Xofigo 1100 Kbq/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xofigo 1100 kBq/ml injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml roztoku obsahuje radium-223 dichloridum 1100 kBq (radium-223 dichlorid, radium
Ra 223 dichloride), což odpovídá radium-223 0,58 ng k referenčnímu datu. Radium je v roztoku jako volný
iont.
Jedna injekční lahvička obsahuje 6 ml roztoku (radium-223 dichloridum 6,6 MBq k referenčnímu datu).
Radium-223 je alfa zářič s poločasem rozpadu 11,4 dne. Specifická aktivita radia-223 je 1,9 MBq/ng.
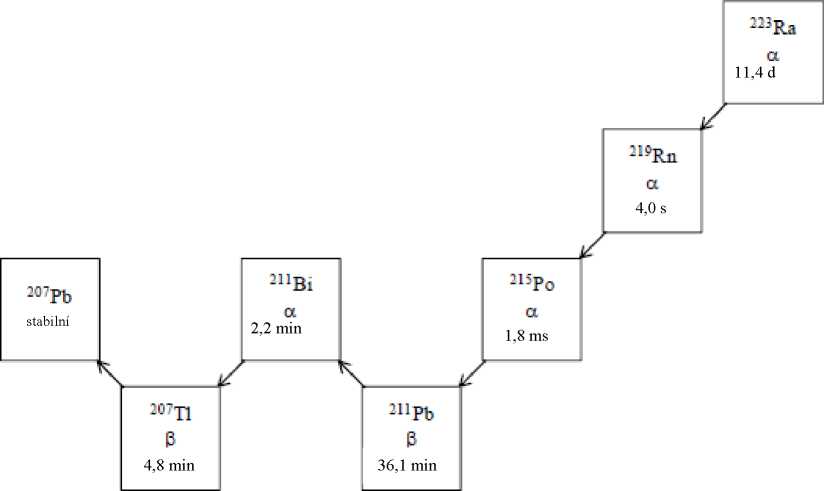
Šestistupňový rozpad radia-223 na olovo-207 nastává přes dceřiné rozpadové produkty s krátkou životností a je doprovázen množstvím alfa, beta a gama záření s různými energiemi a vlastnostmi záření. Frakce energie emitovaná z radia-223 a jeho dceřiných produktů jako alfa částice je 95,3 % (energetické rozmezí 5,0-7,5 MeV). Frakce emitovaná jako beta částice je 3,6 % (průměrné energie jsou 0,445 MeV a 0,492 MeV) a frakce emitovaná jako gama záření je 1,1 % (energetické rozmezí 0,01-1,27 MeV).
Obrázek 1: Řetězec rozpadu radia-223 s fyzikálními poločasy a způsobem rozpadu
Pomocné látky se známým účinkem
Jeden ml roztoku obsahuje 0,194 mmol (odpovídá 4,5 mg) sodíku. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý, bezbarvý isotonický roztok s pH mezi 6,0 a 8,0.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Xofigo je indikován k léčbě dospělých mužů s kastračně rezistentním karcinomem prostaty, symptomatickými metastázami v kostech a bez známých viscerálních metastáz.
4.2 Dávkování a způsob podání
Přípravek Xofigo by měl být podáván pouze osobami oprávněnými k zacházení s radiofarmaky v určených klinických podmínkách (viz bod 6.6) a po vyšetření pacienta kvalifikovaným lékařem.
Dávkování
Přípravek Xofigo je podáván v dávce o aktivitě 55 kBq na kilogram tělesné hmotnosti ve 4 týdenních intervalech. Je podáváno 6 injekcí přípravku Xofigo.
Bezpečnost a účinnost více než 6 injekcí přípravku Xofigo nebyly hodnoceny.
Podrobnosti o výpočtu objemu, který má být podán, jsou uvedeny v bodě 12.
Starší pacienti
Ve studii fáze III nebyly pozorovány mezi staršími (ve věku > 65 let) a mladšími pacienty (ve věku < 65 let) žádné celkové rozdíly v bezpečnosti nebo účinnosti.
U starších pacientů není nutná žádná úprava dávkování.
Pacienti s poruchou funkce jater
Bezpečnost a účinnost přípravku Xofigo nebyla hodnocena u pacientů s poruchou funkce jater.
Protože radium-223 není ani metabolizováno v játrech ani eliminováno žlučí, neočekává se, že porucha funkce jater ovlivní farmakokinetiku radia-223 dichloridu.
U pacientů s poruchou funkce jater není nutná žádná úprava dávkování.
Pacienti s poruchou funkce ledvin
V klinické studii fáze III nebyly pozorovány žádné významné rozdíly v bezpečnosti nebo účinnosti mezi pacienty s mírnou poruchou funkce ledvin (clearance kreatininu [CLCR]: 50 až 80 ml/min) a normální funkcí ledvin. U pacientů se středně závažnou poruchou funkce ledvin (CLCR: 30 až 50 ml/min) jsou k dispozici omezené údaje. Nejsou k dispozici žádné údaje u pacientů se závažnou (CLCR <30 ml/min) poruchou funkce ledvin nebo konečným stadiem onemocnění ledvin.
Protože je však vylučování močí minimální a hlavní cestou eliminace je stolice, neočekává se, že porucha funkce ledvin ovlivní farmakokinetiku radia-223 dichloridu.
U pacientů s poruchou funkce ledvin není nutná žádná úprava dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Xofigo u dětí a dospívajících do 18 let nebyla hodnocena.
Neexistuje žádné relevantní použití tohoto léčivého přípravku u pediatrické populace v indikaci karcinom prostaty.
Způsob podání
Přípravek Xofigo je určen pro intravenózní podání. Musí být podán pomalou injekcí (obvykle do 1 minuty).
Intravenózní sonda nebo kanyla musí být před a po aplikaci injekce přípravku Xofigo propláchnuta isotonickým roztokem chloridu sodného 9 mg/ml (0,9%) určeným pro injekční podání.
Další instrukce o použití tohoto léčivého přípravku jsou uvedeny v bodech 6.6 a 12.
4.3 Kontraindikace
Pro podání přípravku Xofigo neexistují žádné známé kontraindikace.
4.4 Zvláštní upozornění a opatření pro použití
Suprese kostní dřeně
U pacientů léčených přípravkem Xofigo byla hlášena suprese kostní dřeně, konkrétně trombocytopenie, neutropenie, leukopenie a pancytopenie (viz bod 4.8).
Proto musí být na počátku léčby a před každou dávkou přípravku Xofigo provedeno hematologické vyšetření pacientů. Před prvním podáním přípravku by měl být absolutní počet neutrofilů (ANC) > 1,5x109/l, počet trombocytů > 100x109/l a hemoglobin > 10,0 g/dl. Před následným podáním by měl být ANC > 1,0x109/l a trombocyty > 50x109/l. Pokud nedojde k obnovení těchto hodnot během 6 týdnů po posledním podání přípravku Xofigo i přes podávanou standardní péči, měla by další léčba přípravkem Xofigo pokračovat pouze po pečlivém vyhodnocení poměru přínos/riziko.
Pacienti s prokázaným ohrožením rezervy kostní dřeně, např. po předchozí cytotoxické chemoterapii a/nebo ozařování (EBRT) nebo pacienti s karcinomem prostaty s pokročilou difúzní infiltrací kostí (EOD4; “superscan”), by měli být léčeni s opatrností. Během studie fáze III byla u těchto pacientů pozorována zvýšená incidence hematologických nežádoucích účinků jako je neutropenie a trombocytopenie (viz bod 4.8).
Účinnost a bezpečnost cytotoxické chemoterapie podané po léčbě přípravkem Xofigo nebyly stanoveny. Omezené dostupné údaje ukazují, že pacienti dostávající chemoterapii po léčbě přípravkem Xofigo měli ve srovnání s pacienty léčených chemoterapií po podávání placeba podobný hematologický profil (viz také bod 5.1).
Crohnova choroba a ulcerózní kolitida
Bezpečnost a účinnost přípravku Xofigo u pacientů s Crohnovou chorobou a ulcerózní kolitidou nebyly hodnoceny. Vzhledem k tomu, že se přípravek Xofigo vylučuje stolicí, může radiace způsobit zhoršení akutního zánětlivého střevního onemocnění. Pacientům s akutním zánětlivým onemocněním střev by měl být přípravek Xofigo podán pouze po pečlivém vyhodnocení poměru přínos/riziko.
Komprese míchy
U pacientů s neléčenou hrozící nebo přítomnou kompresí míchy by měla být léčba pomocí standardní péče, jak je klinicky indikována, dokončena před zahájením nebo znovuzahájením léčby přípravkem Xofigo.
Fraktury kostí
U pacientů s frakturami kostí by měla být před zahájením nebo znovuzahájením léčby přípravkem Xofigo provedena ortopedická stabilizace fraktur.
Osteonekróza čelisti:
U pacientů léčených bisfosfonáty a přípravkem Xofigo nelze vyloučit zvýšené riziko vzniku osteonekrózy čelisti. Ve studii fáze III byly u 0,67 % pacientů (4/600) v rameni přípravku Xofigo ve srovnání s 0,33 % (1/301)pacientů v rameni placeba hlášeny případy osteonekrózy čelisti. Avšak všichni pacienti s osteonekrózou čelisti dostávali před nebo v průběhu léčby bisfosfonáty (např. kyselinu zoledronovou) a předchozí chemoterapii (např. docetaxel).
Sekundární maligní nádory
Přípravek Xofigo přispívá k celkové dlouhodobé kumulativní radiační expozici u pacienta. Dlouhodobá kumulativní radiační expozice může být spojena se zvýšeným rizikem rakoviny a vrozených defektů. Zejména může být zvýšené riziko osteosarkomu, myelodysplastického syndromu a leukémie. V klinických studiích při sledování po dobu až tří let nebyly hlášeny žádné případy rakoviny vyvolané přípravkem Xofigo.
Pomocné látky se známým účinkem
V závislosti na podaném objemu může tento léčivý přípravek obsahovat až 2,35 mmol (54 mg) sodíku v jedné dávce. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné klinické studie interakcí.
Protože nemůže být vyloučena interakce s vápníkem a fosfáty, podávání přípravků s těmito látkami a/nebo vitaminem D by mělo být několik dnů před zahájením léčby přípravkem Xofigo přerušeno .
Souběžná chemoterapie s přípravkem Xofigo může mít aditivní účinky na supresi kostní dřeně (viz bod 4.4). Bezpečnost a účinnost chemoterapie podávané současně s přípravkem Xofigo nebyly stanoveny.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů
Reprodukční studie u zvířat nebyly s přípravkem Xofigo provedeny.
Vzhledem k možnosti účinků na spermatogenezi související s ozářením by mělo být mužům doporučeno, aby používali účinné metody antikoncepce během léčby a až 6 měsíců po ukončení léčby přípravkem Xofigo.
Těhotenství a kojení
Přípravek Xofigo není indikován u žen. Přípravek Xofigo nemá být používán u žen, které jsou nebo mohou být těhotné nebo u kojících žen.
Fertilita
Nejsou k dispozici žádné údaje u člověka o účinku přípravku Xofigo na fertilitu.
Na základě studií na zvířatech existuje možné riziko, že záření z přípravku Xofigo by mohlo mít nežádoucí účinky na fertilitu (viz bod 5.3). Pacienti by měli zvážit možnost konzervace spermií před léčbou.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nejsou k dispozici žádné důkazy ani se neočekává, že přípravek Xofigo bude mít vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Celkový bezpečnostní profil přípravku Xofigo vychází z dat od 600 pacientů léčených přípravkem Xofigo ve studii fáze III.
Nejčastěji pozorované nežádoucí účinky (> 10 %) u pacientů léčených přípravkem Xofigo byly průjem, nevolnost, zvracení a trombocytopenie.
Nejzávažnější nežádoucí účinky byly trombocytopenie a neutropenie (viz bod 4.4 a „Popis vybraných nežádoucích účinků“ níže).
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky pozorované v souvislosti s podáním přípravku Xofigo jsou uvedeny v tabulce níže (viz Tabulka 1). Jsou klasifikovány podle tříd orgánových systémů. Pro popis určité reakce a jejich synonym a souvisejících stavů je použitý nej vhodnější termín MedDRA.
Nežádoucí účinky z klinických studií jsou klasifikovány podle jejich frekvence. Frekvence jsou definovány jako: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100).
V každé skupině četností jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky hlášené v klinických studiích u pacientů léčených přípravkem Xofigo
|
Třída orgánových systémů (MedDRA) |
Velmi časté |
Časté |
Méně časté |
|
Poruchy krve a lymfatického systému |
Trombocytopenie |
Neutropenie Pancytopenie Leukopenie |
Lymfopenie |
|
Gastrointestinální poruchy | |||
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě aplikace injekce |
Popis vybraných nežádoucích účinků
Trombocytopenie a neutropenie
Trombocytopenie (všechny stupně) se vyskytla u 11,5 % pacientů léčených přípravkem Xofigo a u 5,6 % pacientů na placebu. Trombocytopenie stupně 3 a 4 byla pozorována u 6,3 % pacientů léčených přípravkem Xofigo a u 2 % pacientů užívajících placebo (viz bod 4.4). Frekvence trombocytopenie stupně 3 a 4 byla celkově nižší u pacientů, kteří dosud nedostávali docetaxel (2,8 % pacientů léčených přípravkem Xofigo oproti 0,8 % pacientů užívajících placebo) ve srovnání s pacienty, kteří dříve dostávali docetaxel (8,9 % pacientů léčených přípravkem Xofigo oproti 2,9 % pacientů užívajících placebo). V případě pacientů s EOD4 (“superscan”) byla u 19.6 % pacientů léčených přípravkem Xofigo a u 6,7 % pacientů užívajících placebo hlášena trombocytopenie (všechny stupně). U 5,9 % pacientů léčených přípravkem Xofigo a u 6,7 % pacientů užívajících placebo byl pozorován stupeň trombocytopenie 3 a 4 (viz bod 4.4).
Neutropenie (všechny stupně) byla hlášena u 5 % pacientů léčených přípravkem Xofigo a u 1 % pacientů užívajících placebo. Neutropenie stupně 3 a 4 byla pozorována u 2,2 % pacientů léčených přípravkem Xofigo a u 0,7 % pacientů užívajících placebo. Frekvence neutropenie stupně 3 a 4 byla celkově nižší u pacientů, kteří dosud nedostávali docetaxel (0,8 % pacientů léčených přípravkem Xofigo oproti 0,8 % pacientů užívajících placebo) ve srovnání s pacienty, kteří dříve dostávali docetaxel (3,2 % pacientů léčených přípravkem Xofigo oproti 0,6 % pacientů užívajících placebo).
Ve studii fáze I se minimální hladiny neutrofdů a trombocytů objevily 2 až 3 týdny po intravenózním podání jedné dávky přípravku Xofigo.
Reakce v místě aplikace injekce
Reakce v místě aplikace injekce stupně 1 a 2, např. erytém, bolest a otok, byly hlášeny u 1,2 % pacientů léčených přípravkem Xofigo a 0 % pacientů užívajících placebo.
Sekundární maligní nádory
Přípravek Xofigo přispívá k celkové dlouhodobé kumulativní radiační expozici u pacienta. Dlouhodobá kumulativní expozice může být spojena se zvýšeným rizikem rakoviny a vrozených defektů. Zejména riziko osteosarkomu, myelodysplastického syndromu a leukémie může být zvýšeno. V klinických studiích při sledování po dobu až tří let nebyly hlášeny žádné případy malignity související s přípravkem Xofigo.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Během klinických studií nebyly hlášeny žádné případy neúmyslného předávkování přípravkem Xofigo.
Neexistuje specifické antidotum. V případě neúmyslného předávkování by měla být provedena obecná podpůrná opatření, včetně monitorování možné hematologické a gastrointestinální toxicity.
V klinické studii fáze I při hodnocení jednotlivých dávek přípravku Xofigo o aktivitě až do výše 276 kBq na kilogram tělesné hmotnosti nebylo pozorováno žádné omezení dávky způsobené toxicitou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná různá terapeutická radiofarmaka, ATC kód: V10XX03.
Mechanismus účinku
Přípravek Xofigo je léčivo emitující terapeutické alfa částice.
Aktivní složka radium-223 (jako radium-223 dichlorid) napodobuje vápník a je selektivně cílená na kost, konkrétně na oblasti kostních metastáz tím, že tvoří komplexy s kostním minerálem hydroxyapatitem. Vysoký lineární energetický přenos alfa zářičů (80 keV/mikrometr) vede k vysoké frekvenci zlomů dvojité šroubovice DNA v sousedních nádorových buňkách, což má za následek silný cytotoxický účinek. Další účinky na nádorové mikroprostředí včetně osteoblastů a osteoklastů také přispívají k účinnosti in vivo. Dosah alfa částice z radia-223 je menší než 100 mikrometrů (méně než 10 průměrů buňky), což minimalizuje poškození zdravé sousední tkáně.
Farmakodynamické účinky
Ve srovnání s placebem došlo k významnému rozdílu ve prospěch přípravku Xofigo u všech pěti sérových biomarkerů kostního obratu hodnocených v randomizované studii fáze II (markery novotvorby kostí: kostní alkalická fosfatáza [ALP], celková ALP a prokolagen I N propeptid [PINP], markery kostní resorpce: C-terminální zesítěný telopeptid kolagenu typu I [S-CTX-I] a zesítěný C-telopeptid kolagenu typu I [ICTP]).
Klinická účinnost a bezpečnost
Klinická bezpečnost a účinnost přípravku Xofigo byly hodnoceny ve dvojitě zaslepené, randomizované studii fáze III s vícečetnými dávkami (ALSYMPCA; EudraCT 2007-006195-1) u kastračně rezistentního karcinomu prostaty se symptomatickými kostními metastázami. Pacienti s viscerálními metastázami a maligní lymfadenopatií přesahující velikost 3 cm byli vyloučeni.
Primární cílový ukazatel účinnosti bylo celkové přežití. Sekundární cílové ukazatele zahrnovaly dobu do objevení se symptomatických kostních příhod (SSE) a progresi podle hodnot ALP a PSA.
V mezním datu předem plánované průběžné analýzy (konfirmační analýza) bylo randomizováno celkem 809 pacientů v poměru 2:1 na léčbu přípravkem Xofigo 55 kBq/kg podávaného intravenózně každé 4 týdny po dobu 6 cyklů (N = 541) plus nejlepší standardní péče nebo na léčbu placebem plus nejlepší standardní péče (N = 268). Nejlepší standardní péče zahrnovala např. lokální zevní radioterapii, bisfosfonáty, kortikosteroidy, antiandrogeny, estrogeny, estramustin nebo ketokonazol.
Aktualizovaná deskriptivní analýza bezpečnosti a celkového přežití byla provedena u 921 randomizovaných pacientů před cross-over (tj. pacientům ve skupině placeba bylo nabídnuto převedení na léčbu přípravkem Xofigo).
Demografické a výchozí charakteristiky onemocnění (populace pro průběžnou analýzu) byly podobné mezi skupinami přípravku Xofigo a placeba a jsou uvedeny níže pro přípravek Xofigo:
• průměrný věk pacientů byl 70 let (rozmezí 49 až 90 let),
• 87 % zařazených pacientů mělo skóre výkonnostního stavu ECOG 0-1,
• 41 % dostávalo bisfosfonáty,
• 42 % pacientů nedostávalo předtím docetaxel, protože byli považováni za nevhodné nebo odmítli léčbu docetaxelem,
• 46 % pacientů nemělo žádnou bolest nebo bolest stupně 1 na škále WHO (asymptomatičtí nebo mírně symptomatičtí) a 54 % mělo bolest stupně 2-3 na škále WHO,
• 16 % pacientů mělo < 6 kostních metastáz, 44 % pacientů mělo mezi 6 a 20 kostními metastázami,
40 % pacientů mělo více než 20 kostních metastáz nebo superscan.
Během léčebné fáze dostalo 83 % pacientů agonisty hormonu uvolňujícího luteinizační hormon (LHRH) a 26 % pacientů dostávalo konkomitantně antiandrogeny.
Výsledky jak průběžné, tak aktualizované analýzy odhalily, že celkové přežití bylo významně delší u pacientů léčených přípravkem Xofigo plus nejlepší standardní péčí ve srovnání s pacienty, kteří dostávali placebo plus nejlepší standardní péči (viz tabulka 2 a obrázek 2). Ve skupině s placebem byl pozorován vyšší výskyt úmrtí z jiné příčiny než karcinom prostaty (26/541, 4,8 % v ramenu s přípravkem Xofigo ve srovnání s 23/268, 8,6 % v ramenu s placebem).
|
Xofigo |
Placebo | |
|
Průběžná analýza |
N = 541 |
N = 268 |
|
Počet (%) úmrtí |
191 (35,3 %) |
123 (45,9 %) |
|
Střední doba přežití (měsíce) (95% CI) |
14,0 (12,1-15,8) |
11,2 (9,0-13,2) |
|
Poměr rizikb (95% CI) |
0,695 (0,552-0,875) | |
|
p-hodnotaa (dvoustranná) |
0,00185 | |
|
Aktualizovaná analýza |
N = 614 |
N = 307 |
|
Počet (%) úmrtí |
333 (54,2 %) |
195 (63,5 %) |
|
Střední doba přežití (měsíce) (95% CI) |
14,9 (13,9-16,1) |
11,3 (10,4-12,8) |
|
Poměr rizikb (95% CI) |
0,695 (0,581-0,832) | |
CI = interval spolehlivosti
a Studie ALSYMPCA fáze 3 byla ukončena pro účinnost po průběžné analýze. Aktualizovaná analýza je uvedena pouze pro deskriptivní účely, p-hodnota není uvedena. b Poměr rizik (Xofigo vs placebo) < 1 ve prospěch přípravku Xofigo.
Obrázek 2: Kaplan-Meierovy křivky celkového přežití (aktualizovaná analýza)
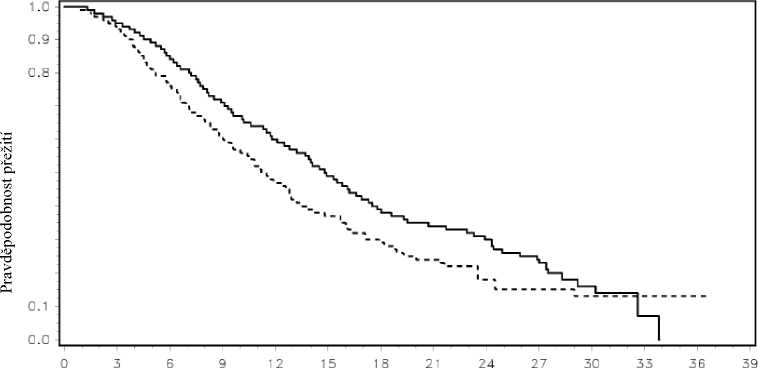
Čas (měsíce)
Léčba_ Xofigo ........Placebo
Počet pacientů v riziku
Xofigo 614 578 504 369 277 178 105 60 41 18 7 1 0 0
Placebo 307 288 228 157 104 67 39 24 14 7 4 2 1 0
Výsledky průběžné analýzy a aktualizovaná analýza také ukázaly významné zlepšení ve všech hlavních sekundárních cílových ukazatelích v rameni s přípravkem Xofigo ve srovnání s ramenem placeba (viz tabulka 3). Údaje doby do příhody pro progresi ALP byly podpořeny statisticky významnou výhodou s ohledem na normalizaci ALP a odpovědi ALP v týdnu 12.
Tabulka 3: Sekundární cílové ukazatele účinnosti z klinické studie fáze III ALSYMPCA (průběžná analýza)
|
Incidence [počet (%) pacientů] Xofigo Placebo N = 541 N = 268 |
Analýza doby do příhody (95% CI) [medián počet měsíců] Poměr Xofigo Placebo rizik N = 541 N = 268 < } ve P^h přípravku Xofigo |
p-hodnota | ||||||
|
Symptomatická kostní příhoda (SSE) |
SSE kompozitní cílový ukazatel a |
132 (24,4 %) |
82 (30,6 %) |
13,5 (122-19,6) |
8,4 (7,2 - NEý |
0,610 (0,461 - 0,807) |
0,00046 | |
|
Zevní radioterapie pro úlevu od bolesti |
122 (22,6 %) |
72 (26,9 %) |
17,0 (129-NE) |
10,8 (7,9 - NE) |
0,649 (0,483 - 0,871) |
0,00375 | ||
|
Složky SSE |
Komprese míchy |
17 (3,1 %) |
16 (6,0 %) |
NE |
NE |
0,443 (0,223 - 0,877) |
0,01647 | |
|
Chirurgická intervence |
9 (1,7 %) |
5 (1,9 %) |
NE |
NE |
0,801 (0,267 - 2,398) |
0,69041 | ||
|
Fraktury kostí |
20 (3,7 %) |
18 (6,7 %) |
NE |
NE |
0,450 (0,236 - 0,856) |
0,01255 | ||
|
Celková progrese dle ALP c |
79 (14,6 %) |
116 (43,3 %) |
NE |
3,7 (3,5 - 4,1) |
0,162 (0,120 - 0,220) |
< 0,00001 | ||
|
Progrese dle PSA d |
288 (53,2 %) |
141 (52,6 %) |
3,6 (3,5 - 3,7) |
3,4 (3,3 - 3,5) |
0,671 (0,546 - 0,826) |
0,00015 | ||
ALP = alkalická fosfatáza; CI = interval spolehlivosti; NE = nelze odhadnout; PSA = prostatický specifický antigen; SSE = symptomatická kostní příhoda
a Definováno j ako výskyt čehokoli z následujících: zevní radioterapie pro úlevu od bolesti nebo patologická fraktura nebo komprese míchy nebo ortopedicko-chirurgická intervence související s nádorem b bez možnosti odhadu díky nedostatečným příhodám po mediánu c Definované j ako > 25% zvýšení ve srovnání s výchozím stavem/nadir.
d Definované jako > 25% zvýšení a zvýšení absolutní hodnoty o > 2 ng/ml ve srovnání s výchozím stavem/nadir.
Analýza přežití u podskupiny
Analýza přežití u podskupiny ukázala konzistentní přínos v přežití u léčby přípravkem Xofigo nezávisle na celkové alkalické fosfatáze (ALP), konkomitantním podávání bisfosfonátů při randomizaci a před použitím docetaxelu.
Kvalita života
Kvalita života související se zdravím (HRQOL) byla hodnocena v klinické studii fáze III ALSYMPCA pomocí specifických dotazníků: EQ-5D (obecný dotazník) a FACT-P (dotazník specifický pro karcinom prostaty). U obou skupin došlo ke zhoršení kvality života. Při léčbě přípravkem Xofigo v porovnání s placebem byl pokles kvality života během léčebného období pomalejší, což bylo hodnoceno pomocí EQ-5D utility index score (-0,040 versus - 0,109; p=0,001), EQ-5D self-reported Visual Analogue health status scores (VAS) (-2,661 versus -5,860; p=0,018) a FACT P total score (-3,880 versus -7,651, p= 0,006), avšak nebylo dosaženo publikovaných minimálně významných rozdílů. Existují pouze omezené důkazy, že pomalejší zhoršování HRQOL přesahuje léčebné období.
Úleva od bolesti
Výsledky z klinické studie fáze III ALSYPMCA týkající se doby do léčby zevní radioterapií pro úlevu od bolesti (EBRT) a menšího množství pacientů udávajících bolest kostí jako nežádoucí příhodu ve skupině přípravku Xofigo ukazují na pozitivní účinek na bolest kostí.
Následná léčba cytotoxickými látkami
V průběhu studie ALSYMPCA s randomizací 2:1 dostalo 93 (17 %) pacientů ve skupině přípravku Xofigo a 54 (16,8 %) pacientů ve skupině placeba cytotoxickou chemoterapii v různých dobách po poslední léčbě. Mezi dvěma skupinami nebyly pozorovány žádné rozdíly v hematologických laboratorních hodnotách.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xofigo u všech podskupin pediatrické populace v indikaci karcinom prostaty (s výjimkou rabdomyosarkomu) (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Obecný úvod
Údaje o farmakokinetice, biodistribuci a dozimetrii byly získány ze tří studií fáze I. Farmakokinetické údaje byly získány od 25 pacientů při aktivitách od 51 do 276 kBq/kg. Údaje o farmakokinetice, biodistribuci a dozimetrii byly získány od 6 pacientů při aktivitě 110 kBq/kg podávané dvakrát s odstupem 6 týdnů a od 10 pacientů při aktivitě 55, 110 nebo 221 kBq/kg.
Absorpce
Přípravek Xofigo se podává v intravenózní injekci a proto má 100 % biologickou dostupnost.
Distribuce a absorpce v orgánech
Po intravenózní injekci je radium-223 rychle eliminováno z krve a ukládá se primárně do kostí a kostních metastáz nebo se vylučuje do střeva.
Patnáct minut po aplikaci injekce zůstalo asi 20 % podané aktivity v krvi. Za 4 hodiny zůstalo v krvi asi 4 % podané aktivity a 24 hodin po aplikaci injekce aktivita klesla na méně než 1 %. Distribuční objem byl vyšší než objem krve, což ukazuje na distribuci do periferních kompartmentů.
10 minut po aplikaci injekce byla aktivita pozorována v kostech a ve střevě. Úroveň aktivity v kostech byla 4 hodiny po injekci 44 % až 77 %.
Za 4 hodiny po aplikaci injekce nebylo pozorováno žádné významné vychytávání v jiných orgánech, jako jsou srdce, játra, ledviny, močový měchýř a slezina.
Biotransformace
Radium-223 je izotop, který se rozpadá a není metabolizován. Eliminace
Eliminace ve stolici je hlavní cestou eliminace z těla. Asi 5 % je vyloučeno v moči a neexistuje žádný důkaz vylučování hepatobiliární cestou.
Celotělové měření 7 dnů po aplikaci injekce (po úpravě vzhledem k rozpadu) ukazuje, že průměrně 76 % podané aktivity bylo vyloučeno z těla. Rychlost eliminace radia-223 dichloridu z gastrointestinálního traktu je ovlivněna vysokou variabilitou intestinálního tranzitu napříč populací s normálním rozmezím vyprazdňování jednou denně až jednou týdně.
Linearita/nelinearita
Farmakokinetika radia-223 dichloridu byla lineární v hodnoceném rozmezí aktivity (51 až 276 kBq/kg). Pediatrická populace
Bezpečnost a účinnost přípravku Xofigo nebyla hodnocena u dětí a dospívajících do 18 let věku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Systémová toxicita
Ve studiích toxicity po jednorázovém a opakovaném podání u potkanů byly hlavními nálezy snížení nárůstu tělesné hmotnosti, hematologické změny, snížení alkalické fosfatázy v séru a mikroskopické nálezy v kostní dřeni (deplece hematopoetických buněk, fibróza), slezině (sekundární extramedulární hematopoeza) a kostech (deplece osteocytů, osteoblastů, osteoklastů, fibro-kostní léze, disrupce/rozpad růstových plotének/růstové linie). Tato zjištění souvisela s poruchou hematopoezy indukovanou radiací a snížením osteogeneze a začínala na nejnižší aktivitě 22 kBq na kilogram tělesné hmotnosti (0,4 násobek klinicky doporučené dávky).
U psů byly pozorovány hematologické změny od nejnižší aktivity 55 kBq/kg, což je klinicky doporučená dávka. Po jednorázovém podání dávky 497 kBq radia-223 dichloridu na kilogram tělesné hmotnosti (9 násobek klinicky doporučené aktivity) byla u psů pozorována myelotoxicita limitující dávku.
Po opakovaném podání klinicky doporučené aktivity 55 kBq na kilogram tělesné hmotnosti jednou za 4 týdny po dobu 6 měsíců vznikly u dvou psů nedislokované fraktury pánve. Vzhledem k přítomnosti osteolýzy trabekulární kosti různého rozsahu v jiných oblastech kostí léčených zvířat nemohou být spontánní fraktury v souvislosti s osteolýzou vyloučeny. Klinický význam těchto nálezů je nejasný.
Po jednorázové injekci aktivity 166 a 497 kBq na kilogram tělesné hmotnosti (3 a 9 násobek klinicky doporučené dávky) bylo u psů pozorováno odchlípení sítnice, ale nikoli po opakovaném podání klinicky doporučené aktivity 55 kBq na kilogram tělesné hmotnosti každé 4 týdny po dobu 6 měsíců. Přesný mechanismus indukce odchlípení sítnice není znám, ale literární údaje naznačují, že radium se specificky vychytává v tapetum lucidum oka psů. Protože u člověka není tapetum lucidum přítomno, klinický význam tohoto zjištění u člověka je nejistý. V klinických studiích nebyl hlášen žádný případ odchlípení sítnice.
Nebyly pozorovány žádné histologické změny v orgánech, které se účastní vylučování radia-223 dichloridu.
Osteosarkomy, známý efekt radionuklidů s afinitou ke kostem, byly pozorovány při klinicky relevantních dávkách u potkanů 7-12 měsíců po zahájení léčby. Osteosarkomy nebyly pozorovány ve studiích u psů. V klinických studiích s přípravkem Xofigo nebyl hlášen žádný případ osteosarkomu. Riziko vývoje osteosarkomu u pacientů exponovaných radiu-223 není v současné době známo. V dlouhodobých (12 až 15 měsíců) studiích toxicity u potkanů byl také hlášen výskyt neoplastických změn jiných než je osteosarkom (viz bod 4.8).
Embryotoxicita/reprodukční toxicita
Studie reprodukční a vývojové toxicity nebyly provedeny. Radionuklidy obecně mají vliv na reprodukci a vývoj.
Minimální počet abnormálních spermatocytů byl pozorován v několika semenotvorných kanálcích varlat u samců potkanů po jednorázovém podání dávky > 2270 kBq radia-223 dichloridu/kg tělesné hmotnosti (> 41 násobek klinicky doporučené aktivity). Jinak se zdálo, že varlata fungují normálně a nadvarlata obsahovala normální množství spermatocytů. Děložní polypy (endometriální stroma) byly pozorovány u samic potkanů po jednorázovém nebo opakovaném podání dávky > 359 kBq radia-223 dichloridu/kg tělesné hmotnosti (> 6,5 násobek klinicky doporučené aktivity).
Protože radium-223 se distribuuje hlavně v kostech, je možné riziko nežádoucích účinků na mužské gonády u onkologických pacientů s kastračně rezistentním karcinomem prostaty velmi nízké, ale nelze jej vyloučit (viz bod 4.6).
Genotoxicita/karcinogenita
Studie mutagenního a karcinogenního potenciálu u přípravku Xofigo nebyly provedeny. Radionuklidy jsou obecně považovány za genotoxické a karcinogenní.
Bezpečnostní farmakologie
Po jednorázovém podání aktivity od 497 do 1100 kBq na kilogram tělesné hmotnosti (9 [pes] až 20 [potkan] násobek klinické doporučené aktivity) nebyly pozorovány žádné významné účinky na životně důležité orgánové systémy, tj. kardiovaskulární (pes), respirační nebo centrální nervový systém (potkan).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Voda na injekci Citrát sodný Chlorid sodný
Kyselina chlorovodíková, zředěná
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
28 dnů
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Uchovávání přípravku Xofigo by mělo být v souladu s národními předpisy pro radioaktivní látky.
6.5 Druh obalu a obsah balení
Bezbarvá skleněná injekční lahvička typu I uzavřená šedým brombutylovým pryžovým uzávěrem s fólií z ethylen-tetrafluorethylenu (ETFE) a hliníkovým pertlem, obsahující 6 ml injekčního roztoku.
Injekční lahvička je uložena v olověné nádobě.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obecná upozornění
Radiofarmaka by měla být přijímána, používána a podávána pouze oprávněnými osobami v určených klinických podmínkách. Jejich příjem, uchovávání, používání, manipulace a likvidace podléhají předpisům a/nebo příslušným povolením kompetentního úředního orgánu.
Manipulace s přípravkem Xofigo by měla být prováděna tak, aby odpovídala jak požadavkům na radiační bezpečnost, tak požadavkům na farmaceutickou kvalitu. Měla by být použita vhodná aseptická opatření.
Radiační ochrana
Gama záření související s rozpadem radia-223 a jeho rozpadových produktů umožňuje měření radioaktivity přípravku Xofigo a detekci kontaminace pomocí standardních nástrojů.
Podávání radiofarmak představuje riziko pro ostatní osoby v důsledku zevního ozáření nebo kontaminace močí, stolicí, zvratky apod. Musí být proto přijata opatření pro radiační ochranu v souladu s národními a lokálními předpisy. Při manipulaci s materiály, jako je ložní prádlo, které přicházejí do kontaktu s takovými tělesnými tekutinami, je třeba postupovat s opatrností. Ačkoli je radium-223 převážně alfa zářič, s rozpadem radia-223 a jeho radioaktivními rozpadovými izotopy je spojeno gama a beta záření. Expozice externímu záření související s manipulací s dávkami pro pacienta je podstatně nižší ve srovnání s dalšími radiofarmaky určenými pro terapeutické použití, protože podávaná radioaktivita bude obvykle nižší než 8 MBq. Avšak pro minimalizaci radiační expozice je v souladu s principem ALARA („As Low As Reasonably Achievable“) doporučováno minimalizovat dobu strávenou v radiačních prostorech, maximalizovat vzdálenost od radiačních zdrojů a používat odpovídající stínění.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. S veškerým materiálem použitým při přípravě nebo podání přípravku Xofigo je třeba nakládat jako s radioaktivním odpadem.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlín Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/873/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. listopad 2013
10. DATUM REVIZE TEXTU
11. DOZIMETRIE
Výpočet absorbované radiační dávky byl proveden na základě klinických údajů o biodistribuci. Výpočty absorbovaných dávek byly provedeny pomocí programu OLINDA/EXM (Organ Level INternal Dose Assessment/EXponential Modeling) využívajícího algoritmus Medical Internal Radiation Dose (MIRD), který je široce používán u stanovených radionuklidů emitujících beta a gama záření. U radia-223, jako primárního alfa zářiče, byly provedeny další výpočty pro střevo, červenou kostní dřeň a kostní/osteogenní buňky s cílem poskytnout nejlepší možný výpočet absorbované dávky pro přípravek Xofigo při zvážení jeho pozorované biodistribuce a specifických vlastností (viz tabulka 4).
Tabulka 4: Vypočtené absorbované radiační dávky pro orgány
|
Cílový orgán |
Alfa1 |
Beta |
Gama |
Celková |
Variační |
|
záření |
záření |
záření |
dávka |
koeficient | |
|
(Gy/MBq) |
(Gy/MBq) |
(Gy/MBq) |
(Gy/MBq) |
(%) | |
|
Nadledviny |
0,00000 |
0,00002 |
0,00009 |
0,00012 |
56 |
|
Mozek |
0,00000 |
0,00002 |
0,00008 |
0,00010 |
80 |
|
Prsa |
0,00000 |
0,00002 |
0,00003 |
0,00005 |
120 |
|
Stěna žlučníku |
0,00000 |
0,00002 |
0,00021 |
0,00023 |
14 |
|
LLI2 stěna |
0,00000 |
0,04560 |
0,00085 |
0,04645 |
83 |
|
Stěna tenkého střeva |
0,00319 |
0,00360 |
0,00047 |
0,00726 |
45 |
|
Stěna žaludku |
0,00000 |
0,00002 |
0,00012 |
0,00014 |
22 |
|
ULI3 stěna |
0,00000 |
0,03150 |
0,00082 |
0,03232 |
50 |
|
Srdeční stěna |
0,00161 |
0,00007 |
0,00005 |
0,00173 |
42 |
|
Ledviny |
0,00299 |
0,00011 |
0,00011 |
0,00320 |
36 |
|
Játra |
0,00279 |
0,00010 |
0,00008 |
0,00298 |
36 |
|
Plíce |
0,00000 |
0,00002 |
0,00005 |
0,00007 |
90 |
|
Sval |
0,00000 |
0,00002 |
0,00010 |
0,00012 |
41 |
|
Vaječníky |
0,00000 |
0,00002 |
0,00046 |
0,00049 |
40 |
|
Slinivka |
0,00000 |
0,00002 |
0,00009 |
0,00011 |
43 |
|
Červená kostní dřeň |
0,13200 |
0,00642 |
0,00020 |
0,13879 |
41 |
|
Osteogenní buňky |
1,14000 |
0,01490 |
0,00030 |
1,15206 |
41 |
|
Kůže |
0,00000 |
0,00002 |
0,00005 |
0,00007 |
79 |
|
Slezina |
0,00000 |
0,00002 |
0,00007 |
0,00009 |
54 |
|
Varlata |
0,00000 |
0,00002 |
0,00006 |
0,00008 |
59 |
|
Brzlík |
0,00000 |
0,00002 |
0,00003 |
0,00006 |
109 |
|
Štítná žláza |
0,00000 |
0,00002 |
0,00005 |
0,00007 |
96 |
|
Stěna močového měchýře |
0,00371 |
0,00016 |
0,00016 |
0,00403 |
63 |
|
Děloha |
0,00000 |
0,00002 |
0,00023 |
0,00026 |
28 |
|
Celé tělo |
0,02220 |
0,00081 |
0,00012 |
0,02311 |
16 |
'Jelikož nebyla pozorována žádná absorpce radia-223 ve většině měkkých tkání, byl podíl alfa záření vzhledem k celkové orgánové dávce nastaven pro tyto orgány na nulu.
2LLI: dolní část tlustého střeva 3ULI: horní část tlustého střeva
Hematologické nežádoucí účinky pozorované v klinických studiích s přípravkem Xofigo jsou mnohem méně četnější a závažnější, než by se dalo očekávat z vypočtené absorbované dávky pro červenou kostní dřeň. To může souviset s prostorovou distribucí záření částic alfa v důsledku nerovnoměrné radiační dávky pro červenou kostní dřeň.
12. NÁVOD PRO PŘÍPRAVU RADIOFARMAK
Tento léčivý přípravek by měl být před použitím vizuálně zkontrolován. Přípravek Xofigo je čirý, bezbarvý roztok a neměl by být použit v případě změny barvy, výskytu částic nebo poškození obalu.
Přípravek Xofigo je roztok k okamžitému použití a neměl by se ředit nebo míchat s jinými roztoky.
Každá injekční lahvička je určena pouze pro jednorázové použití.
Objem, který má být podán danému pacientovi, by měl být vypočten podle:
- Tělesné hmotnosti pacienta (kg)
- Množství dávky (55 kBq/kg tělesné hmotnosti)
Radioaktivní koncentrace přípravku (1100 kBq/ml) k referenčnímu datu. Referenční datum je uvedeno na štítku na injekční lahvičky a na olověné nádobě.
Korekční faktor rozpadu (DK) pro úpravu fyzikálního rozpadu radia-223. Tabulka DK faktorů je uvedena u každé injekční lahvičky jako součást brožury (před příbalovou informací).
Množství radioaktivity v rozpuštěném objemu by mělo být potvrzeno měřením ve správně kalibrovaném měřiči aktivity.
Celkový objem, který má být podán pacientovi, se vypočítá takto:
objem, který má být podán (ml)
tělesná hmotnost (kg) x aktivita (55 kBq/kg tělesné hmotnosti) DK faktor x 1100 kBq/ml
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky na likvidaci radioaktivních látek.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
A.
Název a adresa výrobce odpovědného za propouštění šarží
Bayer AS
Drammensveien 288,
NO-0283 Oslo,
Norsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVADENE NA VNEJSIM OBALU OLOVĚNÁ NÁDOBA
1. NÁZEV LECIVEHO PRIPRAVKU
Xofigo 1100 kBq/ml injekční roztok Radium-223 dichloridum
2. OBSAH LÉČIVÉ LÁTKY
Radium-223 dichloridum
3. SEZNAM POMOCNÝCH LÁTEK
Voda na injekci, citrát sodný, chlorid sodný, kyselina chlorovodíková. Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 6 ml
1100 kBq/ml ve 12 hodin (SEČ) k referenčnímu datu: [DD/MM/RRRR]
6,6 MBq/injekční lahvičku ve 12 hodin (SEČ) k referenčnímu datu: [DD/MM/RRRR]
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ

8. POUŽITELNOST
EXP
9. zvláštní podmínky pro uchováváni
Uchovávání by mělo být v souladu s národními předpisy pro radioaktivní látky.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LECIVYCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlín Německo
[Bayer logo]
12. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/13/873/001
13. ČÍSLO ŠARŽE
č.š.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Xofigo 1100 kBq/ml injekční roztok Radium-223 dichloridum Intravenózní podání.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6 ml
6,6 MBq/injekční lahvičku ve 12 hodin (SEČ) k referenčnímu datu: [DD/MM/RRRR]
6. JINÉ

[Bayer logo]
B. PŘÍBALOVÁ INFORMACE
Xofigo 1100 kBq/ml injekční roztok.
Radium-223 dichloridum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, který dohlíží na proceduru.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Xofigo a k čemu se používá
2. Čemu musíte věnovat pozornost, než bude přípravek Xofigo použit
3. Jak se přípravek Xofigo používá
4. Možné nežádoucí účinky
5. Jak přípravek Xofigo uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xofigo a k čemu se používá
Tento léčivý přípravek obsahuje léčivou látku radium-223 dichloridum (radium-223 dichlorid, radium Ra 223 dichloride).
Přípravek Xofigo se používá pro léčbu dospělých s pokročilým kastračně rezistentním karcinomem prostaty. Jedná se o nádor prostaty (žláza, která je součástí pohlavního ústrojí mužů), který neodpovídá na léčbu, která snižuje množství mužských pohlavních hormonů. Přípravek Xofigo se používá pouze, když se onemocnění rozšířilo do kostí, ale není známo, zda se rozšířilo do jiných vnitřních orgánů a vyvolává příznaky (např. bolest).
Přípravek Xofigo obsahuje radioaktivní látku radium-223, která napodobuje vápník, který se nachází v kostech. Pokud je radium-223 podáno injekcí pacientovi, dostává se do kostí, kam se rozšířil nádor a vysílá záření krátkého dosahu (alfa částice), které usmrcuje okolní nádorové buňky.
2. Čemu musíte věnovat pozornost, než bude přípravek Xofigo použit
Přípravek Xofigo nesmí být podáván
Nejsou známa žádná onemocnění, při kterých by Vám nemohl být podán přípravek Xofigo.
Upozornění a opatření
Předtím, než Vám bude přípravek Xofigo podán, se poraďte se svým lékařem.
- Přípravek Xofigo může vést ke snížení počtu krvinek a krevních destiček. Před zahájením léčby a před každou následnou dávkou provede Váš lékař vyšetření krve. V závislosti na výsledcích těchto vyšetření Váš lékař rozhodne, zda může být léčba zahájena, zda může pokračovat nebo zda je nutné ji odložit nebo přerušit. Pokud trpíte sníženou tvorbou krevních buněk v kostní dřeni, např. pokud jste byli dříve léčeni chemoterapií (jinými léky používanými k ničení nádorových buněk) a/nebo ozařováním, může být u Vás riziko vyšší a Váš lékař Vám bude přípravek Xofigo podávat s opatrností.
- Pokud se nádor rozšířil do kostí ve velké míře, může být také pravděpodobnější, že máte snížené množství krvinek a krevních destiček a tak Vám Váš lékař bude přípravek Xofigo podávat s opatrností.
- Omezené dostupné údaje nenaznačují žádné větší rozdíly ve tvorbě krevních buněk u pacientů, kteří po léčbě přípravkem Xofigo dostávali chemoterapii, ve srovnání s těmi, kteří nebyli léčeni přípravkem Xofigo.
- Pokud máte neléčený útlak míchy nebo pokud se předpokládá, že se u Vás vyvinul útlak míchy (tlak na míšní nervy, což může být způsobeno nádorem či jiným poškozením), bude Váš lékař nejprve léčit toto onemocnění pomocí standardní léčby před zahájením nebo pokračováním léčby přípravkem Xofigo.
- Pokud se u Vás objeví zlomenina kosti, Váš lékař nejprve stabilizuje zlomenou kost před zahájením nebo pokračováním léčby přípravkem Xofigo.
- Nejsou k dispozici žádné údaje o použití přípravku Xofigo u pacientů s Crohnovou chorobou (dlouhodobé zánětlivé střevní onemocnění) a s ulcerózní kolitidou (dlouhodobý zánět tlustého střeva). Vzhledem k tomu, že se přípravek Xofigo vylučuje stolicí, může dojít ke zhoršení akutního zánětu střev. Pokud trpíte tímto onemocněním, Váš lékař pečlivě zváží, zda můžete být léčen přípravkem Xofigo.
- Pokud užíváte nebo jste užíval bisfosfonáty nebo Vám před léčbou přípravkem Xofigo byla podávána chemoterapie, sdělte to svému lékaři. Nelze vyloučit riziko osteonekrózy čelisti (odumřelá tkáň v čelistní kosti, která je pozorována především u pacientů, kteří byli léčeni bisfosfonáty) (viz bod 4).
- Přípravek Xofigo přispívá k celkové dlouhodobé kumulativní radiační expozici. Dlouhodobá kumulativní radiační expozice může zvyšovat riziko rozvoje zhoubného nádoru (především zhoubného nádoru kostí a leukémie) a vrozených abnormalit. V klinických studiích při sledování po dobu až tří let nebyly hlášeny žádné případy rakoviny způsobené přípravkem Xofigo.
Děti a dospívající
Tento přípravek není určen k použití u dětí a dospívajících.
Další léčivé přípravky a přípravek Xofigo
Nebyly provedeny žádné studie interakcí s jinými léčivými přípravky.
Pokud užíváte vápník, fosfáty a/nebo vitamin D, Váš lékař pečlivě zváží, zda je nutné, abyste před léčbou přípravkem Xofigo přestal dočasně užívat tyto látky.
Nejsou k dispozici žádné údaje o použití přípravku Xofigo zároveň s chemoterapií (jiné léky používané k ničení nádorových buněk).
Přípravek Xofigo a chemoterapie používané společně mohou způsobit větší snížení počtu krevních buněk a krevních destiček. Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval nebo které možná budete užívat, včetně léků dostupných bez lékařského předpisu.
Těhotenství a kojení
Přípravek Xofigo nemá být používán u žen a nesmí být podáván ženám, které jsou nebo mohou být těhotné nebo u kojících žen.
Antikoncepce u mužů a žen
Pokud jste sexuálně aktivní se ženou, která by mohla otěhotnět, doporučuje se, abyste použil účinnou antikoncepční metodu během a až 6 měsíců po léčbě přípravkem Xofigo.
Plodnost
Existuje možné riziko, že záření z přípravku Xofigo by mohlo ovlivnit Vaši plodnost. Zeptejte se prosím svého lékaře, jaký to může mít pro Vás vliv, zejména pokud plánujete mít v budoucnu děti. Možná se budete chtít poradit o možnosti uchování spermatu před zahájením léčby.
Řízení dopravních prostředků a obsluha strojů
Je považováno za nepravděpodobné, že přípravek Xofigo ovlivní Vaši schopnost řídit nebo obsluhovat stroje.
Přípravek Xofigo obsahuje sodík
V závislosti na podaném objemu může tento léčivý přípravek obsahovat až 54 mg sodíku v jedné dávce. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak se přípravek Xofigo používá
Pro používání, manipulaci a likvidaci léčivých přípravků jako je Xofigo existují přísné předpisy. Přípravek Xofigo bude používán pouze ve zvláštních kontrolovaných prostorech. S tímto přípravkem budou manipulovat a bude Vám podáván pouze osobami, které jsou proškolené a kvalifikované pro jeho bezpečné používání. Tyto osoby budou věnovat zvláštní pozornost bezpečnému použití tohoto přípravku a budou Vás informovat o své činnosti.
Dávka, kterou dostanete, závisí na Vaší tělesné hmotnosti. Lékař, který dohlíží na proceduru, vypočte množství přípravku Xofigo, které se ve Vašem případě použije.
Doporučená dávka přípravku Xofigo je 55 kBq (becquerel je jednotka radioaktivity) na kilogram tělesné hmotnosti.
Pokud je Vám 65 let nebo více nebo pokud máte sníženou funkci ledvin nebo jater, není nutná žádná úprava dávkování.
Podání přípravku Xofigo a postup
Přípravek Xofigo se podává pomalou injekcí jehlou do žíly (intravenózně). Zdravotnický pracovník propláchne intravenózní sondu nebo kanylu před a po aplikaci injekce fyziologickým roztokem.
Délka léčby
- Přípravek Xofigo se podává jednou za 4 týdny a celkem Vám bude podáno 6 injekcí.
- Nejsou k dispozici žádné údaje o bezpečnosti a účinnosti léčby přípravkem Xofigo s více než 6 injekcemi.
Po podání přípravku Xofigo
Při manipulaci s materiály, jako je ložní prádlo, které přicházejí do kontaktu s tělesnými tekutinami (např. s močí, stolicí, zvratky apod.), je třeba postupovat s opatrností
Přípravek Xofigo je vylučován převážně stolicí. Lékař Vám řekne, zda jsou nutná zvláštní opatření po podání tohoto léčivého přípravku. Pokud budete mít jakékoli otázky, zeptejte se svého lékaře.
Jestliže Vám bylo podáno více přípravku Xofigo, než by mělo
Předávkování je nepravděpodobné.
V případě náhodného předávkování lékař zahájí vhodnou podpůrnou léčbu a provede kontrolu počtu krevních buněk a příznaků týkajících se trávicího traktu (např. průjem, nauzea (nevolnost), zvracení).
Máte-li jakékoli další otázky týkající se používání přípravku Xofigo, zeptejte se svého lékaře, který dohlíží na proceduru.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nejzávažnějšími nežádoucími účinky u pacientů léčených přípravkem Xofigo jsou
- snížení počtu krevních destiček (trombocytopenie),
- snížení počtu neutrofilů, typu bílých krvinek (neutropenie, která může vést k zvýšenému riziku infekce).
Váš lékař provede vyšetření krve před zahájením léčby a před každou injekcí, aby zkontroloval počet krevních buněk a krevních destiček (viz také část 2).
Kontaktujte okamžitě svého lékaře, jakmile si všimnete následujících příznaků, protože by se mohlo jednat o známky trombocytopenie nebo neutropenie (viz výše):
- jakékoli neobvyklé modřiny,
- větší krvácení než je obvyklé po poranění,
- horečka,
- nebo pokud budete mít pocit, že jste náchylnější k infekcím.
Nejčastější nežádoucí účinky u pacientů léčených přípravkem Xofigo (velmi časté [mohou postihnout více než 1 osobu z 10]) jsou:
- průjem, nauzea (nevolnost), zvracení a trombocytopenie (snížení počtu krevních destiček).
Další možné nežádoucí účinky jsou uvedeny níže podle toho, jak jsou pravděpodobné:
Časté (mohou postihnout až 1 osobu z 10)
- snížení počtu bílých krvinek (leukopenie)
- snížení počtu neutrofilů, typu bílých krvinek (neutropenie, která může vést ke zvýšenému riziku infekce)
- snížení počtu červených a bílých krvinek a krevních destiček (pancytopenie)
- reakce v místě aplikace injekce (např. zarudnutí kůže, bolest a otok)
Méně časté (mohou postihnout až 1 osobu ze 100)
- snížení počtu lymfocytů, typu bílých krvinek (lymfopenie).
Přípravek Xofigo přispívá k celkové dlouhodobé kumulativní radiační expozici. Dlouhodobá kumulativní radiační expozice může zvyšovat riziko rozvoje zhoubného nádoru (především zhoubného nádoru kostí a leukémie) a vrozených abnormalit. V klinických studiích při sledování po dobu až tří let nebyly hlášeny žádné případy rakoviny způsobené přípravkem Xofigo.
Pokud máte příznaky bolesti, otoku nebo znecitlivění čelisti, “pocit těžké čelisti” nebo ztrátu zubu, prosím, kontaktujte svého lékaře. U pacientů léčených přípravkem Xofigo se vyskytly případy osteonekrózy čelisti (odumírání tkáně v čelistních kostech, které je převážně pozorováno u pacientů, kteří byli léčeni bisfosfonáty). Všechny tyto případy byly pozorovány pouze u pacientů, kteří užívali bisfosfonáty před nebo současně s léčbou přípravkem Xofigo a chemoterapií, která předcházela léčbě přípravkem Xofigo.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Xofigo uchovávat
Tento léčivý přípravek nebudete muset uchovávat. Za uchovávání tohoto léčivého přípravku ve vhodných prostorách je zodpovědný odborník. Uchovávání radiofarmak musí být v souladu s národními předpisy pro radioaktivní látky.
Následující informace jsou určeny pouze pro odborníka:
Přípravek Xofigo nesmí být používán po uplynutí doby použitelnosti, která je uvedena na injekční lahvičce a olověné nádobě.
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Přípravek Xofigo nesmí být použit, jestliže je zaznamenána změna barvy, výskyt částic nebo poškození obalu.
Co přípravek Xofigo obsahuje
Léčivou látkou je: radium-223 dichloridum (radium-223 dichlorid, radium Ra 223 dichloride).
Jeden ml roztoku obsahuje radium-223 dichloridum 1100 kBq (radium-223 dichlorid, radium Ra 223 dichloride), což odpovídá radium-223 0,58 ng k referenčnímu datu.
Jedna injekční lahvička obsahuje 6 ml roztoku (radium-223 dichloridum 6600 kBq k referenčnímu datu).
Dalšími složkami jsou: voda na injekci, citrát sodný, chlorid sodný a kyselina chlorovodíková (viz konec bodu 2 pro další informace o sodíku).
Jak přípravek Xofigo vypadá a co obsahuje toto balení
Přípravek Xofigo je čirý a bezbarvý injekční roztok. Je dodáván v bezbarvé skleněné injekční lahvičce s šedým brombutylovým pryžovým uzávěrem s fólií a hliníkovým pertlem. Injekční lahvička obsahuje 6 ml roztoku. Je uložena v olověné nádobě.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlín Německo
Výrobce
Bayer AS
Drammensveien 288,
NO-0283 Oslo,
Norsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie / Belgique / Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Btnrapnn Eaňep Etnrapna EOOfl Ten. +359 02 81 401 01 Česká republika Bayer s.r.o. Tel: +420 266 101 111 Danmark Bayer A/S Tlf: +45-45 23 50 00 Deutschland Bayer Vital GmbH Tel: +49 (0)214-30 513 48 Eesti Bayer OU Tel: +372 655 8565 EXlába Bayer Ekkág ABEE Tqk: +30 210 61 87 500 Espaňa Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél(N° vert): +33-(0)800 87 54 54 Hrvatska Bayer d.o.o. Tel: + 385-(0)1-6599 900 Ireland Bayer Limited Tel: +353 1 2999313 Island Icepharma hf. Sími: +354 540 8000 Italia Bayer S.p.A. Tel: +39 02 397 81 Kónpoq NOVAGEM Limited Tnk: +357 22 48 38 58 Latvija SIA Bayer Tel: +371 67 84 55 63 |
Lietuva UAB Bayer Tel. +37 05 23 36 868 Luxembourg / Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel.:+36 14 87-41 00 Malta Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf. +47 23 13 05 00 Osterreich Bayer Austria Ges. m. b. H. Tel: +43-(0)1-711 46-0 Polska Bayer Sp. z o.o. Tel.: +48 22 572 35 00 Portugal Bayer Portugal, Lda. Tel: +351 21 416 42 00 Románia SC Bayer SRL Tel: +40 21 529 59 00 Slovenija Bayer d. o. o. Tel.: +386 (0)1 58 14 400 Slovenská republika Bayer, spol. s r.o. Tel: +421 2 59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358 20 785 21 Sverige Bayer AB Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc Tel: +44 (0)1635 563000 |
Tato příbalová informace byla naposledy revidována{ MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Kompletní Souhrn údajů o přípravku Xofigo je dodáván jako oddělitelná součást na konci tištěného letáku v balení s přípravkem s cílem poskytnout odborným zdravotnickým pracovníkům další odborné a praktické informace o podávání a použití tohoto radiofarmaka.
31