Xeomin 200 Jednotek Prášek Pro Injekční Roztok
sp.zn.sukls116668/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
XEOMIN 200 jednotek prášek pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje Clostridium botulini neurotoxinum typus A 200 jednotek (150 kD), bez komplexotvorných proteinů*.
* Botulini toxinum typus A sine complex proteine, očištěný od kultur Clostridium botulinum (Hall kmen)
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro injekční roztok Bílý prášek
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
XEOMIN je indikován u dospělých k symptomatické léčbě blefarospasmu, cervikální dystonie převážně rotační formy (torticollis spastica) a spasticity horních končetin po cévní mozkové příhodě projevující se flektovaným zápěstím a sevřenou pěstí.
4.2 Dávkování a způsob podání
Kvůli rozdílům v jednotkách v analýze účinnosti nejsou jednotkové dávky pro přípravek XEOMIN zaměnitelné s jinými přípravky obsahujícími botulotoxin.
Podrobné informace týkající se klinických studií porovnávajících přípravek XEOMIN s konvenčním botulotoxinovým komplexem typu A (900 kD) jsou uvedeny v bodě 5.1.
Obecné
XEOMIN smí podávat pouze lékaři s vhodnou kvalifikací a potřebnou zkušeností s aplikací botulotoxinu.
Optimální dávku a počet míst vpichu v léčeném svalu určí lékař u každého pacienta individuálně. Je nutné provést titraci dávky.
Dávkování
Blefarospasmus
Doporučená počáteční dávka je 1,25 až 2,5 jednotek do jednoho injekční místo. Počáteční dávka nesmí překročit 25 jednotek na oko. Celková dávka nesmí překročit 100 jednotek každých 12 týdnů. Intervaly mezi léčebnými sezeními mají být určeny na základě aktuální klinické potřeby u daného pacienta.
V průměru je první nástup účinku pozorován během čtyř dnů po aplikaci. Účinek léčby přípravkem XEOMIN trvá obvykle 3 až 4 měsíce, ovšem může trvat významně delší nebo kratší dobu. Léčbu lze v případě potřeby opakovat.
Při opakovaných léčebných sezeních lze dávku zvyšovat až dvojnásobně, pokud je odezva na počáteční léčbu považována za nedostatečnou. Přesto se zdá, že aplikování více než 5,0 jednotek do jednoho místa vpichu nepřináší další výhody.
Torticollis spastica
Při léčbě spastické tortikolis musí být dávkování přípravku XEOMIN přizpůsobené konkrétnímu pacientovi na základě polohy hlavy a krku pacienta, místa možné bolesti, svalové hypertrofie, tělesné hmotnosti pacienta a odezvy na injekci.
V prvním cyklu léčby nemá být podáno více než 200 jednotek, přičemž dávka se upraví v dalších cyklech dle odpovědi. Při jednom sezení se nesmí překročit celková dávka 300 jednotek. Do žádného místa vpichu se nesmí aplikovat více než 50 jednotek.
V průměru je první nástup účinku pozorován během sedmi dnů po aplikaci. Účinek léčby přípravkem XEOMIN trvá obvykle 3 až 4 měsíce, ovšem může trvat významně delší nebo kratší dobu. Nedoporučují se kratší intervaly mezi léčebnými sezeními než 10 týdnů. Intervaly mezi léčebnými sezeními mají být určeny na základě aktuální klinické potřeby u daného pacienta.
Spasticita horních končetin po cévní mozkové příhodě
Velikost dávky a počet vpichů je třeba individuálně upravit na základě velikosti, počtu a lokalizace postižených svalů, míry spasticity a přítomnosti lokální svalové slabosti.
Doporučené úvodní dávky:
|
Klinický symptom |
Jednotky |
|
Sval | |
|
Flektované zápěstí | |
|
M. flexor carpi radialis |
50 |
|
M. flexor carpi ulnaris |
40 |
|
Zaťatá pěst | |
|
M. flexor digitorum superficialis |
40 |
|
M. fflexor digitorum profundus |
40 |
|
Flektovaný loket | |
|
M. brachioradialis |
60 |
|
M. biceps brachii |
80 |
|
M. brachialis |
50 |
|
Pronace předloktí | |
|
M. pronator quadratus |
25 |
|
M. pronator teres |
40 |
|
Palec v dlani | |
|
M. flexor pollicis longus |
20 |
|
M. adductor pollicis |
10 |
|
M. flexor pollicis brevis/ |
10 |
|
M. opponens pollicis |
V pivotních klinických studiích byly použity minimální celkové dávky 170 jednotek a maximální celkové dávky 400 jednotek na sezení.
Doporučené dávky pro opakovanou léčbu:
|
Klinický symptom |
Jednotky |
Počet míst |
|
Sval |
(rozmezí) |
vpichu na sval |
|
Flektované zápěstí | ||
|
M. flexor carpi radialis |
25-100 |
1-2 |
|
M. f flexor carpi ulnaris |
20-100 |
1-2 |
|
Zaťatá pěst | ||
|
M. flexor digitorum superficialis |
40-100 |
2 |
|
M. flexor digitorum profundus |
40-100 |
2 |
|
Flektovaný loket | ||
|
M. brachioradialis |
25-100 |
1-3 |
|
M. biceps brachii |
75-200 |
1-4 |
|
M. brachialis |
25-100 |
1-2 |
|
Pronace předloktí | ||
|
M. pronator quadratus |
10-50 |
1 |
|
M. pronator teres |
25-75 |
1-2 |
|
Palec v dlani | ||
|
M. flexor pollicis longus |
10-50 |
1 |
|
M. adductor pollicis |
5-30 |
1 |
|
M. flexor pollicis brevis/ |
5-30 |
1 |
|
M. opponens pollicis |
Maximální doporučená celková dávka je 400 jednotek na sezení.
Pacienti hlásili zlepšení 4 dny po aplikaci. Maximální účinek zlepšení svalového tonu byl pozorován během 4 týdnů. Obvykle léčebný účinek přetrvává 12 týdnů. Opakovaná léčba nemá být prováděna častěji než každých 12 týdnů.
Všechny indikace
Jestliže se neobjeví žádný léčebný účinek během jednoho měsíce po první injekci, je nutné přijmou následující opatření:
- Klinické ověření účinku neurotoxinu na příslušný sval: např. Elektromyografické vyšetření ve specializovaném zařízení
- Analýza příčin selhání, např. Špatný výběr svalů, nedostatečná dávka, špatná injekční technika, výskyt fixní kontraktury, příliš oslabený svalový antagonista, tvorba toxin-neutralizujících protilátek
- Přehodnocení vhodnosti léčby botulotoxinem typu A
- Pokud první léčebná kúra nebyla spojena s výskytem nežádoucích účinků, lze zahájit další léčbu podle následujícího doporučení: 1) úprava dávky se zohledněním příčin selhání předcházející léčby; 2) použití EMG a 3) zachování minimálního intervalu mezi dvěma léčebnými kúrami.
Pediatrická populace
Bezpečnost a účinnost přípravku XEOMIN u dětí ve věku 0-17 let nebyly ověřeny. Proto není přípravek XEOMIN doporučen pro pediatrickou populaci, dokud nebudou údaje doplněny.
Způsob podání
Rekonstituovaný XEOMIN je určen k intramuskulární injekci.
Blefarospasmus
Po rekonstituci se roztok přípravku XEOMIN injikuje vhodnou sterilní jehlou (např. 27-30 gauge / 0,30-0,40 mm). Aplikaci není nutno provádět pod elektromyografickou kontrolou. Doporučuje se objem injekce přibližně 0,05 až 0,1 ml.
XEOMIN se aplikuje do m. orbicularis oculi, mediálně a laterálně na horním víčku a laterálně na dolním víčku. Může se aplikovat také do oblasti obočí, do laterální části m. orbicularis oculi nebo do horní části tváře, pokud zde spasmy ruší vidění.
Torticollis spastica
Vhodná sterilní jehla (např. 25-30 gauge (0,30-0,50 mm) se používá pro injekce do povrchových svalů a např. jehla 22 gauge / 0,70 mm může být použita pro injekce do hlubší svaloviny. Doporučuje se objem injekce přibližně 0,1 až 0,5 ml na jedno místo vpichu.
Při léčbě spastické tortikolis se XEOMIN aplikuje do m. stemocleidomastoideus, m. levator scapulae, m. scalenus, m. splenius capitis a/nebo m. trapezius Tento seznam není úplný a nezahrnuje všechny svaly zodpovědné za polohu hlavy, které může být třeba léčit. V případě potíží při lokalizaci jednotlivých svalů je třeba injekce aplikovat pod elektromyografickou kontrolou. Svalová hmota a stupeň hypertrofie či atrofie jsou faktory, které je nutno vzít v úvahu při volbě vhodné dávky.
Více míst vpichu umožňuje přípravku XEOMIN rovnoměrnější působení v inervovaných oblastech dystonického svalu, to je zvláště užitečné u větších svalů. Optimální počet míst vpichu závisí na velikosti svalu, který má být chemicky denervován.
Injekce se nemá podávat bilaterálně do m. stemocleidomastoideus, protože při bilaterární aplikaci nebo aplikaci dávek celkově překračujících 100 jednotek se zvyšuje riziko nežádoucích účinků (zejména dysfagie).
Spasticita horních končetin po cévní mozkové příhodě
Rekonstituovaný XEOMIN se aplikuje vhodnou sterilní jehlou (např. 26 gauge / 0,45 mm průměr /
37 mm délka pro povrchové svaly a např. 22 gauge / 0,7 mm průměr / 75 mm délka pro hlubší muskulaturu).
V případě obtíží při izolaci jednotlivých svalů mají být injekce podávány s použitím elektromyografické kontroly. Aplikace injekcí přípravku XEOMIN do více míst umožní rovnoměrný kontakt s inervovanou oblastí svalu, což je zvláště užitečné u velkých svalů.
Všechny indikace
Pokyny pro rekonstituci léčivého přípravku před podáním a pokyny pro likvidaci injekčních lahviček jsou uvedeny v bodě 6.6. Po rekonstituci je nutné použít XEOMIN pouze pro jedno sezení a pouze pro jednoho pacienta.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Generalizované poruchy svalové aktivity (např. myasthenia gravis, Lambert-Eatonův syndrom).
• Infekce či zánět v navrhovaném místě aplikace.
4.4 Zvláštní upozornění a opatření pro použití
Obecné
Před podáním přípravku XEOMIN se musí lékař nejprve seznámit s anatomií pacienta a změnami vyvolanými předchozími chirurgickými výkony.
Je třeba dbát na to, aby XEOMIN nebyl podán do cévy. Při léčbě cervikální dystonie a spasticity po cévní mozkové příhodě je zapotřebí zvláštní opatrnosti, jsou-li místa vpichu v blízkosti citlivých struktur, například karotid, plicních hrotů a jícnu.
XEOMIN se má používat s opatrností:
• jestliže se objeví poruchy krvácivosti jakéhokoliv typu
• u pacientů léčených antikoagulancii nebo jinými látkami, které by mohly mít antikoagulační účinek.
Doporučené jednotlivé dávky přípravku XEOMIN se nemají překračovat.
Dříve nepohyblivé pacienty a pacienty, kteří před léčbou vedli sedavý způsob života, je třeba upozornit, aby aktivitu po aplikaci přípravku XEOMIN obnovovali postupně.
Klinické účinky botulotoxinu typu A mohou být zvýšeny nebo sníženy opakovanými injekcemi. Možnými důvody jsou různé postupy rekonstituce, zvolené intervaly mezi aplikacemi, použitý sval, lehké odchylky v aktivitě toxinu dané použitými metodami biologického testování nebo sekundární ztráta odpovědi.
Účinky lokálního a vzdáleného šíření toxinu
Nežádoucí účinky mohou nastat u chybně umístěných injekcí botulotoxinu typu A s dočasnou paralýzou okolních skupin svalů. Velké dávky mohou způsobit paralýzu svalů vzdálených od místa vpichu.
Byly hlášeny nežádoucí účinky, které mohly souviset se šířením botulotoxinu do vzdálených míst od místa vpichu injekce (viz bod 4.8). Některé z těchto účinků byly život ohrožující a byly hlášeny i případy úmrtí, které byly v některých případech spojeny s dysfagií, pneumonií a/nebo značnou tělesnou slabostí.
Dysfagie byla hlášena též po aplikaci do jiných míst než krčních svalů.
Stávající neuromuskulární poruchy
U pacientů léčených terapeutickými dávkami se může projevit nadměrná svalová slabost. Pacienti s neuromuskulárními poruchami mohou být ve zvýšené míře ohroženi nadměrnou svalovou slabostí.
U takových pacientů je nutné botulotoxinový přípravek používat pouze za dohledu specialisty a pouze pokud prospěch z léčby převáží riziko. Pacienty s dysfagií a aspirací v anamnéze je nutno léčit se zvláštní opatrností.
Pacienti nebo jejich ošetřovatelé mají být upozorněni, aby vyhledali okamžitě lékařskou pomoc, pokud se vyskytnou poruchy polykání, řeči nebo dýchání.
XEOMIN se má používat s opatrností:
• u pacientů trpících amyotrofickou laterální sklerózou
• u pacientů s jinými onemocněními, která způsobují periferní neuromuskulární dysfunkci
• v cílových svalech, které vykazují značnou slabost nebo atrofii.
Hypersenzitivní reakce
Při použití přípravků obsahujících botulotoxin byly hlášeny hypersenzitivní reakce. Pokud se vyskytnou závažné (např. anafylaktické reakce) a/nebo okamžité hypersenzitivní reakce, je třeba nasadit vhodnou léčbu.
Tvorba protilátek
Příliš časté dávky mohou zvýšit riziko tvorby protilátek, což může vést k selhání léčby (viz bod 4.2). Potenciál pro tvorbu protilátek je možno minimalizovat podáváním nejnižší účinné dávky v co nejdelších intervalech mezi injekcemi tak, jak je klinicky indikováno.
Indikace
Blefarospasmus
Je třeba se vyvarovat aplikace injekce do blízkosti m. levator palpebrae superioris, aby se snížil výskyt ptózy. V důsledku difuze botulotoxinu typu A do m. obliquus inferior může dojít k diplopii. Vyvarováním se injekce do mediální části dolního víčka lze snížit riziko tohoto nežádoucího účinku.
Kvůli anticholinergnímu účinku botulotoxinu typu A se má přípravek XEOMIN používat s opatrností u pacientů s rizikem rozvoje glaukomu s uzavřeným úhlem.
Aby se zabránilo ektropiu, je nutné se vyhnout aplikaci do dolní části víčka a je nezbytná důkladná léčba každého defektu rohovkového epitelu. Tato léčba může zahrnovat aplikaci ochranných kapek, mastí, terapeutických měkkých kontaktních čoček nebo zakrytí oka přelepením nebo jiným způsobem.
Snížená četnost mrkání po injekci přípravku XEOMIN do m. orbicularis oculi může vést ke snížení ochrany rohovky, trvalému poškození epitelu a ulceraci rohovky, obzvláště u pacientů s poruchou hlavového nervu (n. facialis). U pacientů po operaci očí je třeba zajistit pečlivé vyšetření citlivosti rohovky.
V měkkých tkáních víčka snadno vznikají ekchymózy. Ty mohou být minimalizovány působením jemného tlaku na místo vpichu bezprostředně po injekci.
Torticollis spastica
Pacienti s cervikální dystonií léčení přípravkem XEOMIN mají být informováni o možném vzniku dysfagie, která může být mírná, ale i závažná, s rizikem aspirace a dyspnoe. Může být nutná lékařská intervence (např. gastrická sonda) (viz také bod 4.8). Riziko vzniku dysfagie může být sníženo omezením dávky aplikované do m. sternocleidomastoideus na méně než 100 jednotek. U pacientů s nižší svalovou hmotou v oblasti krku nebo u pacientů, kterým je aplikována injekce bilaterálně do musculus sternocleidomastoideus, je vyšší riziko dysfagie. Dysfagie se přisuzuje šíření farmakologického účinku přípravku XEOMIN do esofageálních svalů.
Spasticita horních končetin po cévní mozkové příhodě
Při léčbě fokální spasticity byl přípravek XEOMIN sledován pouze ve spojení se standardní péčí a není určen jako náhrada těchto léčebných metod. Není pravděpodobné, že by přípravek XEOMIN zlepšoval rozsah pohybu v kloubu postiženém fixní kontrakturou.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Účinek botulotoxinu může být teoreticky potencován aminoglykosidovými antibiotiky nebo jinými léčivými přípravky, které zasahují do nervosvalového přenosu, např. tubokurarinovými svalovými relaxancii.
Proto je při souběžném použití přípravku XEOMIN s aminoglykosidy nebo spektinomycinem nutná zvláštní opatrnost. Periferní svalová relaxancia je nutné používat opatrně, v případě potřeby snížit počáteční dávku relaxancia nebo používat látku působící střednědobě, například vekuronium nebo atrakurium, spíše než látky s dlouhotrvajícími účinky.
Účinek přípravku XEOMIN mohou snižovat 4-aminochinoliny.
4.6 Fertilita, těhotenství a kojení
Nejsou k dispozici žádné adekvátní údaje o použití botulotoxinu typu A u těhotných žen. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Není známé možné riziko pro člověka. Přípravek XEOMIN proto má být používán během těhotenství pouze, pokud to je nezbytně nutné a potenciální přínos léčby převáží rizika.
Kojení
Není známo, je-li botulotoxin typu A vylučován do mateřského mléka. Proto XEOMIN nemá být používán během kojení.
Fertilita
Nejsou k dispozici klinické údaje o účincích používání botulotoxinu typu A. Studie na samcích a samicích králíků neprokázaly žádný vliv na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
XEOMIN má mírný až středně silný vliv na schopnost řídit a obsluhovat stroje.
Pacienti mají být poučeni, aby neřídili či nevykonávali jiné potenciálně rizikové činnosti, pokud se u nich vyskytne astenie, svalová slabost, závratě, poruchy vidění či pokles očních víček.
4.8 Nežádoucí účinky
Obvykle jsou nežádoucí účinky pozorovány během prvního týdne po léčbě a mají přechodnou povahu. Nežádoucí účinky mohou souviset s léčivou látkou, podáním injekce nebo obojím.
Nežádoucí účinky nezávislé na indikaci
Nežádoucí účinky související s aplikací
Výkon může být spojen s lokální bolestí, zánětem, parestézií, hypoestezií, citlivostí, otokem / edémem, erytémem, svěděním, lokalizovanou infekcí, hematomem, krvácením a/nebo modřinami v důsledku injekčního podání.
Bolest a/nebo strach související s injekcí mohou způsobit vasovagální reakce, včetně přechodného symptomatického snížení krevního tlaku nebo synkopy.
Nežádoucí účinky spojené s botulotoxinem typu A
Očekávaným farmakologickým účinkem botulotoxinu je lokalizovaná svalová slabost.
Šíření toxinu
Vzácně byly hlášeny nežádoucí účinky v důsledku šíření toxinu do vzdálených míst od místa podání (nadměrná svalová slabost, dysfagie a aspirační pneumonitida někdy se smrtelnými následky) (viz bod 4.4).
Hypersenzitivní reakce
Vzácně byly hlášeny závažné a/nebo okamžité hypersenzitivní reakce včetně anafylaxe, sérové nemoci, kopřivky, edému měkkých tkání a dušnosti. Některé z těchto reakcí byly hlášeny po použití běžného botulotoxinu typu A buď samotného, nebo v kombinaci s jinými látkami, o nichž je známo, že způsobují podobné reakce.
Nežádoucí účinky závislé na indikaci
Torticollis spastica
Léčba spastické tortikolis může způsobit dysfagii s různými stupni závažnosti s potenciální aspirací, která si může vyžádat lékařský zásah. Dysfagie může přetrvávat až dva či tři týdny po aplikaci, ale v jednom případě bylo hlášeno trvání až po dobu pěti měsíců.
Nežádoucí účinky z klinických zkušeností
Níže jsou uvedeny frekvence nežádoucích účinků pro každou indikaci vycházející z klinických studií. Tyto frekvence jsou definovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
Blefarospasmus
U přípravku XEOMIN byly hlášeny následující nežádoucí účinky:
Poruchy nervového systému
Časté: bolest hlavy, faciální paréza
Poruchy oka
Velmi časté: ptóza očního víčka, suché oči
Časté: rozostřené vidění, porucha vidění, diplopie, zvýšené slzení
Gastrointestinální poruchy
Časté: sucho v ústech, dysfagie
Poruchy kůže a podkožní tkáně Časté: vyrážka
Celkové poruchy a reakce v místě aplikace Časté: bolest v místě injekce, únava
Poruchy svalové a kosterní soustavy a pojivové tkáně Časté: svalová slabost
Torticollis spastica
U přípravku XEOMIN byly hlášeny následující nežádoucí účinky:
Poruchy nervového systému
Časté: bolest hlavy, presynkopa, závrať
Méně časté: porucha řeči
Respirační, hrudní a mediastinální poruchy Méně časté: dysfonie, dušnost
Gastrointestinální poruchy
Velmi časté: dysfagie
Časté: sucho v ústech, nauzea
Poruchy kůže a podkožní tkáně Časté: hyperhidróza
Méně časté: vyrážka
Poruchy svalové a kosterní soustavy a pojivové tkáně
Časté: bolest krku, svalová slabost, myalgie, svalová křeč, ztuhlost svalů a kostí
Celkové poruchy a reakce v místě aplikace Časté: bolest v místě injekce, astenie
Infekce a infestace
Časté: infekce horních cest dýchacích
Spasticita horních končetin po cévní mozkové příhodě U přípravku XEOMIN byly hlášeny následující nežádoucí účinky:
Poruchy nervového systému
Časté: bolest hlavy, dysestezie, hypoestezie
Gastrointestinální poruchy Časté: dysfagie
Poruchy svalové a kosterní soustavy a pojivové tkáně Časté: svalová slabost, bolest v končetinách
Méně časté: myalgie
Celkové poruchy a reakce v místě aplikace Méně časté: astenie
Časté: pocit horka, bolest v místě vpichu injekce
Některé z uvedených nežádoucích účinků mohou souviset s onemocněním.
Zkušenosti po uvedení na trh
Byly hlášeny chřipkové příznaky a hypersenzitivní reakce jako je otok, edémy (také mimo místo vpichu), erytém, svědění, vyrážka (lokální nebo celková) a dušnost.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování Příznaky předávkování:
Zvýšené dávky botulotoxinu typu A mohou mít za následek silnou nervosvalovou paralýzu vzdálenou od místa vpichu s různými symptomy. Příznaky mohou zahrnovat celkovou slabost, ptózu, diplopii, dýchací obtíže, obtíže s řečí, paralýzu respiračních svalů nebo obtíže s polykáním, které mohou způsobit aspirační pneumonií.
Opatření při předávkování:
V případě předávkování má být pacient pod lékařským dohledem se sledováním symptomů nadměrné svalové slabosti nebo svalové paralýzy. Může být zapotřebí symptomatická léčba. Jestliže nastane paralýza dýchacích svalů, může být nezbytné nasadit respirační podporu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná periferně působící myorelaxancia ATC kód: M03AX01
Botulotoxin typu A blokuje cholinergní přenos na nervosvalové ploténce prostřednictvím inhibice uvolňování acetylcholinu. Nervová zakončení neuromuskulárních spojení dále neodpovídají na nervové impulzy a sekrece neurotransmiteru na motorických ploténkách je blokována (chemická denervace). Mechanizmus obnovení přenosu vzruchu spočívá v tvorbě nových nervových zakončení a opětovném vytvoření motorických plotének.
Mechanismus účinku
Mechanizmus účinku botulotoxinu typu A na cholinergní nervová zakončení může být popsán ve 4 následných krocích:
• Vazba: těžký řetězec botulotoxinu typu A se váže s výjimečně vysokou selektivitou a afinitou
na receptory nacházející se pouze na cholinergních zakončeních.
• Internalizace: konstrikce membrány na nervových zakončeních a absorpce toxinu nervového zakončení (endocytóza).
• Translokace: Konečný amino-terminální segment neurotoxinového těžkého řetězce vytvoří pór ve vesikulární membráně, disulfidické vazby jsou rozštěpeny a lehký neurotoxinový řetězec prochází pórem do cytosolu.
• Účinek: Po uvolnění lehký neurotoxinový řetězec se velmi specificky štěpí cílový protein (SNAP 25), který je nezbytný pro uvolnění acetylcholinu.
K úplnému obnovení funkce nervosvalových plotének / přenosu vzruchu dochází obvykle během
3-4 měsíců po intramuskulární injekci, kdy nervová zakončení dorostou a znovu vytvoří nervosvalovou
ploténku.
Výsledky klinických studií
Ve 2 srovnávacích studiích fáze III s jednotlivou dávkou byla prokázána rovnocennost účinnosti přípravku XEOMIN ve srovnání se srovnávacím přípravkem obsahujícím konvenční botulotoxinový komplex typu A onabotulotoxinA (900 kD), v jedné u pacientů s blefarospasmem (studie MRZ 602010003, n=300) a ve druhé u pacientů s cervikální dystonií (studie MRZ 60201-0013, n=463). Výsledky studií také prokázaly, že přípravek XEOMIN a srovnávací přípravek měly při použití v dávkovacím poměru 1:1 podobnou účinnost a bezpečnostní profil u pacientů s blefarospasmem a s cervikální dystonií (viz bod 4.2).
Blefarospasmus
XEOMIN byl hodnocen v randomizované, dvojitě zaslepené, placebem kontrolované, multicentrické studii fáze III, která zařadila celkem 109 pacientů s blefarospasmem. Pacienti měli klinickou diagnózu benigního esenciálního blefarospasmu s úvodním sub-skóre na Jankovicově hodnotící stupnici závažnosti (JRS) > 2 a stabilní uspokojivou terapeutickou odpovědí na předchozí podání onabotulotoxinu A (Botox). Pacienti byli randomizováni (2:1) k podání jedné dávky přípravku XEOMIN (n=75) nebo placeba (n=34) v dávce podobné (+/- 10%) jako při dvou posledních sezení s podáním injekce onabotulotoxinu A před zařazením do studie. Nejvyšší dávkou povolenou v této studii bylo 50 jednotek na jedno oko, průměrná dávka přípravku XEOMIN byla 32 jednotek na jedno oko.
Primárním parametrem účinnosti byla změna sub-skóre závažnosti na stupnici JRS od počátku do 6. týdne po injekci v populaci zamýšlené léčit (ITT), kdy chybějící hodnoty byly nahrazeny poslední dostupnou hodnotou u daného pacienta (poslední provedené pozorování). V ITT populaci činil rozdíl mezi skupinou léčenou přípravkem XEOMIN a placebo skupinou ve změně sub-skóre závažnosti na stupnici JRS od počátku do 6. týdne po injekci -1,0 (95 % CI -1,4; -0,5) bod a byl statisticky signifikantní (p<0,001).
Pokud byla zapotřebí další injekce, pacienti mohli pokračovat v prodloužení studie. Pacientům bylo aplikováno až pět injekcí přípravku XEOMIN s minimálním intervalem mezi dvěma injekcemi nejméně šest týdnů (48 - 69 týdnů celková délka studie a maximální dávka 50 jednotek na každé oko).
V průběhu celé studie se průměrný interval mezi injekcemi u subjektů léčených NT 201 pohyboval mezi 10,14 (1. interval) a 12,00 (2. až 5. interval) týdny.
Torticollis spastica
XEOMIN byl hodnocen v randomizované, dvojitě zaslepené, placebem kontrolované, multicentrické studii fáze III, která zařadila celkem 233 pacientů s cervikální dystonií. Pacienti měli klinickou diagnózu převážně rotační cervikální dystonie s úvodním celkovým skóre na Torontské západní hodnotící stupnici spastické tortikolis (TWSTRS) > 20. Pacienti byli randomizováni (1:1:1) k podání jedné dávky přípravku XEOMIN 240 jednotek (n=81), přípravku XEOMIN 120 jednotek (n=78) nebo placeba (n=74). Počet a místa injekcí určoval zkoušející lékař.
Primárním parametrem účinnosti byla průměrná změna LS od počátku do 4. týdne po injekci v celkovém skóre TWSTRS v populaci zamýšlené léčit (ITT), kdy chybějící hodnoty byly nahrazeny vstupní hodnotou pacienta (úplný statistický model). Změna celkového skóre TWSTRS od počátku do 4. týdne byla signifikantně větší ve skupinách s NT 201 v porovnání se změnou v placebo skupině (p<0,001) ve všech statistických modelech). Tyto rozdíly byly též klinicky významné: např. -0,9 bodů u 240 jednotek vs. placebo a -7,5 bodů u 120 jednotek vs. placebo v úplném statistickém modelu. Pokud byla zapotřebí další injekce, pacienti mohli pokračovat v prodloužení studie. Pacientům bylo aplikováno až pět injekcí přípravku XEOMIN v dávce 120 jednotek nebo 240 jednotek s minimálním intervalem mezi dvěma injekcemi nejméně šest týdnů (48 - 69 týdnů celková délka studie). V průběhu celé studie se průměrný interval mezi injekcemi u subjektů léčených NT 201 pohyboval mezi 10,00 (1. interval) a 13,14 (3. až 6. interval) týdny.
Spasticita horních končetin po cévní mozkové příhodě
V pivotní studii (dvojitě zaslepená, placebem kontrolovaná, multicentrická) provedené u pacientů se spasticitou horních končetin po cévní mozkové příhodě bylo randomizováno 148 pacientů k aplikaci přípravku XEOMIN (n=73) a placeba (n=75), v souladu s dávkovacími doporučeními pro zahájení léčby uvedenými v bodě 4.2 SPC. Kumulativní dávka po 6 opakovaných léčebných sezeních byla v klinické studii v průměru 1 333 jednotek (maximálně 2 395 jednotek) v průběhu až 89 týdnů.
Jak stanovil primární parametr účinnosti (výskyt odpovědi flexorů zápěstí dle Ashworth Scale skóre v týdnu 4, odpověď definovaná jako zlepšení alespoň v 1 bodu z 5 stupňového skóre), pacienti léčení přípravkem XEOMIN (výskyt odpovědi: 68,5 %) měli 3,97 krát vyšší pravděpodobnost odpovědi na léčbu ve srovnání s pacienty s placebem (výskyt odpovědi: 37,3 %; 95 % Cl: 1,90 až 8,30; p < 0,001,
ITT populace).
Tato studie s fixní dávkou nebyla navržena, aby rozlišovala mezi ženami a muži, nicméně v post-hoc analýze byl výskyt odpovědi na léčbu vyšší u žen (89,3 %) než u mužů (55,6 %), tento rozdíl byl statisticky významný pouze u žen. Přesto, u mužů léčených přípravkem XEOMIN ve srovnání s placebem byl výskyt odpovědi v Ashworth Scale skóre po 4 týdnech trvale vyšší u všech svalových skupin.
V prodlouženém otevřeném pokračování pivotní studie (v této fázi bylo možné flexibilní dávkování) byl poměr pacientů odpovídajících na léčbu podobný u mužů i žen. Tohoto pokračování se zúčastnilo 145 pacientů a bylo aplikováno až 5 cyklů injekcí, stejně jako v zaslepené studii (EudraCT číslo 2006003036-30), ve které byla účinnost a bezpečnost přípravku XEOMIN hodnocena ve 2 různých ředěních u 192 pacientů se spasticitou horních končetin různé etiologie.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem XEOMIN u všech podskupin pediatrické populace v léčbě dystonie a u kojenců a batolat od 0 do 24 měsíců věku v léčbě svalové spasticity (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Obecná charakteristika léčivé látky
Klasické kinetické a distribuční studie nelze s botulotoxinem typu A provádět, protože se léčivá látka aplikuje v malých množstvích (pikogramy na injekci) a váže se rychle a ireverzibilně na cholinergní nervová zakončení.
Nativní botulotoxin je komplex o vysoké molekulární hmotnosti, který kromě neurotoxinu (150 kD) obsahuje další netoxické proteiny jako hemaglutininy a non-hemaglutininy. Na rozdíl od konvenčních přípravků obsahujících botulotoxinový komplex typu A obsahuje přípravek XEOMIN čistý (150 kD) neurotoxin bez komplexotvorných proteinů a má tudíž nízký obsah cizorodých bílkovin. Obsah cizorodých bílkovin je považován za jeden z faktorů sekundárního selhání léčby.
Podobně jako u jiných proteinů se i u botulotoxinu typu A ukázalo, že podléhá po intramuskulární injekci retrográdnímu axonálnímu transportu. Retrográdní transsynaptický průchod aktivního botulotoxinu typu A do centrálního nervového systému však zjištěn nebyl.
Receptorově vázaný botulotoxin typu A je endocytován do nervového zakončení před dosažením svého cíle (SNAP 25) a poté je intracelulárně degradován. Volně cirkulující molekuly botulotoxinu typu A, které nebyly vázány na presynaptické cholinergní nervové terminální receptory, jsou fagocytovány nebo pinocytovány a jsou degradovány podobně jako jiné volně cirkulující proteiny.
Distribuce léčivé látky v organismu pacienta
Humánní farmakokinetické studie s přípravkem XEOMIN nebyly provedeny ze shora uvedených důvodů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních studií farmakologie kardiovaskulární bezpečnosti neodhalily žádné zvláštní riziko pro člověka.
Zjištění ze studií chronické toxicity hodnotící systémovou toxicitu přípravku XEOMIN u zvířat se převážně týkala jeho farmakodynamického působení, tj. atonie, parézy a atrofie svalu, do něhož byl aplikován.
Nebyl zaznamenán žádný důkaz o lokální nesnášenlivosti. Studie reprodukční toxicity s přípravkem XEOMIN neprokázaly nežádoucí účinky na fertilitu samců ani samic u králíků, ani přímé účinky na vývoj embrya a plodu či na pre- a postnatální vývoj u potkanů a/nebo králíků. Nicméně ve studii prenatální toxicity podávání přípravku XEOMIN v týdenních či dvoutýdenních intervalech v dávkových úrovních vykazujících snížení hmotnosti u matky zvýšilo počet potratů u králíků a mírné snížení tělesné hmotnosti plodu u potkanů. Není nezbytné v těchto studiích předpokládat trvalou systémovou expozici samic během (neznámé) senzitivní fáze organogeneze jakožto nezbytný předpoklad k indukci teratogenních účinků.
Podobně i bezpečnostní hranice s ohledem na klinickou terapii byly obecně nízké z hlediska vysokých klinických dávek.
S přípravkem XEOMIN nebyly prováděny žádné studie genotoxicity či kancerogenity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Lidský albumin Sacharóza
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
XEOMIN 200 jednotek prášek pro injekční roztok: 3 roky Rekonstituovaný roztok:
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 24 hodin při teplotě 2 °C-8 °C.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2 °C až 8 °C, pokud rekonstituce neproběhla za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička (sklo třídy I) se zátkou (brombutylová pryž) a uzávěr garantující neporušenost obalu (hliník).
XEOMIN 200 jednotek prášek pro injekční roztok: Velikost balení: 1, 2, 3, 4 a 6 lahviček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rekonstituce
XEOMIN se před použitím rekonstituuje roztokem chloridu sodného 9 mg/ml (0,9 %) na injekce. Rekonstituce a ředění musejí probíhat v souladu s pravidly správné klinické praxe, zvláště pak s ohledem na aseptické podmínky.
Při správné praxi se obsah injekční lahvičky rekonstituuje a injekční stříkačka plní nad plastem potaženou papírovou utěrkou, která zachytí případně rozlitý roztok. Správné množství rozpouštědla se vtáhne do injekční stříkačky (viz tabulka ředění). Po vertikálním nasazení jehly přes pryžovou zátku se rozpouštědlo aplikuje zvolna do injekční lahvičky, aby se zabránilo tvorbě pěny. Doporučuje se krátká zkosená jehla velikosti 20-27 gauge. Injekční lahvička musí být zlikvidována, pokud rozpouštědlo není vtaženo vakuem do lahvičky. Vyjměte injekční stříkačku z injekční lahvičky a přípravek XEOMIN smíchejte s rozpouštědlem opatrným kroužením injekční lahvičky a jejím otáčením nahoru a dolů -netřepejte silně. V případě potřeby ponechte jehlu použitou k rekonstituci v injekční lahvičce a požadované množství roztoku natáhněte novou sterilní injekční stříkačkou vhodnou k podání injekce.
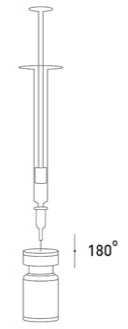
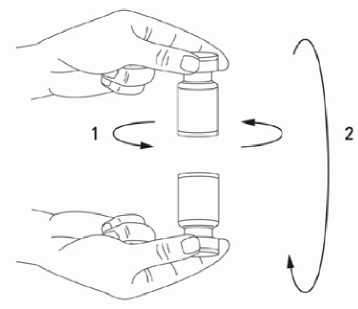
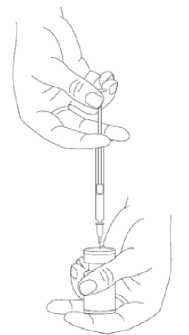
Rekonstituovaný XEOMIN je čirý, bezbarvý roztok bez viditelných částic.
XEOMIN se nesmí používat, pokud je rekonstituovaný roztok zakalený nebo obsahuje vločky či částice.
Je zapotřebí dbát na použití správného objemu rozpouštědla pro zvolenou formu k prevenci náhodného předávkování. Pokud se při jedné injekci používají rozdílné velikosti injekčních lahviček přípravku XEOMIN, je zapotřebí dbát na použití správného množství rozpouštědla při rekonstituci příslušného počtu jednotek na 0,1 ml. Množství rozpouštědla se liší u síly XEOMIN 50 jednotek, XEOMIN 100 jednotek a XEOMIN 200 jednotek. Každá injekční stříkačka má být adekvátně označena.
Doporučená naředění síly XEOMIN 50, 100 a 200 j ednotek j sou uvedena v následuj ící tabulce:
|
Výsledná dávka (v jednotkách na 0,1 ml) |
Přidané rozpouštědlo (chlorid sodný 9 mg/ml (0,9 %) roztok na injekci) | ||
|
Injekční lahvička obsahující 50 jednotek |
Injekční lahvička obsahující 100 jednotek |
Injekční lahvička obsahující 200 jednotek | |
|
20 jednotek |
0,25 ml |
0,5 ml |
1 ml |
|
10 jednotek |
0,5 ml |
1 ml |
2 ml |
|
5 jednotek |
1 ml |
2 ml |
4 ml |
|
2,5 jednotek |
2 ml |
4 ml |
8 ml |
|
1,25 jednotek |
4 ml |
8 ml |
Neuplatňuje se |
Veškerý injekční roztok, který je uchováván více než 24 hodin, i veškerý nepoužitý injekční roztok musí být zlikvidován.
Pokyny pro bezpečnou likvidaci injekčních lahviček, stříkaček a použitého materiálu Všechny použité injekční lahvičky, zbývající rekonstituovaný roztok v injekční lahvičce a/nebo stříkačky je nutné autoklávovat. Alternativně lze zbývající XEOMIN deaktivovat přidáním jednoho z následujících roztoků: 70% ethanol, 50% isopropanol, 0,1% SDS (aniontový detergent), naředěný roztok hydroxidu sodného (0,1 N NaOH) nebo naředěným roztokem chlornanu sodného (nejméně 0,1 % NaOCl).
Po deaktivaci použitých injekčních lahviček, stříkaček a materiálu tyto nesmí být vyprázdněny, a musí být vyhozeny do příslušných nádob a zlikvidovány v souladu s místními požadavky.
Doporučení v případě nehody při manipulaci s botulotoxinem
• Veškerý vysypaný nebo vylitý přípravek je nutné otřít: buď se použije absorpční materiál napuštěný některým z uvedených roztoků v případě prášku, nebo suchým absorpčním materiálem v případě vylití rekonstituovaného přípravku.
• Kontaminované povrchy se musí vyčistit absorpčním materiálem napuštěným některým z uvedených roztoků a poté osušit.
• Jestliže je injekční lahvička rozbitá, opatrně posbírejte kusy skla a vytřete přípravek, jak je výše uvedeno, dejte pozor, abyste se nepořezali.
• Při kontaktu přípravku s pokožkou opláchněte postiženou plochu velkým množstvím vody.
• Při vniknutí do očí důkladně oči propláchněte velkým množstvím vody nebo roztokem pro výplach očí.
• Jestliže se přípravek dostane do kontaktu s poraněním, s pořezanou nebo porušenou pokožkou, omyjte ránu velkým množstvím vody a podnikněte příslušná lékařská opatření podle podané dávky.
Tyto pokyny pro zacházení a likvidaci přípravku musí být přísně dodržovány.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merz Pharmaceuticals GmbH Eckenheimer LandstraBe 100 60318 Frankfurt/Main PO. Box 11 13 53 60048 Frankfurt/Main Německo
Tel.: +49-69/15 03-1 Fax: +49-69/15 03-200
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
63/257/16-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
8.6.2016
10. DATUM REVIZE TEXTU
8.6.2016
15