatu.
Těhotenství a kojení
Xolair by Vám neměl být podáván, jestliže jste těhotná, ledaže by to Váš lékař považoval za nezbytné.
Pokud plánujete otěhotnět, informujte o tom lékaře, než zahájíte léčbu Xolairem. Váš Lékař s Vámi prodiskutuje přínos i potenciální rizika, která mohou nastat při podání tohoto léku během těhotenství.
Pokud jste při léčbě Xolairem otěhotněla, okamžitě to oznamte lékaři.
Xolair by Vám neměl být podáván, jestliže kojíte.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že Xolair bude ovlivňovat Vaši schopnost řídit nebo obsluhovat stroje.
3. Jak se Xolair podává
Pokyny k užívání Xolairu jsou uvedeny v bodě “Informace pro zdravotnické pracovníky”.
Xolair je podán lékařem nebo zdravotní sestrou jako injekce rovnou pod kůži (podkožně).
Pečlivě se držte instrukcí svého lékaře nebo zdravotní sestry.
Jaká dávka Vám bude podána
Alergické astma
Váš lékař rozhodne, kolik Xolairu potřebujete a jak často Vám bude podáván. Závisí to na Vaší tělesné hmotnosti a výsledcích vyšetření krve provedeného před zahájením léčby ke změření množství IgE ve Vaší krvi.
Bude Vám podána 1-4 injekce najednou, každé dva nebo čtyři týdny.
Během léčby Xolairem pokračujte v užívání současných léků na astma. Nepřestávejte užívat žádné léky na astma, aniž byste informoval(a) svého lékaře.
Po zahájení léčby Xolairem nemusíte zpozorovat okamžité zlepšení astmatu. Obvykle to trvá mezi 12 a 16 týdny, než pocítíte plný účinek.
Chronická spontánní kopřivka (CSU)
Budou Vám podávány dvě 150 mg injekce najednou každé čtyři týdny.
Pokračujte v užívání Vašeho současného léku na CSU během léčby Xolairem. Nepřestávejte užívat jakýkoli lék bez předchozí porady s Vaším lékařem.
Použití u dětí a dospívajících
Alergické astma
Xolair může být podán dětem a dospívajícím ve věku 6 let nebo starším, které již užívají léky na astma, ale u kterých nejsou astmatické příznaky dobře kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů. Váš lékař rozhodne, jakou dávku Xolairu Vaše dítě potřebuje a jak často bude podávána. Bude to záviset na váze Vašeho dítěte a výsledcích krevního vyšetření, provedeného před zahájením léčby kvůli změření množství IgE v jeho/její krvi.
Chronická spontánní kopřivka (CSU)
Xolair může být podáván dospívajícím ve věku 12 let a starším, kteří již užívají antihistaminika, ale jejichž příznaky CSU nejsou těmito léky dobře kontrolovány.
Jestliže jste zmeškal(a) podání dávky přípravku Xolair
Kontaktujte svého lékaře nebo nemocnici ihned, jakmile je to možné, abyste znovu naplánoval(a) návštěvu lékaře.
Jestliže přerušíte léčbu Xolairem
Nepřerušujte léčbu přípravkem Xolair, pokud Vám to nedoporučí Váš lékař. Přerušení nebo ukončení léčby Xolairem může způsobit návrat příznaků astmatu nebo kopřivky.
Jste-li však v současné době léčen(a) pro CSU, Váš lékař může léčbu Xolairem občas přerušit, aby mohly být příznaky vyhodnoceny. Postupujte podle pokynů Vašeho lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého. Nežádoucí účinky způsobené Xolairem jsou obvykle mírné až středně závažné,
ale příležitostně mohou být závažné.
Závažné nežádoucí účinky jsou:
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• Náhlé těžké alergické reakce: pokud zpozorujete náhlé příznaky alergické reakce nebo kombinace příznaků jako například vyrážku, svědění nebo kopřivku na kůži, otok obličeje, rtů, jazyka, hrtanu, průdušnice nebo jiných částí těla, rychlý srdeční tep, závratě nebo mírné točení hlavy, zkrácení dechu, sípání nebo dýchací potíže, anebo jakékoliv další jiné příznaky, řekněte to okamžitě svému lékaři nebo zdravotní sestře. Pokud máte v anamnéze závažné alergické reakce (anafylaxi) nesouvisející s podáním Xolairu, můžete mít větší riziko vývoje závažné alergické reakce po podání Xolairu.
• Systémový lupus erythematodes (SLE). Příznaky mohou zahrnovat bolest svalů, bolest kloubů a otok, a vyrážku. Mohou se u Vás objevit také další příznaky jako je horečka, úbytek hmotnosti a únava.
Není známo (četnost z dostupných údajů nelze určit)
• Výskyt jednoho nebo více z následujících příznaků: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (významná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (neprůchodnost nosu), srdeční problémy, bolest, znecitlivění, brnění rukou a nohou (příznaky tzv. syndromu „Churga-Straussové nebo hypereozinofilního syndromu“).
• Nízký počet krevních destiček s příznaky jako např. krvácení nebo podlitiny, které vznikají velmi snadno oproti normálnímu stavu.
• Rozvoj jakýchkoli následujících příznaků, obzvláště pokud se vyskytnou v kombinaci: bolest kloubů s otokem nebo bez otoku nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, bolest svalů (známky sérové nemoci).
Jestliže se u Vás cokoli z toho vyskytne, řekněte to ihned svému lékaři nebo zdravotní sestře.
Další možné nežádoucí účinky jsou:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 lidí)
• horečka (u dětí)
Časté nežádoucí účinky (mohou postihovat až 1 z 10 lidí)
• reakce v místě injekce včetně bolesti, zduření, svědění a zčervenání
• bolest v nadbřišku (u dětí)
• bolest hlavy (velmi časté u dětí)
• infekce horních cest dýchacích, j ako j e zánět hltanu a nachlazení
• pocit tlaku nebo bolesti ve tvářích a na čele (zánět dutin, bolest hlavy při zánětu dutin)
• bolest v kloubech (artralgie)
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 lidí)
• pocit závratě, ospalosti nebo únavy
• brnění nebo znecitlivění rukou nebo nohou
• mdloby, nízký krevní tlak při sezení nebo ve stoji (posturální hypotenze), zčervenání
• bolesti v krku, kašel, akutní potíže s dechem
• pocit na zvracení (nausea), průjem, zažívací potíže
• svědění, kopřivka, vyrážka, zvýšená citlivost kůže na slunce
• zvýšení hmotnosti
• symptomy podobné chřipce
• oteklé paže
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• parazitární infekce
Není známo (četnost z dostupných údajů nelze určit)
• bolesti svalů a otoky kloubů
• vypadávání vlasů
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xolair uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
6. Obsah balení a další informace Co Xolair obsahuje
- Léčivou látkou je omalizumabum. Jedna injekční lahvička obsahuje omalizumabum 150 mg. Po rozpuštění obsahuje jedna injekční lahvička omalizumabum 125 mg/ml (150 mg v 1,2 ml).
- Pomocnými látkami jsou: sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 20.
Jak Xolair vypadá a co obsahuje toto balení
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok se dodává jako bílý až téměř bílý prášek v malé skleněné injekční lahvičce spolu s ampulkou obsahující 2 ml vody na injekci. Prášek se musí rozpustit ve vodě dříve, než je aplikován lékařem nebo zdravotní sestrou.
Xolair 150 mg prášek a rozpouštědlo pro injekční roztok je dostupný v baleních obsahujících jednu injekční lahvičku prášku pro přípravu injekčního roztoku a jednu ampulku s 2 ml vody na injekci a vícečetná balení obsahující čtyři nebo deset vnitřních balení, z nichž každé obsahuje jednu injekční lahvičku prášku pro přípravu injekčního roztoku a jednu ampulku s 2 ml vody na injekci. Na trhu nemusí být všechny velikosti balení.
Xolair je také dostupný v injekčních lahvičkách se 75 mg omalizumabu.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Bt^rapnn
Novartis Pharma Services Inc. Ten.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
EXlába
Novartis (Hellas) A.E.B.E. Tp^: +30 210 281 17 12
Espaňa
Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 375 4888
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kónpoq Novartis Pharma Services Inc. TpA,: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema.europa.eu
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Rozpuštění lyofdizovaného léčivého přípravku trvá 15-20 minut, ačkoliv v některých případech to může trvat déle. Zcela rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý a může v něm být několik malých bublinek nebo pěna okolo okraje injekční lahvičky. Vzhledem k viskozitě rekonstituovaného léčivého přípravku se musí věnovat péče tomu, aby se odebral celý obsah léčivého přípravku z injekční lahvičky za účelem získání 1,2 ml před tím, než je vytlačen vzduch nebo přebytečné množství roztoku ze stříkačky.
Pro přípravu Xolairu 150 mg v injekční lahvičce pro subkutánní podání se prosím řiďte následujícími instrukcemi:
1. Odeberte z ampulky 1,4 ml vody na injekci do injekční stříkačky opatřené jehlou o velikosti průměru 18
2. Za použití standardních aseptických postupů zasuňte jehlu do injekční lahvičky umístěné kolmo na rovný povrch, přidejte vodu na injekci do lahvičky obsahující lyofilizovaný prášek tak, aby voda na injekci směřovala přímo na prášek.
3. Držte lahvičku ve svislé poloze a intenzivně s ní kružte (netřepejte) po dobu přibližně 1 minuty, aby se prášek rovnoměrně namočil.
4. Aby se napomohlo rozpouštění po dokončení kroku 3, jemně kružte lahvičkou po dobu 5-10 sekund přibližně každých 5 minut, aby se rozpustil všechen zbývající prášek.
Pamatujte, že v některých případech to může trvat déle než 20 minut, aby se prášek zcela rozpustil. Jestliže jde o tento případ, opakujte krok 4, dokud jsou v roztoku viditelné částečky podobné gelu.
Když je léčivý přípravek zcela rozpuštěný, neměly by být v roztoku viditelné žádné částečky podobné gelu. Malé bublinky nebo pěna okolo okraje lahvičky jsou obvyklé. Rekonstituovaný léčivý přípravek se jeví jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý. Nepoužívejte jej, jestliže jsou přítomny pevné částečky.
5. Obraťte injekční lahvičku dnem vzhůru na dobu nejméně 15 sekund, aby roztok natekl až k zátce. Použijte novou 3ml stříkačku opatřenou jehlou o velikosti průměru 18, zasuňte jehlu do obrácené injekční lahvičky. Držte injekční lahvičku v obrácené poloze, jehlu postrčte až na samé dno roztoku v injekční lahvičce, když odebíráte roztok do stříkačky. Před vyjmutím jehly z injekční lahvičky táhněte píst zpátky až na konec těla stříkačky, aby se nasál celý obsah roztoku z obrácené lahvičky.
6. Pro subkutánní injekci nahraďte jehlu o velikosti 18 jehlou velikosti 25.
7. Vytlačte vzduch, velké bubliny a přebytečný roztok tak, aby se získala požadovaná dávka
1,2 ml. Nepatrná vrstva malých bublinek může zůstat v horní části roztoku v injekční stříkačce. Protože je roztok mírně viskózní, může aplikace subkutánně podaného roztoku trvat 5-10 sekund.
Z injekční lahvičky se získá 1,2 ml (150 mg) Xolairu. Pro dávku 75 mg odeberte do injekční stříkačky 0,6 ml a zbylý roztok zlikvidujte.
8. Injekce se podávájí subkutánně do oblasti deltového svalu ramene nebo do stehna.
Příbalová informace: informace pro uživatele Xolair 75 mg injekční roztok
omalizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Xolair a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán
3. Jak se Xolair podává
4. Možné nežádoucí účinky
5. Jak Xolair uchovávat
6. Obsah balení a další informace
1. Co je Xolair a k čemu se používá
Léčivou látkou v Xolairu je omalizumab. Omalizumab je člověkem vyrobená bílkovina, která se podobá přírodním bílkovinám produkovaným tělem; patří do skupiny léčivých přípravků zvaných monoklonální protilátky. Užívá se k prevenci zhoršení astmatu tím, že kontroluje astmatické příznaky u dospělých a dospívajících (12 let a starší) a dětí (6 až 11 let), kteří již užívají lék proti astmatu, ale u kterých příznaky astmatu nejsou dostatečně kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů.
Xolair způsobuje zablokování látky, která se nazývá imunoglobulin E (IgE) a která se v organismu vytváří. IgE hraje klíčovou roli při vyvolání alergického astmatu.
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán Xolair Vám nesmí být podán
- jestliže jste alergický(á) na omalizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže si myslíte, že můžete být alergický(á) na kteroukoli složku přípravku, řekněte to svému lékaři, protože Xolair by Vám neměl být podán.
Upozornění a opatření
Xolair obsahuje bílkovinu a bílkoviny mohou u některých lidí způsobit závažné alergické reakce. Projevy zahrnují vyrážku, obtíže při dýchání, otok nebo pocit na omdlení. Jestliže se u Vás vyskytnou po užití Xolairu jakékoli alergické reakce, kontaktujte lékaře ihned, jak budete moci.
U pacientů léčených Xolairem byl pozorován určitý druh alergické reakce zvaný sérová nemoc. Projevy sérové nemoci mohou být jeden nebo více z následujících příznaků: bolest kloubu s otokem nebo bez něj nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, svalová bolest. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
Při užívání přípravku Xolair dbejte zvláštní opatrnosti, pokud jste někdy měli alergickou reakci na latex (kryt jehly injekční stříkačky může obsahovat suchou gumu (latex)).
U pacientů léčených Xolairem byly pozorovány syndrom Churga-Straussové a hypereozinofilní syndrom. Příznaky mohou zahrnovat jeden nebo více z následujících: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (výrazná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (nosní neprůchodnost), srdeční problémy, bolest, pocit necitlivosti, brnění v rukou a nohou. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
Poraďte se se svým lékařem, než Vám bude podán Xolair:
- jestliže máte problémy s ledvinami nebo játry.
- jestliže máte onemocnění, při kterém Váš imunitní systém napadá části Vašeho těla (autoimunitní onemocnění).
- jestliže žijete v oblasti, kde je častý výskyt parazitárních infekcí, nebo cestujete do takového místa, jelikož Xolair může oslabit Vaši odolnost vůči těmto infekcím.
Xolair neléčí příznaky akutního astmatu, jako je náhlý astmatický záchvat. Proto by Xolair neměl být užíván k léčbě takových příznaků.
Xolair není určen k prevenci nebo léčbě jiných typů alergických stavů, jako jsou náhlé alergické reakce, syndrom hyperimunoglobulinemie E (dědičné imunitní onemocnění), aspergilóza (onemocnění plic vyvolané houbou), alergické jídlo, ekzém nebo senná rýma, protože nebyl u těchto stavů studován.
Děti (mladší 6 let)
Xolair se nemá podávat dětem, které j sou mladší než 6 let. U této věkové skupiny nej sou k dispozici dostatečná data.
Další léčivé přípravky a Xolair
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Toto je obvzlášť důležité, jestliže užíváte:
- léky používané k léčbě parazitární infekce, protože Xolair může oslabit účinek Vaší léčby.
- inhalační kortikosteroidy a jiné léky používané k léčbě astmatu.
Těhotenství a kojení
Xolair by Vám neměl být podáván, jestliže jste těhotná, ledaže by to Váš lékař považoval za nezbytné.
Pokud plánujete otěhotnět, informujte o tom lékaře, než zahájíte léčbu Xolairem. Váš Lékař s Vámi prodiskutuje přínos i potenciální rizika, která mohou nastat při podání tohoto léku během těhotenství.
Pokud jste při léčbě Xolairem otěhotněla, okamžitě to oznamte lékaři.
Xolair by Vám neměl být podáván, jestliže kojíte.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že Xolair bude ovlivňovat Vaši schopnost řídit nebo obsluhovat stroje.
3. Jak se Xolair podává
Pokyny k užívání Xolairu jsou uvedeny v bodě “Informace pro zdravotnické pracovníky”.
Váš lékař rozhodne, jakou dávku Xolairu potřebujete a jak často Vám bude podána. To záleží na Vaší tělesné hmotnosti a na výsledcích krevních testů stanovujících množství IgE v krvi ještě před tím, než zahájíte léčbu Xolairem.
Xolair je podán lékařem nebo zdravotní sestrou jako injekce rovnou pod kůži (podkožně).
Pečlivě se držte instrukcí svého lékaře nebo zdravotní sestry.
Jaká dávka Vám bude podána
Bude Vám podána 1-4 injekce najednou, každé dva nebo čtyři týdny.
Během léčby Xolairem pokračujte v užívání současných léků na astma. Nepřestávejte užívat žádné léky na astma, aniž byste informoval(a) svého lékaře.
Po zahájení léčby Xolairem nemusíte zpozorovat okamžité zlepšení astmatu. Obvykle to trvá mezi 12 a 16 týdny, než pocítíte plný účinek.
Použití u dětí a dospívajících
Xolair může být podán dětem a dospívajícím ve věku 6 let nebo starším, které již užívají léky na astma, ale u kterých nejsou astmatické příznaky dobře kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů. Váš lékař rozhodne, jakou dávku Xolairu Vaše dítě potřebuje a jak často bude podávána. Bude to záviset na váze Vašeho dítěte a výsledcích krevního vyšetření, provedeného před zahájením léčby kvůli změření množství IgE v jeho/její krvi.
Jestliže jste zmeškal(a) podání dávky přípravku Xolair
Kontaktujte svého lékaře nebo nemocnici ihned, jakmile je to možné, abyste znovu naplánoval(a) návštěvu lékaře.
Jestliže přerušíte léčbu Xolairem
Nepřerušujte léčbu přípravkem Xolair, pokud Vám to nedoporučí Váš lékař. Přerušení nebo ukončení léčby Xolairem může způsobit návrat astmatických symptomů.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Nežádoucí účinky způsobené Xolairem jsou obvykle mírné až středně závažné, ale příležitostně mohou být závažné.
Závažné nežádoucí účinky jsou:
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• Náhlé těžké alergické reakce: pokud zpozorujete náhlé příznaky alergické reakce nebo kombinace příznaků jako například vyrážku, svědění nebo kopřivku na kůži, otok obličeje, rtů, jazyka, hrtanu, průdušnice nebo jiných částí těla, rychlý srdeční tep, závratě nebo mírné točení hlavy, zkrácení dechu, sípání nebo dýchací potíže, anebo jakékoliv další jiné příznaky, řekněte to okamžitě svému lékaři nebo zdravotní sestře. Pokud máte v anamnéze závažné alergické reakce (anafylaxi) nesouvisející s podáním Xolairu, můžete mít větší riziko vývoje závažné alergické reakce po podání Xolairu.
• Systémový lupus erythematodes (SLE). Příznaky mohou zahrnovat bolest svalů, bolest kloubů a otok, a vyrážku. Mohou se u Vás objevit také další příznaky jako je horečka, úbytek hmotnosti a únava.
Není známo (četnost z dostupných údajů nelze určit)
• Výskyt jednoho nebo více z následujících příznaků: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (významná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (neprůchodnost nosu), srdeční problémy, bolest, znecitlivění, brnění rukou a nohou (příznaky tzv. syndromu „Churga-Straussové nebo hypereozinofilního syndromu“).
• Nízký počet krevních destiček s příznaky jako např. krvácení nebo podlitiny, které vznikají velmi snadno oproti normálnímu stavu.
• Rozvoj jakýchkoli následujících příznaků, obzvláště pokud se vyskytnou v kombinaci: bolest kloubů s otokem nebo bez otoku nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, bolest svalů (známky sérové nemoci).
Jestliže se u Vás cokoli z toho vyskytne, řekněte to ihned svému lékaři nebo zdravotní sestře.
Další možné nežádoucí účinky jsou:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 lidí)
• horečka (u dětí)
Časté nežádoucí účinky (mohou postihovat až 1 z 10 lidí)
• reakce v místě injekce včetně bolesti, zduření, svědění a zčervenání
• bolest v nadbřišku (u dětí)
• bolest hlavy (velmi časté u dětí)
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 lidí)
• pocit závratě, ospalosti nebo únavy
• brnění nebo znecitlivění rukou nebo nohou
• mdloby, nízký krevní tlak při sezení nebo ve stoji (posturální hypotenze), zčervenání
• bolesti v krku, kašel, akutní potíže s dechem
• pocit na zvracení (nausea), průjem, zažívací potíže
• svědění, kopřivka, vyrážka, zvýšená citlivost kůže na slunce
• zvýšení hmotnosti
• symptomy podobné chřipce
• oteklé paže
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• parazitární infekce
Není známo (četnost z dostupných údajů nelze určit)
• bolesti kloubů a svalů, otoky kloubů
• vypadávání vlasů
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xolair uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
- Nepoužívejte přípravek, pokud má poškozený obal nebo pokud si všimnete jakýchkoli známek předchozí manipulace.
6. Obsah balení a další informace Co Xolair obsahuje
- Léčivou látkou je omalizumabum. Jedna injekční stříkačka s objemem 0,5 ml roztoku obsahuje omalizumabum 75 mg.
- Pomocné látky jsou: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20 a voda na injekci.
- Kryt jehly injekční stříkačky může obsahovat suchou gumu (latex).
Jak Xolair vypadá a co obsahuje toto balení
Xolair injekční roztok je dodáván jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý injekční roztok v předplněné injekční stříkačce.
Xolair 75 mg injekční roztok je dostupný v baleních obsahujících 1 předplněnou injekční stříkačku a ve vícečetném balení, které obsahuje 4 nebo 10 balení, z nichž každé obsahuje 1 předplněnou injekční stříkačku.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Btarapna Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EkláSa Novartis (Hellas) A.E.B.E. Tqk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema.europa.eu
INFORMACE PRO ZDRAVOTNICKÉ PRACOVNÍKY
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Před použitím injekční stříkačky si, prosím, pečlivě přečtěte následující informaci.
Každé balení Xolairu obsahuje předplněnou injekční stříkačku samostatně zatavenou v plastovém obalu.
Jednotlivé části předplněné injekční stříkačky
Kryt jehly
Ochrana jehly
Obruba pro prsty
Aktivační
svorky
Píst
Kontrolní okénko Štítek a doba použitelnosti


Objemová ryska
Injekce Xolairu jsou určené pouze pro použití zdravotnickým pracovníkem.
Kryt jehly injekční stříkačky může obsahovat suchou gumu (latex). Osoby přecitlivělé na tuto látku by s ním neměly manipulovat.
Příprava injekce k použití
Před dokončením přípravy injekce se vyvarujte kontaktu s aktivačními svorkami zařízení, abyste
zabránili předčasnému zakrytí jehly ochranou jehly (viz první obrázek).
1. Vyjměte krabičku, která obsahuje injekční stříkačku, z lednice a ponechejte ji po dobu asi 20 minut tak, aby dosáhla teploty místnosti (ponechte injekční stříkačku v krabičce, aby byla chráněna před světlem).
2. Pokud je to nutné, můžete vrátit injekční stříkačku do lednice pro pozdější použití, ale tento krok nesmí být proveden více než jednou. Celková doba, po kterou je injekční stříkačka ponechána při pokojové teplotě (25 °C) nesmí překročit 4 hodiny.
3. Jakmile j ste připraveni k aplikaci injekce, umyjte si důkladně ruce mýdlem a vodou.
4. Očistěte místo vpichu.
5. Vyjměte plastikový zásobník z krabičky, sloupněte papírový kryt a vyjměte injekční stříkačku.
6. Zkontrolujte injekční stříkačku. NEPOUŽÍVEJTE, jestliže je rozbitá nebo náplň vypadá zakaleně nebo jestliže obsahuje částice. Ve všech těchto případech vraťte celé balení do lékárny.
7. Držte injekční stříkačku v horizontální poloze (jak je ukázáno níže), podívejte se skrze ní do kontrolního okénka, abyste zkontroloval(a) dávku léčivého přípravku (75 mg) a dobu použitelnosti vytištěnou na štítku. Poznámka: Otáčejte vnitřní částí kompletu injekční stříkačky, jak je ukázáno níže, abyste mohl(a) kontrolním okénkem přečíst štítek.
NEPOUŽÍVEJTE, jestliže doba použitelnosti přípravku uplynula nebo jestliže dávka není správná. V obou těchto případech vraťte celé balení do lékárny.

8. Držte injekční stříkačku ve vertikální poloze s pístem nahoře a poklepejte prstem na stěnu stříkačky, abyste umožnil(a) vzduchovým bublinám vystoupat vzhůru.
9. Zkontrolujte, že hladina tekutiny je na minimální hranici objemu nebo nad ní. Jestliže je roztok pod objemovou čárkou, vraťte celé balení do lékárny.

Použití injekce

Držte injekční stříkačku s jehlou směřující vzhůru, opatrně sejměte kryt z jehly na injekční stříkačce a vyhoďte ho. Nedotýkejte se odkryté jehly. Potom prstem zlehka poklepejte na injekční stříkačku, dokud stoupá vzduchová bublina k vrcholu injekční stříkačky. Pomalu tlačte píst vzhůru tak, aby byly vzduchové bubliny vytlačeny ze stříkačky bez zbytečné ztráty roztoku.

Jemně uchopte kožní řasu v místě vpichu a zasuňte jehlu.

Za přidržení obruby prsty pomalu stlačujte píst injekce, dokud to půjde.
Jestliže dochází k úniku roztoku v místě vpichu, zasuňte jehlu do kůže hlouběji.
|
□ |
Držte píst zcela stlačený a přitom opatrně a přímo vytáhněte jehlu z místa vpichu. | |
|
□ -- _A —1 ■ -- |
Pomalu pusťte píst, tím umožníte automatické zakrytí jehly. Přidržte gázu na místě vpichu po dobu přibližně 30 sekund. |
Instrukce pro likvidaci
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Příbalová informace: informace pro uživatele
Xolair 150 mg injekční roztok
omalizumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Xolair a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán
3. Jak se Xolair podává
4. Možné nežádoucí účinky
5. Jak Xolair uchovávat
6. Obsah balení a další informace
1. Co je Xolair a k čemu se používá
Xolair se používá k léčbě alergického astmatu a chronické spontánní kopřivky (CSU).
Léčivou látkou v Xolairu je omalizumab. Omalizumab je člověkem vyrobená bílkovina, která se podobá přírodním bílkovinám produkovaným tělem; patří do skupiny léčivých přípravků zvaných monoklonální protilátky. Xolair způsobuje zablokování látky, která se nazývá imunoglobulin E (IgE) a která se v organismu vytváří. IgE hraje klíčovou roli při vyvolání alergického astmatu nebo CSU.
Alergické astma
Tento léčivý přípravek se užívá k prevenci zhoršení astmatu tím, že kontroluje astmatické příznaky u dospělých a dospívajících (12 let a starší) a dětí (6 až 11 let), kteří již užívají lék proti astmatu, ale u kterých příznaky astmatu nejsou dostatečně kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů.
Chronická spontánní kopřivka (CSU)
Tento léčivý přípravek se užívá k léčbě chronické spontánní kopřivky u dospělých a dospívajících (ve věku 12 let a starších), kteří již užívají antihistaminika, ale jejichž příznaky CSU nejsou těmito léky dobře kontrolovány.
2. Čemu musíte věnovat pozornost, než Vám bude Xolair podán Xolair Vám nesmí být podán
- jestliže jste alergický(á) na omalizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Jestliže si myslíte, že můžete být alergický(á) na kteroukoli složku přípravku, řekněte to svému lékaři, protože Xolair by Vám neměl být podán.
Upozornění a opatření
Xolair obsahuje bílkovinu a bílkoviny mohou u některých lidí způsobit závažné alergické reakce. Projevy zahrnují vyrážku, obtíže při dýchání, otok nebo pocit na omdlení. Jestliže se u Vás vyskytnou po užití Xolairu jakékoli alergické reakce, kontaktujte lékaře ihned, jak budete moci.
U pacientů léčených Xolairem byl pozorován určitý druh alergické reakce zvaný sérová nemoc. Projevy sérové nemoci mohou být jeden nebo více z následujících příznaků: bolest kloubu s otokem nebo bez něj nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, svalová bolest. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
Při užívání přípravku Xolair dbejte zvláštní opatrnosti, pokud jste někdy měli alergickou reakci na latex (kryt jehly injekční stříkačky může obsahovat suchou gumu (latex)).
U alergických pacientů s astmatem léčených Xolairem byly pozorovány syndrom Churga-Straussové a hypereozinofilní syndrom. Příznaky mohou zahrnovat jeden nebo více z následujících: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (výrazná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (nosní neprůchodnost), srdeční problémy, bolest, pocit necitlivosti, brnění v rukou a nohou. Pokud pocítíte jakýkoliv z těchto příznaků nebo zvláště pokud se u Vás objeví kombinace takových příznaků, kontaktujte ihned svého lékaře.
Poraďte se se svým lékařem, než Vám bude podán Xolair:
- jestliže máte problémy s ledvinami nebo játry.
- jestliže máte onemocnění, při kterém Váš imunitní systém napadá části Vašeho těla (autoimunitní onemocnění).
- jestliže žijete v oblasti, kde je častý výskyt parazitárních infekcí, nebo cestujete do takového místa, jelikož Xolair může oslabit Vaši odolnost vůči těmto infekcím.
Xolair neléčí příznaky akutního astmatu, jako je náhlý astmatický záchvat. Proto by Xolair neměl být užíván k léčbě takových příznaků.
Xolair není určen k prevenci nebo léčbě jiných typů alergických stavů, jako jsou náhlé alergické reakce, syndrom hyperimunoglobulinemie E (dědičné imunitní onemocnění), aspergilóza (onemocnění plic vyvolané houbou), alergické jídlo, ekzém nebo senná rýma, protože nebyl u těchto stavů studován.
Děti a dospívající
Alergické astma
Xolair se nedoporučuje podávat dětem mladším 6 let.
Chronická spontánní kopřivka (CSU)
Nepodávejte Xolair dětem do 12 let věku. Jeho použití u dětí do 12 let nebylo studováno.
Další léčivé přípravky a Xolair
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Toto je obvzlášť důležité, jestliže užíváte:
- léky používané k léčbě parazitární infekce, protože Xolair může oslabit účinek Vaší léčby.
- inhalační kortikosteroidy a jiné léky používané k léčbě astmatu.
Těhotenství a kojení
Xolair by Vám neměl být podáván, jestliže jste těhotná, ledaže by to Váš lékař považoval za nezbytné.
Pokud plánujete otěhotnět, informujte o tom lékaře, než zahájíte léčbu Xolairem. Váš Lékař s Vámi prodiskutuje přínos i potenciální rizika, která mohou nastat při podání tohoto léku během těhotenství.
Pokud jste při léčbě Xolairem otěhotněla, okamžitě to oznamte lékaři.
Xolair by Vám neměl být podáván, jestliže kojíte.
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že Xolair bude ovlivňovat Vaši schopnost řídit nebo obsluhovat stroje.
3. Jak se Xolair podává
Pokyny k užívání Xolairu jsou uvedeny v bodě “Informace pro zdravotnické pracovníky”.
Xolair je podán lékařem nebo zdravotní sestrou jako injekce rovnou pod kůži (podkožně).
Pečlivě se držte instrukcí svého lékaře nebo zdravotní sestry.
Jaká dávka Vám bude podána
Alergické astma
Váš lékař rozhodne, kolik Xolairu potřebujete a jak často Vám bude podáván. Závisí to na Vaší tělesné hmotnosti a výsledcích vyšetření krve provedeného před zahájením léčby ke změření množství IgE ve Vaší krvi.
Bude Vám podána 1-4 injekce najednou, každé dva nebo čtyři týdny.
Během léčby Xolairem pokračujte v užívání současných léků na astma. Nepřestávejte užívat žádné léky na astma, aniž byste informoval(a) svého lékaře.
Po zahájení léčby Xolairem nemusíte zpozorovat okamžité zlepšení astmatu. Obvykle to trvá mezi 12 a 16 týdny, než pocítíte plný účinek.
Chronická spontánní kopřivka (CSU)
Budou Vám podávány dvě 150 mg injekce najednou každé čtyři týdny.
Pokračujte v užívání Vašeho současného léku na CSU během léčby Xolairem. Nepřestávejte užívat jakýkoli lék bez předchozí porady s Vaším lékařem.
Použití u dětí a dospívajících
Alergické astma
Xolair může být podán dětem a dospívajícím ve věku 6 let nebo starším, které již užívají léky na astma, ale u kterých nejsou astmatické příznaky dobře kontrolovány léčivými přípravky, jako jsou vysoké dávky inhalačních steroidů nebo inhalačních beta-agonistů. Váš lékař rozhodne, jakou dávku Xolairu Vaše dítě potřebuje a jak často bude podávána. Bude to záviset na váze Vašeho dítěte a výsledcích krevního vyšetření, provedeného před zahájením léčby kvůli změření množství IgE v jeho/její krvi.
Chronická spontánní kopřivka (CSU)
Xolair může být podáván dospívajícím ve věku 12 let a starším, kteří již užívají antihistaminika, ale jejichž příznaky CSU nejsou těmito léky dobře kontrolovány.
Jestliže jste zmeškal(a) podání dávky přípravku Xolair
Kontaktujte svého lékaře nebo nemocnici ihned, jakmile je to možné, abyste znovu naplánoval(a) návštěvu lékaře.
Jestliže přerušíte léčbu Xolairem
Nepřerušujte léčbu přípravkem Xolair, pokud Vám to nedoporučí Váš lékař. Přerušení nebo ukončení léčby Xolairem může způsobit návrat příznaků astmatu nebo kopřivky.
Jste-li však v současné době léčen(a) pro CSU, Váš lékař může léčbu Xolairem občas přerušit, aby mohly být příznaky vyhodnoceny. Postupujte podle pokynů Vašeho lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého. Nežádoucí účinky způsobené Xolairem jsou obvykle mírné až středně závažné,
ale příležitostně mohou být závažné.
Závažné nežádoucí účinky jsou:
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• Náhlé těžké alergické reakce: pokud zpozorujete náhlé příznaky alergické reakce nebo kombinace příznaků jako například vyrážku, svědění nebo kopřivku na kůži, otok obličeje, rtů, jazyka, hrtanu, průdušnice nebo jiných částí těla, rychlý srdeční tep, závratě nebo mírné točení hlavy, zkrácení dechu, sípání nebo dýchací potíže, anebo jakékoliv další jiné příznaky, řekněte to okamžitě svému lékaři nebo zdravotní sestře. Pokud máte v anamnéze závažné alergické reakce (anafylaxi) nesouvisející s podáním Xolairu, můžete mít větší riziko vývoje závažné alergické reakce po podání Xolairu.
• Systémový lupus erythematodes (SLE). Příznaky mohou zahrnovat bolest svalů, bolest kloubů a otok, a vyrážku. Mohou se u Vás objevit také další příznaky jako je horečka, úbytek hmotnosti a únava.
Není známo (četnost z dostupných údajů nelze určit)
• Výskyt jednoho nebo více z následujících příznaků: otok, bolest nebo vyrážka kolem krevních nebo mízních cév, vysoká hladina určitého typu bílých krvinek (významná eozinofilie), zhoršení problémů s dýcháním, zduření nosní sliznice (neprůchodnost nosu), srdeční problémy, bolest, znecitlivění, brnění rukou a nohou (příznaky tzv. syndromu „Churga-Straussové nebo hypereozinofilního syndromu“).
• Nízký počet krevních destiček s příznaky jako např. krvácení nebo podlitiny, které vznikají velmi snadno oproti normálnímu stavu.
• Rozvoj jakýchkoli následujících příznaků, obzvláště pokud se vyskytnou v kombinaci: bolest kloubů s otokem nebo bez otoku nebo ztuhlost, vyrážka, horečka, zduření lymfatických uzlin, bolest svalů (známky sérové nemoci).
Jestliže se u Vás cokoli z toho vyskytne, řekněte to ihned svému lékaři nebo zdravotní sestře.
Další možné nežádoucí účinky jsou:
Velmi časté nežádoucí účinky (mohou postihovat více než 1 z 10 lidí)
• horečka (u dětí)
Časté nežádoucí účinky (mohou postihovat až 1 z 10 lidí)
• reakce v místě injekce včetně bolesti, zduření, svědění a zčervenání
• bolest v nadbřišku (u dětí)
• bolest hlavy (velmi časté u dětí)
• infekce horních cest dýchacích, j ako j e zánět hltanu a nachlazení
• pocit tlaku nebo bolesti ve tvářích a na čele (zánět dutin, bolest hlavy při zánětu dutin)
• bolest v kloubech (artralgie)
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 lidí)
• pocit závratě, ospalosti nebo únavy
• brnění nebo znecitlivění rukou nebo nohou
• mdloby, nízký krevní tlak při sezení nebo ve stoji (posturální hypotenze), zčervenání
• bolesti v krku, kašel, akutní potíže s dechem
• pocit na zvracení (nausea), průjem, zažívací potíže
• svědění, kopřivka, vyrážka, zvýšená citlivost kůže na slunce
• zvýšení hmotnosti
• symptomy podobné chřipce
• oteklé paže
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí)
• parazitární infekce
Není známo (četnost z dostupných údajů nelze určit)
• bolesti svalů a otoky kloubů
• vypadávání vlasů
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xolair uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
- Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
- Nepoužívejte přípravek, pokud má poškozený obal nebo pokud si všimnete jakýchkoli známek předchozí manipulace.
6. Obsah balení a další informace
Co Xolair obsahuje
- Léčivou látkou je omalizumabum. Jedna injekční stříkačka s objemem 1 ml roztoku obsahuje omalizumabum 150 mg.
- Pomocné látky jsou: arginin-hydrochlorid, histidin-hydrochlorid, histidin, polysorbát 20 a voda na injekci.
- Kryt jehly injekční stříkačky může obsahovat suchou gumu (latex).
Jak Xolair vypadá a co obsahuje toto balení
Xolair injekční roztok je dodáván jako čirý až mírně opalescentní, bezbarvý až světle hnědavě-žlutý injekční roztok v předplněné injekční stříkačce.
Xolair 150 mg injekční roztok je dostupný v baleních obsahujících 1 předplněnou injekční stříkačku a ve vícečetném balení, které obsahuje 4 nebo 10 balení, z nichž každé obsahuje 1 předplněnou injekční stříkačku.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Btarapun
Novartis Pharma Services Inc. Ten.: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Eklába
Novartis (Hellas) A.E.B.E. Tqk: +30 210 281 17 12
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúrcpog Novartis Pharma Services Inc. TpA,: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http: //www .ema.europa.eu
INFORMACE PRO ZDRAVOTNICKÉ PRACOVNÍKY
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Před použitím injekční stříkačky si, prosím, pečlivě přečtěte následující informaci.
Každé balení Xolairu obsahuje předplněnou injekční stříkačku samostatně zatavenou v plastovém obalu.
Jednotlivé části předplněné injekční stříkačky
Kryt jehly
Ochrana jehly
Obruba pro prsty
Aktivační
svorky
Píst
Kontrolní okénko Štítek a doba použitelnosti


Objemová ryska
Injekce Xolairu jsou určené pouze pro použití zdravotnickým pracovníkem.
Kryt jehly injekční stříkačky může obsahovat suchou gumu (latex). Osoby přecitlivělé na tuto látku by s ním neměly manipulovat.
Příprava injekce k použití
Před dokončením přípravy injekce se vyvarujte kontaktu s aktivačními svorkami zařízení, abyste
zabránili předčasnému zakrytí jehly ochranou jehly (viz první obrázek).
1. Vyjměte krabičku, která obsahuje injekční stříkačku, z lednice a ponechejte ji po dobu asi 20 minut tak, aby dosáhla teploty místnosti (ponechte injekční stříkačku v krabičce, aby byla chráněna před světlem).
2. Pokud je to nutné, můžete vrátit injekční stříkačku do lednice pro pozdější použití, ale tento krok nesmí být proveden více než jednou. Celková doba, po kterou je injekční stříkačka ponechána při pokojové teplotě (25 °C) nesmí překročit 4 hodiny.
3. Jakmile j ste připraveni k aplikaci injekce, umyjte si důkladně ruce mýdlem a vodou.
4. Očistěte místo vpichu.
5. Vyjměte plastikový zásobník z krabičky, sloupněte papírový kryt a vyjměte injekční stříkačku.
6. Zkontrolujte injekční stříkačku. NEPOUŽÍVEJTE, jestliže je rozbitá nebo náplň vypadá zakaleně nebo jestliže obsahuje částice. Ve všech těchto případech vraťte celé balení do lékárny.
7. Držte injekční stříkačku v horizontální poloze (jak je ukázáno níže), podívejte se skrze ní do kontrolního okénka, abyste zkontroloval(a) dávku léčivého přípravku (150 mg) a dobu použitelnosti vytištěnou na štítku. Poznámka: Otáčejte vnitřní částí kompletu injekční stříkačky, jak je ukázáno níže, abyste mohl(a) kontrolním okénkem přečíst štítek.
NEPOUŽÍVEJTE, jestliže doba použitelnosti přípravku uplynula nebo jestliže dávka není správná. V obou těchto případech vraťte celé balení do lékárny.

8. Držte injekční stříkačku ve vertikální poloze s pístem nahoře a poklepejte prstem na stěnu stříkačky, abyste umožnil(a) vzduchovým bublinám vystoupat vzhůru.
9. Zkontrolujte, že hladina tekutiny je na minimální hranici objemu nebo nad ní. Jestliže je roztok pod objemovou čárkou, vraťte celé balení do lékárny.

Použití injekce

Držte injekční stříkačku s jehlou směřující vzhůru, opatrně sejměte kryt z jehly na injekční stříkačce a vyhoďte ho. Nedotýkejte se odkryté jehly. Potom prstem zlehka poklepejte na injekční stříkačku, dokud stoupá vzduchová bublina k vrcholu injekční stříkačky. Pomalu tlačte píst vzhůru tak, aby byly vzduchové bubliny vytlačeny ze stříkačky bez zbytečné ztráty roztoku.

Jemně uchopte kožní řasu v místě vpichu a zasuňte jehlu.

Za přidržení obruby prsty pomalu stlačujte píst injekce, dokud to půjde.
Jestliže dochází k úniku roztoku v místě vpichu, zasuňte jehlu do kůže hlouběji.
|
□ |
Držte píst zcela stlačený a přitom opatrně a přímo vytáhněte jehlu z místa vpichu. | |
|
□ -- _A —1 ■ -- |
Pomalu pusťte píst, tím umožníte automatické zakrytí jehly. Přidržte gázu na místě vpichu po dobu přibližně 30 sekund. |
Instrukce pro likvidaci
Ihned zlikvidujte použitou injekční stříkačku do k tomuto účelu vyhrazeného kontejneru. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizované zprávy o bezpečnosti (PSUR) pro omalizumab dospěl výbor CHMP k těmto vědeckým závěrům:
Byly hlášeny případy systémového lupus erythematodes (SLE) ve spojení s léčbou přípravkem Xolair, včetně dvou případů s pozitivním de-challenge a jedním případem s pozitivním de-challenge/re-challenge. I když byly informace k posouzení kauzality ve většině případů velmi omezené, v mnoha ze zbývajících případů byly přítomny zavádějící faktory jako je preexistující lupus , včetně potenciálního počínajícího SLE a patogeneze SLE/lékem vyvolaného lupusu je dosud spatně známa a je pravděpodobně multifaktoriální, nezdá se bezdůvodné, že by Xolair, lék, který vytváří imunokomplexy s IgE s potenciálem indukovat poškození způsobené imunokomplexy, a u kterého byly vzácně hlášeny příhody jako je sérová nemoc, mohl hrát roli v patogenezi SLE/lékem vyvolaného lupusu. Po důkladném zhodnocení dostupných dat se podpora možnosti příčinné souvislosti mezi Xolairem a systémovým lupus erythematodes jeví jako rozumná.
Proto s ohledem na data prezentovaná ve zhodnoceném PSURu považuje PRAC změny v informacích o přípravku u léčivých přípravků obsahujících omalizumab za opodstatněné.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se omalizumabu výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících omalizumab zůstává nezměněný, a to pod podmínkou, že v informacích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
148