Xarelto 10 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xarelto 2,5 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rivaroxabanum 2,5 mg.
Pomocná látka se známým účinkem:
Jedna potahovaná tableta obsahuje 33,92 mg laktózy (jako monohydrátu) viz bod 4.4. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Světle žluté, kulaté, bikonvexní tablety (průměr 6 mm, poloměr zakřivení 9 mm) označené logem (kříž) BAYER na jedné straně a číslem „2,5“ a trojúhelníkem na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Xarelto, podávaný společně s kyselinou acetylsalicylovou (ASA) samotnou nebo s kombinací ASA plus klopidogrel nebo tiklopidin, je indikován k prevenci aterotrombotických příhod u dospělých pacientů po akutním koronárním syndromu (AKS) se zvýšenými hladinami srdečních biomarkerů (viz body 4.3, 4.4 a 5.1).
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je 2,5 mg dvakrát denně.
Pacienti by měli rovněž užívat denní dávku 75 - 100 mg ASA nebo denní dávku 75 - 100 mg ASA současně s denní dávkou 75 mg klopidogrelu nebo se standardní denní dávkou tiklopidinu.
Léčba by měla být u jednotlivých pacientů pravidelně hodnocena zvážením rizika ischemické příhody oproti riziku krvácení. Rozhodnutí o prodloužení léčby nad 12 měsíců by mělo být provedeno individuálně u každého jednotlivého pacienta, protože zkušenosti s léčbou trvající déle než 24 měsíců jsou omezené (viz bod 5.1).
Léčbu přípravkem Xarelto je třeba zahájit co nejdříve po stabilizaci akutního koronárního syndromu (včetně revaskularizačních zákroků); nejdříve za 24 hodin po přijetí do nemocnice a v době, kdy by normálně byla ukončena parenterální antikoagulační léčba.
Pokud dojde k vynechání dávky, měl by pacient pokračovat užitím příští pravidelné dávky dle doporučeného dávkovacího schématu. Dávka se nezdvojnásobuje, aby se nahradila vynechaná dávka.
Převod z antagonistů vitaminu K (VKA) na přípravek Xarelto
Při převodu pacientů z antagonistů vitaminu K na přípravek Xarelto budou po užití přípravku Xarelto hodnoty mezinárodního normalizovaného poměru (INR) falešně zvýšeny. Test INR není pro měření antikoagulační aktivity přípravku Xarelto validní a proto by neměl být používán (viz bod 4.5).
Převod z přípravku Xarelto na antagonisty vitaminu K (VKA)
Během přechodu z přípravku Xarelto na antagonisty vitaminu K existuje možnost neadekvátní antikoagulace. Během jakéhokoli převodu na alternativní antikoagulancia by měla být zajištěna kontinuální adekvátní antikoagulace. Je třeba uvést, že přípravek Xarelto může přispět ke zvýšení INR.
U pacientů, kteří jsou převáděni z přípravku Xarelto na antagonisty vitaminu K, by měli být tito antagonisté podáváni současně, dokud není hladina INR > 2,0. Po dobu prvních dvou dnů fáze převodu by mělo být použito standardní úvodní dávkování antagonistů vitaminu K s následným dávkováním těchto antagonistů na základě testování INR. Během doby, kdy pacienti užívají jak přípravek Xarelto tak antagonisty vitaminu K, by nemělo být prováděno testování INR dříve než 24 hodin po předchozí dávce, ale před další dávkou přípravku Xarelto. Jakmile je přípravek Xarelto vysazen, může být testování INR spolehlivě provedeno minimálně 24 hodin po poslední dávce (viz body 4.5 a 5.2).
Převod z parenterálních antikoagulancií na přípravek Xarelto
U pacientů, kteří dostávají parenterální antikoagulancia, přerušte podávání parenterálního antikoagulancia a začněte léčbu přípravkem Xarelto v rozmezí 0 až 2 hodiny před tím, než by mělo dojít k dalšímu plánovanému podání parenterálního přípravku (např. nízkomolekulární hepariny) nebo v době vysazení kontinuálně podávaného parenterálního přípravku (např. intravenózní nefrakciovaný heparin).
Převod z přípravku Xarelto na parenterálněpodávaná antikoagulancia
První dávku parenterálního antikoagulancia podejte v době, kdy by měla být užita další dávka přípravku Xarelto.
Zvláštní populace
Porucha funkce ledvin
Omezené klinické údaje u nemocných s těžkou poruchou funkce ledvin (clearance kreatininu 15 - 29 ml/min) signalizují, že jsou plazmatické koncentrace rivaroxabanu významně zvýšeny. Xarelto je proto u těchto pacientů nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.4 a 5.2).
Úprava dávky není nutná u pacientů s lehkou poruchou funkce ledvin (clearance kreatininu 50 - 80 ml/min) nebo středně těžkou poruchou funkce ledvin (clearance kreatininu 30 - 49 ml/min) (viz bod 5.2).
Porucha funkce jater
Xarelto je kontraindikováno u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child-Pugh B a C (viz body 4.3 a 5.2).
Starší populace
Dávky bez úprav (viz body 4.4 a 5.2).
Tělesná hmotnost
Dávky bez úprav (viz body 4.4 a 5.2).
Pohlaví
Dávky bez úprav (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Xarelto u dětí ve věku 0 až 18 let nebyla stanovena. Nej sou dostupné žádné údaje. Podávání přípravku Xarelto dětem do 18 let se proto nedoporučuje.
Způsob podání Perorální podání.
Přípravek Xarelto se může užívat s jídlem nebo nezávisle na jídle (viz body 4.5 a 5.2).
Pacientům, kteří nej sou schopni polykat celé tablety, může být tableta přípravku Xarelto těsně před užitím rozdrcena a smíchána s vodou nebo s jablečným pyré a poté podána perorálně.
Rozdrcená tableta přípravku Xarelto může být také podána gastrickou sondou poté, co je potvrzeno správné umístění sondy v žaludku. Rozdrcená tableta by měla být podána žaludeční sondou v malém množství vody a sonda by poté měla být propláchnuta vodou (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku. Aktivní klinicky významné krvácení.
Léze nebo stavy, které jsou považovány za významné riziko závažného krvácení. Mohou mezi ně patřit současné nebo nedávno prodělané ulcerace gastrointestinálního traktu, přítomnost maligních nádorů s vysokým rizikem krvácení, nedávno prodělané poranění mozku nebo míchy, operace mozku, míchy nebo oka v nedávné době, intrakraniální krvácení v nedávné době, jícnové varixy nebo podezření na ně, arteriovenózní malformace, cévní aneurysma nebo významné cévní abnormality v míše nebo mozku.
Souběžná léčba jinými antikoagulačními přípravky, např. nefrakcionovaným heparinem (UFH), nízkomolekulárními hepariny (enoxaparin, dalteparin atd.), heparinovými deriváty (fondaparinux atd), perorálními antikoagulancii (warfarin, dabigatran etexilát, apixaban, atd.) se nedoporučuje s výjimkou specifické situace, kdy je pacient převáděn z antikoagulační léčby (viz bod 4.2) nebo když je podáván UFH v dávkách nezbytných pro udržení průchodnosti centrálního žilního nebo arteriálního katetru (viz bod 4.5).
Souběžná léčba akutního koronárního syndromu (AKS) protidestičkovou léčbou u pacientů s anamnézou cévní mozkové příhody nebo trazitorní ischemické ataky (TIA) (viz bod 4.4).
Jaterní onemocnění, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení včetně cirhotických pacientů s klasifikací Child-Pugh B a C (viz bod 5.2).
Těhotenstí a kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Účinnost a bezpečnost přípravku Xarelto byla hodnocena v kombinaci s protidestičkovými látkami aspirinem a klopidogrelem/tiklopidinem. Léčba v kombinaci s jinými protidestičkovými látkami, např. prasugrelem nebo tikagrelorem, nebyla studována a nedoporučuje se.
V průběhu léčby se doporučuje pacienta klinicky sledovat v souladu s praxí běžnou při podávání antikoagulační léčby.
Riziko krvácení
Jako v případě jiných antikoagulancií, u pacientů užívajících přípravek Xarelto mají být pečlivě sledovány známky krvácení. Doporučuje se opatrnost při použití přípravku v případě zvýšeného rizika krvácení. Pokud se objeví závažné krvácení, podávání přípravku Xarelto je třeba přerušit.
V klinických studiích bylo během dlouhodobé léčby rivaroxabanem podávaným spolu s monoterapií nebo duální protidestičkovou léčbou častěji pozorováno slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální, genitourinární krvácení) a anémie. Proto kromě adekvátního klinického sledování, pokud je to vhodné, může být laboratorní vyšetření hemoglobinu/hematokritu přínosem pro detekci okultního krvácení.
U několika podskupin pacientů (podrobně uvedených dále) hrozí zvýšené riziko krvácení. Proto při použití přípravku Xarelto spolu s dvojkombinační léčbou antiagregancii u pacientů se známým zvýšeným rizikem krvácení je třeba zvažovat zvýšené riziko krvácení oproti přínosům léčby v prevenci aterotrombotických příhod. Tyto pacienty je navíc třeba pečlivě sledovat, zda se po zahájení léčby neobjeví známky a příznaky krvácivých komplikací a anémie (viz bod 4.8).
Při jakémkoli nevysvětlitelném poklesu hladin hemoglobinu nebo krevního tlaku je třeba hledat místo krvácení.
Přestože léčba rivaroxabanem nevyžaduje rutinní monitorování expozice, hladiny rivaroxabanu měřené kalibrovanou kvantitativní analýzou anti-faktoru Xa mohou být užitečné ve výjimečných situacích, kdy znalost expozice rivaroxabanu může pomoci při klinickém rozhodování, např. při předávkování nebo při urgentních chirurgických zákrocích (viz body 5.1 a 5.2).
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) mohou být plazmatické hladiny rivaroxabanu významně zvýšeny (v průměru 1,6násobně), což může vést ke zvýšenému riziku krvácení. Přípravek Xarelto musí být u pacientů s clearance kreatininu 15 - 29 ml/min užíván s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.2 a 5.2). U pacientů se středně těžkou poruchou funkce ledvin (clearance kreatininu 30 - 49 ml/min), kteří současně užívají jiné léčivé přípravky, zvyšující koncentraci rivaroxabanu v plazmě, musí být přípravek Xarelto používán s opatrností (viz bod 4.5).
Interakce s jinými léčivými přípravky
Použití přípravku Xarelto se nedoporučuje u pacientů současně léčených systémově podávanými azolovými antimykotiky (jako jsou ketokonazol, itrakonazol, vorikonazol a posakonazol) nebo inhibitory HIV proteáz (například ritonavir). Tyto léčivé látky jsou silnými inhibitory systémů CYP3A4 a současně P-gp, a proto mohou klinicky významně zvyšovat plazmatické koncentrace rivaroxabanu (v průměru 2,6 násobek), což může vést ke zvýšenému riziku krvácení (viz bod 4.5).
Postupujte opatrně, pokud jsou pacienti současně léčeni léčivými přípravky ovlivňujícími krevní srážlivost, jako jsou například nesteroidní antirevmatika (NSAID), kyselina acetylsalicylová (ASA) a inhibitory agregace trombocytů. U pacientů s rizikem vředové gastrointestinální choroby lze zvážit vhodnou profylaktickou léčbu (viz bod 4.5).
Po akutním koronárním syndromu by pacienti léčení přípravkem Xarelto a ASA nebo kombinací Xarelto a ASA plus klopidogrel/tiklopidin měli užívat souběžnou léčbu NSAID pouze tehdy, jestliže výhody převáží riziko krvácení.
Jiné rizikové faktory krvácení
Rivaroxaban, podobně jako jiná antitrombotika, není doporučen u pacientů se zvýšeným rizikem krvácení, například:
• vrozené nebo získané poruchy krvácení
• léčbou nekorigovaná těžká arteriální hypertenze
• jiné gastrointestinální onemocnění bez aktivní ulcerace, které může potenciálně vést ke krvácivým komplikacím (např. zánětlivé střevní onemocnění, esofagitida, gastritida a gastroesofageální refluxní choroba)
• cévní retinopatie
• bronchiektázie nebo plicní krvácení v anamnéze
Přípravek musí být používán s opatrností u pacientů s akutním koronárním syndromem:
• ve věku > 75 let, pokud je podáván společně s kyselinou acetylsalicylovou (ASA) samotnou nebo s kombinací ASA plus klopidogrel nebo tiklopidin
• s nižší tělesnou hmotností (< 60 kg), pokud je podáván společně s kyselinou acetylsalicylovou (ASA) samotnou nebo s kombinací ASA plus klopidogrel nebo tiklopidin
Pacienti s anamnézou cévní mozkové příhody nebo trazitomí ischemické ataky (TIA)
Podávání přípravku Xarelto 2,5 mg je v léčbě akutního koronárního syndromu kontraindikováno u pacientů s anamnézou cévní mozkové příhody nebo trazitomí ischemické ataky (TIA) (viz bod 4.3). Pacientů s akutním koronárním syndromem a anamnézou cévní mozkové příhody nebo TIA byl studován malý počet, omezené dostupné údaje o účinnosti však ukazují, že tito pacienti nemají z léčby prospěch.
Spinální / epidurální anestezie nebo punkce
Pokud je u pacienta provedena anestezie (spinální či epidurální anestezie) nebo spinální resp. epidurální punkce, u pacientů preventivně léčených antitrombotiky pro prevenci tromboembolických komplikací hrozí riziko vývinu epidurálního či spinálního hematomu, který může vyústit v dlouhodobou nebo trvalou paralýzu. Riziko těchto příhod může dále zvýšit epidurální katetr dlouhodobě zavedený po operaci, nebo současné použití léčivých přípravků ovlivňujících krevní srážlivost. Riziko může také zvýšit provedení traumatické nebo opakované epidurální či spinální punkce. Pacienty je třeba často monitorovat, zda nejeví známky a příznaky neurologického poškození (například necitlivost nebo slabost dolních končetin, dysfunkce střev nebo močového měchýře). Pokud se zjistí neurologické potíže, je nutno urgentně stanovit diagnózu a zajistit léčbu. Před neuroaxiální intervencí lékař zváží potenciální přínos a riziko u pacientů na antikoagulační terapii i u pacientů, kde hodlá antikoagulační léčbu podat v rámci tromboprofylaxe.
S použitím 2,5 mg s ASA samotnou nebo s ASA plus klopidogrel nebo ticlopidin v těchto situacích nejsou klinické zkušenosti.
Ke snížení možného rizika krvácení během současného užívání rivaroxabanu při neuroaxiální (spinální nebo epidurální) anestezii nebo spinální punkci se bere v úvahu farmakokinetický profil rivaroxabanu. Zavedení nebo odstranění epidurálního katetru nebo lumbální punkci je nejlépe provést, když je odhadovaný antikoagulační účinek rivaroxabanu nízký (viz bod 5.2). Přesný čas, kdy je u každého pacienta antikoagulační účinek dostatečně nízký, však není znám. Inhibitory agregace krevních destiček je třeba vysadit podle pokynů výrobce pro předepisování přípravku.
Doporučení pro dávkování před a po invazivních procedurách a chirurgickém výkonu Pokud je nutná invazivní procedura nebo chirurgický zákrok, měl by být přípravek Xarelto 2,5 mg vysazen minimálně 12 hodin před zákrokem, pokud je to možné a na základě klinického posouzení lékařem. Pokud má pacient podstoupit elektivní operaci a antiagregační účinek není žádoucí, je třeba inhibitory agregace krevních destiček vysadit podle pokynů výrobce k předepisování přípravku.
Pokud není možné výkon odložit, je třeba posoudit zvýšené riziko krvácení oproti neodkladnosti zákroku. Léčba přípravkem Xarelto má být znovu zahájena po invazivní proceduře nebo chirurgickém zákroku co nejdříve, pokud to situace umožní a pokud je podle úsudku ošetřujícího lékaře dosaženo odpovídající hemostázy (viz bod 5.2).
Starší populace
Se zvyšujícím se věkem se může zvyšovat riziko krvácení (viz bod 5.2).
Informace o pomocných látkách
Xarelto obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a P-gp
Současné podávání rivaroxabanu s ketokonazolem (400 mg jednou denně) nebo ritonavirem (600 mg dvakrát denně) vedlo k 2,6 resp. 2,5násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,7 resp. 1,6násobnému nárůstu jeho střední hodnoty Cmax, s významným zesílením farmakodynamických účinků, což může vést ke zvýšenému riziku krvácení. Proto se použití přípravku Xarelto nedoporučuje u pacientů užívajících současně a systémově azolová antimykotika, jako je ketokonazol, itrakonazol, vorikonazol a posakonazol, nebo inhibitory proteáz HIV. Tyto léčivé látky jsou silnými inhibitory systémů CYP3A4 a současně P-gp (viz bod 4.4).
Léčivé látky silně inhibující pouze jednu z metabolických cest eliminace rivaroxabanu (buď CYP3A4, nebo P-gp) budou pravděpodobně zvyšovat plazmatické koncentrace rivaroxabanu méně. Například klaritromycin (500 mg dvakrát denně), který je považován za silný inhibitor CYP3A4 a středně silný inhibitor P-gp, způsobuje 1,5násobný nárůst středních hodnot AUC rivaroxabanu a 1,4násobný nárůst Cmax. Tento nárůst není považován za klinicky relevantní. (Pacienti s poruchou funkce ledvin: viz bod 4.4).
Erythromycin (500 mg třikrát denně), který středně silně inhibuje CYP3A4 a P-gp, způsobuje 1,3násobný nárůst středních hodnot AUC a Cmax rivaroxabanu. Tento nárůst není považován za klinicky relevantní.
U pacientů se sníženou funkcí ledvin vedl erythromycin (500 mg třikrát denně) k 1,8 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu Cmax u pacientů s mírným renálním poškozením ve srovnání s pacienty s normální renální funkcí. U pacientů se středně těžkým renálním poškozením vedl erythromycin k 2,0 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu v Cmax ve srovnání s pacienty s normální renální funkcí. Účinek erythromycinu je aditivní k renálnímu poškození (viz bod 4.4).
Flukonazol (400 mg jednou denně), který je považován za středně silný inhibitor CYP3A4, vedl k 1,4násobnému zvýšení středních hodnot AUC rivaroxabanu a k 1,3násobnému zvýšení průměrné Cmax. Toto zvýšení není považováno za klinicky významné. (Pacienti se sníženou funkcí ledvin: viz bod 4.4).
Dronedaron by neměl být podáván spolu s rivaroxabanem, vzhledem k omezeným klinickým údajům, které jsou k dispozici.
Antikoagulační přípravky
Po kombinovaném podávání enoxaparinu (40 mg, jednorázová dávka) s rivaroxabanem (10 mg, jednorázová dávka) byl zjištěn aditivní vliv na inhibici faktoru Xa, a to bez dalších účinků na výsledky testů srážení krve (PT, apTT). Enoxaparin neovlivňoval farmakokinetiku rivaroxabanu.
Vzhledem ke zvýšenému riziku krvácení je třeba postupovat opatrně, pokud jsou pacienti současně léčeni jinými antikoagulačními přípravky (viz body 4.3 a 4.4).
NSAID / inhibitory agregace trombocytů
Při současném podávání rivaroxabanu (15 mg) a 500 mg naproxenu nebylo zjištěno klinicky relevantní prodloužení doby krvácení. Některé osoby však mohou mít silnější farmakodynamickou odezvu.
Žádné klinicky významné farmakokinetické ani farmakodynamické interakce nebyly zjištěny při současném podání rivaroxabanu s 500 mg kyseliny acetylsalicylové.
Klopidogrel (úvodní dávka 300 mg, poté udržovací dávka 75 mg) nevykazoval farmakokinetické interakce s rivaroxabanem (15 mg), ale u části populace pacientů došlo k relevantnímu nárůstu doby krvácení, který nekoreloval s agregací trombocytů, ani hladinami P-selektinu nebo receptoru GPIIb/IIIa.
Postupovat opatrně je třeba, pokud jsou pacienti současně léčeni NSAID (včetně kyseliny acetylsalicylové) a inhibitory agregace trombocytů, protože tyto léčivé přípravky obvykle zvyšují riziko krváceni (viz bod 4.4).
Warfarin
Převod pacientů z antagonisty vitaminu K warfarinu (INR 2,0 až 3,0) na rivaroxaban (20 mg) nebo z rivaroxabanu (20 mg) na warfarin (INR 2,0 až 3,0) zvýšil protrombinový čas/INR (Neoplastin) více než aditivně (mohou být pozorovány jednotlivé hodnoty INR až 12), přičemž vliv na aPTT, inhibici aktivity faktoru Xa a potenciál endogenního trombinu byl aditivní.
Pokud je potřeba testovat farmakodynamické účinky rivaroxabanu během fáze převodu, mohou se použít testy anti-Xa aktivity, PiCT a Heptest, protože tyto testy nebyly warfarinem ovlivněny. Čtvrtý den po poslední dávce warfarinu odrážely všechny testy (včetně testů PT, aPTT, inhibice aktivity faktoru Xa a ETP) pouze účinek rivaroxabanu.
Pokud je potřeba testovat farmakodynamické účinky warfarinu během fáze převodu lze použít měření INR při Cmin rivaroxabanu (24 hodin po předchozím užití rivaroxabanu), protože tento test je v tento okamžik minimálně ovlivněn rivaroxabanem.
Mezi warfarinem a rivaroxabanem nebyla pozorována žádná farmakokinetická interakce.
Induktory CYP3A4
Současné podávání rivaroxabanu se silným induktorem CYP3A4 rifampicinem vedlo k přibližně 50% poklesu střední hodnoty AUC rivaroxabanu, s odpovídajícím poklesem farmakodynamického účinku.
Současné podání rivaroxabanu s jinými silnými induktory CYP3A4 (například fenytoinem, karbamazepinem, fenobarbitalem nebo třezalkou tečkovanou (Hypericum perforatum)) může také vést ke snížení plazmatických koncentrací rivaroxabanu. Proto je třeba se vyhnou současnému podávání silných induktorů CYP3A4,, pokud není pacient pozorně sledován kvůli známkám a příznakům trombózy.
Jiné současně podávané léky
Žádné klinicky významné farmakokinetické nebo farmakodynamické interakce nebyly zjištěny při současném podávání rivaroxabanu s midazolamem (substrát CYP3A4), digoxinem (substrát P-gp), atorvastatinem (substrát CYP3A4 a P-gp) nebo omeprazolem (inhibitor protonové pumpy). Rivaroxaban neinhibuje ani neindukuje významné izoformy CYP jako je CYP3A4.
Žádné klinicky relevantní interakce s jídlem nebyly zjištěny (viz bod 4.2).
Laboratorní parametry
Parametry srážení krve (například PT, aPTT, Heptest) j sou ovlivněny podle očekávání na základě mechanismu působení rivaroxabanu (viz bod 5.1).
4.6 Fertilita, těhotenství a kojení
Bezpečnost a účinnost přípravku Xarelto u těhotných žen nebyly stanoveny. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Vzhledem k možné reprodukční toxicitě, známému riziku krvácení a důkazu, že rivaroxaban prochází placentou, je přípravek Xarelto kontraindikován v těhotenství (viz bod 4.3). Ženy ve fertilním věku musí během léčby rivaroxabanem zabránit otěhotnění.
Kojení
Bezpečnost a účinnost přípravku Xarelto u kojících žen nebyly stanoveny. Údaje z experimentů na zvířatech signalizují, že je rivaroxaban vylučován do mléka. Podávání přípravku Xarelto je během kojení kontraindikováno (viz bod 4.3). Je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit léčbu.
Fertilita
Nebyly provedeny žádné specifické studie užívání rivaroxabanu u lidí s cílem vyhodnotit účinky na fertilitu. Ve studii samčí a samičí fertility na potkanech nebyly pozorovány žádné účinky (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Xarelto má malý vliv na schopnost řídit a obsluhovat stroje. Byly hlášeny nežádoucí účinky jako synkopa (méně časté) a závrať (časté) ( viz bod 4.8). Pacienti, kteří zaznamenali tyto nežádoucí účinky, by neměli řídit vozidla a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost rivaroxabanu byla hodnocena v jedenácti studiích fáze III, kterých se účastnilo 32 625 pacientů léčených rivaroxabanem (viz tabulka 1).
Tabulka 1: Počet hodnocených pacientů, maximální ^ denní dávka a doba léčby ve studiích fáze III
|
Indikace |
Počet pacientů* |
Maximální denní dávka |
Maximální délka léčby |
|
Prevence žilního tromboembolismu (TEN) u dospělých pacientů podstupujících elektivní náhradu kyčelního nebo kolenního kloubu |
6 097 |
10 mg |
39 dnů |
|
Prevence žilního tromboembolismu u nechirurgických pacientů |
3 997 |
10 mg |
39 dnů |
|
Léčba hluboké žilní trombózy a plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie |
4 556 |
1. - 21. den: 30 mg 22. den a dále: 20 mg |
21 měsíců |
|
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní |
7 750 |
20 mg |
41 měsíců |
|
Prevence aterotrombotických příhod u pacientů po AKS |
10 225 |
5 mg nebo 10 mg, podávaných společně s ASA nebo s kombinací ASA plus klopidogrel či tiklopidin |
31 měsíců |
*Pacienti, kteří dostali alespoň jednu dávku rivaroxabanu
Nejčastěji hlášenými nežádoucí účinky u pacientů, kteří dostávali rivaroxaban, bylo krvácení (viz bod 4.4 a níže uvedený „Popis vybraných nežádoucích účinků“). Nejčastěji hlášeným krvácením (> 4 %) byla epistaxe (5,9 %) a gastrointestinální krvácení (4,2 %).
Celkem asi u 67 % pacientů exponovaných minimálně jedné dávce rivaroxabanu byl hlášen výskyt nežádoucích příhod. Asi u 22 % pacientů se vyskytly nežádoucí příhody, které byly považovány za související s léčbou podle posouzení zkoušejícími. U pacientů léčených 10 mg přípravku Xarelto a podstupujících náhradu kyčelního kloubu nebo kolenního kloubu a u hospitalizovaných nechirurgických pacientů se krvácivé příhody objevily přibližně u 6,8 % resp. 12,6 % pacientů a anémie se objevila přibližně u 5,9 % resp. 2,1 % pacientů. U pacientů, kteří dostávali buď 15 mg přípravku Xarelto dvakrát denně a následně 20 mg jednou denně z důvodu léčby hluboké žilní trombózy nebo plicní embolie, nebo 20 mg jednou denně jako prevenci recidivující hluboké žilní trombózy a plicní embolie se krvácivé příhody objevily přibližně u 27,8 % pacientů a anémie se objevila přibližně u 2,2 % pacientů. U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace bylo hlášeno krvácení jakéhokoli typu nebo stupně závažnosti s frekvencí 28 na 100 paciento-roků a anémie s frekvencí 2,5 na 100 paciento-roků. U pacientů léčených z důvodu prevence aterotrombotických příhod po akutním koronárním syndromu (AKS) bylo hlášeno krvácení jakéhokoli typu nebo stupně závažnosti s frekvencí 22 na 100 paciento-roků. Anémie byla hlášena s frekvencí 1,4 na 100 paciento-roků.
Seznam nežádoucích účinků v tabulce
Výskyt nežádoucích účinků hlášený u přípravku Xarelto je shrnutý v tabulce 2 níže podle tříd orgánových systémů (v MedDRA) a podle frekvence výskytu.
Frekvence jsou definovány takto:
velmi časté (> 1/10)
časté (> 1/100 až < 1/10)
méně časté ( > 1/1 000 až < 1/100)
vzácné (> 1/10 000 až < 1/1 000)
velmi vzácné (< 1/10 000)
není známo (z dostupných údajů nelze určit)
Tabulka 2: Všechny nežádoucí účinky při léčbě, hlášené u pacientů ve studiích fáze III
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (vč. příslušných laboratorních parametrů) |
Trombocytémie (včetně zvýšeného počtu trombocytů)A | ||
|
Poruchy imunitního systému | |||
|
Alergické reakce, alergická | |||
|
Poruchy nervového systému | |||
|
Závratě, bolest hlavy |
Mozkové a intrakraniální krvácení, synkopa | ||
|
Poruchy oka | |||
|
Oční krvácení (včetně krvácení do spojivek) | |||
|
Srdeční poruchy | |||
|
Cévní poruchy | |||
|
hematom | |||
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe, hemoptýza | |||
|
Gastrointestinální poruchy | |||
|
Gingivální krvácení, krvácení z gastrointe stinálního traktu (včetně a rektálního krvácení), gastrointestinální a abdominální bolest, dyspepsie, nauzea, A |
Sucho v ústech | ||
|
Poruchy jater a žlučových cest | |||
|
Abnormity j aterní funkce |
Žloutenka | ||
|
Poruchy kůže a por |
kožní tkáně | ||
|
Pruritus (včetně vzácných případů generalizovaného pruritu), vyrážka, ekchymóza, kožní a podkožní krvácení |
Kopřivka | ||
|
Poruchy svalové a kosterní soustavy a pojivové tká ně | |||
|
Bolest v končetináchA |
Hemartros |
Krvácení do svalů |
Kompartment syndrom sekundárně po krvácení |
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy ledvin a močových cest | |||
|
Urogenitální krvácení (včetně hematurie a menorhagieB), porucha funkce ledvin (včetně zvýšení hladin kreatininu a močoviny v krvi)A |
Renální selhání/akutní renální selhání vzniklé sekundárně po krvácení natolik silném, aby způsobilo hypoperfúzi | ||
|
Celkové poruchy a reakce v místě aplikace | |||
|
HorečkaA, periferní edém, pokles celkové síly a energie (včetně únavy, tělesné slabosti) |
Pocit indispozice (včetně malátnosti) |
Lokalizovaný edémA | |
|
Vyšetření | |||
|
Zvýšená hladina transamináz |
Zvýšená hladina bilirubinu, alkalické fosfatázyA, LDHA, lipázyA, amylázyA, GMTA |
Zvýšení konjugovaného bilirubinu (s přidruženým zvýšením ALT nebo bez zvýšení ALT) | |
|
Poranění, otravy a |
procedurální komplikace | ||
|
Pooperační krvácení (včetně pooperační anémie a krvácení z rány), pohmožděnina, sekrece z ranA |
Cévní pseudoaneuryzmaC | ||
A: pozorováno u prevence žilního tromboembolismu u dospělých pacientů po elektivní náhradě kyčelního nebo kolenního kloubu
B: pozorováno u léčby hluboké žilní trombózy a plicní embolie a jejich recidivy jako velmi časté u žen < 55 let
C: pozorováno jako méně časté u prevence aterotrombotických příhod u pacientů po akutním koronárním syndromu (po perkutánní koronární intervenci)
Popis vybraných nežádoucích účinků
Vzhledem k farmakologickému mechanismu působení může být užívání přípravku Xarelto spojeno se zvýšeným rizikem okultního nebo zjevného krvácení z jakékoli tkáně nebo orgánu s možným následkem posthemoragické anémie. Známky, příznaky a závažnost (včetně fatálního zakončení) se mohou různit podle místa a stupně nebo rozsahu krvácení a/nebo anémie (viz bod 4.9 Léčba krvácení). V klinických studiích bylo během dlouhodobé léčby rivaroxabanem v porovnání s léčbou VKA mnohem častěji pozorováno slizniční krvácení (tj. epistaxe, krvácení z dásní, gastrointestinální krvácení, urogenitální krvácení) a anémie. Při posuzování stavu může být potřeba, pokud je to shledáno vhodným, kromě adekvátního klinického sledování pacientů provést laboratorní vyšetření hemoglobinu/hematokritu pro detekci okultního krvácení. Riziko krvácení bude možná zvýšeno u některých skupin pacientů, například osob s nekontrolovanou těžkou arteriální hypertenzí a/nebo souběžnou léčbou ovlivňující krevní srážlivost (viz Riziko krvácení v bodu 4.4). Menstruační krvácení může být intenzivnější a/nebo prodloužené. Hemoragické komplikace se mohou projevovat jako celková slabost, bledost, závratě, bolest hlavy nebo nevysvětlitelné otoky, dušnost a nevysvětlitelný šok. V některých případech byly v důsledku anémie pozorovány příznaky srdeční ischémie, jako je například bolest na hrudníku nebo angina pectoris.
V souvislosti s užíváním přípravku Xarelto byly hlášeny známé sekundární komplikace závažného krvácení, jako je například kompartment syndrom a renální selhání v důsledku hypoperfúze. Možnost krvácení je proto třeba zvážit při posuzování stavu pacientů s jakoukoli antikoagulační léčbou.
Postmarketingové sledování
V postmarketingovém sledování byly v časové souvislosti s užitím přípravku Xarelto hlášeny následující nežádoucí účinky. Frekvenci těchto nežádoucích účinků hlášených po uvedení na trh nelze určit.
Poruchy imunitního systému: angioedém a alergický edém (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Poruchy jater a žlučových cest: cholestáza, hepatitida (včetně hepatocelulárního poškození) (v souhrnných studiích fáze III byly tyto příhody vzácné (>1/10 000 až <1/1000)).
Poruchy krve a lymfatického systému: trombocytopenie (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny vzácné případy předávkování dávkou až 600 mg bez krvácivých komplikací nebo jiných nežádoucích reakcí. Z důvodu omezené absorpce se očekává efekt stropu bez dalšího zvýšení průměrné plazmatické expozice v případě supraterapeutické dávky 50 mg rivaroxabanu nebo dávek vyšších.
Specifické antidotum blokující farmakodynamický účinek rivaroxabanu není k dispozici.
Lze zvážit podání aktivního uhlí ke snížení absorpce v případě předávkování rivaroxabanem.
Léčba krvácení
Pokud dojde ke krvácivým komplikacím u pacienta léčeného rivaroxabanem, musí se podání další dávky rivaroxabanu odložit nebo se léčba musí ukončit, dle potřeby. Rivaroxaban má biologický poločas asi 5 až 13 hodin (viz bod 5.2). Léčba by měla být individuální podle závažnosti a lokalizace krvácení. Podle potřeby je třeba použít vhodnou symptomatickou léčbu, jako je mechanická komprese (např. u závažné epistaxe), chirurgická hemostáza se zajištěním kontroly krvácení, náhradou tekutin a zajištěním hemodynamické podpory, krevní deriváty (erytrocyty nebo čerstvá zmrazená plasma, v závislosti na související anémii nebo koagulopatii) nebo trombocyty.
Pokud krvácení nelze kontrolovat výše uvedenými opatřeními, lze zvážit podávání specifické prokoagulační reverzní látky, jako je koncentrát protrombinového komplexu (PCC), aktivovaný koncentrát protrombinového komplexu (APCC) nebo rekombinantní faktor VIIa (r-FVIIa). V současnosti jsou však k dispozici velmi omezené klinické zkušenosti s použitím těchto přípravků u osob užívajících rivaroxaban. Doporučení je též podloženo omezenými neklinickými údaji. Opakované podání rekombinantního faktoru VIIa je třeba zvážit a titrovat v závislosti na zlepšování krvácení. V případě závažného krvácení je třeba konzultovat odborníka na koagulaci, pokud je odborník v místě dostupný (viz bod 5.1).
Protamin sulfát a vitamin K podle všeho nebudou ovlivňovat antikoagulační aktivitu rivaroxabanu. U osob užívajících rivaroxaban jsou omezené zkušenosti s použítím kyseliny tranexamové a neexistují zkušenosti s použitím kyseliny aminokaproové a aprotininu. Neexistují ani vědecké důvody přínosu ani zkušenosti s použitím systémového hemostatika desmopressinu u osob užívajících rivaroxaban. Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: přímé inhibitory faktoru Xa, ATC kód: B01AF01
Mechanismus účinku
Rivaroxaban je vysoce selektivní přímý inhibitor faktoru Xa biologicky dostupný při perorálním podání. Inhibice faktoru Xa blokuje vnitřní a vnější cestu koagulační kaskády a inhibuje vznik trombinu i vytváření trombů. Rivaroxaban neinhibuje trombin (aktivovaný faktor II) a nebyly prokázány žádné účinky na trombocyty.
Farmakodynamické účinky
U lidí byla zjištěna inhibice faktoru Xa přímo úměrná dávce. Protrombinový čas (PT) je rivaroxabanem ovlivňován úměrně dávce, a pokud je při testu použit Neoplastin, objevuje se vysoká korelace s plazmatickými koncentracemi (hodnota R je 0,98). Jiné reagenty by mohly přinést jiné výsledky. Hodnotu PT je nutno odečíst v sekundách, protože INR (mezinárodní normalizovaný poměr) je kalibrován a validován pouze pro kumariny, a nelze jej používat pro jiná antikoagulancia.
V klinické farmakologické studii sledující reverzi farmakodynamického účinku rivaroxabanu u zdravých dospělých osob (n=22) byl hodnocen účinek jednotlivé dávky (50 IU/kg) u dvou rozdílných typů PCC, 3-faktorového PCC (faktory II, IX a X) a 4-faktorového PCC (II, VII, IX a X). 3-faktorový PCC redukoval průměrnou hodnotu PT času (protrombinového času), hodnocenou neoplastinem, přibližně o 1,0 sekundy během 30 minut, ve srovnání s přibližně 3,5 sekundami pozorovanými u 4-faktorového PCC. Naproti tomu, 3-faktorový PCC měl větší a rychlejší celkový efekt na zvrácení změny tvorby endogenního trombinu než 4-faktorový PCC (viz bod 4.9).
Aktivovaný parciální tromboplastinový čas (aPTT) a hodnoty analýzy HepTest jsou také prodlouženy úměrně dávce; nedoporučuje se však tyto metody používat k hodnocení farmakodynamických účinků rivaroxabanu. Během léčby rivaroxabanem v běžné klinické praxi není třeba monitorovat parametry koagulace. Pokud však je to klinicky indikováno, lze hladiny rivaroxabanu měřit pomocí kalibrovaných kvantitativních testů anti-Xa (viz bod 5.2).
Klinická účinnost a bezpečnost
Klinický program přípravku Xarelto byl koncipován tak, aby byla prokázána účinnost přípravku Xarelto při prevenci kardiovaskulárního úmrtí, infarktu myokardu nebo cévní mozkové příhody u pacientů s nedávným akutním koronárním syndromem -AKS (infarkt myokardu s elevací segmentu ST [STEMI], infarkt myokardu bez elevace segmentu ST [NSTEMI] nebo nestabilní angina pectoris [NSAP]). V pivotní dvojitě zaslepené klinické studii ATLAS ACS 2 TIMI 51 bylo 15 526 pacientů náhodně zařazeno v poměru 1:1:1 do jedné ze tří léčebných skupin: skupiny užívající přípravek Xarelto 2,5 mg perorálně dvakrát denně, skupiny užívající přípravek Xarelto 5 mg perorálně dvakrát denně nebo do skupiny užívající placebo dvakrát denně spolu s ASA samotnou nebo s ASA plus thienopyridin (klopidogrel nebo tiklopidin). Pacienti s akutním koronárním syndromem ve věku do 55 let měli buď diabetes mellitus, nebo prodělali infarkt myokardu. Medián léčby byl 13 měsíců a celková doba trvání léčby byla téměř 3 roky. 93,2 % pacientů užívalo souběžně ASA plus thienopyridin a 6,8 % pouze ASA. Z pacientů, kteří užívali duální protidestičkovou léčbu, 98,8 % užívalo klopidogrel, 0,9 % užívalo tiklopidin a 0,3 % užívalo prasugrel. Pacienti obdrželi první dávku přípravku Xarelto nejméně za 24 hodin a nejvýše za 7 dní (medián 4,7 dne) po přijetí do nemocnice, vždy však co nejdříve po stabilizaci akutního koronárního syndromu (AKS ), včetně revaskularizačních zákroků, a v době obvyklého ukončení parenterální antikoagulační léčby.
Oba dávkovací režimy, 2,5 mg rivaroxabanu dvakrát denně a 5 mg rivaroxabanu dvakrát denně, byly účinné při dalším snižování incidence kardiovaskulárních příhod na pozadí standardní protidestičkové léčby. V režimu s podáváním 2,5 mg dvakrát denně byla snížena mortalita a prokázalo se, že při nižším dávkování bylo riziko krvácení nižší; proto je dávka rivaroxabanu 2,5 mg dvakrát denně souběžně podávaných s kyselinou acetylsalicylovou (ASA) samotnou nebo s kombinací ASA plus klopidogrel nebo tiklopidin doporučována jako prevence aterotrombotických příhod u dospělých pacientů po akutním koronárním syndromu se zvýšenými hladinami srdečních biomarkerů.
Ve srovnání s placebem přípravek Xarelto signifikantně snížil primární kompozitní cílový parametr kardiovaskulárního úmrtí, infarktu myokardu nebo cévní mozkové příhody. Prospěch byl dán redukcí výskytu kardiovaskulárního úmrtí a infarktu myokardu a objevil se brzy s konstantním léčebným účinkem během celého sledovaného období (viz tabulka 3 a graf1). Také první sekundární cílový parametr (úmrtí z jakékoli příčiny, infarkt myokardu nebo cévní mozková příhoda) byl signifikantně snížen. Další retrospektivní analýza prokázala nominální signifikantní snížení incidence trombózy stentu v porovnání s placebem (viz tabulka 3). Incidence pro hlavní bezpečnostní ukazatel (závažné krvácivé příhody nesouvisející s CABG TIMI) byly vyšší u pacientů léčených přípravkem Xarelto než u pacientů užívajících placebo (viz tabulka 5). Incidence pro komponenty fatální krvácivé příhody, hypotenze vyžadující léčbu intravenózními inotropními látkami a chirurgický zákrok kvůli probíhajícímu krvácení byly u přípravku Xarelto a placeba srovnatelné.
V tabulce 4 jsou uvedeny výsledky účinnosti u pacientů podstupujících perkutánní koronární intervenci (PCI). Bezpečnostní výsledky v této podskupině pacientů podstupujících PCI byly srovnatelné s celkovými bezpečnostními výsledky.
80% studijní populace tvořili pacienti se zvýšenými biomarkery (troponin nebo CK-MB) a bez předchozí cerebrovaskulární příhody/tranzitorní ischemické ataky. Výsledky této skupiny byly také konsistentní s celkovými výsledky účinnosti a bezpečnosti.
Tabulka 3: Výsledky účinnosti ze studie fáze III ATLAS ACS 2 TIMI 51
|
Populace studie |
Pacienti s nedávným akutním koronárním syndromem a) | |
|
Dávkování |
Xarelto 2,5 mg, dvakrát denně, N=5 114 n (%) Poměr rizik HR (95% CI) p-hodnota b) |
Placebo N=5 113 n (%) |
|
Kardiovaskulární úmrtí, infarkt myokardu nebo cévní mozková příhoda |
313 (6,1 %) 0,84 (0,72; 0,97) p = 0,020* |
376 (7,4 %) |
|
Úmrtí z jakékoli příčiny, infarkt myokardu nebo cévní mozková příhoda |
320 (6,3 %) 0,83 (0,72; 0,97) p = 0,016* |
386 (7,5 %) |
|
Kardiovaskulární úmrtí |
94 (1,8 %) 0,66 (0,51; 0,86) p = 0,002** |
143 (2,8 %) |
|
Úmrtí z jakékoli příčiny |
103 (2,0 %) 0,68 (0,53; 0,87) p = 0,002** |
153 (3,0 %) |
|
Infarkt myokardu |
205 (4,0 %) 0,90 (0,75; 1,09) p = 0,270 |
229 (4,5 %) |
|
Cévní mozková příhoda |
46 (0,9 %) 1,13 (0,74; 1,73) p = 0,562 |
41 (0,8 %) |
|
Trombóza stentu |
61 (1,2 %) 0,70 (0,51; 0,97) p = 0,033** |
87 (1,7 %) |
|
a) modifikovaná intent-to-treat analýza (intent-to-treat celková analýza pro trom |
bózu stentu) | |
b) oproti placebu; Log-Rank p-hodnota * statisticky superiorní ** nominálně významné
Tabulka 4: Výsledky účinnosti ze studie fáze III ATLAS ACS 2 TIMI 51 u pacientů podstupujících PCI
|
Populace studie |
Pacienti s nedávným akutním koronárním syndromem podstupující PCIa) | |
|
Dávkování |
Xarelto 2,5 mg, dvakrát denně, N=3114, n (%) Poměr rizik (95% CI) p-hodnota b) |
Placebo N=3096 n (%) |
|
Kardiovaskulární úmrtí, infarkt myokardu nebo cévní mozková příhoda |
153 (4,9 %) 0,94 (0,75; 1,17) p = 0,572 |
165 (5,3 %) |
|
Kardiovaskulární úmrtí |
24 (0,8 %) 0,54 (0,33; 0,89) p = 0,013** |
45 (1,5 %) |
|
Úmrtí z jakékoli příčiny |
31 (1,0 %) 0,64 (0,41; 1,01) p = 0,053 |
49 (1,6 %) |
|
Infarkt myokardu |
115 (3,7 %) 1,03 (0,79; 1,33) p = 0,829 |
113 (3,6 %) |
|
Cévní mozková příhoda |
27 (0,9 %) 1,30 (0,74; 2,31) p = 0,360 |
21 (0,7 %) |
|
Trombóza stentu |
47 (1,5 %) 0,66 (0,46; 0,95) p = 0,026** |
71 (2,3 %) |
a) modifikovaná intent-to-treat analýza (intent-to-treat celková analýza pro trombózu stentu)
b) oproti placebu; Log-Rank p-hodnota
nominálně významné
|
Populace studie |
Pacienti s nedávným akutním koronárním syndromem a) | |
|
Dávkování |
Xarelto 2,5 mg, dvakrát denně, N=5,115, n (%) Poměr rizik (95% CI) p-hodnota b) |
Placebo N=5,125 n (%) |
|
Závažné TIMI krvácivé příhody nesouvisející s CABG |
65 (1,3 %) 3,46 (2,08; 5,77) p = < 0,001* |
19 (0,4 %) |
|
Fatální krvácivé příhody |
6 (0,1 %) 0,67 (0,24; 1,89) p = 0,450 |
9 (0,2 %) |
|
Symptomatické intrakraniální krvácení |
14 (0,3 %) 2,83 (1,02; 7,86) p = 0,037 |
5 (0,1 %) |
|
Hypotenze vyžadující léčbu intravenózními inotropními látkami |
3 (0,1 %) |
3 (0,1 %) |
|
Chirurgický zákrok kvůli pokračujícímu krvácení |
7 (0,1 %) |
9 (0,2 %) |
|
Transfuze 4 nebo více krevních jednotek za 48 hodin |
19 (0,4 %) |
6 (0,1 %) |
a) “Safety“ populace, „on treatment“ (populace, ve které byla hodnocena bezpečnost, po dobu léčby)
b) oproti placebu; Log-Rank p-hodnota statisticky významné
Obrázek 1: Doba do prvního výskytu primárního cílového parametru účinnosti (kardiovaskulární úmrtí, infarkt myokardu nebo cévní mozková příhoda)
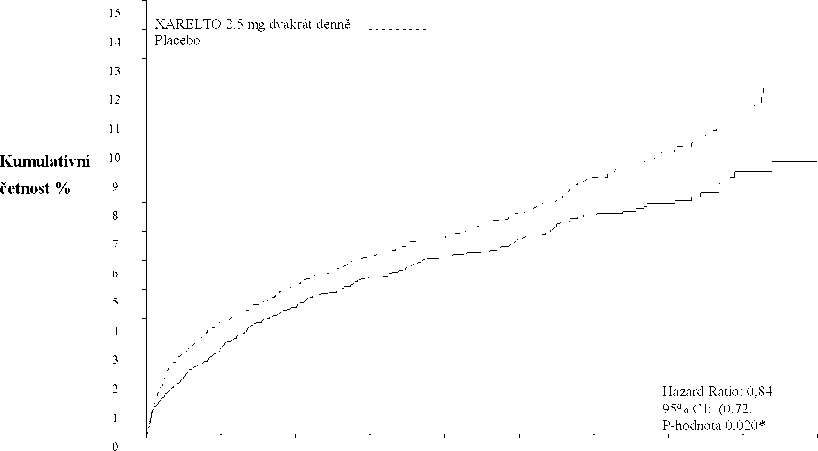
|
0 |
90 |
180 |
270 360 450 |
540 |
630 |
720 |
810 |
|
Počet pacientů s rizikem |
Relativní dny od randomizace | ||||||
|
XARELTO 5114 |
4431 |
3943 |
3199 2609 2005 |
1425 |
878 |
415 |
89 |
|
Placebo 5113 |
4437 |
3974 |
3253 2664 2059 |
1460 |
878 |
421 |
87 |
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xarelto v léčbě tromboembolických příhod u jedné nebo více podskupin pediatrické populace. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xarelto v prevenci tromboembolických příhod u všech podskupin pediatrické populace (informace o použití u pediatrické populace viz bod 4.2.).
5.2 Farmakokinetické vlastnosti
Absorpce
Rivaroxaban je rychle absorbován; maximální koncentrace (Cmax) se objeví 2 - 4 hodiny po užití tablety.
Bez ohledu na stav na lačno nebo po jídle je u dávky 2,5 mg a 10 mg rivaroxabanu tablety perorální absorpce téměř kompletní a perorální biologická dostupnost vysoká (80 - 100 %). Užívání při jídle neovlivňuje při dávce 2,5 mg a 10 mg AUC ani Cmax rivaroxabanu. Rivaroxaban 2,5 mg a 10 mg tablety lze užívat při jídle nebo nezávisle na jídle.
Farmakokinetické vlastnosti rivaroxabanu jsou až do dávky 15 mg jednou denně přibližně lineární. Ve vyšších dávkách je absorbce rivaroxabanu omezena disolucí, dochází ke snížení biologické dostupností a míra absorbce se snižuje se zvyšující se dávkou. To se výrazněji projevuje ve stavu na lačno než po jídle. Variabilita farmakokinetiky rivaroxabanu je střední, s interindividuální variabilitou v rozmezí od 30 % do 40 %.
Absorpce rivaroxabanu je závislá na místě jeho uvolnění v gastrointestinálním traktu. Bylo hlášeno 29% a 56% snížení AUC a Cmax ve srovnání s tabletou, pokud byl rivaroxaban v granulátu uvolněn v proximální časti tenkého střeva. Expozice je dále snížena, když je rivaroxaban uvolněn v distální části tenkého střeva nebo ve vzestupné části tračníku. Proto by se mělo zabránit podání rivaroxabanu distálně od žaludku, jelikož to může vést ke snížení absorpce a související expozice rivaroxabanu.
Biologická dostupnost (AUC a Cmax) pro podání 20 mg rivaroxabanu per os ve formě rozdrcené tablety vmíchané do jablečného pyré nebo rozpuštěné ve vodě a pro podání žaludeční sondou následované tekutou stravou, ve srovnání s celou tabletou byla srovnatelná. Vzhledem k předvídatelnému dávce úměrnému farmakokinetickému profilu rivaroxabanu odpovídají výsledky biologické dostupnosti z této studie pravděpodobně pro nižší dávky rivaroxabanu.
Distribuce v organismu
Vazba na plazmatické proteiny u lidí je vysoká, přibližně 92 % - 95 %, přičemž hlavní část se váže na sérový albumin. Distribuční objem je střední, Vss činí přibližně 50 litrů.
Biotransformace a eliminace z organismu
Z podané dávky rivaroxabanu se přibližně 2/3 metabolicky degradují, z čehož je polovina vylučována ledvinami a druhá polovina stolicí. Zbývající 1/3 podané dávky je vylučována ledvinami přímo jako nezměněná léčivá látka, hlavně prostřednictvím aktivní ledvinové sekrece.
Rivaroxaban je metabolizován prostřednictvím systémů CYP3A4 a CYP2J2 i mechanismy na CYP nezávislými. Hlavními cestami transformace je oxidativní degradace morfolinonové části a hydrolýza amidových vazeb. Na základě in vitro experimentů je zřejmé, že rivaroxaban slouží jako substrát transportních proteinů - P-gp (P-glykoprotein) a BCRP (breast cancer resistance protein).
Nezměněný rivaroxaban je nejvýznamnější formou přípravku v lidské plazmě; v krevním oběhu nejsou žádné významné nebo aktivní metabolity. Rivaroxaban lze vzhledem ke systémové clearance asi 10 l/h klasifikovat jako látku s nízkou clearance. Po intravenózním podání dávky 1 mg je eliminační poločas asi 4,5 hodiny. Po perorálním podání je eliminace omezována mírou absorpce. K eliminaci rivaroxabanu z plazmy dochází s terminálním poločasem 5 až 9 hodin u mladších osob a s terminálním poločasem 11 až 13 hodin u starších osob.
Zvláštní populace
Pohlaví
Mezi muži a ženami nebyl žádný klinicky relevantní rozdíl ve farmakokinetice a farmakodynamice přípravku.
Starší populace
Starší pacienti vykazovali vyšší plazmatické koncentrace než mladší, se střední hodnotou AUC přibližně 1,5 x vyšší, hlavně vzhledem ke snížené (zdánlivé) celkové a ledvinové clearance. Žádná úprava dávky není nutná.
Různé váhové kategorie
Extrémy v tělesné hmotnosti (< 50 kg nebo > 120 kg) měly pouze malý vliv na plazmatické koncentrace rivaroxabanu (méně než 25 %). Žádná úprava není dávky nutná.
Rozdíly mezi etniky
Žádné klinicky relevantní rozdíly mezi etniky nebyly ve farmakokinetice a farmakodynamice rivaroxabanu zjištěny u pacientů z řad bělochů, Afroameričanů, Hispánců, Japonců ani Číňanů.
Porucha funkce jater
Pacienti s cirhózou s lehkou poruchou funkce jater (Child-Pugh A) vykazovali pouze menší změny ve farmakokinetice rivaroxabanu (v průměru 1,2x nárůst AUC rivaroxabanu), a výsledky byly téměř srovnatelné s kontrolní skupinou zdravých pacientů. U pacientů trpících cirhózou se středně těžkou poruchou funkce jater (Child-Pugh B) střední AUC rivaroxabanu významně stoupla - 2,3x v porovnání se zdravými dobrovolníky. AUC nevázané látky stoupla 2,6x. U těchto pacientů dochází ke snížení renální eliminace rivaroxabanu, podobně jako u pacientů se středně těžkou poruchou funkce ledvin. O účinku u pacientů s těžkou poruchou funkce jater nejsou k dispozici žádné údaje.
Inhibice aktivity faktoru Xa byla u pacientů se středně těžkou poruchou funkce jater zvýšena ve srovnání se zdravými dobrovolníky 2,6x; prodloužení PT bylo obdobně zvýšeno 2,1x. Pacienti se středně těžkou poruchou funkce jater byli na rivaroxaban citlivější, a vztah mezi koncentrací a PT měl tak strmější průběh. Přípravek Xarelto je kontraindikován u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child-Pugh B a C (viz bod 4.3).
Porucha funkce ledvin
Byl zjištěn nárůst expozice rivaroxabanu související s poklesem funkce ledvin, která byla posuzována prostřednictvím hodnot clearance kreatininu. U osob s lehkou (clearance kreatininu 50 - 80 ml/min), středně těžkou (clearance kreatininu 30 - 49 ml/min) a těžkou (clearance kreatininu 15 - 29 ml/min) poruchou funkce ledvin byly plazmatické koncentrace rivaroxabanu (AUC) zvýšeny 1,4, 1,5 resp. 1,6 x. Odpovídající zesílení farmakodynamických účinků bylo výraznější. U osob s lehkou, střední a těžkou ledvinovou nedostatečností byla celková inhibice faktoru Xa ve srovnání se zdravými dobrovolníky zvýšena 1,5, 1,9 resp. 2,0 x; prodloužení PT bylo obdobně zvýšeno 1,3, 2,2 a 2,4 x. O použití u pacientů s clearance kreatininu < 15 ml/min nejsou žádné údaje.
Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min. Xarelto je proto u pacientů s clearance kreatininu 15 - 29 ml/min nutno používat s opatrností (viz bod 4.4).
Farmakokinetické údaje u pacientů
U pacientů užívajících rivaroxaban 2,5 mg dvakrát denně jako prevenci aterotrombotických příhod u pacientů s akutním koronárním syndromem (AKS) byl geometrický průměr koncentrace (90% interval odpovědi) 2 - 4 h a asi 12 h po podání dávky (představující zhruba maximální a minimální koncentrace během dávkovacího intervalu) 47 (13 - 123) a 9,2 (4,4 - 18) pg/l.
F armakokinetické/farmakodynamické vztahy
Po podání velkého rozmezí dávek (5 - 30 mg dvakrát denně) byl hodnocen farmakokinetický a farmakodynamický (PK/PD) vztah mezi plazmatickou koncentrací rivaroxabanu a několika cílovými parametry PD (inhibice faktoru Xa , PT, aPTT, Heptest). Vztah mezi plazmatickou koncentrací rivaroxabanu a aktivitou faktoru Xa byl nejlépe popsán pomocí modelu Emax. U PT byly údaje lépe vyjádřeny pomocí lineárního ohraničeného modelu. Hodnoty PT se významně lišily v závislosti na použitých reagenciích. Při použití Neoplastinu byl výchozí PT asi 13 sekund a odchylka hodnot přibližně 3 až 4 s/(100 ^g/l). Výsledky analýz PK/PD ve studii fáze II a III byly v souladu s údaji získanými u zdravých jedinců.
Pediatrická populace
Bezpečnost a účinnost nebyly stanoveny u dětí a dospívajících do 18 let věku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po jednorázovém podání, fototoxicity, genotoxicity a kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studii toxicity při opakovaném podání byly způsobeny hlavně zesílenou farmakologickou aktivitou rivaroxabanu. Při klinicky relevantních úrovních expozice byly u potkanů pozorovány zvýšené plazmatické hladiny IgG a IgA.
U potkanů nebyly pozorovány žádné účinky na fertilitu samců nebo samic. Studie na zvířatech prokázaly reprodukční toxicitu související s farmakologickým mechanismem působení rivaroxabanu (např. hemoragickými komplikacemi). V klinicky relevantních plazmatických koncentracích byla pozorována embryonální a fetální toxicita (post-implantační ztráty, opožděná nebo progredující osifikace, hepatální mnohočetné světle zbarvené skvrny) a zvýšený výskyt malformací a také placentárních změn. V prenatálních a postnatálních experimentech u potkanů byla zjištěna snížená životaschopnost potomků, a to v dávkách toxických pro matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Sodná sůl kroskarmelózy Monohydrát laktózy Hypromelóza Natrium-lauryl-sulfát Magnesium-stearát.
Potah tablety:
Makrogol 3350
Hypromelóza
Oxid titaničitý (E 171)
Žlutý oxid železitý (E 172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Blistry z PP/Al fólie v krabičkách po 14, 28, 30, 56, 60, 98, 168 nebo 196 potahovaných tabletách nebo perforované jednodávkové blistry obsahující 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních obsahujících 100 potahovaných tablet (10 balení po 10 x 1 tabletě).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/472/025-035
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. září 2008
Datum posledního prodloužení registrace: 22. května 2013
10. DATUM REVIZE TEXTU {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xarelto 10 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rivaroxabanum 10 mg.
Pomocná látka se známým účinkem:
Jedna potahovaná tableta obsahuje 26,51 mg laktózy (jako monohydrátu), viz bod 4.4. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Světle červené, kulaté, bikonvexní tablety (průměr 6 mm, poloměr zakřivení 9 mm) označené logem (kříž) BAYER na jedné straně a číslem „10“ a trojúhelníkem na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence žilního tromboembolismu (VTE) u dospělých pacientů podstupujících elektivní operativní náhradu kyčelního nebo kolenního kloubu.
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je 10 mg rivaroxabanu perorálně jednou denně. První dávka se podává 6 až 10 hodin po operaci, pokud byla nastolena hemostáza.
Délka léčby závisí na individuálním riziku žilního tromboembolismu u pacienta, které je dáno typem operace.
• U pacientů absolvujících velkou operaci kyčle doporučujeme užívání po dobu 5 týdnů.
• U pacientů absolvujících velkou operaci kolena doporučujeme užívání po dobu 2 týdnů.
Pokud pacient vynechá dávku, musí ji užít hned po zjištění a potom pokračovat následující den jednou tabletou denně jako předtím.
Převod z antagonistů vitamínu K (VKA) na přípravek Xarelto
Při převodu pacientů z antagonistů vitaminu K na přípravek Xarelto, budou po užití přípravku Xarelto hladiny mezinárodního normalizačního poměru (INR) falešně zvýšeny. Test INR není pro měření antikoagulační aktivity přípravku Xarelto validní a proto by neměl být používán (viz bod 4.5).
Převod z přípravku Xarelto na antagonisty vitamínu K (VKA)
Během přechodu z přípravku Xarelto na antagonisty vitaminu K existuje možnost neadekvátní antikoagulace. Během jakéhokoli převodu na alternativní antikoagulancia by měla být zajištěna kontinuální adekvátní antikoagulace. Je třeba uvést, že přípravek Xarelto může přispět ke zvýšení INR.
U pacientů, kteří jsou převáděni z přípravku Xarelto na antagonisty vitaminu K by měli být tito antagonisté podáváni současně, dokud není hladina INR > 2,0. Po dobu prvních dvou dnů fáze převodu by mělo být použito standardní úvodní dávkování antagonistů vitaminu K s následným dávkováním těchto antagonistů na základě testování INR. Během doby, kdy pacienti užívají jak přípravek Xarelto tak antagonisty vitaminu K by nemělo být prováděno testování INR dříve než 24 hodin po předchozí dávce, ale před další dávkou přípravku Xarelto. Jakmile je přípravek Xarelto vysazen, může být testování INR spolehlivě provedeno minimálně 24 hodin po poslední dávce (viz body 4.5 a 5.2).
Převod z parenterálních antikoagulancií na přípravek Xarelto
U pacientů, kteří dostávají parenterální antikoagulancia, přerušte podávání parenterálního antikoagulancia a začněte léčbu přípravkem Xarelto v rozmezí 0 až 2 hodiny před tím, než by mělo dojít k dalšímu plánovanému podání parenterálního přípravku (např. nízkomolekulární hepariny) nebo v době vysazení kontinuálně podávaného parenterálního přípravku (např. intravenózní nefrakciovaný heparin).
Převod z přípravku Xarelto na parenterálně podávaná antikoagulancia
První dávku parenterálního antikoagulancia podejte v době, kdy by měla být užita další dávka přípravku Xarelto.
Speciální populace
Ledvinová nedostatečnost
Omezené klinické údaje u nemocných s těžkou renální nedostatečností (clearance kreatininu 15 - 29 ml/min) signalizují, že jsou plazmatické koncentrace rivaroxabanu významně zvýšeny. Xarelto je proto u těchto pacientů nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.4 a 5.2).
Úprava dávky není nutná u pacientů s mírnou renální nedostatečností (clearance kreatininu 50 - 80 ml/min) nebo střední renální nedostatečností (clearance kreatininu 30 - 49 ml/min) (viz bod 5.2).
Jaterní nedostatečnost
Xarelto je kontraindikováno u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child-Pugh B a C (viz body 4.3 a 5.2).
Starší populace
Dávky bez úprav (viz bod 5.2).
Tělesná hmotnost
Dávky bez úprav (viz bod 5.2).
Pohlaví
Dávky bez úprav (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Xarelto u dětí ve věku 0 až 18 let nebyla ještě stanovena. Nejsou dostupné žádné údaje. Podávání přípravku Xarelto dětem nebo do 18 let se proto nedoporučuje.
Způsob podání
Perorální podání. Přípravek Xarelto se může užívat s jídlem nebo nezávisle na jídle (viz body 4.5 a 5.2).
Pacientům, kteří nejsou schopni polykat celé tablety, může být tableta přípravku Xarelto těsně před užitím rozdrcena a smíchána s vodou nebo s jablečným pyré a poté podána perorálně.
Rozdrcená tableta přípravku Xarelto může být také podána gastrickou sondou poté, co je potvrzeno správné umístění sondy v žaludku. Rozdrcená tableta by měla být podána žaludeční sondou v malém množství vody a sonda by poté měla být propláchnuta vodou (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku. Aktivní klinicky významné krvácení.
Léze nebo stavy, které jsou považovány za významné riziko závažného krvácení. Mohou mezi ně patřit současné nebo nedávno prodělané ulcerace gastrointestinálního traktu, přítomnost maligních nádorů s vysokým rizikem krvácení, nedávno prodělané poranění mozku nebo míchy, operace mozku, míchy nebo oka v nedávné době, intrakraniální krvácení v nedávné době, jícnové varixy nebo podezření na ně, arteriovenózní malformace, cévní aneurysma nebo významné cévní abnormality v míše nebo mozku.
Souběžná léčba jinými antikoagulačními přípravky, např. nefrakcionovaným heparinem (UFH), nízkomolekulárními hepariny (enoxaparin, dalteparin atd.), heparinovými deriváty (fondaparinux atd), perorálními antikoagulancii (warfarin, dabigatran etexilát, apixaban, atd.) se nedoporučuje s výjimkou specifické situace, kdy je pacient převáděn z antikoagulační léčby (viz bod 4.2) nebo když je podáván UFH v dávkách nezbytných pro udržení průchodnosti centrálního žilního nebo arteriálního katetru (viz bod 4.5).
Jaterní onemocnění, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení včetně cirhotických pacientů s klasifikací Child-Pugh B a C (viz bod 5.2).
Těhotenstí a kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Riziko krvácení
U několika podskupin pacientů (podrobně uvedených dále) hrozí zvýšené riziko krvácení. Tyto pacienty je třeba pečlivě sledovat, zda se po zahájení léčby neobjeví známky a příznaky krvácivé komplikace a anemie (viz bod 4.8). Sledování lze zajistit pravidelným klinickým vyšetřováním pacientů, pečlivým sledováním stavu drenáže operační rány a pravidelným měřením hemoglobinu.
Při jakémkoli nevysvětlitelném poklesu hladin hemoglobinu nebo krevního tlaku je třeba hledat místo krvácení.
Přestože léčba rivaroxabanem nevyžaduje rutinní monitorování expozice, hladiny rivaroxabanu měřené kalibrovanou kvantitativní analýzou anti-faktoru Xa mohou být užitečné ve výjimečných situacích, kdy znalost expozice rivaroxabanu může pomoci při klinickém rozhodování, např. při předávkování nebo při urgentních chirurgických zákrocích (viz body 5.1 a 5.2).
Ledvinová nedostatečnost
U pacientů s těžkou ledvinovou nedostatečností (clearance kreatininu < 30 ml/min) mohou být plazmatické hladiny rivaroxabanu významně zvýšeny (1,6 násobek průměru), což může vést ke zvýšenému riziku krvácení. Xarelto je proto u pacientů s clearance kreatininu 15 - 29 ml/min nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.4 a 5.2).
U pacientů se středně závažnou renální nedostatečností (clearance kreatininu 30 - 49 ml/min) užívajících současně léky zvyšující plazmatické koncentrace rivaroxabanu, musí být přípravek Xarelto používán s opatrností (viz bod 4.5).
Interakce s jinými léčivými přípravky
Použití přípravku Xarelto se nedoporučuje u pacientů současně léčených systémovými azolovými antimykotiky (jako jsou ketokonazol, itrakonazol, vorikonazol a posakonazol) nebo inhibitory proteáz HIV (například ritonavir). Tyto léčivé látky jsou silnými inhibitory systémů CYP3A4 a současně P-gp, a proto mohou zvyšovat plazmatické koncentrace rivaroxabanu v klinicky významném rozsahu (v průměru 2,6 násobek), což může vést ke zvýšenému riziku krvácení (viz bod 4.5).
Postupujte opatrně, pokud jsou pacienti současně léčeni léčivými přípravky ovlivňujícími krevní srážlivost, jako jsou například nesteroidní protizánětlivé léčivé přípravky (NSAID), kyselina acetylsalicylová (ASA) a inhibitory agregace trombocytů. U pacientů s rizikem vředové gastrointestinální choroby lze zvážit vhodnou profylaktickou léčbu (viz bod 4.5).
Jiné rizikové faktory krvácení
Rivaroxaban, podobně jako jiná antitrombotika, je nutno používat s opatrností u pacientů se zvýšeným rizikem krvácení, například:
• vrozené nebo získané poruchy krvácení
• léčbou neupravená těžká arteriální hypertenze
• jiné gastrointestinální onemocnění bez aktivní ulcerace, které může potenciálně vést ke krvácivým komplikacím (např. zánětlivé střevní onemocnění, esofagitida, gastritida a gastroesofageální refluxní choroba)
• cévní retinopatie
• bronchiektázie nebo plicní krvácení v anamnéze.
Operace fraktury krčku kosti stehenní
Rivaroxaban nebyl hodnocen z hlediska účinnosti a bezpečnosti léčby v intervenčních klinických studiích u pacientů absolvujících operace pro frakturu krčku kosti stehenní.
Spinální / epidurální anestezie nebo punkce
Pokud je u pacienta provedena anestezie (spinální či epidurální anestezie) nebo spinální resp. epidurální punkce, u pacientů preventivně léčených antitrombotiky pro prevenci tromboembolických komplikací hrozí riziko vývinu epidurálního či spinálního hematomu, který může vyústit v dlouhodobou nebo trvalou paralýzu. Riziko těchto příhod může dále zvýšit epidurální katetr dlouhodobě zavedený po operaci, nebo současné použití léčivých přípravků ovlivňujících krevní srážlivost. Riziko může také zvýšit provedení traumatické nebo opakované epidurální či spinální punkce. Pacienty je třeba často monitorovat, zda nejeví známky a příznaky neurologického poškození (například necitlivost nebo slabost dolních končetin, dysfunkce střev nebo močového měchýře). Pokud zjistíte neurologické potíže, je nutno urgentně stanovit diagnózu a zajistit léčbu. Před neuroaxiální intervencí lékař zváží potenciální přínos a riziko u pacientů na antikoagulační terapii i u pacientů, kde hodlá toto ošetření provést v rámci tromboprofylaxe.
Ke snížení možného rizika krvácení během současného užívání rivaroxabanu při neuroaxiální (spinální nebo epidurální) anestezii nebo spinální punkci se bere v úvahu farmakokinetický profil rivaroxabanu. Zavedení nebo odstranění epidurálního katetru nebo lumbální punkci je nejlépe provést, když je odhadovaný antikoagulační účinek rivaroxabanu nízký (viz bod 5.2).
Epidurální katetr se neodstraňuje dříve než 18 hodin po posledním podání rivaroxabanu. Další dávka rivaroxabanu se nepodává dříve než 6 hodin po vyjmutí katetru.
Pokud dojde k traumatické punkci, podávání rivaroxabanu se odloží o 24 hodin.
Doporučení pro dávkování před a po invazivních procedurách a chirurgickém výkonu jiném než elektivní náhradě kyčelního nebo kolenního kloubu
Pokud je nutná invazivní procedura nebo chirurgický zákrok, měl by být přípravek Xarelto 10 mg vysazen minimálně 24 hodin před zákrokem, pokud je to možné a na základě klinického posouzení lékařem.
Pokud není možné výkon odložit, je třeba posoudit zvýšené riziko krvácení oproti neodkladnosti zákroku. Léčba přípravkem Xarelto má být znovu zahájena po invazivní proceduře nebo chirurgickém zákroku co nejdříve, pokud to situace umožní a pokud je podle úsudku ošetřujícího lékaře dosaženo odpovídající hemostázy (viz bod 5.2).
Starší populace
Se zvyšujícím se věkem se může zvyšovat riziko krvácení (viz bod 5.2).
Informace o pomocných látkách
Xarelto obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a P-gp
Současné podávání rivaroxabanu s ketokonazolem (400 mg jednou denně) nebo ritonavirem (600 mg dvakrát denně) vedlo k 2,6 resp. 2,5 násobnému nárůstu střední hodnoty AUC rivaroxabanu a
1,7 resp. 1,6 násobnému nárůstu jeho střední hodnoty Cmax, s významným zesílením farmakodynamických účinků, což může vést ke zvýšenému riziku krvácení. Proto se použití přípravku Xarelto nedoporučuje u pacientů užívajících současně a systémově azolová antimykotika, jako je ketokonazol, itrakonazol, vorikonazol a posakonazol, nebo inhibitory proteáz HIV. Tyto léčivé látky jsou silnými inhibitory systémů CYP3A4 a současně P-gp (viz bod 4.4).
Léčivé látky silně inhibující pouze jednu z metabolických cest eliminace rivaroxabanu (buď CYP3A4, nebo P-gp) podle všeho zvyšují plazmatické koncentrace rivaroxabanu méně. Například klaritromycin (500 mg dvakrát denně), který zřejmě silně inhibuje CYP3A4 a středně P-gp, způsobuje 1,5 násobný nárůst středních hodnot AUC rivaroxabanu a 1,4 násobný nárůst Cmax. Tento nárůst není považován za klinicky relevantní. (Pacienti s poruchou funkce ledvin: viz bod 4.4).
Erythromycin (500 mg třikrát denně), který mírně inhibuje 3A4 a P-gp, způsobuje 1,3 násobný nárůst středních hodnot AUC a Cmax rivaroxabanu. Tento nárůst není považován za klinicky relevantní.
U pacientů s mírnou insuficiencí ledvin vedl erythromycin (500 mg třikrát denně) k1,8 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu Cmax ve srovnání s pacienty s normální renální funkcí. U pacientů se středně těžkým renálním poškozením vedl erythromycin k 2,0 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu v Cmax ve srovnání s pacienty s normální renální funkcí. Účinek erythromycinu je aditivní k renálnímu poškození (viz bod 4.4).
Flukonazol (400 mg jednou denně), který je považován za středně silný inhibitor CYP3A4, vedl k 1,4 násobnému zvýšení středních hodnot AUC rivaroxabanu a k 1,3 násobnému zvýšení průměrné Cmax. Toto zvýšení není považováno za klinicky významné. (Pacienti se sníženou funkcí ledvin: viz bod 4.4).
Dronedaron by neměl být podáván spolu s rivaroxabanem, vzhledem k omezeným klinickým údajům, které jsou k dispozici.
Antikoagulační přípravky
Po kombinovaném podávání enoxaparinu (40 mg, jednorázová dávka) s rivaroxabanem (10 mg, jednorázová dávka) byl zjištěn aditivní vliv na inhibici faktoru Xa, a to bez dalších účinků na výsledky testů srážení krve (PT, apTT). Enoxaparin neovlivňoval farmakokinetiku rivaroxabanu.
Vzhledem ke zvýšenému riziku krvácení je třeba postupovat opatrně, pokud jsou pacienti současně léčeni jinými antikoagulačními přípravky (viz body 4.3 a 4.4).
NSAID / inhibitory srážení trombocytů
Při současném podávání rivaroxabanu (15 mg) a 500 mg naproxenu nebylo zjištěno klinicky relevantní prodloužení doby krvácení. Některé osoby však mohou mít silnější farmakodynamickou odezvu.
Žádné klinicky významné farmakokinetické ani farmakodynamické interakce nebyly zjištěny při současném podání rivaroxabanu s 500 mg kyseliny acetylsalicylové.
Clopidogrel (úvodní dávka 300 mg, poté udržovací dávka 75 mg) nevykazoval farmakokinetické interakce s rivaroxabanem (15 mg), ale u části populace pacientů došlo k relevantnímu nárůstu doby krvácení, který nekoreloval s agregací trombocytů, ani hladinami P-selektinu nebo receptoru GPIIb/IIIa.
Postupovat opatrně je třeba, pokud jsou pacienti současně léčeni NSAID (včetně kyseliny acetylsalicylové) a inhibitory agregace trombocytů, protože tyto léčivé přípravky obvykle zvyšují riziko krváceni (viz bod 4.4).
Warfarin
Převod pacientů z antagonisty vitamínu K warfarinu (INR 2,0 až 3,0) na rivaroxaban (20 mg) nebo z rivaroxabanu (20 mg) na warfarin (INR 2,0 až 3,0) zvýšil protrombinový čas/INR (Neoplastin) více než aditivně (mohou být pozorovány jednotlivé hodnoty INR až 12), přičemž vliv na aPTT, inhibici aktivity faktoru Xa a potenciál endogenního trombinu byl aditivní.
Pokud je potřeba testovat farmakodynamické účinky rivaroxabanu během fáze převodu, mohou se použít testy anti-faktor Xa aktivity, PiCT a Heptest, protože tyto testy nebyly warfarinem ovlivněny. Čtvrtý den po poslední dávce warfarinu odrážely všechny testy (včetně testů PT, aPTT, inhibice aktivity faktoru Xa a ETP) pouze účinek rivaroxabanu.
Pokud je potřeba testovat farmakodynamické účinky warfarinu během fáze převodu lze použít měření INR při Cmin h rivaroxabanu (24 hodin po předchozím užití rivaroxabanu), protože tento test je v tento okamžik minimálně ovlivněn rivaroxabanem.
Mezi warfarinem a rivaroxabanem nebyla pozorována žádná farmakokinetická interakce.
Induktory CYP3A4
Současné podávání rivaroxabanu se silným induktorem CYP3A4 rifampicinem vedlo k přibližně 50% poklesu střední hodnoty AUC rivaroxabanu, s odpovídajícím poklesem farmakodynamického účinku. Současné použití rivaroxabanu s jinými silnými induktory CYP3A4 (například fenytoinem, karbamazepinem, fenobarbitalem nebo třezalkou (Hypericum perforatum)) může také vést ke snížení plazmatických koncentrací rivaroxabanu. Proto je třeba se vyhnou současnému podávání silných induktorů CYP3A4, pokud není pacient pozorně sledován kvůli známkám a příznakům trombózy.
Jiné současně podávané léky
Žádné klinicky významné farmakokinetické nebo farmakodynamické interakce nebyly zjištěny při současném podávání rivaroxabanu s midazolamem (substrát CYP3A4), digoxinem (substrát P-gp), atorvastatinem (substrát CYP3A4 a P-gp) nebo omeprazolem (inhibitor protonové pumpy). Rivaroxaban neinhibuje ani neindukuje významné izoformy CYP jako je CYP3A4.
Žádné klinicky relevantní interakce s jídlem nebyly zjištěny (viz bod 4.2).
Laboratorní parametry
Parametry srážení krve (například PT, aPTT, Heptest) jsou ovlivněny podle očekávání na základě mechanismu působení rivaroxabanu (viz bod 5.1).
4.6 Fertilita, těhotenství a kojení
Bezpečnost a účinnost přípravku Xarelto u těhotných žen nebyly stanoveny. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Vzhledem k možné reprodukční toxicitě, známému riziku krvácení a důkazu, že rivaroxaban prochází placentou, je přípravek Xarelto kontraindikován v těhotenství (viz bod 4.3). Ženy ve fertilním věku musí během léčby rivaroxabanem zabránit otěhotnění.
Kojení
Bezpečnost a účinnost přípravku Xarelto u kojících žen nebyly stanoveny. Údaje z experimentů na zvířatech signalizují, že je rivaroxaban vylučován do mléka. Podávání přípravku Xarelto je během kojení kontraindikováno (viz bod 4.3). Je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit léčbu.
Fertilita
Nebyly provedeny žádné specifické studie užívání rivaroxabanu u lidí s cílem vyhodnotit účinky na fertilitu. Ve studii samčí a samičí fertility na potkanech nebyly pozorovány žádné účinky (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Xarelto má malý vliv na schopnost řídit a obsluhovat stroje. Byly hlášeny nežádoucí účinky jako synkopa (frekvence výskytu: méně časté) a závrať (frekvence výskytu: časté) (viz bod 4.8). Pacienti, kteří zaznamenali tyto nežádoucí účinky, by neměli řídit vozidla a obsluhovat stroje.
Souhrn bezpečnostních informací
Bezpečnost rivaroxabanu byla hodnocena v jedenácti studiích fáze III, kterých se účastnilo 32 625 pacientů dostávajících rivaroxaban (viz tabulka 1).
Tabulka 1: Počet hodnocených pacientů, maximální ^ denní dávka a doba léčby ve studiích fáze III
|
Indikace |
Počet pacientů* |
Maximální denní dávka |
Maximální délka léčby |
|
Prevence žilního tromboembolismu (VTE) u dospělých pacientů podstupujících elektivní operativní náhradu kyčelního nebo kolenního kloubu |
6 097 |
10 mg |
39 dnů |
|
Prevence žilního tromboembolismu u nechirurgických pacientů |
3 997 |
10 mg |
39 dnů |
|
Léčba hluboké žilní trombózy, plicní embolie a prevence recidivy |
4 566 |
1. - 21. den: 30 mg 22. den a dále: 20 mg |
21 měsíců |
|
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní |
7 750 |
20 mg |
41 měsíců |
|
Prevence aterotrombotických příhod u pacientů po AKS |
10 225 |
5 mg nebo 10 mg, podávaných společně s ASA nebo s kombinací ASA plus klopidogrel či tiklopidin |
31 měsíců |
*Pacienti, kteří dostali alespoň jednu dávku rivaroxabanu
Nejčastěji hlášenými nežádoucí účinky u pacientů, kteří dostávali rivaroxaban, bylo krvácení (viz bod 4.4 a níže uvedený „Popis vybraných nežádoucích účinků“). Nejčastěji hlášeným krvácením (>4 %) byla epistaxe (5,9 %) a gastrointestinální krvácení (4,2 %).
Celkem asi u 67 % pacientů exponovaných minimálně jedné dávce rivaroxabanu byl hlášen výskyt nežádoucích příhod. Asi u 22 % pacientů se vyskytly nežádoucí příhody, které byly považovány za související s léčbou podle posouzení zkoušejícími. U pacientů léčených 10 mg přípravku Xarelto a podstupujících náhradu kyčelního kloubu nebo kolenního kloubu a u hospitalizovaných nechirurgických pacientů se krvácivé příhody objevily přibližně u 6,8 %, resp. 12,6 % pacientů a anémie se objevila přibližně u 5,9 %, resp. 2,1 % pacientů. U pacientů, kteří dostávali buď 15 mg přípravku Xarelto dvakrát denně a následně 20 mg jednou denně z důvodu léčby hluboké žilní trombózy nebo plicní embolie, nebo 20 mg jednou denně jako prevenci recidivující hluboké žilní trombózy a plicní embolie se krvácivé příhody objevily přibližně u 27,8 % pacientů a anémie se objevila přibližně u 2,2 % pacientů. U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace bylo hlášeno krvácení jakéhokoli typu nebo stupně závažnosti s frekvencí 28 na 100 paciento-roků a anémie s frekvencí 2,5 na 100 paciento-roků.
U pacientů léčených z důvodu prevence aterotrombotických příhod po akutním koronárním syndromu (AKS) bylo hlášeno krvácení jakéhokoli typu nebo stupně závažnosti s frekvencí 22 na 100 paciento-roků. Anémie byla hlášena s frekvencí 1,4 na 100 paciento-roků.
Tabulkový seznam nežádoucích účinků
Výskyt nežádoucích účinků hlášený u přípravku Xarelto je shrnutý v tabulce 2 níže podle orgánové klasifikace (v MedDRA) a podle frekvence výskytu.
Frekvence jsou definovány takto: velmi časté (> 1/10) časté (> 1/100 až < 1/10) méně časté ( > 1/1 000 až < 1/100)
vzácné (> 1/10 000 až < 1/1 000)
velmi vzácné ( < 1/10,000)
není známo ( z dostupných údajů nelze určit)
Tabulka 2: Všechny s léčbou související nežádoucí účinky, hlášené u pacientů ve studiích fáze III
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (vč. příslušných laboratorních parametrů) |
Trombocytémie (včetně zvýšeného počtu trombocytů)A | ||
|
Poruchy imunitního systému | |||
|
Alergické reakce, alergická | |||
|
Poruchy nervového systému | |||
|
Závratě, bolest hlavy |
Cerebrální a intrakraniální krvácení, synkopa | ||
|
Poruchy oka | |||
|
Oční krvácení (včetně krvácení do spojivek) | |||
|
Srdeční poruchy | |||
|
Cévní poruchy | |||
|
hematomy | |||
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe, hemoptýza | |||
|
Gastrointestinální poruchy | |||
|
Gingivální krvácení, krvácení z gastrointestinálního traktu (včetně rektálního krvácení), gastrointestinální a abdominální bolest, dyspepsie, nausea, A |
Sucho v ústech | ||
|
Poruchy jater a žlučových cest | |||
|
Abnormity j aterní funkce |
Žloutenka | ||
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy kůže a por |
kožní tkáně | ||
|
Pruritus (včetně vzácných případů generalizovaného pruritu), vyrážka, ekchymóza, kožní a podkožní krvácení |
Kopřivka | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |||
|
Bolest v končetináchA |
Hemartróza |
Krvácení do svalů |
Kompartment syndrom sekundárně po krvácení |
|
Poruchy ledvin a močových cest | |||
|
Urogenitální krvácení (včetně hematurie a menorhagieB), poškození ledvin (včetně zvýšení hladin kreatininu a močoviny v krvi)A |
Renální selhání/akutní renální selhání vzniklé sekundárně po krvácení natolik silném, aby způsobilo hypoperfúzi | ||
|
Celkové poruchy a reakce v místě aplikace | |||
|
HorečkaA, periferní edém, pokles celkové síly a energie (včetně únavy, tělesné slabosti) |
Pocit indispozice (včetně malátnosti) |
Lokalizovaný edémA | |
|
Vyšetření | |||
|
Zvýšená hladina transamináz |
Zvýšená hladina bilirubinu, alkalické fosfatázyA, LDHA, lipázyA, amylázyA, GMTA |
Zvýšení konjugovaného bilirubinu (s přidruženým zvýšením ALT nebo bez zvýšení ALT) | |
|
Poranění, otravy a |
procedurální komplikace | ||
|
Pooperační krvácení (včetně pooperační anémie a krvácení z rány), kontuze, sekrece z ranA |
Vaskulární pseudoaneurysmaC | ||
A: pozorováno u prevence žilního tromboembolismu u dospělých pacientů po plánované náhradě kyčelního nebo kolenního kloubu
B: pozorováno u léčby hluboké žilní trombózy, plicní embolie a u prevence recidivy jako velmi časté u žen < 55 let
C: pozorováno jako méně časté u prevence aterotrombotických příhod u pacientů po akutním koronárním syndromu (po perkutánní koronární intervenci)
Popis vybraných nežádoucích účinků
Vzhledem k farmakologickému mechanismu působení může být užívání přípravku Xarelto spojeno se zvýšeným rizikem okultního nebo zjevného krvácení z jakékoli tkáně nebo orgánu s možným následkem posthemoragické anémie. Známky, příznaky a závažnost (včetně fatálního zakončení) se mohou různit podle místa a stupně nebo rozsahu krvácení a/nebo anémie (viz bod 4.9 Léčba krvácení). V klinických studiích bylo během dlouhodobé léčby rivaroxabanem v porovnání s léčbou VKA mnohem častěji pozorováno slizniční krvácení (tj. epistaxe, krvácení z dásní, gastrointestinální krvácení, krvácení v urogenitálním traktu) a anémie. Při posuzování stavu může být potřeba kromě adekvátního klinického sledování pacientů provést laboratorní vyšetření hemoglobinu/hematokritu pro detekci okultního krvácení. Riziko krvácení bude možná zvýšeno u některých skupin pacientů, například osob s nekontrolovanou těžkou arteriální hypertenzí a/nebo souběžnou léčbou ovlivňující krevní srážlivost (viz Riziko krvácení v bodu 4.4). Menstruační krvácení může být intenzivnější a/nebo prodloužené. Hemoragické komplikace se mohou projevovat jako celková slabost, bledost, závratě, bolesti hlavy nebo nevysvětlitelné otoky, dušnost a nevysvětlitelný šok. V některých případech byly v důsledku anémie pozorovány příznaky srdeční ischémie, jako je například bolest na hrudníku nebo angina pectoris.
V souvislosti s užíváním přípravku Xarelto byly hlášeny známé sekundární komplikace závažného krvácení, jako je například kompartment syndrom a renální selhání v důsledku hypoperfúze. Možnost krvácení je proto třeba zvážit při posuzování stavu pacientů s jakoukoli antikoagulační léčbou.
Post-marketingové sledování
V postmarketingovém sledování byly v časové souvislosti s užitím přípravku Xarelto hlášeny následující nežádoucí účinky. Frekvenci těchto nežádoucích účinků hlášených po uvedení na trh nelze určit.
Poruchy imunitního systému: angioedém a alergický edém (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Poruchy jater a žlučových cest: cholestáza, hepatitida (včetně hepatocelulárního poškození) (v souhrnných studiích fáze III byly tyto příhody vzácné (>1/10 000 až <1/1000)).
Poruchy krve a lymfatického systému: trombocytopenie (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny vzácné případy předávkování dávkou až 600 mg bez krvácivých komplikací nebo jiných nežádoucích reakcí. Z důvodu omezené absorpce se očekává strop účinku bez dalšího zvýšení průměrné plazmatické hladiny v případě vyšší než terapeutické dávky 50 mg rivaroxabanu nebo dávek vyšších. Specifické antidotum blokující farmakodynamický účinek rivaroxabanu není k dispozici.
Lze zvážit podání aktivního uhlí ke snížení absorpce v případě předávkování rivaroxabanem.
Léčba krvácení
Pokud dojde ke krvácivým komplikacím u pacienta léčeného rivaroxabanem, musí se podání další dávky rivaroxabanu odložit nebo se léčba musí ukončit, dle potřeby. Rivaroxaban má biologický poločas asi 5 až 13 hodin (viz bod 5.2). Léčba by měla být individuální podle závažnosti a lokalizace krvácení. Podle potřeby je třeba použít vhodnou symptomatickou léčbu, jako je mechanická komprese (např. u závažné epistaxe), chirurgická hemostáza se zajištěním kontroly krvácení, náhradou tekutin a zajištěním hemodynamické podpory, krevní deriváty (erytrocyty nebo čerstvá zmrazená plasma, v závislosti na související anémii nebo koagulopatii) nebo trombocyty.
Pokud krvácení nelze kontrolovat výše uvedenými opatřeními, lze zvážit podávání specifické prokoagulační reverzní látky, jako je koncentrát protrombinového komplexu (PCC), aktivovaný koncentrát protrombinového komplexu (APCC) nebo rekombinantní faktor VIIa (r-FVIIa). V současnosti jsou však k dispozici velmi omezené klinické zkušenosti s použitím těchto přípravků u osob užívajících rivaroxaban. Doporučení je též podloženo omezenými neklinickými údaji. Opakované podání rekombinantního faktoru VIIa je třeba zvážit a titrovat v závislosti na zlepšování krvácení. V případě závažného krvácení je třeba konzultovat odborníka na koagulaci, pokud je odborník v místě dostupný (viz bod 5.1).
Protamin sulfát a vitamin K podle všeho nebudou ovlivňovat antikoagulační aktivitu rivaroxabanu. U osob užívajících rivaroxaban jsou omezené zkušenosti s použítím kyseliny tranexamové a neexistují zkušenosti s použitím kyseliny aminokaproové a aprotininu. Neexistují ani vědecké důvody přínosu ani zkušenosti
s použitím systémového hemostatika desmopressinu u osob užívajících rivaroxaban. Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: přímé inhibitory faktoru Xa, ATC kód: B01AF01 Mechanismus účinku
Rivaroxaban je vysoce selektivní přímý inhibitor faktoru Xa biologicky dostupný při perorálním podání. Inhibice faktoru Xa blokuje vnitřní a vnější metabolické cesty koagulační kaskády, a inhibuje vznik trombinu i vytváření trombů. Rivaroxaban neinhibuje trombin (aktivovaný faktor II) a nebyly prokázány žádné účinky na trombocyty.
Farmakodynamické účinky
U lidí byla zjištěna inhibice faktoru Xa přímo úměrná dávce. Protrombinový čas (PT) je rivaroxabanem ovlivňován úměrně dávce, a pokud je při testu použit Neoplastin, objevuje se vysoká korelace s plazmatickými koncentracemi (hodnota R je 0,98). Jiné reagenty mohou přinést jiné výsledky. Hodnotu PT je nutno odečíst v sekundách, protože INR (mezinárodní normalizovaný poměr) je kalibrován a validován pouze pro kumariny, a nelze jej využívat pro jiné antikoagulanty. U pacientů absolvujících velkou ortopedickou operaci se v 5/95 percentilu hodnoty PT (Neoplastin) za 2 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 13 až 25 s (výchozí hodnoty před operací byly 12 až 15 s).
V klinické farmakologické studii sledující reverzi farmakodynamického účinku rivaroxabanu u zdravých dospělých osob (n=22) byl hodnocen účinek jednotlivé dávky (50 IU/kg) u dvou rozdílných typů PCC, 3-faktorového PCC (faktory II, IX a X) a 4-faktorového PCC (II, VII, IX a X). 3-faktorový PCC redukoval průměrnou hodnotu PT času (protrombinového času), hodnocenou neoplastinem, přibližně o 1,0 sekundy během 30 minut, ve srovnání s přibližně 3,5 sekundami pozorovanými u 4-faktorového PCC. Naproti tomu, 3-faktorový PCC měl větší a rychlejší celkový efekt na zvrácení změny tvorby endogenního trombinu než 4-faktorový PCC (viz bod 4.9).
Aktivovaný parciální tromboplastinový čas (aPTT) a hodnoty analýzy HepTest jsou také prodlouženy úměrně dávce; nedoporučuje se však tyto metody používat k hodnocení farmakodynamických účinků rivaroxabanu. Během léčby rivaroxabanem v klinické praxi není třeba monitorovat parametry koagulace. Pokud však je klinicky indikováno, lze hladiny rivaroxabanu měřit pomocí kalibrovaných kvantitativních testů anti-faktoru Xa (viz bod 5.2).
Klinická účinnost a bezpečnost
Klinický program ověření rivaroxabanu měl prokázat účinnost rivaroxabanu při prevenci VTE, tedy proximální a distální hluboké žilní trombózy (DVT) a plicní embolie (PE) u pacientů absolvujících rozsáhlé ortopedické operace dolních končetin. Přes 9 500 pacientů (7 050 absolvujících totální náhradu kyčelního kloubu a 2 531 implantaci totální endoprotézy kolenního kloubu) bylo sledováno v kontrolovaných randomizovaných dvojitě slepých studiích fáze III - programu RECORD.
Rivaroxaban 10 mg jednou denně, podávaný ne dříve než 6 hodin po operaci byl porovnáván s enoxaparinem (40 mg jednou denně, podávaný od 12 hodin před operací).
Ve všech třech studiích fáze III (viz tabulka 3) rivaroxaban významně snížil výskyt všech VTE (jakákoli venograficky zjištěná nebo symptomatická DVT, nefatální PE a smrt) a závažných typů VTE (proximální DVT, nefatální PE a smrt vinou VTE), a také předem stanovené primární a hlavní sekundární výstupy v oblasti účinnosti. Kromě toho byl ve všech třech studiích výskyt symptomatické VTE (symptomatická DVT, nefatální PE, úmrtí vinou VTE) u rivaroxabanem léčených pacientů oproti enoxaparinu nižší.
Hlavní bezpečnostní výstup, rozsáhlé krvácení, vykazoval srovnatelnou četnost u pacientů léčených rivaroxabanem (10 mg) i enoxaparinem (40 mg).
|
Tabulka 3: Účinnost a bezpečnost - výsledky z klinických studií fáze I |
I | ||
|
RECORD 1 |
RECORD 2 |
RECORD 3 | |
|
Populace studie: |
4 541 pacientů absolvujících totální náhradu kyčelního kloubu |
2 509 pacientů absolvujících totální náhradu kyčelního kloubu |
2 531 pacientů absolvujících implantaci totální endoprotézy kolenního kloubu |
|
Dávkování přípravků a délka podávání po operaci |
Rivaroxaban Enoxaparin p 10 mg jednou 40 mg jednou denně denně 35 ± 4 dny 35 ± 4 dny |
Rivaroxaban Enoxaparin p 10 mg jednou 40 mg jednou denně denně 35 ± 4 dny 12 ± 2 dny |
Rivaroxaban Enoxaparin p 10 mg jednou 40 mg jednou denně denně 12 ± 2 dny 12 ± 2 dny |
|
Všechny VTE |
18 (1,1%) 58 (3,7%) <0,001 |
17 (2,0%) 81 (9,3%) <0,001 |
79 (9,6%) 166 (18,9%) <0,001 |
|
Závažné VTE |
4 (0,2%) 33 (2,0%) <0,001 |
6 (0,6%) 49 (5,1%) <0,001 |
9 (1,0%) 24 (2,6%) 0,01 |
|
Symptomatické VTE |
6 (0,4%) 11 (0,7%) |
3 (0,4%) 15 (1,7 %) |
8 (1,0%) 24 (2,7%) |
|
Rozsáhlé krvácení |
6 (0,3 %) 2 (0,1%) |
1 (0,1%) 1 (0,1%) |
7 (0,6%) 6 (0,5%) |
Analýza spojených výsledků z uvedených studií fáze III potvrdila údaje získané v jednotlivých studiích ohledně snížení celkových VTE, závažných VTE a symptomatických VTE při užívání rivaroxabanu 10 mg jednou denně, a to v porovnání s dávkami 40 mg enoxaparinu jednou denně.
Kromě fáze III RECORD programu byla provedena poregistrační neintervenční otevřená kohortová studie (XAMOS) u 17 413 pacientů, kteří podstoupili rozsáhlou ortopedickou operaci kyčle nebo kolena, k porovnání rivaroxabanu a ostatní farmakologické tromboprofylaxe (standardní péče) v reálné klinické praxi. Symptomatická hluboká žilní trombóza se vyskytla u 57 (0,6 %) pacientů ve skupině dostávající rivaroxaban (n=8 778) a u 88 (1,0 %) pacientů dostávajících standardní léčbu (n=8 635; HR 0,63; 95% CI 0,43-0,91); bezpečnostní populace). Závažné krvácení se vyskytlo u 35 (0,4 %) pacientů ze skupiny dostávající rivaroxaban a u 29 (0,3 %) pacientů dostávajících standardní léčbu (HR 1,10; 95% CI 0,67-1,80). Tudíž výsledky byly v souladu s výsledky z pivotních randomizovaných studií.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xarelto v léčbě tromboembolických příhod u jedné nebo více podskupin pediatrické populace. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xarelto v prevenci tromboembolických příhod u všech podskupin pediatrické populace (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Rivaroxaban je rychle absorbován; maximální koncentrace (Cmax) se objeví 2 - 4 hodiny po užití tablety.
Bez ohledu na stav na lačno nebo po jídle je u dávky 2,5 mg a 10 mg rivaroxabanu tablety perorální absorpce téměř kompletní a perorální biologická dostupnost vysoká (80 - 100 %). Rivaroxaban 2,5 mg a 10 mg tablety lze užívat při jídle nebo nezávisle na jídle.Užívání při jídle neovlivňuje při 2,5 mg a 10 mg dávce AUC ani Cmax rivaroxabanu. Farmakokinetické vlastnosti rivaroxabanu jsou až do denní dávky 15 mg přibližně lineární. Ve vyšších dávkách je absorbce rivaroxabanu omezena disolucí, dochází ke snížení biologické dostupností a míra absorbce se snižuje se zvyšující se dávkou. To se výrazněji projevuje ve stavu na lačno než po jídle. Variabilita farmakokinetiky rivaroxabanu je střední, s interindividuální variabilitou v rozmezí od 30 % do 40 %, kromě dne operace a následujícího dne, kdy je variabilita expozice vysoká (70 %).
Absorpce rivaroxabanu je závislá na místě jeho uvolnění v gastrointestinálním traktu. Bylo hlášeno 29% a 56% snížení AUC a Cmax ve srovnání s tabletou, pokud byl rivaroxaban v granulátu uvolněn v proximální časti tenkého střeva. Expozice je dále snížena, když je rivaroxaban uvolněn v distální části tenkého střeva nebo ve vzestupné části tračníku. Podání rivaroxabanu distálně od žaludku by se mělo zabránit, jelikož to může vést ke snížení absorpce a související expozice rivaroxabanu.
Biologická dostupnost (AUC a Cmax) byla pro podání 20 mg rivaroxabanu per os ve formě rozdrcené tablety vmíchané do jablečného pyré nebo rozpuštěné ve vodě a pro podání žaludeční sondou následované tekutou stravou, v porovnání s celou tabletou srovnatelná. Vzhledem k předvídatelnému dávce úměrnému farmakokinetickému profilu rivaroxabanu odpovídají výsledky biologické dostupnosti z této studie pravděpodobně nižším dávkám rivaroxabanu.
Distribuce
Vazba na plazmatické proteiny u lidí je vysoká, přibližně 92% - 95%, přičemž hlavní část se váže na sérový albumin. Distribuční objem je střední, Vss činí přibližně 50 litrů.
Biotransformace a eliminace
Z podané dávky rivaroxabanu se přibližně 2/3 metabolicky degradují, z čehož je polovina vylučována ledvinami a druhá polovina stolicí. Zbývající 1/3 podané dávky je vylučována ledvinami přímo jako nezměněná léčivá látka, hlavně prostřednictvím aktivní ledvinové sekrece.
Rivaroxaban je metabolizován prostřednictvím systémů CYP3A4 a CYP2J2 i mechanismy na CYP nezávislými. Hlavními cestami transformace je oxidativní degradace morfolinonové části a hydrolýza amidových vazeb. Na základě in vitro experimentů je zřejmé, že rivaroxaban slouží jako substrát transportních proteinů - P-gp (P-glykoprotein) a BCRP (breast cancer resistance protein).
Nezměněný rivaroxaban je nejvýznamnější formou přípravku v lidské plazmě; v krevním oběhu nejsou žádné významné nebo aktivní metabolity. Rivaroxaban lze vzhledem ke systémové clearance asi 10 l/h klasifikovat jako látku s nízkou clearance. Po intravenózním podání dávky 1 mg je eliminační poločas asi
4.5 hodiny. Po perorálním podání je eliminace omezována mírou absorpce. K eliminaci rivaroxabanu z plazmy dochází s terminálním poločasem 5 až 9 hodin u mladších osob a s terminálním poločasem 11 až 13 hodin u starších osob.
Zvláštní skupiny
Pohlaví
Mezi muži a ženami nebyl žádný klinicky relevantní rozdíl ve farmakokinetice a farmakodynamice přípravku.
Starší populace
Starší pacienti vykazovali vyšší plazmatické koncentrace než mladší, se střední hodnotou AUC přibližně
1.5 x vyšší, hlavně vzhledem ke snížené (zdánlivé) celkové a ledvinové clearance. Žádná úprava dávky není nutná.
Různé váhové kategorie
Extrémy v tělesné hmotnosti (< 50 kg nebo > 120 kg) měly pouze malý vliv na plazmatické koncentrace rivaroxabanu (méně než 25%). Žádná úprava není dávky nutná.
Rozdíly mezi etniky
Žádné klinicky relevantní rozdíly mezi etniky nebyly ve farmakokinetice a farmakodynamice rivaroxabanu zjištěny u pacientů z řad bělochů, Afroameričanů, Hispánců, Japonců ani Číňanů.
Jaterní nedostatečnost
Pacienti s cirhózou s mírnou jaterní nedostatečností (Child-Pugh A) vykazovali pouze menší změny ve farmakokinetice rivaroxabanu (v průměru 1,2x nárůst AUC rivaroxabanu), a výsledky byly téměř srovnatelné s kontrolní skupinou pacientů se srovnatelným zdravotním stavem. U pacientů trpících cirhózou se středně závažnou jaterní nedostatečností (Child-Pugh B) střední AUC rivaroxabanu významně stoupla -2,3x v porovnání se zdravými dobrovolníky. AUC nevázané látky stoupla 2,6x. U těchto pacientů dochází ke snížení renální eliminace rivaroxabanu, podobně jako u pacientů se středně těžkou ledvinovou nedostatečností. O účinku u pacientů s těžkým jaterním poškozením nejsou k dispozici žádné údaje.
Inhibice aktivity faktoru Xa byla u pacientů se střední jaterní nedostatečností zvýšena ve srovnání se zdravými dobrovolníky 2,6x; prodloužení PT bylo obdobně zvýšeno 2,1x. Pacienti se střední jaterní nedostatečností byli na rivaroxaban citlivější, a vztah mezi koncentrací a PT měl tak strmější průběh.
Přípravek Xarelto je kontraindikován u pacientů s jatemím onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child-Pugh B a C (viz bod 4.3).
Ledvinová nedostatečnost
Byl zjištěn nárůst expozice rivaroxabanu související s poklesem funkce ledvin, která byla posuzována prostřednictvím hodnot clearance kreatininu. U osob s lehkou (clearance kreatininu 50 - 80 ml/min), střední (clearance kreatininu 30 - 49 ml/min) a těžkou (clearance kreatininu 15 - 29 ml/min) ledvinovou nedostatečností byly plazmatické koncentrace rivaroxabanu (AUC) zvýšeny 1,4, 1,5 resp. 1,6 x.
Odpovídající zesílení farmakodynamických účinků bylo výraznější. U osob s lehkou, střední a těžkou ledvinovou nedostatečností byla celková inhibice faktoru Xa ve srovnání se zdravými dobrovolníky zvýšena 1,5, 1,9 resp. 2,0 x; prodloužení PT bylo obdobně zvýšeno 1,3, 2,2 a 2,4 x. O použití u pacientů s clearance kreatininu < 15 ml/min nejsou žádné údaje.
Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min. Xarelto je proto u pacientů s clearance kreatininu 15 - 29 ml/min nutno používat s opatrností (viz bod 4.4).
Farmakokinetické údaje u pacientů
U pacientů užívajících rivaroxaban jako prevenci žilního tromboembolismu (VTE) 10 mg jednou denně byl geometrický průměr koncentrace (90 % interval odpovědi) 2 - 4 h a asi 24 h po podání dávky (představující zhruba maximální a minimální koncentrace během dávkovacího intervalu) 101 (7 - 273) a 14 (4 - 51) pg/l.
Farmakokinetické a farmakodynamické vztahy
Po podání různě velkých dávek (5 - 30 mg dvakrát denně) byl hodnocen farmakokinetický a farmakodynamický (PK/PD) vztah mezi plazmatickou koncentrací rivaroxabanu a několika konečnými cílovými ukazateli PD (inhibice faktoru Xa , PT, aPTT, Heptest). Vztah mezi plazmatickou koncentrací rivaroxabanu a aktivitou faktoru Xa byl nejlépe popsán pomocí modelu Emax. U PT byly údaje lépe vyjádřeny pomocí lineárního ohraničeného modelu. Hodnoty PT se významně lišily v závislosti na použitých reagenciích. Při použití Neoplastinu byl výchozí PT asi 13 sekund a odchylka hodnot přibližně 3 až 4 s/(100 ^g/l). Výsledek analýz PK/PD ve studii fáze II a III byl v souladu s údaji získanými u zdravých jedinců. U pacientů byly výchozí hodnoty faktoru Xa a PT ovlivněny operací, následkem toho byl zjištěn rozdíl v počátečních hodnotách a v hodnotách zaznamenaných den po operaci.
Pediatrická populace
Bezpečnost a účinnost nebyly stanoveny u dětí a dospívajících do 18 let věku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po jednorázovém podání, fototoxicity, genotoxicity, kancerogenního potenciálu a juvenilní toxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studii toxicity při opakovaném podání byly způsobeny hlavně zesílenou farmakologickou aktivitou rivaroxabanu. Při klinicky relevantních úrovních expozice byly u potkanů pozorovány zvýšené plazmatické hladiny IgG a IgA.
U potkanů nebyly pozorovány žádné účinky na fertilitu samců nebo samic. Studie na zvířatech prokázaly reprodukční toxicitu související s farmakologickým mechanismem působení rivaroxabanu (např. hemoragickými komplikacemi). V klinicky relevantních plazmatických koncentracích byla pozorována embryonální a fetální toxicita (post-implantační pokles, opožděná nebo progredující osifikace, hepatální mnohočetné světle zbarvené skvrny) a zvýšený výskyt malformací a také placentárních změn. V prenatálních a postnatálních experimentech u potkanů byla zjištěna snížená životaschopnost potomků, a to v dávkách toxických pro matky.
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Sodná sůl kroskarmelózy Monohydrát laktózy Hypromelóza Natrium-lauryl-sulfát Magnesium-stearát.
Potah tablety:
Makrogol 3350
Hypromelóza
Oxid titaničitý (E 171)
Červený oxid železitý (E 172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Blistry z Al/PP fólie nebo PVC/PVDC/Al, v krabičkách po 5, 10 nebo 30 potahovaných tabletách nebo perforované jednodávkové blistry obsahující 10 x 1 nebo 100 x 1 potahovanou tabletu v krabičkách. Perforované jednodávkové blistry z PP/Al fólie v multibaleních obsahujících 100 potahovaných tablet (10 balení po 10 x 1 tabletě).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/472/001-010, EU/1/08/472/022
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. září 2008
Datum posledního prodloužení registrace: 22. května 2013
10. DATUM REVIZE TEXTU {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xarelto 15 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rivaroxabanum 15 mg.
Pomocná látka se známým účinkem:
Jedna potahovaná tableta obsahuje 24,13 mg laktózy (jako monohydrátu), viz bod 4.4. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Červené, kulaté, bikonvexní tablety (o průměru 6 mm, poloměru zakřivení 9 mm) označené logem (kříž) BAYER na jedné straně a číslem „15“ a trojúhelníkem na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence cévní mozkové příhody a systémové embolizace u dospělých pacientů s nevalvulární fibrilací síní s jedním nebo více rizikovými faktory, jako je městnavé srdeční selhání, hypertenze, věk 75 let a vyšší, diabetes mellitus, prodělaná cévní mozková příhoda nebo tranzitorní ischemická ataka.
Léčba hluboké žilní trombózy (HŽT) a plicní embolie (PE) a prevence recidivující hluboké žilní trombózy a plicní embolie u dospělých (hemodynamicky nestabilní pacienti s PE viz bod 4.4).
4.2 Dávkování a způsob podání
Dávkování
Prevence cévní mozkové příhody a systémové embolizace
Doporučená dávka je 20 mg jednou denně, což je také doporučená maximální dávka.
Léčba přípravkem Xarelto by měla být dlouhodobá za předpokladu, že přínos prevence cévní mozkové příhody a systémové embolizace převáží riziko krvácení (viz bod 4.4).
Pokud dojde k vynechání dávky, měl by pacient užít přípravek Xarelto co nejdříve a pokračovat v užívání jednou denně následující den podle doporučení. Dávka by neměla být tentýž den zdvojnásobena, aby se nahradila vynechaná dávka.
Léčba hluboké žilní trombózy, léčba plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie
Doporučená dávka pro úvodní léčbu akutní hluboké žilní trombózy nebo plicní embolie je 15 mg dvakrát denně po dobu prvních tří týdnů a dále 20 mg jednou denně jako udržovací léčba a prevence hluboké žilní trombózy a plicní embolie podle tabulky uvedené níže.
|
Dávkování |
Maximální denní dávka | |
|
1. až 21. den |
15 mg dvakrát denně |
30 mg |
|
22. den a dále |
20 mg jednou denně |
20 mg |
K usnadnění změny dávkování po 21. dnu léčby z 15 mg na 20 mg při léčbě HZT/PE je dostupné balení pro zahájení léčby přípravkem Xarelto pro první 4 týdny léčby (viz bod 6.5).
Délka léčby by měla být stanovena individuálně po pečlivém zhodnocení přínosu léčby a rizika krvácení (viz bod 4.4). Krátkodobá léčba (nejméně 3 měsíce) by měla být indikována na základě přechodných rizikových faktorů (např. operace prodělaná v nedávné době, úraz, imobilizace) a dlouhodobá léčba by měla být indikována na základě trvalých rizikových faktorů nebo idiopatické HZT nebo PE.
Pokud dojde k vynechání dávky během té fáze léčby, kdy je přípravek podáván v dávce 15 mg dvakrát denně (1. - 21. den), měl by pacient užít přípravek Xarelto co nejdříve, aby se zajistilo dávkování 30 mg přípravku Xarelto denně. V tomto případě mohou být užity dvě 15 mg tablety najednou. Pacient by měl pokračovat s pravidelným užíváním dávky 15 mg dvakrát denně následující den podle doporučení.
Pokud dojde k vynechání dávky během té fáze léčby, kdy je přípravek podáván v jedné denní dávce (22. den a dále), měl by pacient užít přípravek Xarelto co nejdříve a pokračovat s užíváním jednou denně následující den podle doporučení. Dávka by neměla být pro nahrazení vynechané dávky ve stejný den zdvojnásobena.
Převod z antagonistů vitaminu K (VKA) na přípravek Xarelto
U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace by měly být antagonisté vitaminu K vysazeny a léčba přípravkem Xarelto by měla být zahájena při hladině mezinárodního normalizačního poměru (INR) < 3,0.
U pacientů léčených pro hlubokou žilní trombózu, plicní embolii a pro prevenci recidivy by měly být antagonisté vitaminu K vysazeny a léčba přípravkem Xarelto by měla být zahájena při hladině INR < 2,5.
Při převodu pacientů z antagonistů vitaminu K na přípravek Xarelto, budou po užití přípravku Xarelto hladiny INR falešně zvýšeny. Test INR není pro měření antikoagulační aktivity přípravku Xarelto validní a proto by neměl být používán (viz bod 4.5).
Převod z přípravku Xarelto na antagonisty vitaminu K (VKA)
Během přechodu z přípravku Xarelto na antagonisty vitaminu K existuje možnost neadekvátní antikoagulace. Během jakéhokoli převodu na alternativní antikoagulancia by měla být zajištěna kontinuální adekvátní antikoagulace. Je třeba uvést, že přípravek Xarelto může přispět ke zvýšení INR.
U pacientů, kteří jsou převáděni z přípravku Xarelto na antagonisty vitaminu K by měli být tito antagonisté podáváni současně, dokud není hladina INR > 2,0. Po dobu prvních dvou dnů fáze převodu by mělo být použito standardní úvodní dávkování antagonistů vitaminu K s následným dávkováním těchto antagonistů na základě testování INR. Během doby, kdy pacienti užívají jak přípravek Xarelto tak antagonisty vitaminu K by nemělo být prováděno testování INR dříve než 24 hodin po předchozí dávce, ale před další dávkou přípravku Xarelto. Jakmile je přípravek Xarelto vysazen, může být testování INR spolehlivě provedeno minimálně 24 hodin po poslední dávce (viz body 4.5 a 5.2).
Převod z parenterálních antikoagulancií přípravků na přípravek Xarelto
U pacientů, kteří dostávají parenterální antikoagulancia, přerušte podávání parenterálního antikoagulancia a začněte léčbu přípravkem Xarelto v rozmezí 0 až 2 hodiny před tím, než by mělo dojít k dalšímu plánovanému podání parenterálního přípravku (např. nízkomolekulární hepariny) nebo v době vysazení kontinuálně podávaného parenterálního přípravku (např. intravenózní nefrakciovaný heparin).
Převod z přípravku Xarelto na parenterální antikoagulancia
Podejte první dávku parenterálního antikoagulancia v době, kdy by měla být užita další dávka přípravku Xarelto.
Speciální populace
Ledvinová nedostatečnost
Omezené klinické údaje u nemocných s těžkou renální nedostatečností (clearance kreatininu 15 - 29 ml/min) signalizují, že u této populace pacientů jsou plazmatické koncentrace rivaroxabanu významně zvýšeny. Xarelto je proto u těchto pacientů nutno užívat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.4 a 5.2).
U pacientů se středně závažnou (clearance kreatininu 30 - 49 ml/min) nebo závažnou (clearance kreatininu 15-29 ml/min) renální nedostatečností platí následující doporučení pro dávkování:
- pro prevenci cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní je doporučené dávkování 15 mg jednou denně (viz bod 5.2),
- pro léčbu hluboké žilní trombózy, léčbu plicní embolie a prevenci recidivující hluboké žilní trombózy a plicní embolie: pacienti by měli být léčeni dávkou 15 mg dvakrát denně po dobu prvních tří týdnů. Poté je doporučená dávka 20 mg jednou denně. Snížení dávky z 20 mg jednou denně na 15 mg jednou denně je třeba zvážit, pokud u pacienta riziko krvácení převáží riziko vzniku recidivující HZT a PE. Doporučení pro použití dávky 15 mg je založeno na farmakokinetickém modelu a nebylo v těchto klinických podmínkách studováno (viz body 4.4, 5.1 a 5.2).
Úprava dávky není nutná u pacientů s mírnou renální nedostatečností (clearance kreatininu 50 - 80 ml/min) (viz bod 5.2).
Jaterní nedostatečnost
Xarelto je kontraindikováno u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child Pugh B a C (viz body 4.3 a 5.2).
Starší populace
Dávky bez úprav (viz bod 5.2).
Tělesná hmotnost
Dávky bez úprav (viz bod 5.2).
Pohlaví
Dávky bez úprav (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Xarelto u dětí ve věku 0 až 18 let nebyla ještě stanovena. Nejsou dostupné žádné údaje. Podávání přípravku Xarelto dětem do 18 let se proto nedoporučuje.
Pacienti podstupující kardioverzi
Léčba přípravkem Xarelto může být zahájena nebo v ní lze pokračovat u pacientů, jejichž stav vyžaduje provedení kardioverze.
U pacientů podstupujících transezofageální echokardiografií (TEE) řízenou kardioverzi, kteří nebyli předem léčeni antikoagulancii, má být léčba přípravkem Xarelto zahájena nejméně 4 hodiny před kardioverzí, aby byla zajištěna odpovídající antikoagulace (viz body 5.1 a 5.2). Před provedením kardioverze je třeba u všech pacientů usilovat o potvrzení, že pacient užíval Xarelto, jak bylo předepsáno. Při rozhodování o zahájení léčby a o jejím trvání se musí vzít v úvahu pokyny dané doporučením pro antikoagulační léčbu pacientů podstupujících kardioverzi.
Způsob podání
Perorální podání. Tablety se mají užívat s jídlem (viz bod 5.2).
Pacientům, kteří nej sou schopni polykat celé tablety, může být tableta přípravku Xarelto před užitím rozdrcena a smíchána s vodou nebo s jablečným pyré, a poté podána perorálně. Po podání rozdrcené potahované tablety Xarelto 15 mg nebo 20 mg musí být dávka okamžitě následována jídlem.
Rozdrcená tableta přípravku Xarelto může být také podána gastrickou sondou poté, co je potvrzeno správné umístění sondy v žaludku. Umístění sondy v žaludku by mělo být před podáním přípravku Xarelto potvrzeno. Rozdrcená tableta by měla být podána žaludeční sondou v malém množství vody a sonda by poté měla být propláchnuta vodou. Po podání rozdrcené potahované tablety Xarelto 15 mg nebo 20 mg musí být poté dávka okamžitě následována enterální výživou (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku. Aktivní klinicky významné krvácení.
Léze nebo stavy, které jsou považovány za významné riziko závažného krvácení. Mohou mezi ně patřit současné nebo nedávno prodělané ulcerace gastrointestinálního traktu, přítomnost maligních nádorů s vysokým rizikem krvácení, nedávno prodělané poranění mozku nebo míchy, operace mozku, míchy nebo oka v nedávné době, intrakraniální krvácení v nedávné době, jícnové varixy nebo podezření na ně, arteriovenózní malformace, cévní aneurysma nebo významné cévní abnormality v míše nebo mozku.
Souběžná léčba jinými antikoagulačními přípravky, např. nefrakcionovaným heparinem (UFH), nízkomolekulárními hepariny (enoxaparin, dalteparin atd), heparinovými deriváty (fondaparinux atd), orálními antikoagulancii (warfarin, dabigatran etexilát, apixaban, atd.), se nedoporučuje s výjimkou specifické situace, kdy je pacient převáděn z antikoagulační léčby (viz bod 4.2), nebo když je podáván UFH v dávkách nezbytných pro udržení průchodnosti centrálního žilního nebo arteriálního katetru (viz bod 4.5).
Jaterní onemocnění, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně pacientů s cirhózou s klasifikací Child Pugh B a C (viz bod 5.2).
Těhotenstí a kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
V průběhu léčby se doporučuje pacienta klinicky sledovat v souladu s praxí běžnou při podávání antikoagulační léčby.
Riziko krvácení
Jako v případě jiných antikoagulancií, u pacientů užívajících přípravek Xarelto mají být pečlivě sledovány známky krvácení. Doporučuje se opatrnost při použití přípravku v případě zvýšeného rizika krvácení. Pokud se objeví závažné krvácení, podávání přípravku Xarelto je třeba přerušit.
V klinických studiích bylo během dlouhodobé léčby rivaroxabanem ve srovnání s léčbou VKA častěji pozorováno slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Proto, kromě adekvátního klinického sledování, pokud je shledáno vhodným, může být přínosem pro detekci okultního krvácení laboratorní vyšetření hemoglobinu/hematokritu.
U několika podskupin pacientů (podrobně uvedených dále) hrozí zvýšené riziko krvácení. Tyto pacienty je třeba pečlivě sledovat, zda se po zahájení léčby neobjeví známky a příznaky krvácivých komplikací a anémie (viz bod 4.8).
Při jakémkoli nevysvětlitelném poklesu hladin hemoglobinu nebo krevního tlaku je třeba hledat místo krvácení.
Přestože léčba rivaroxabanem nevyžaduje rutinní monitorování expozice, hladiny rivaroxabanu měřené kalibrovanou kvantitativní analýzou anti-faktoru Xa mohou být užitečné ve výjimečných situacích, kdy znalost expozice rivaroxabanu může pomoci při klinických rozhodováních, např. při předávkování nebo při urgentních chirurgických zákrocích (viz body 5.1 a 5.2).
Ledvinová nedostatečnost
U pacientů s těžkou ledvinovou nedostatečností (clearance kreatininu < 30 ml/min) mohou být plazmatické hladiny rivaroxabanu významně zvýšeny (1,6 násobek průměru), což může vést ke zvýšenému riziku krvácení. Xarelto je u pacientů s clearance kreatininu 15 - 29 ml/min nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.2 a 5.2).
Xarelto musí být používáno s opatrností u pacientů s renálním poškozením, kteří současně užívají jiné léčivé přípravky, které zvyšují koncentraci rivaroxabanu v plazmě (viz bod 4.5).
Interakce s jinými léčivými přípravky
Použití přípravku Xarelto se nedoporučuje u pacientů současně léčených systémovými azolovými antimykotiky (jako jsou ketokonazol, itrakonazol, vorikonazol a posakonazol) nebo inhibitory proteáz HIV (například ritonavir). Tyto léčivé látky jsou silnými inhibitory současně obou systémů CYP3A4 a P-gp, a proto mohou zvyšovat plazmatické koncentrace rivaroxabanu v klinicky významném rozsahu (v průměru 2,6 násobek), což může vést ke zvýšenému riziku krvácení (viz bod 4.5).
Postupujte opatrně, pokud jsou pacienti současně léčeni léčivými přípravky ovlivňujícími krevní srážlivost, jako jsou například nesteroidní antirevmatika (NSAID), kyselina acetylsalicylová a inhibitory agregace trombocytů. U pacientů s rizikem vředové gastrointestinální choroby lze zvážit vhodnou profylaktickou léčbu (viz bod 4.5).
Jiné rizikové faktory krvácení
Podobně jako v případě jiných antitrombotik, se použití rivaroxabanu nedoporučuje u pacientů se zvýšeným rizikem krvácení, například:
• vrozené nebo získané krvácivé poruchy
• léčbou neupravená těžká arteriální hypertenze
• jiné gastrointestinální onemocnění bez aktivní ulcerace, které může potenciálně vést ke krvácivým komplikacím (např. zánětlivé střevní onemocnění, esofagitida, gastritida a gastroesofageální refluxní choroba)
• cévní retinopatie
• bronchiektázie nebo plicní krvácní v anamnéze Pacienti s chlopenními náhradami
Bezpečnost a účinnost přípravku Xarelto nebyly hodnoceny u pacientů s implantovanými srdečními chlopněmi; proto neexistují žádné údaje podporující tvrzení, že Xarelto 20 mg (15 mg u pacientů se středně závažnou nebo závažnou ledvinovou nedostatečností) poskytuje odpovídající antikoagulaci u této skupiny pacientů. Léčba přípravkem Xarelto se u těchto pacientů nedoporučuje.
Hemodynamicky nestabilní pacienti s plicní embolií nebo pacienti, kteří vyžadují trombolýzu nebo plicní embolektomii
Přípravek Xarelto se nedoporučuje používat jako alternativní léčbu k nefrakcionovanému heparinu u pacientů s plicní embolií, kteří jsou hemodynamicky nestabilní nebo kteří mohou podstoupit trombolýzu nebo plicní embolektomii, protože bezpečnost a účinnost přípravku Xarelto nebyla pro tyto klinické situace stanovena.
Spinální / epidurální anestezie nebo punkce
Pokud je u pacienta provedena anestezie (spinální či epidurální anestezie) nebo spinální resp. epidurální punkce, u pacientů preventivně léčených antitrombotiky pro prevenci tromboembolických komplikací hrozí riziko vývinu epidurálního či spinálního hematomu, který může vyústit v dlouhodobou nebo trvalou paralýzu. Riziko těchto příhod může dále zvýšit epidurální katetr dlouhodobě zavedený po operaci, nebo současné použití léčivých přípravků ovlivňujících krevní srážlivost. Riziko může také zvýšit provedení traumatické nebo opakované epidurální či spinální punkce. Pacienty je třeba často monitorovat, zda nejeví známky a příznaky neurologického poškození (například necitlivost nebo slabost dolních končetin, dysfunkce střev nebo močového měchýře). Pokud se zjistí neurologické potíže, je nutno urgentně stanovit diagnózu a zajistit léčbu. Před neuroaxiální intervencí lékař zváží potenciální přínos a riziko u pacientů na antikoagulační terapii i u pacientů, kde hodlá antikoagulační léčbu podat v rámci tromboprofylaxe.
S použitím 15 mg rivaroxabanu v těchto situacích nejsou klinické zkušenosti.
Ke snížení možného rizika krvácení během současného užívání rivaroxabanu při neuroaxiální (spinální nebo epidurální) anestezii nebo spinální punkci se bere v úvahu farmakokinetický profil rivaroxabanu. Zavedení nebo odstranění epidurálního katetru nebo lumbální punkci je nejlépe provést, když je odhadovaný antikoagulační účinek rivaroxabanu nízký (viz bod 5.2). Přesný čas, kdy je u každého pacienta antikoagulační účinek dostatečně nízký, však není znám.
Odstranění epidurálního katetru by mělo být na základě farmakokinetických vlastností nejméně za dobu představující 2x poločas, to je nejméně 18 hodin u mladých pacientů a 26 hodin u starších pacientů po posledním podání rivaroxabanu (viz bod 5.2).
Další dávka rivaroxabanu se nepodává dříve než 6 hodin po vyjmutí katetru.
Pokud dojde k traumatické punkci, podávání rivaroxabanu se odloží o 24 hodin.
Doporučení pro dávkování před a po invazivních procedurách a chirurgickém výkonu
Pokud je nutná invazivní procedura nebo chirurgický zákrok, měl by být přípravek Xarelto 15 mg vysazen
minimálně 24 hodin před zákrokem, pokud je to podle posouzení lékaře možné.
Pokud není možné výkon odložit, je třeba posoudit zvýšené riziko krvácení s ohledem na neodkladnost zákroku.
Léčba přípravkem Xarelto má být znovu zahájena po invazivní proceduře nebo chirurgickém zákroku co nejdříve, pokud to situace umožní a pokud je podle úsudku ošetřujícího lékaře dosaženo odpovídající hemostázy (viz bod 5.2).
Starší populace
Se zvyšujícím se věkem se může zvyšovat riziko krvácení (viz bod 5.2).
Informace o pomocných látkách
Xarelto obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a P-gp
Současné podávání rivaroxabanu s ketokonazolem (400 mg jednou denně) nebo ritonavirem (600 mg dvakrát denně) vedlo k 2,6 resp. 2,5 násobnému nárůstu střední hodnoty AUC rivaroxabanu a
1,7 resp. 1,6 násobnému nárůstu jeho střední hodnoty Cmax, s významným zesílením farmakodynamických účinků, což může vést ke zvýšenému riziku krvácení. Proto se použití přípravku Xarelto nedoporučuje u pacientů užívajících současně a systémově azolová antimykotika, jako je ketokonazol, itrakonazol, vorikonazol a posakonazol, nebo inhibitory proteáz HIV. Tyto léčivé látky jsou silnými inhibitory obou systémů CYP3A4 a současně P-gp (viz bod 4.4).
Léčivé látky silně inhibující pouze jednu z metabolických cest eliminace rivaroxabanu (buď CYP3A4, nebo P-gp) podle všeho zvyšují plazmatické koncentrace rivaroxabanu méně. Například klaritromycin (500 mg dvakrát denně), který je považován za silného inhibitora CYP3A4 a středně silného inhibitora P-gp, způsobuje 1,5 násobný nárůst středních hodnot AUC rivaroxabanu a 1,4 násobný nárůst Cmax. Tento nárůst není považován za klinicky relevantní. (Pacienti s poruchou funkce ledvin: viz bod 4.4).
Erythromycin (500 mg třikrát denně), který středně silně inhibuje 3A4 a P-gp, způsobuje 1,3násobný nárůst středních hodnot AUC a Cmax rivaroxabanu. Tento nárůst není považován za klinicky relevantní.
U pacientů s mírnou renální insuficiencí vedlo podávání erythromycinu (500 mg třikrát denně) k 1,8 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu Cmax ve srovnání s pacienty s normální renální funkcí. U pacientů se středně těžkým renálním poškozením vedl erythromycin k 2,0 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu v Cmax ve srovnání s pacienty s normální renální funkcí. Účinek erythromycinu je aditivní k renálnímu poškození (viz bod 4.4).
Flukonazol (400 mg jednou denně), který je považován za středně silný inhibitor CYP3A4, vedl k
1,4 násobnému zvýšení průměrné AUC rivaroxabanu a k 1,3 násobnému zvýšení průměrné Cmax. Toto zvýšení není považováno za klinicky významné. (Pacienti se sníženou funkcí ledvin: viz bod 4.4).
Dronedaron by neměl být podáván spolu s rivaroxabanem, vzhledem k omezeným klinickým údajům, které jsou k dispozici.
Antikoagulační přípravky
Po kombinovaném podávání enoxaparinu (40 mg, jednorázová dávka) s rivaroxabanem (10 mg, jednorázová dávka) byl zjištěn aditivní vliv na inhibici faktoru Xa, a to bez dalších účinků na výsledky testů srážení krve (PT, apTT). Enoxaparin neovlivňoval farmakokinetiku rivaroxabanu.
Vzhledem ke zvýšenému riziku krvácení je třeba postupovat opatrně, pokud jsou pacienti současně léčeni jinými antikoagulačními přípravky (viz body 4.3 a 4.4).
NSAID / inhibitory srážení trombocytů
Při současném podávání rivaroxabanu (15 mg) a 500 mg naproxenu nebylo zjištěno klinicky relevantní prodloužení doby krvácení. Některé osoby však mohou mít silnější farmakodynamickou odezvu.
Žádné klinicky významné farmakokinetické ani farmakodynamické interakce nebyly zjištěny při současném podání rivaroxabanu s 500 mg kyseliny acetylsalicylové.
Clopidogrel (úvodní dávka 300 mg, poté udržovací dávka 75 mg) nevykazoval farmakokinetické interakce s rivaroxabanem (15 mg), ale u části populace pacientů došlo k relevantnímu nárůstu doby krvácení, který nekoreloval s agregací trombocytů, ani hladinami P-selektinu nebo receptoru GPIIb/IIIa.
Postupovat opatrně je třeba, pokud jsou pacienti současně léčeni NSAID (včetně kyseliny acetylsalicylové) a inhibitory agregace trombocytů, protože tyto léčivé přípravky obvykle zvyšují riziko krvácení (viz bod 4.4).
Warfarin
Konverze pacientů z antagonisty vitaminu K warfarinu (INR 2,0 až 3,0) na rivaroxaban (20 mg) nebo z rivaroxabanu (20 mg) na warfarin (INR 2,0 až 3,0) vedla ke zvýšení protrombinového času/INR (Neoplastin) více než aditivně (mohou být pozorovány jednotlivé hladiny INR až 12), zatímco účinky na aPTT, inhibici aktivity faktoru Xa a potenciál endogenního trombinu byly aditivní.
Pokud je třeba testovat farmakodynamické účinky rivaroxabanu během fáze konverze, mohou být použity anti Xa aktivita, PiCT a Heptest, protože tyto testy nebyly ovlivněny warfarinem. Čtvrtý den po poslední dávce warfarinu odráží všechny testy (včetně PT, aPTT, inhibice aktivity faktoru Xa a ETP) pouze účinek rivaroxabanu.
Pokud je třeba testovat farmakodynamické účinky warfarinu během fáze převodu, může být použito měření INR při Cmin rivaroxabanu (24 hodin po předchozím užití rivaroxabanu), protože tento test je v tento okamžik minimálně ovlivněný rivaroxabanem.
Mezi warfarinem a rivaroxabanem nebyla pozorována žádná farmakokinetická interakce.
Induktory CYP3A4
Současné podávání rivaroxabanu se silným induktorem CYP3A4 rifampicinem vedlo k přibližně 50% poklesu střední hodnoty AUC rivaroxabanu, s odpovídajícím poklesem farmakodynamického účinku. Současné použití rivaroxabanu s jinými silnými induktory CYP3A4 (například fenytoinem, karbamazepinem, fenobarbitalem nebo třezalkou (Hypericum perforatum)) může také vést ke snížení plazmatických koncentrací rivaroxabanu. Proto je třeba se vyhnou současnému podávání silných induktorů CYP3A4, pokud není pacient pozorně sledován kvůli známkám a příznakům trombózy.
Jiné současně podávané léky
Žádné klinicky významné farmakokinetické nebo farmakodynamické interakce nebyly zjištěny při současném podávání rivaroxabanu s midazolamem (substrát CYP3A4), digoxinem (substrát P-gp), atorvastatinem (substrát CYP3A4 a P-gp) nebo omeprazolem (inhibitor protonové pumpy). Rivaroxaban neinhibuje ani neindukuje významné izoformy CYP jako je CYP3A4.
Laboratorní parametry
Parametry srážení krve (například PT, aPTT, Heptest) j sou ovlivněny podle očekávání na základě mechanismu působení rivaroxabanu (viz bod 5.1).
4.6 Fertilita, těhotenství a kojení
Bezpečnost a účinnost přípravku Xarelto nebyly u těhotných žen stanoveny. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Vzhledem k možné reprodukční toxicitě, známému riziku krvácení a důkazu, že rivaroxaban prochází placentou, je přípravek Xarelto kontraindikován v těhotenství (viz bod 4.3). Ženy ve fertilním věku musí během léčby rivaroxabanem zabránit otěhotnění.
Kojení
Bezpečnost a účinnost přípravku Xarelto nebyly u kojících žen stanoveny. Údaje z experimentů na zvířatech signalizují, že je rivaroxaban vylučován do mléka. Podávání přípravku Xarelto je během kojení kontraindikováno (viz bod 4.3). Je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit léčbu.
Fertilita
Nebyly provedeny žádné specifické studie užívání rivaroxabanu u lidí s cílem vyhodnotit účinky na fertilitu. Ve studii samčí a samičí fertility na potkanech nebyly pozorovány žádné účinky (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xarelto má malý vliv na schopnost řídit a obsluhovat stroje. Byly hlášeny nežádoucí účinky jako synkopa (frekvence výskytu: méně časté) a závrať (frekvence výskytu: časté) (viz bod 4.8). Pacienti, kteří zaznamenali tyto nežádoucí účinky, by neměli řídit vozidla a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostních informací
Bezpečnost rivaroxabanu byla hodnocena v jedenácti studiích fáze III u 32 625 pacientů, kteří byli vystaveni rivaroxabanu (viz tabulka 1).
Tabulka 1: Počet hodnocených pacientů, maximální ^ denní dávka a délka léčby ve studiích fáze II
|
Indikace |
Počet pacientů* |
Maximální denní dávka |
Maximální délka léčby |
|
Prevence žilního tromboembolismu u dospělých pacientů podstupujících elektivní operativní náhradu kyčelního nebo kolenního kloubu |
6 097 |
10 mg |
39 dnů |
|
Prevence žilního tromboembolismu u hospitalizovaných nechirurgických pacientů |
3 997 |
10 mg |
39 dnů |
|
Léčba hluboké žilní trombózy a plicní embolie a prevence jejich recidivy |
4 556 |
Den 1-21: 30 mg Den 22 a dále: 20 mg |
21 měsíců |
|
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní |
7 750 |
20 mg |
41 měsíců |
|
Prevence aterotrombotických příhod u pacientů po AKS |
10 225 |
5 mg nebo 10 mg, podávaných společně s ASA nebo s kombinací ASA plus klopidogrel či tiklopidin |
31 měsíců |
*Pacienti exponovaní minimálně jedné dávce rivaroxabanu
Nejčastěji hlášenými nežádoucí účinky u pacientů, kteří dostávali rivaroxaban, bylo krvácení (viz bod 4.4 a níže uvedený „Popis vybraných nežádoucích účinků“). Nejčastěji hlášeným krvácením (>4 %) byla epistaxe (5,9 %) a gastrointestinální krvácení (4,2 %).
Celkem asi u 67% pacientů exponovaných minimálně jedné dávce rivaroxabanu byl hlášen výskyt nežádoucích příhod. Asi u 22% pacientů se vyskytly nežádoucí příhody, které byly považovány za související s léčbou podle posouzení zkoušejícími. U pacientů léčených přípravkem Xarelto v dávce 10 mg podstupujících chirurgickou náhradu kyčelního nebo kolenního kloubu a u nechirurgických pacientů hospitalizovaných pro jiné onemocnění se krvácivé příhody vyskytly asi u 6,8% a 12,6% pacientů a anémie se objevila asi u 5,9% a 2,1% pacientů. U pacientů léčených přípravkem Xarelto buď v dávce 15 mg dvakrát denně s následným podáváním 20 mg jednou denně pro léčbu hluboké žilní trombózy nebo plicní embolie nebo dávkou 20 mg jednou denně z důvodu prevence recidivující hluboké žilní trombózy a plicní embolie se krvácivé příhody vyskytly asi u 27,8% pacientů a anémie se objevila asi u 2,2% pacientů. U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace bylo krvácení jakéhokoli typu a závažnosti hlášeno s frekvencí 28 na 100 paciento-roků a anémie s frekvencí 2,5 na 100 paciento-roků. U pacientů léčených z důvodu prevence kardiovaskulárního úmrtí a infarktu myokardu po akutním koronárním syndromu (ACS), bylo krvácení jakéhokoli typu a závažnosti hlášeno s frekvencí 22 na 100 paciento-roků. Anémie byla hlášena s frekvencí 1,4 na 100 paciento-roků.
Seznam nežádoucích účinků uvedený v tabulce
Výskyt nežádoucích účinků hlášený u přípravku Xarelto je shrnutý v tabulce 2 níže podle orgánové klasifikace (v MedDRA) a podle frekvence výskytu.
Četnosti j sou definovány takto:
velmi časté (> 1/10)
časté (> 1/100 až < 1/10)
méně časté (> 1/1 000 až < 1/100)
vzácné (> 1/10 000 až < 1/1 000)
velmi vzácné ( < 1/10,000)
není známo (z dostupných údajů nelze určit)
Tabulka 2: Všechny s léčbou související nežádoucí účinky hlášené u pacientů ve studiích fáze III
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (včetně příslušných laboratorních parametrů) |
Trombocytémie (včetně zvýšeného počtu trombocytů)A | ||
|
Poruchy imunitního systému | |||
|
Alergická reakce, alergická dermatitida | |||
|
Poruchy nervového systému | |||
|
Závratě, bolesti hlavy |
Cerebrální a intrakraniální krvácení, synkopa | ||
|
Poruchy oka | |||
|
Oční krvácení (včetně krvácení do spojivek) | |||
|
Srdeční poruchy | |||
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Cévní poruchy | |||
|
Hypotenze, hematom | |||
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe, hemoptýza | |||
|
Gastrointestinální poruchy | |||
|
Krvácení z dásní, krvácení z gastrointestinálního traktu (včetně rektálního krvácení), gastrointestinální a abdominální bolest, dyspepsie, nausea, zácpaA, průjem, zvraceníA |
Sucho v ústech | ||
|
Poruchy jater a žlučových cest | |||
|
Abnormity jaterní funkce |
Žloutenka | ||
|
Poruchy kůže a podkožní tkáně | |||
|
Pruritus (včetně méně častých případů generalizovaného pruritu), vyrážka, ekchymóza, kožní a podkožní krvácení |
Kopřivka | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |||
|
Bolest v končetináchA |
Hemartróza |
Krvácení do svalů |
Kompartment syndrom sekundárně po krvácení |
|
Poruchy ledvin a močových cest | |||
|
Urogenitální krvácení (včetně hematurie a menorhagieB), poškození ledvin (včetně zvýšení hladin kreatininu a močoviny v krvi)A |
Renální selhání/akutní renální selhání vzniklé sekundárně po krvácení natolik silném, aby způsobilo hypoperfúzi | ||
|
Celkové poruchy a reakce v místě aplikace | |||
|
HorečkaA, periferní edém, pokles celkové síly a energie (včetně únavy a tělesné slabosti) |
Pocit indispozice (včetně malátnosti) |
Lokalizovaný edémA | |
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Vyšetření | |||
|
Zvýšená hladina transamináz |
Zvýšená hladina bilirubinu, alkalické fosfatázyA, LDHA, lipázyA, amylázyA, GMTA |
Zvýšení konjugovaného bilirubinu (s přidruženým zvýšením ALT nebo bez zvýšení ALT) | |
|
Poranění, otravy a procedurální komplikace | |||
|
Pooperační krvácení (včetně pooperační anémie a krvácení z rány), kontuze, sekrece z ranA |
Cévní pseudoaneurysmaC | ||
A: pozorováno u prevence žilního tromboembolismu u dospělých pacientů, kteří podstoupili chirurgickou náhradu kyčelního nebo kolenního kloubu
B: pozorováno u léčby hluboké žilní trombózy, plicní embolie a u prevence recidivy jako velmi časté u žen do 55 let
C: pozorováno méně často u prevence aterotrombotických příhod u pacientů po akutním koronárním syndromu (po perkutánní koronární intervenci)
Popis vybraných nežádoucích účinků
Vzhledem k farmakologickému mechanismu působení může být užívání přípravku Xarelto spojeno se zvýšeným rizikem okultního nebo zjevného krvácení z jakékoli tkáně nebo orgánu s možným následkem posthemoragické anémie. Známky, příznaky a závažnost (včetně možného fatálního zakončení) se mohou různit podle místa a stupně nebo rozsahu krvácení a/nebo anémie (viz bod 4.9 Léčba krvácení). V klinických studiích bylo během dlouhodobé léčby rivaroxabanem ve srovnání s léčbou VKA častěji pozorováno slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Proto, kromě adekvátního klinického sledování, pokud je shledáno vhodným, může laboratorní vyšetření hemoglobinu/hematokritu být přínosem pro detekci okultního krvácení. Riziko krvácení bude možná zvýšeno u některých skupin pacientů, například osob s těžkou arteriální hypertenzí neupravenou léčbou, a/nebo souběžnou léčbou ovlivňující krevní srážlivost (viz Riziko krvácení v bodu 4.4). Menstruační krvácení může být zesíleno a/nebo prodlouženo. Hemoragické komplikace se mohou projevovat jako celková slabost, bledost, závratě, bolesti hlavy nebo nevysvětlitelné otoky, dušnost a nevysvětlitelný šok. V některých případech byly v důsledku anémie pozorovány příznaky srdeční ischémie, jako je například bolest na hrudníku nebo angina pectoris.
V souvislosti s užíváním přípravku Xarelto byly hlášeny známé sekundární komplikace závažného krvácení, jako je například kompartment syndrom a renální selhání v důsledku hypoperfúze. Možnost krvácení je proto třeba zvážit při posuzování stavu pacientů s jakoukoli antikoagulační léčbou.
Post-marketingové sledování
V postmarketingovém sledování byly v časové souvislosti s užitím přípravku Xarelto hlášeny následující nežádoucí účinky. Frekvenci těchto nežádoucích účinků hlášených po uvedení na trh nelze určit.
Poruchy imunitního systému: angioedém a alergický edém (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Poruchy jater a žlučových cest: cholestáza, hepatitida (včetně hepatocelulárního poškození) (v souhrnných studiích fáze III byly tyto příhody vzácné (>1/10 000 až <1/1000)).
Poruchy krve a lymfatického systému: trombocytopenie (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili
podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny vzácné případy předávkování dávkou až 600 mg bez krvácivých komplikací nebo jiných nežádoucích reakcí. Z důvodu omezené absorpce se očekává strop účinku bez dalšího zvýšení průměrné plazmatické hladiny v případě vyšší než terapeutické dávky 50 mg rivaroxabanu nebo dávek vyšších. Specifické antidotum blokující farmakodynamický účinek rivaroxabanu není k dispozici.
Lze zvážit podání aktivního uhlí ke snížení absorpce v případě předávkování rivaroxabanem.
Léčba krvácení
Pokud dojde ke krvácivým komplikacím u pacienta léčeného rivaroxabanem, musí se podání další dávky rivaroxabanu odložit nebo se léčba musí ukončit, dle potřeby. Rivaroxaban má biologický poločas asi 5 až 13 hodin (viz bod 5.2). Léčba by měla být individuální podle závažnosti a lokalizace krvácení. Podle potřeby je třeba použít vhodnou symptomatickou léčbu, jako je mechanická komprese (např. u závažné epistaxe), chirurgická hemostáza se zajištěním kontroly krvácení, náhradou tekutin a zajištěním hemodynamické podpory, krevní deriváty (erytrocyty nebo čerstvá zmrazená plasma, v závislosti na související anémii nebo koagulopatii) nebo trombocyty.
Pokud krvácení nelze kontrolovat výše uvedenými opatřeními, lze zvážit podávání specifické prokoagulační reverzní látky, jako je koncentrát protrombinového komplexu (PCC), aktivovaný koncentrát protrombinového komplexu (APCC) nebo rekombinantní faktor VIIa (r-FVIIa). V současnosti jsou však k dispozici velmi omezené klinické zkušenosti s použitím těchto přípravků u osob užívajících rivaroxaban. Doporučení je též podloženo omezenými neklinickými údaji. Opakované podání rekombinantního faktoru VIIa je třeba zvážit a titrovat v závislosti na zlepšování krvácení. V případě závažného krvácení je třeba konzultovat odborníka na koagulaci, pokud je odborník v místě dostupný (viz bod 5.1).
Protamin sulfát a vitamin K podle všeho nebudou ovlivňovat antikoagulační aktivitu rivaroxabanu. U osob užívajících rivaroxaban jsou omezené zkušenosti s použítím kyseliny tranexamové a neexistují zkušenosti s použitím kyseliny aminokaproové a aprotininu. Neexistují ani vědecké důvody přínosu ani zkušenosti s použitím systémového hemostatika desmopressinu u osob užívajících rivaroxaban. Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: přímé inhibitory faktoru Xa, ATC kód: B01AF01 Mechanismus účinku
Rivaroxaban je vysoce selektivní přímý inhibitor faktoru Xa biologicky dostupný při perorálním podání. Inhibice faktoru Xa blokuje vnitřní a vnější cesty koagulační kaskády, a inhibuje vznik trombinu i vytváření trombů. Rivaroxaban neinhibuje trombin (aktivovaný faktor II) a nebyly prokázány žádné účinky na trombocyty.
Farmakodynamické účinky
U lidí byla zjištěna inhibice faktoru Xa přímo úměrná dávce. Protrombinový čas (PT) je rivaroxabanem ovlivňován úměrně dávce, objevuje se vysoká korelace s plazmatickými koncentracemi (hodnota R je 0,98), pokud je při testu jako assay použit Neoplastin. Jiné reagenty mohou přinést jiné výsledky. Hodnotu PT je nutno stanovit v sekundách, protože INR (mezinárodní normalizovaný poměr) je kalibrován a validován pouze pro kumariny, a nelze jej využívat pro jiné antikoagulanty.
U pacientů užívajících rivaroxaban v léčbě hluboké žilní trombózy a plicní embolie a k prevenci jejich recidivy se v 5/95 percentilu hodnoty PT (Neoplastin) za 2 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 17 až 32 s pro dávku 15 mg rivaroxabanu dvakrát denně a od 15 do 30 s pro dávku 20 mg rivaroxabanu jednou denně. Nejnižší hodnoty se v 5/95 percentilu pohybovaly od 14 do 24 s pro dávku 15 mg dvakrát denně (8 - 16 hodin po požití) a od 13 do 20 s pro dávku 20 mg jednou denně (18 - 30 hodin po požití). U pacientů s nevalvulární fibrilací síní užívajících rivaroxaban v prevenci cévní mozkové příhody a systémové embolizace se v 5/95 percentilu hodnoty PT (Neoplastin) za 1 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 14 až 40 s pro dávku 20 mg rivaroxabanu jednou denně a od 10 do 50 s u pacientů se středně závažným poškozením renálních funkcí léčených dávkou 15 mg jednou denně. Nejnižší hodnoty (16 - 36 hodin po požití) se v 5/95 percentilu pohybovaly od 12 do 26 s pro dávku 20 mg jednou denně a u pacientů se středně závažným poškozením renálních funkcí léčených 15 mg jednou denně se hodnoty pohybovaly od 12 do 26 s.
V klinické farmakologické studii sledující reverzi farmakodynamického účinku rivaroxabanu u zdravých dospělých osob (n=22) byl hodnocen účinek jednotlivé dávky (50 IU/kg) u dvou rozdílných typů PCC, 3-faktorového PCC (faktory II, IX a X) a 4-faktorového PCC (II, VII, IX a X). 3-faktorový PCC redukoval průměrnou hodnotu PT času (protrombinového času), hodnocenou neoplastinem, přibližně o 1,0 sekundy během 30 minut, ve srovnání s přibližně 3,5 sekundami pozorovanými u 4-faktorového PCC. Naproti tomu, 3-faktorový PCC měl větší a rychlejší celkový efekt na reverzní změny generace endogenního trombinu než 4-faktorový PCC (viz bod 4.9).
Aktivovaný parciální tromboplastinový čas (aPTT) a hodnoty analýzy HepTest jsou také prodlouženy úměrně dávce; nedoporučuje se však tyto metody používat k hodnocení farmakodynamických účinků rivaroxabanu. Během léčby rivaroxabanem v běžné klinické praxi není třeba monitorovat parametry koagulace. Pokud však je klinicky indikováno, lze hladiny rivaroxabanu měřit pomocí kalibrovaných kvantitativních testů anti-faktoru Xa (viz bod 5.2).
Klinická účinnost a bezpečnost
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní Klinický program přípravku Xarelto byl navržen tak, aby prokázal účinnost přípravku Xarelto v prevenci cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní.
V pivotní dvojitě zaslepené studii ROCKET AF bylo 14 264 pacientů přiřazeno buď do léčby přípravkem Xarelto 20 mg jednou denně (15 mg jednou denně u pacientů s clearance kreatininu 30 - 49 ml/min) nebo léčby warfarinem titrovaným na cílovou hodnotu INR 2,5 (terapeutické rozmezí 2,0 až 3,0). Střední doba léčby byla 19 měsíců a celková doba léčby byla až 41 měsíců.
34,9% pacientů bylo léčeno kyselinou acetylsalicylovou a 11,4% bylo léčeno pomocí antiarytmik třídy III, včetně amiodaronu.
V porovnání s warfarinem dosáhl přípravek non-inferiority co do primárního kompozitního cílového ukazatele cévní mozkové příhody a systémové embolizace nepostihující CNS. U populace “per protocol“ (dle protokolu) v období sledování „on treatment“ (po dobu léčby), se cévní mozková příhoda nebo systémová embolizace vyskytla u 188 pacientů na rivaroxabanu (1,71% za rok) a u 241 pacientů na warfarinu (2,16% za rok) (HR 0,79; 95% CI, 0,66 - 0,96; P<0,001 pro non-inferioritu). Mezi všemi randomizovanými pacienty analyzovanými podle ITT, se primární cílový parametr vyskytl u 269 pacientů na rivaroxabanu (2,12% za rok) a u 306 pacientů na warfarinu (2,42% za rok) (HR 0,88; 95% CI, 0,74 - 1,03; P<0,001 pro non-inferioritu; P=0,117 pro superioritu). Výsledky sekundárních cílových ukazatelů, v pořadí, jak byly testovány v ITT analýze, jsou ukázány v tabulce 3.
Mezi pacienty na léčbě warfarinem, byly hodnoty INR uvnitř terapeutického rozmezí (2,0 až 3,0) v průměru 55% doby (median 58%; rozsah mezi kvartily byl 43 až 71). Účinek rivaroxabanu se nelišil napříč úrovněmi TTR v centru (čas v cílovém INR rozmezí 2,0 až 3,0) ve stejnoměrně velkých kvartilech (P=0,74 pro interakci). V centrech v nejvyšším kvartilu bylo HR rivaroxaban versus warfarin 0,74 (95% CI, 0,49 -1,12). Četnost incidence pro hlavní bezpečnostní ukazatel (závažné a klinicky významné méně závažné krvácivé příhody) byla pro obě léčebné skupiny podobná (viz tabulka 4).
|
Populace studie |
ITT analýzy účinnosti u pacientů s nevalvulární fibrilací síní | ||
|
Xarelto |
Warfarin |
Poměr rizik HR | |
|
20 mg jednou denně |
titrovaný na cílovou |
(95% CI) | |
|
(15 mg jednou denně u |
hladinu INR 2,5 |
p-hodnota, test pro | |
|
pacientů se středně |
(terapeutické rozmezí |
superioritu | |
|
Dávkování |
závažnou renální insuficiencí |
2,0 až 3,0) | |
|
Výskyt příhod |
Výskyt příhod | ||
|
(100 paciento-roků) |
(100 paciento-roků) | ||
|
Cévní mozková příhoda a |
269 |
306 |
0,88 (0,74 - 1,03) |
|
systémová embolizace nepostihující CNS |
(2,12) |
(2,42) |
0,117 |
|
Cévní mozková příhoda, |
572 |
609 |
0,94 (0,84 - 1,05) |
|
systémová embolizace nepostihující CNS a vaskulámí úmrtí |
(4,51) |
(4,81) |
0,265 |
|
Cévní mozková příhoda, |
659 |
709 |
0,93 (0,83 - 1,03) |
|
systémová embolizace nepostihující CNS, vaskulámí úmrtí a infarkt myokardu |
(5,24) |
(5,65) |
0,158 |
|
Cévní mozková příhoda |
253 |
281 |
0,90 (0,76 - 1,07) |
|
(1,99) |
(2,22) |
0,221 | |
|
Systémová embolizace |
20 |
27 |
0,74 (0,42 - 1,32) |
|
nepostihující CNS |
(0,16) |
(0,21) |
0,308 |
|
Infarkt myokardu |
130 |
142 |
0,91 (0,72 - 1,16) |
|
(1,02) |
(1,11) |
0,464 | |
|
Populace studie |
Pacienti s nevalvulární fibrilací sínía) | ||
|
Dávkování |
Xarelto 20 mg jednou denně (15 mg jednou denně u pacientů se středně závažnou renální insuficiencí |
Warfarin titrovaný na cílovou hladinu INR 2,5 (terapeutické rozmezí 2,0 až 3,0) |
Poměr rizik (95% CI) p-hodnota |
|
Výskyt příhod (100 paciento-roků) |
Výskyt příhod (100 paciento-roků) | ||
|
Závažné a méně závažné klinicky významné příhody krvácení |
1 475 (14,91) |
1 449 (14,52) |
1,03 (0,96 - 1,11) 0,442 |
|
Závažné příhody krvácení |
395 (3,60) |
386 (3,45) |
1,04 (0,90 - 1,20) 0,576 |
|
Úmrtí v důsledku krvácení1 |
27 (0,24) |
55 (0,48) |
0,50 (0,31 - 0,79) 0,003 |
|
Krvácení do kritického orgánu1 |
91 (0,82) |
133 (1,18) |
0,69 (0,53 - 0,91) 0,007 |
|
Intrakraniální krvácení1 |
55 (0,49) |
84 (0,74) |
0,67 (0,47 - 0,93) 0,019 |
|
Pokles hemoglobinu1 |
305 (2,77) |
254 (2,26) |
1,22 (1,03 - 1,44) 0,019 |
|
Transfuze 2 nebo více jednotek erytrocytů nebo plné krve1 |
183 (1,65) |
149 (1,32) |
1,25 (1,01 - 1,55) 0,044 |
|
Méně závažné klinicky významné krvácivé příhody |
1 185 (11,80) |
1 151 (11,37) |
1,04 (0,96 - 1,13) 0,345 |
|
Úmrtí z jakékoli příčiny |
208 (1,87) |
250 (2,21) |
0,85 (0,70 - 1,02) 0,073 |
a) “Safety“ populace, „on treatment“ (populace, ve které byla hodnocena bezpečnost, po dobu léčby)
Pacienti podstupující kardioverzi
Prospektivní, randomizovaná, otevřená, multicentrická, analytická studie se zaslepeným hodnocením cílů (X-VERT) byla provedena u 1504 pacientů (bez předchozí léčby perorálními antikoagulancii nebo předléčených) s nevalvulární fibrilací síní naplánovaných ke kardioverzi, srovnávající rivaroxaban s adjustovanou dávkou VKA (randomizovaných v poměru 2:1) v prevenci kardiovaskulárních příhod.
Byly sledovány buď kardioverze s provedenou TEE (1-5 dní léčby) nebo konvenční kardioverze (nejméně tři týdny léčby). Primární cíl účinnosti (všechny CMP, transitorní ischemická ataka, systémová embolie mimo CNS, IM a úmrtí z kardiovaskulárních příčin) nastal u 5 (0,5%) pacientů užívajících rivaroxaban (n=978) a u 5 (1,0%) pacientů užívajících VKA (n=492; RR% 0,5; CI 0,15-1,73; modifikovaná ITT populace). Hlavní bezpečnostní ukazatel (závažné krvácení) se vyskytl u 6 (0,6%) pacientů léčených rivaroxabanem (n= 988) a u 4 (0,8%) pacientů léčených VKA (n=499), (RR 0,76; 95% CI 0,21-2,67; safety populace). Tato analytická studie ukázala srovnatelnou účinnost a bezpečnost mezi skupinami s rivaroxabanem a VKA v případě kardioverze.
Léčba hluboké žilní trombózy, plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie Klinický program přípravku Xarelto byl navržen tak, aby prokázal účinnost přípravku Xarelto v úvodní a pokračující léčbě hluboké žilní trombózy a plicní embolie a prevenci jejich recidivy.
Více než 9 400 pacientů bylo hodnoceno ve třech randomizovaných kontrolovaných studiích fáze III (Einstein DVT, Einstein PE a Einstein Extension) a poté byla provedena predefinovaná poolovaná analýza studií Einstein DVT a Einstein PE. Celková kombinovaná délka léčby ve všech studiích byla až 21 měsíců.
Ve studii Einstein DVT bylo hodnoceno 3 449 pacientů s akutní hlubokou žilní trombózou v léčbě hluboké žilní trombózy a prevenci recidivující hluboké žilní trombózy a plicní embolie (pacienti, kteří měli symptomatickou plicní embolii, byli z této studie vyřazeni). Délka léčby byla 3, 6 nebo 12 měsíců v závislosti na klinickém posouzení zkoušejícím.
V úvodní 3-týdenní léčbě akutní hluboké žilní trombózy byl podáván rivaroxaban v dávce 15 mg dvakrát denně. Poté následovalo podávání dávky 20 mg rivaroxabanu jednou denně.
Ve studii Einstein PE bylo hodnoceno 4 832 pacientů s akutní plicní embolií v léčbě plicní embolie a v prevenci recidivující hluboké žilní trombózy a plicní embolie. Délka léčby byla 3, 6 nebo 12 měsíců v závislosti na klinickém posouzení zkoušejícím.
V úvodní léčbě akutní PE bylo podáváno 15 mg rivaroxabanu dvakrát denně 3 týdny. Poté následovalo podávání dávky 20 mg rivaroxabanu jednou denně.
V obou studiích Einstein DVT a Einstein PE zahrnoval srovnávaný léčebný režim enoxaparin podávaný minimálně 5 dnů v kombinaci s antagonisty vitaminu K do dosažení terapeutického rozmezí PT/INR (> 2,0). Léčba pokračovala antagonistou vitaminu K, jehož dávka byla upravena pro udržení hodnot PT/INR v terapeutickém rozmezí 2,0 až 3,0.
Ve studii Einstein Extension bylo hodnoceno 1 197 pacientů s hlubokou žilní trombózou nebo plicní embolií v prevenci recidivující hluboké žilní trombózy a plicní embolie. Trvání léčby bylo dalších 6 nebo 12 měsíců u pacientů, kteří dokončili 6 až 12 měsíců léčby pro žilní tromboembolismus v závislosti na klinickém posouzení zkoušejícím. Přípravek Xarelto 20 mg jednou denně byl srovnáván s placebem.
Všechny studie fáze III využívaly stejné předem definované primární a sekundární parametry účinnosti. Primární parametr účinnosti byl symptomatický recidivující žilní tromboembolismus definovaný jako kompozit recidivující hluboké žilní trombózy nebo fatální či nefatální plicní embolie. Sekundární parametr účinnosti byl definovaný jako kompozit recidivující hluboké žilní trombózy, nefatální plicní embolie a mortality ze všech příčin.
Ve studii Einstein DVT (viz tabulka 5) prokázal rivaroxaban non-inferioritu proti enoxaparinu/antagonistům vitaminu K v primárním parametru účinnosti (p < 0,0001 (test non-inferiority); poměr rizik: 0,680 (0,443 - 1,042), p = 0,076 (test superiority)). Předem definovaný čistý klinický přínos (primární parametr účinnosti plus závažná krvácivá příhoda) byl hlášen s poměrem rizik 0,67 ((95% CI: 0,47- 0,95), s nominální hodnotou p = 0,027) ve prospěch rivaroxabanu. Hodnoty INR byly uvnitř terapeutického rozmezí
s průměrem 60,3% pro průměrnou dobu léčby 189 dní a 55,4%, 60,1% a 62,8% doby pro skupiny s plánovanou léčbou 3, 6 a 12 měsíců. Ve skupině enoxaparin/VKA nebyl jasný vztah mezi hladinou TTR v centru (doba v cílovém INR rozmezí 2,0 -3,0) ve stejně velkých tertilech a incidencí recidivující HZT (P=0,932 pro interakci). V centrech v nejvyšším tertilu bylo HR rivaroxaban versus warfarin 0,69 (95% CI: 0,35 - 1,35).
Výskyt primárního bezpečnostního ukazatele (závažné nebo klinicky významné méně závažné krvácivé příhody) stejně jako sekundárního bezpečnostního ukazatele (závažné krvácivé příhody) byl podobný v obou léčebných skupinách.
Tabulka 5: Výsledky účinnosti a bezpečnosti ze studie fáze III Einstein DVT (hluboká žilní trombóza)
|
Populace studie |
3 449 pacientů se symptomatickou hlubokou žilní trombózou | |
|
Dávkování a délka léčby |
Xareltoa) |
Enoxaparin/VKAb) |
|
3, 6 nebo 12 měsíců |
3, 6 nebo 12 měsíců | |
|
N = 1 731 |
N = 1 718 | |
|
Symptomatický recidivující žilní |
36 |
51 |
|
tromboembolismus* |
(2,1%) |
(3,0%) |
|
Symptomatická recidivující |
20 |
18 |
|
(1,2%) |
(1,0%) | |
|
Symptomatická recidivující |
14 |
28 |
|
hluboká žilní trombóza |
(0,8%) |
(1,6%) |
|
Symptomatická plicní embolie a |
1 |
0 |
|
hluboká žilní trombóza |
(0,1%) | |
|
Fatální plicní embolie/úmrtí, |
4 |
6 |
|
kde plicní embolie nemůže být vyloučena |
(0,2%) |
(0,3%) |
|
Závažné nebo klinicky významné |
139 |
138 |
|
méně závažné krvácení |
(8,1%) |
(8,1%) |
|
Závažné krvácivé příhody |
14 |
20 |
|
(0,8%) |
(1,2%) | |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů vitaminu K
* p < 0,0001 (non-inferiorita k stanovenému poměru rizik 2,0); poměr rizik: 0,680 (0,443 - 1,042), p = 0,076 (superiorita)
Ve studii Einstein PE (viz tabulka 6) prokázal rivaroxaban non-inferioritu proti enoxaparinu/antagonistům vitaminu K v primárním parametru účinnosti (p = 0,0026 (test non-inferiority); poměr rizik: 1,123 (0,749 - 1,684)). Předem definovaný čistý klinický přínos (primární parametr účinnosti plus závažná krvácivá příhoda) byl hlášen s poměrem rizik 0,849 ((95% CI: 0,633-1,139), s nominální hodnotou p = 0,275). Hodnoty INR byly uvnitř terapeutického rozmezí s průměrem 63% pro průměrnou dobu léčby 215 dní a 57%, 62% a 65% doby pro skupiny s plánovanou léčbou 3, 6 a 12 měsíců. Ve skupině enoxaparin/VKA nebyl jasný vztah mezi hladinou TTR v centru (doba v cílovém INR rozmezí 2,0 - 3,0) ve stejně velkých tertilech a incidencí recidivující HZT (P=0,082 pro interakci). V centrech v nejvyšším tertilu bylo HR rivaroxaban versus warfarin 0,642 (95% CI: 0,277 - 1,484).
Výskyt primárního bezpečnostního ukazatele (závažné nebo klinicky významné méně závažné krvácivé příhody) byl lehce nižší ve skupině léčené rivaroxabanem (10,3% (249/2412)) než ve skupině léčené enoxaparinem/antagonisty vitaminu K (11,4% (274/2405)). Výskyt sekundárního bezpečnostního ukazatele (závažné krvácivé příhody) byl nižší ve skupině léčené rivaroxabanem (1.1% (26/2412)) než ve skupině enoxaparin/antagonisté vitaminu K (2.2% (52/2405)) s poměrem rizik 0,493 (95% CI: 0,308 - 0,789).
|
Populace studie |
4 832 pacientů s akutní symptomatickou PE | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Dávkování a doba léčby |
3, 6 nebo 12 měsíců |
3, 6 nebo 12 měsíců |
|
N=2 419 |
N=2 413 | |
|
Symptomatický recidivující žilní |
50 |
44 |
|
tromboembolismus* |
(2,1%) |
(1,8%) |
|
Symptomatická recidivující PE |
23 (1,0%) |
20 (0,8%) |
|
Symptomatická recidivující |
18 |
17 |
|
hluboká žilní trombóza |
(0,7%) |
(0,7%) |
|
Symptomatická recidivující plicní embolie a hluboká žilní trombóza |
0 |
2 (<0,1%) |
|
Fatální plicní embolie/úmrtí, kde plicní embolie nemůže být vyloučena |
11 (0,5%) |
7 (0,3%) |
|
Závažné nebo klinicky významné |
249 |
274 |
|
méně závažné krvácení |
(10,3%) |
(11,4%) |
|
Závažné krvácivé příhody |
26 (1,1%) |
52 (2,2%) |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů
vitaminu K
p < 0,0026 (non-inferiorita k predefinovanému poměru rizik 2,0); poměr rizik: 1,123 (0,749 - 1,684)
Byla provedena predefinovaná poolovaná analýza výsledku studií Einstein DVT a PE (viz tabulka 7). Tabulka 7: Výsledky účinnosti a bezpečnosti z poolované analýzy studií fáze III Einstein DVT
a Einstein PE
|
Populace studie |
8281 pacientů s akutní symptomatickou HZT nebo PE | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Dávkování a doba léčby |
3, 6 nebo 12 měsíců |
3, 6 nebo12 měsíců |
|
N=4150 |
N=4131 | |
|
Symptomatický recidivující žilní |
86 |
95 |
|
tromboembolismus* |
(2,1%) |
(2,3%) |
|
Symptomatická recidivující |
43 |
38 |
|
(1,0%) |
(0,9%) | |
|
Symptomatická recidivující |
32 |
45 |
|
hluboká žilní trombóza |
(0,8%) |
(1,1%) |
|
Symptomatická recidivující plicní embolie a hluboká žilní trombóza |
1 (<0,1%) |
2 (<0,1%) |
|
Fatální plicní embolie/úmrtí, kde |
15 (0,4%) |
13 (0,3%) |
|
plicní embolie nemůže být vyloučena | ||
|
Závažné nebo klinicky významné |
388 |
412 |
|
méně závažné krvácení |
(9,4%) |
(10,0%) |
|
Závažné krvácivé příhody |
40 (1,0%) |
72 (1,7%) |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou
denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním
antagonistů vitaminu K
* p < 0.0001 (non-inferiorita k předefinovanému poměru rizik 1,75); poměr rizik: 0,886
(0,661 - 1,186)
Předefinovaný čistý klinický prospěch (výsledek primární účinnosti plus závažné krvácivé příhody) poolované analýzy byl hlášen s poměrem rizik 0,771 ((95% CI: 0,614 - 0,967), nominální hodnota p= 0,0244).
Ve studii Einstein Extension (viz tabulka 8) byl rivaroxaban lepší než placebo v primárních a sekundárních parametrech účinnosti. U primárního bezpečnostního ukazatele (závažné krvácivé příhody) byl nevýznamný numericky vyšší výskyt u pacientů léčených rivaroxabanem v dávce 20 mg jednou denně v srovnání s placebem. Sekundární bezpečnostní ukazatel (závažné nebo klinicky významné méně závažné krvácivé příhody) prokázal vyšší výskyt u pacientů léčených rivaroxabanem 20 mg jednou denně ve srovnání s placebem.
Tabulka 8: Výsledky účinnosti a bezpečnosti ze studie fáze III Einstein Extension
|
Populace studie |
pokračování léčby u 1 197 pacientů, u nichž byla podávána léčba a prevence recidivujícího žilního tromboembolismu | |
|
Dávkování a doba léčby |
Xareltoa) |
Placebo |
|
6 nebo 12 měsíců |
6 nebo 12 měsíců | |
|
N = 602 |
N = 594 | |
|
Symptomatický recidivující žilní |
8 |
42 |
|
tromboembolismus* |
(1,3%) |
(7,1%) |
|
Symptomatická recidivující |
2 |
13 |
|
(0,3%) |
(2,2%) | |
|
Symptomatická recidivující |
5 |
31 |
|
hluboká žilní trombóza |
(0,8%) |
(5,2%) |
|
Fatální plicní embolie/úmrtí, |
1 |
1 |
|
kde plicní embolie nemůže být vyloučena |
(0,2%) |
(0,2%) |
|
Závažné krvácivé příhody |
4 |
0 |
|
(0,7%) |
(0,0%) | |
|
Klinicky významné méně závažné |
32 |
7 |
|
krvácení |
(5,4%) |
(1,2%) |
a) Rivaroxaban 20 mg j ednou denně
* p < 0,0001 (superiorita), poměr rizik: 0,185 (0,087 - 0,393)
Kromě studií fáze III programu EINSTEIN byla provedena prospektivní, neintervenční, otevřená kohortová studie (XALIA) s centrálním vyhodnocováním sledovaných ukazatelů zahrnujících recidivující žilní tromboembolismus, závažné krvácení a úmrtí. Bylo zařazeno 5 142 pacientů s akutní hlubokou žilní trombózou za účelem posoudit dlouhodobou bezpečnost rivaroxabanu v porovnání se standardní antikoagulační terapií v klinické praxi. Výskyt závažného krvácení, recidivujícího žilního tromboembolismu a úmrtí ze všech příčin byly v rivaroxabanové větvi 0,7 %, 1,4 % a 0,5 %.
Ve vstupních charakteristikách pacientů byly rozdíly včetně věku, výskytu nádorových onemocnění a ledvinové nedostatečnosti. Přestože byla pro úpravu získaných základních rozdílů použita předem stanovená analýza stratifikovaná dle propensity skóre, mohou reziduální zavádějící faktory tyto výsledky ovlivnit. Upravené poměry rizik srovnávající rivaroxaban a standardní léčbu pro závažné krvácení, recidivující žilní tromboembolismus a mortalitu ze všech příčin byly 0,77 (95% CI: 0,40 - 1,50), 0,91 (95% CI: 0,54 - 1,54) a 0,51 (95% CI: 0,24 - 1,07).
Tato pozorování z klinické praxe jsou v souladu s potvrzeným bezpečnostním profilem v této indikaci. Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xarelto v léčbě tromboembolických příhod u jedné nebo více podskupin pediatrické populace. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xarelto v prevenci tromboembolických příhod u všech podskupin pediatrické populace (informace o použití u pediatrické populace viz bod 4.2)
5.2 Farmakokinetické vlastnosti
Absorpce
Rivaroxaban je rychle absorbován; maximální koncentrace (Cmax) se objeví 2 - 4 hodiny po užití tablety.
Bez ohledu na stav na lačno nebo po jídle je u dávky 2,5 mg a 10 mg rivaroxabanu tablety perorální absorpce téměř kompletní a perorální biologická dostupnost vysoká (80 - 100 %). Užívání při jídle neovlivňuje při 2,5 mg a 10 mg dávce AUC ani Cmax rivaroxabanu.
Pro 20 mg tabletu byla v důsledku sníženého rozsahu absorpce stanovena biologická dostupnost 66% při stavu nalačno. Když byly užívány 20 mg tablety přípravku Xarelto společně s jídlem, došlo ke zvýšení průměrné AUC o 39% ve srovnání s užíváním tablety nalačno, což ukazuje na téměř kompletní absorpci a vysokou biologickou dostupnost po perorálním podání. Přípravek Xarelto 15 mg a 20 mg se má užívat s jídlem (viz bod 4.2).
Farmakokinetické vlastnosti rivaroxabanu jsou až do denní dávky 15 mg nalačno přibližně lineární. Po jídle byla u přípravku Xarelto 10 mg, 15 mg a 20 mg prokázána farmakokinetika závislá na dávce. Ve vyšších dávkách je absorbce rivaroxabanu omezena disolucí, dochází ke snížení biologické dostupností a stupeň absorbce se snižuje se zvyšující se dávkou.
Variabilita farmakokinetiky rivaroxabanu je střední, s interindividuální variabilitou v rozmezí od 30% do 40%.
Absorpce rivaroxabanu je závislá na místě jeho uvolnění v gastrointestinálním traktu. Bylo hlášeno 29% a 56% snížení AUC a Cmax ve srovnání s tabletou, pokud byl rivaroxaban v granulátu uvolněn v proximální časti tenkého střeva. Expozice je dále snížena, když je rivaroxaban uvolněn v distální části tenkého střeva nebo ve vzestupné části tračníku. Podání rivaroxabanu distálně od žaludku by se mělo zabránit, jelikož to může vést ke snížení absorpce a související expozice rivaroxabanu.
Biologická dostupnost (AUC and Cmax) byla pro podání 20 mg rivaroxabanu per os ve formě rozdrcené tablety vmíchané do jablečného pyré nebo rozpuštěné ve vodě a pro podání žaludeční sondou následované tekutou stravou, v porovnání s celou tabletou srovnatelná. Vzhledem k předvídatelnému dávce úměrnému farmakokinetickému profilu rivaroxabanu odpovídají výsledky biologické dostupnosti z této studie pravděpodobně nižším dávkám rivaroxabanu.
Distribuce
Vazba na plazmatické proteiny u lidí je vysoká, přibližně 92% - 95%, přičemž hlavní část se váže na sérový albumin. Distribuční objem je střední, Vss činí přibližně 50 litrů.
Biotransformace a eliminace
Z podané dávky rivaroxabanu se přibližně 2/3 metabolicky degradují, z čehož je polovina vylučována ledvinami a druhá polovina stolicí. Zbývající 1/3 podané dávky je vylučována ledvinami přímo jako nezměněná léčivá látka, hlavně prostřednictvím aktivní ledvinové sekrece.
Rivaroxaban je metabolizován prostřednictvím systémů CYP3A4 a CYP2J2 i mechanismy na CYP nezávislými. Hlavními cestami transformace je oxidativní degradace morfolinonové části a hydrolýza amidových vazeb. Na základě in vitro experimentů je zřejmé, že rivaroxaban slouží jako substrát transportních proteinů - P-gp (P-glykoprotein) a Bcrp (breast cancer resistance protein).
Nezměněný rivaroxaban je nejvýznamnější formou přípravku v lidské plazmě; v krevním oběhu nejsou žádné významné nebo aktivní metabolity. Rivaroxaban lze vzhledem ke systémové clearance asi 10 l/h klasifikovat jako látku s nízkou clearance. Po intravenózním podání dávky 1 mg je eliminační poločas asi
4,5 hodiny. Po perorálním podání je eliminace limitována stupněm absorpce. K eliminaci rivaroxabanu z plazmy dochází s terminálním poločasem 5 až 9 hodin u mladších osob a s terminálním poločasem 11 -13 hodin u starších osob.
Zvláštní skupiny
Pohlaví
Mezi muži a ženami nebyl žádný klinicky relevantní rozdíl ve farmakokinetice a farmakodynamice přípravku.
Starší populace
Starší pacienti vykazovali vyšší plazmatické koncentrace než mladší, s průměrnou hodnotou AUC přibližně
1,5 x vyšší, hlavně vzhledem ke snížené (zdánlivé) celkové a ledvinové clearance. Žádná úprava dávky není nutná.
Různé váhové kategorie
Extrémy v tělesné hmotnosti (< 50 kg nebo > 120 kg) měly pouze malý vliv na plazmatické koncentrace rivaroxabanu (méně než 25%). Žádná úprava není dávky nutná.
Rozdíly mezi etniky
Žádné klinicky relevantní rozdíly mezi etniky nebyly ve farmakokinetice a farmakodynamice rivaroxabanu zjištěny u pacientů z řad bělochů, Afroameričanů, Hispánců, Japonců ani Číňanů.
Jaterní nedostatečnost
Pacienti s cirhózou s mírnou jaterní nedostatečností (Child-Pugh A) vykazovali pouze menší změny ve farmakokinetice rivaroxabanu (v průměru 1,2x nárůst AUC rivaroxabanu), a výsledky byly téměř srovnatelné s kontrolní skupinou zdravých dobrovolníků. U pacientů trpících cirhózou se středně závažnou jaterní nedostatečností (Child-Pugh B) průměrná AUC rivaroxabanu významně stoupla - 2,3x v porovnání se zdravými dobrovolníky. AUC nevázané látky stoupla 2,6x. Tito pacienti měli současně sníženou renální eliminaci rivaroxabanu, podobně jako pacienti se středně závažnou ledvinovou nedostatečností. O farmakokinetice u pacientů s těžkým jaterním poškozením nejsou k dispozici žádné údaje.
Inhibice aktivity faktoru Xa byla u pacientů se středně závažnou jaterní nedostatečností zvýšena ve srovnání se zdravými dobrovolníky 2,6x; prodloužení PT bylo obdobně zvýšeno 2,1x. Pacienti se středně závažnou jaterní nedostatečností byli na rivaroxaban citlivější, a vztah mezi koncentrací a PT měl tak strmější průběh. Přípravek Xarelto je kontraindikován u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child Pugh B a C (viz bod 4.3).
Ledvinová nedostatečnost
Byl zjištěn nárůst expozice rivaroxabanu související s poklesem funkce ledvin, která byla posuzována prostřednictvím hodnot clearance kreatininu. U osob s lehkou (clearance kreatininu 50 - 80 ml/min), střední (clearance kreatininu 30 - 49 ml/min) a těžkou (clearance kreatininu 15 - 29 ml/min) ledvinovou nedostatečností byly plazmatické koncentrace rivaroxabanu (AUC) zvýšeny 1,4, 1,5 resp. 1,6 x. Odpovídající zesílení farmakodynamických účinků bylo výraznější. U osob s lehkou, střední a těžkou ledvinovou nedostatečností byla celková inhibice faktoru Xa ve srovnání se zdravými dobrovolníky zvýšena 1,5, 1,9 resp. 2,0 x; prodloužení PT bylo obdobně zvýšeno 1,3, 2,2 a 2,4 x. O použití u pacientů s clearance kreatininu < 15 ml/min nejsou žádné údaje.
Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min. U pacientů s clearance kreatininu 15 - 29 ml/min je nutno Xarelto používat s opatrností (viz bod 4.4).
Farmakokinetické údaje u pacientů
U pacientů užívajících rivaroxaban 20 mg jednou denně k léčbě akutní hluboké žilní trombózy (HŽT) byl geometrický průměr koncentrace (90% interval přepovědi) 2 - 4 hodiny a přibližně 24 hodin po podání dávky (zhruba představující maximální a minimální koncentrace během dávkovacího intervalu) 215 (22535) a 32 (6-239) ^g/l.
Farmakokinetické a farmakodynamické vztahy
Po podání různě velkých dávek (5 - 30 mg dvakrát denně) byl hodnocen farmakokinetický a farmakodynamický (PK/PD) vztah mezi plazmatickou koncentrací rivaroxabanu a několika konečnými cílovými ukazateli PD (inhibice faktoru Xa, PT, aPTT, Heptest). Vztah mezi plazmatickou koncentrací rivaroxabanu a aktivitou faktoru Xa byl nejlépe popsán pomocí modelu Emax. U PT byly údaje lépe vyjádřeny pomocí lineárního ohraničeného modelu. Hodnoty PT se významně lišily v závislosti na použitých reagenciích. Při použití Neoplastinu byl výchozí PT asi 13 sekund a odchylka hodnot přibližně 3 až 4 s/(100 ^g/l). Výsledek analýz PK/PD ve studiích fáze II a III byl v souladu s údaji získanými u zdravých jedinců.
Pediatrická populace
Bezpečnost a účinnost nebyly stanoveny u dětí a dospívajících do 18 let.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po jednorázovém podání, fototoxicity, genotoxicity, kancerogenního potenciálu a juvenilní toxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studii toxicity při opakovaném podání byly způsobeny hlavně zesílenou farmakologickou aktivitou rivaroxabanu. Při klinicky relevantních úrovních expozice byly u potkanů pozorovány zvýšené plazmatické hladiny IgG a IgA.
U potkanů nebyly pozorovány žádné účinky na fertilitu samců nebo samic. Studie na zvířatech prokázaly reprodukční toxicitu související s farmakologickým mechanismem působení rivaroxabanu (např. hemoragickými komplikacemi). V klinicky relevantních plazmatických koncentracích byla pozorována embryonální a fetální toxicita (post-implantační ztráta, opožděná nebo progredující osifikace, hepatální mnohočetné světle zbarvené skvrny) a zvýšený výskyt běžných malformací, a také placentárních změn.
V prenatálních a postnatálních experimentech u potkanů byla zjištěna snížená životaschopnost potomků, a to v dávkách toxických pro matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Sodná sůl kroskarmelózy Monohydrát laktózy Hypromelóza Natrium-lauryl-sulfát Magnesium-stearát.
Potah tablety:
Makrogol 3350
Hypromelóza
Oxid titaničitý (E171)
Červený oxid železitý (E172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Blistry z fólie Al/PP, v krabičkách po 10, 14, 28, 42 nebo 98 potahovaných tabletách nebo perforované jednodávkové blistry v krabičkách obsahující 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních obsahujících 100 potahovaných tablet (10 balení po 10 x 1 tabletě).
HDPE lahvičky obsahující 100 potahovaných tablet se šroubovacím PP uzávěrem.
Blistry z fólie Al/PP v pouzdru obsahující 42 potahovaných tablet přípravku Xarelto 15 mg a 7 potahovaných tablet přípravku Xarelto 20 mg (balení pro zahájení léčby).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/472/011-016, EU/1/08/472/023, EU/1/08/472/036, EU/1/08/472/038, EU/1/08/472/040
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. září 2008
Datum posledního prodloužení registrace: 22. května 2013
10. DATUM REVIZE TEXTU {MM.RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xarelto 20 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rivaroxabanum 20 mg.
Pomocná látka se známým účinkem:
Jedna potahovaná tableta obsahuje 21,76 mg laktózy (jako monohydrátu), viz bod 4.4. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Hnědo-červené, kulaté, bikonvexní tablety (o průměru 6 mm, poloměru zakřivení 9 mm) označené logem (kříž) BAYER na jedné straně a číslem „20“ a trojúhelníkem na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence cévní mozkové příhody a systémové embolizace u dospělých pacientů s nevalvulární fibrilací síní s jedním nebo více rizikovými faktory, jako je městnavé srdeční selhání, hypertenze, věk 75 let a vyšší, diabetes mellitus, prodělaná cévní mozková příhoda nebo tranzitorní ischemická ataka.
Léčba hluboké žilní trombózy (HŽT) a plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolizace u dospělých (hemodynamicky nestabilní pacienti s PE viz bod 4.4).
4.2 Dávkování a způsob podání
Dávkování
Prevence cévní mozkové příhody a systémové embolizace
Doporučená dávka je 20 mg jednou denně, což je také doporučená maximální dávka.
Léčba přípravkem Xarelto by měla být dlouhodobá za předpokladu, že přínos prevence cévní mozkové příhody a systémové embolizace převáží riziko krvácení (viz bod 4.4).
Pokud dojde k vynechání dávky, měl by pacient užít přípravek Xarelto co nejdříve a pokračovat v užívání jednou denně následující den podle doporučení. Dávka by neměla být tentýž den zdvojnásobena, aby se nahradila vynechaná dávka.
Léčba hluboké žilní trombózy, léčba plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie
Doporučená dávka pro úvodní léčbu akutní hluboké žilní trombózy nebo plicní embolie je 15 mg dvakrát denně po dobu prvních tří týdnů a dále 20 mg jednou denně jako udržovací léčba a prevence hluboké žilní trombózy a plicní embolie podle tabulky uvedené níže.
|
Dávkování |
Maximální denní dávka | |
|
1. až 21. Den |
15 mg dvakrát denně |
30 mg |
|
22. den a dále |
20 mg jednou denně |
20 mg |
K usnadnění změny dávkování po 21. dnu léčby z 15 mg na 20 mg při léčbě HZT/PE je dostupné balení pro zahájení léčby přípravkem Xarelto pro první 4 týdny léčby (viz bod 6.5).
Délka léčby by měla být stanovena individuálně po pečlivém zhodnocení přínosu léčby a rizika krvácení (viz bod 4.4). Krátkodobá léčba (nejméně 3 měsíce) by měla být indikována na základě přechodných rizikových faktorů (např. operace prodělaná v nedávné době, úraz, imobilizace) a dlouhodobá léčba by měla být indikována na základě trvalých rizikových faktorů nebo idiopatické HZT nebo PE.
Pokud dojde k vynechání dávky během té fáze léčby, kdy je přípravek podáván v dávce 15 mg dvakrát denně (1. - 21. den), měl by pacient užít přípravek Xarelto co nejdříve, aby se zajistilo dávkování 30 mg přípravku Xarelto denně. V tomto případě mohou být užity dvě 15 mg tablety najednou. Pacient by měl pokračovat s pravidelným užíváním dávky 15 mg dvakrát denně následující den podle doporučení.
Pokud dojde k vynechání dávky během té fáze léčby, kdy je přípravek podáván v jedné denní dávce (22. den a dále), měl by pacient užít přípravek Xarelto co nejdříve a pokračovat s užíváním jednou denně následující den podle doporučení. Dávka by neměla být pro nahrazení vynechané dávky ve stejný zdvojnásobena.
Převod z antagonistů vitaminu K (VKA) na přípravek Xarelto
U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace by měly být antagonisté vitaminu K vysazeny a léčba přípravkem Xarelto by měla být zahájena při hladině mezinárodního normalizačního faktoru (INR) < 3,0.
U pacientů léčených pro hlubokou žilní trombózu, plicní embolii a pro prevenci recidivy by měly být antagonisté vitaminu K vysazeny a léčba přípravkem Xarelto by měla být zahájena při hladině INR < 2,5.
Při převodu pacientů z antagonistů vitaminu K na přípravek Xarelto, budou po užití přípravku Xarelto hladiny INR falešně zvýšeny. Test INR není pro měření antikoagulační aktivity přípravku Xarelto validní a proto by neměl být používán (viz bod 4.5).
Převod z přípravku Xarelto na antagonisty vitaminu K (VKA)
Během přechodu z přípravku Xarelto na antagonisty vitaminu K existuje možnost neadekvátní antikoagulace. Během jakéhokoli převodu na alternativní antikoagulancia by měla být zajištěna kontinuální adekvátní antikoagulace. Je třeba uvést, že přípravek Xarelto může přispět ke zvýšení INR.
U pacientů, kteří jsou převáděni z přípravku Xarelto na antagonisty vitaminu K by měli být tito antagonisté podáváni současně, dokud není hladina INR > 2,0. Po dobu prvních dvou dnů fáze převodu by mělo být použito standardní úvodní dávkování antagonistů vitaminu K s následným dávkováním těchto antagonistů na základě testování INR. Během doby, kdy pacienti užívají jak přípravek Xarelto tak antagonisty vitaminu K by nemělo být prováděno testování INR dříve než 24 hodin po předchozí dávce, ale před další dávkou přípravku Xarelto. Jakmile je přípravek Xarelto vysazen, může být testování INR spolehlivě provedeno minimálně 24 hodin po poslední dávce (viz body 4.5 a 5.2).
Převod z parenterálních antikoagulancií na přípravek Xarelto
U pacientů, kteří dostávají parenterální antikoagulancia, přerušte podávání parenterálního antikoagulancia a začněte léčbu přípravkem Xarelto v rozmezí 0 až 2 hodiny před tím, než by mělo dojít k dalšímu plánovanému podání parenterálního přípravku (např. nízkomolekulární hepariny) nebo v době vysazení kontinuálně podávaného parenterálního přípravku (např. intravenózní nefrakciovaný heparin).
Převod z přípravku Xarelto na parenterální antikoagulancia
Podejte první dávku parenterálního antikoagulancia v době, kdy by měla být užita další dávka přípravku Xarelto.
Speciální populace
Ledvinová nedostatečnost
Omezené klinické údaje u nemocných s těžkou renální nedostatečností (clearance kreatininu 15 - 29 ml/min) signalizují, že u této populace pacientů jsou plazmatické koncentrace rivaroxabanu významně zvýšeny. Xarelto je proto u těchto pacientů nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.4 a 5.2).
U pacientů se středně závažnou (clearance kreatininu 30 - 49 ml/min) nebo závažnou (clearance kreatininu 15-29 ml/min) renální nedostatečností platí následující doporučení pro dávkování:
- pro prevenci cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní je doporučené dávkování 15 mg jednou denně (viz bod 5.2),
- pro léčbu hluboké žilní trombózy, léčbu plicní embolie a prevenci recidivující hluboké žilní trombózy a plicní embolie: pacienti by měli být léčeni dávkou 15 mg dvakrát denně po dobu prvních tří týdnů. Poté je doporučená dávka 20 mg jednou denně. Snížení dávky z 20 mg jednou denně na 15 mg jednou denně je třeba zvážit, pokud u pacienta riziko krvácení převáží riziko vzniku recidivující HZT a PE. Doporučení pro použití dávky 15 mg je založeno na farmakokinetickém modelu a nebylo v těchto klinických podmínkách studováno (viz body 4.4, 5.1 a 5.2).
Úprava dávky není nutná u pacientů s mírnou renální nedostatečností (clearance kreatininu 50 - 80 ml/min) (viz bod 5.2).
Jaterní nedostatečnost
Xarelto je kontraindikováno u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child Pugh B a C (viz body 4.3 a 5.2).
Starší populace
Dávky bez úprav (viz bod 5.2).
Tělesná hmotnost
Dávky bez úprav (viz bod 5.2).
Pohlaví
Dávky bez úprav (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Xarelto u dětí ve věku 0 až 18 let nebyla ještě stanovena. Nejsou dostupné žádné údaje. Podávání přípravku Xarelto dětem do 18 let se proto nedoporučuje.
Pacienti podstupující kardioverzi
Léčba přípravkem Xarelto může být zahájena nebo v ní lze pokračovat u pacientů, jejichž stav vyžaduje provedení kardioverze.
U pacientů podstupujících transezofageální echokardiografií (TEE) řízenou kardioverzi, kteří nebyli předem léčeni antikoagulancii, má být léčba přípravkem Xarelto zahájena nejméně 4 hodiny před kardioverzí, aby byla zajištěna odpovídající antikoagulace (viz body 5.1 a 5.2). Před provedením kardioverze je třeba u všech pacientů usilovat o potvrzení, že pacient užíval Xarelto, jak bylo předepsáno. Při rozhodování o zahájení léčby a o jejím trvání se musí vzít v úvahu pokyny dané doporučením pro antikoagulační léčbu pacientů podstupujících kardioverzi.
Způsob podání Perorální podání.
Tablety se mají užívat s jídlem (viz bod 5.2).
Pacientům, kteří nej sou schopni polykat celé tablety, může být tableta přípravku Xarelto těsně před užitím rozdrcena a smíchána s vodou nebo s jablečným pyré a poté podána perorálně. Po podání rozdrcené potahované tablety Xarelto 15 mg nebo 20 mg musí být dávka okamžitě následována jídlem.
Rozdrcená tableta přípravku Xarelto může být také podána gastrickou sondou poté, co je potvrzeno správné umístění sondy v žaludku. Rozdrcená tableta by měla být podána žaludeční sondou v malém množství vody a sonda by poté měla být propláchnuta vodou. Po podání rozdrcené potahované tablety Xarelto 15 mg nebo 20 mg musí být poté dávka okamžitě následována enterální výživou (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku. Aktivní klinicky významné krvácení.
Léze nebo stavy, které jsou považovány za významné riziko závažného krvácení. Mohou mezi ně patřit současné nebo nedávno prodělané ulcerace gastrointestinálního traktu, přítomnost maligních nádorů s vysokým rizikem krvácení, nedávno prodělané poranění mozku nebo míchy, operace mozku, míchy nebo oka v nedávné době, intrakraniální krvácení v nedávné době, jícnové varixy nebo podezření na ně, arteriovenózní malformace, cévní aneurysma nebo významné cévní abnormality v míše nebo mozku.
Souběžná léčba jinými antikoagulačními přípravky, např. nefrakcionovaným heparinem (UFH), nízkomolekulárními hepariny (enoxaparin, dalteparin atd), heparinovými deriváty (fondaparinux atd), orálními antikoagulancii (warfarin, dabigatran etexilát, apixaban, atd.) se nedoporučuje s výjimkou specifické situace, kdy je pacient převáděn z antikoagulační léčby (viz bod 4.2) nebo když je podáván UFH v dávkách nezbytných pro udržení průchodnosti centrálního žilního nebo arteriálního katetru (viz bod 4.5).
Jaterní onemocnění, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně pacientů s cirhózou s klasifikací Child Pugh B a C (viz bod 5.2).
Těhotenstí a kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
V průběhu léčby se doporučuje pacienta klinicky sledovat v souladu s praxí běžnou při podávání antikoagulační léčby.
Riziko krvácení
Jako v případě jiných antikoagulancií, u pacientů užívajících přípravek Xarelto mají být pečlivě sledovány známky krvácení. Doporučuje se opatrnost při použití přípravku v případě zvýšeného rizika krvácení. Pokud se objeví závažné krvácení, podávání přípravku Xarelto je třeba přerušit.
V klinických studiích bylo během dlouhodobé léčby rivaroxabanem ve srovnání s léčbou VKA častěji pozorovány slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Proto, kromě adekvátního klinického sledování, pokud je shledáno vhodným, může být přínosem pro detekci okultního krvácení laboratorní vyšetření hemoglobinu/hematokritu.
U několika podskupin pacientů (podrobně uvedených dále) hrozí zvýšené riziko krvácení. Tyto pacienty je třeba pečlivě sledovat, zda se po zahájení léčby neobjeví známky a příznaky krvácivých komplikací a anémie (viz bod 4.8).
Při jakémkoli nevysvětlitelném poklesu hladin hemoglobinu nebo krevního tlaku je třeba hledat místo krvácení.
Přestože léčba rivaroxabanem nevyžaduje rutinní monitorování expozice, hladiny rivaroxabanu měřené kalibrovanou kvantitativní analýzou anti-faktoru Xa mohou být užitečné ve výjimečných situacích, kdy znalost expozice rivaroxabanu může pomoci při klinických rozhodováních, např. při předávkování nebo při urgentních chirurgických zákrocích (viz body 5.1 a 5.2).
Ledvinová nedostatečnost
U pacientů s těžkou ledvinovou nedostatečností (clearance kreatininu < 30 ml/min) mohou být plazmatické hladiny rivaroxabanu významně zvýšeny (1,6 násobek průměru), což může vést ke zvýšenému riziku krvácení. Xarelto je u pacientů s clearance kreatininu 15 - 29 ml/min nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.2 a 5.2).
Xarelto musí být používáno s opatrností u pacientů s renálním poškozením, kteří současně užívají jiné léčivé přípravky, které zvyšují koncentraci rivaroxabinu v plazmě (viz bod 4.5).
Interakce s jinými léčivými přípravky
Použití přípravku Xarelto se nedoporučuje u pacientů současně léčených systémovými azolovými antimykotiky (jako jsou ketokonazol, itrakonazol, vorikonazol a posakonazol) nebo inhibitory proteáz HIV (například ritonavir). Tyto léčivé látky jsou silnými inhibitory současně obou systémů CYP3A4 a P-gp, a proto mohou zvyšovat plazmatické koncentrace rivaroxabanu v klinicky významném rozsahu (v průměru
2,6 násobek), což může vést ke zvýšenému riziku krvácení (viz bod 4.5).
Postupujte opatrně, pokud jsou pacienti současně léčeni léčivými přípravky ovlivňujícími krevní srážlivost, jako jsou například nesteroidní antirevmatika (NSAID), kyselina acetylsalicylová a inhibitory agregace trombocytů. U pacientů s rizikem vředové gastrointestinální choroby lze zvážit vhodnou profylaktickou léčbu (viz bod 4.5).
Jiné rizikové faktory krvácení
Podobně jako v případě jiných antitrombotik, se použití rivaroxabanu nedoporučuje u u pacientů se zvýšeným rizikem krvácení, například:
• vrozené nebo získané krvácivé poruchy
• léčbou neupravená těžká arteriální hypertenze
• jiné gastrointestinální onemocnění bez aktivní ulcerace, které může potenciálně vést ke krvácivým komplikacím (např. zánětlivé střevní onemocnění, esofagitida, gastritida a gastroesofageální refluxní choroba)
• cévní retinopatie
• bronchiektázie nebo plicní krvácní v anamnéze.
Pacienti s chlopenními náhradami
Bezpečnost a účinnost přípravku Xarelto nebyly hodnoceny u pacientů s implantovanými srdečními chlopněmi; proto neexistují žádné údaje podporující tvrzení, že Xarelto 20 mg (15 mg u pacientů se středně závažnou nebo závažnou ledvinovou nedostatečností) poskytuje odpovídající antikoagulaci u této skupiny pacientů. Léčba přípravkem Xarelto se u těchto pacientů nedoporučuje.
Hemodynamicky nestabilní pacienti s plicní embolií nebo pacienti, kteří vyžadují trombolýzu nebo plicní embolektomii
Přípravek Xarelto se nedoporučuje používat jako alternativní léčbu k nefrakcionovanému heparinu u pacientů s plicní embolií, kteří jsou hemodynamicky nestabilní nebo kteří mohou podstoupit trombolýzu nebo plicní embolektomii, protože bezpečnost a účinnost přípravku Xarelto nebyla pro tyto klinické situace stanovena.
Spinální / epidurální anestezie nebo punkce
Pokud je u pacienta provedena anestezie (spinální či epidurální anestezie) nebo spinální resp. epidurální punkce, u pacientů preventivně léčených antitrombotiky pro prevenci tromboembolických komplikací hrozí riziko vývinu epidurálního či spinálního hematomu, který může vyústit v dlouhodobou nebo trvalou
paralýzu. Riziko těchto příhod může dále zvýšit epidurální katetr dlouhodobě zavedený po operaci, nebo současné použití léčivých přípravků ovlivňujících krevní srážlivost. Riziko může také zvýšit provedení traumatické nebo opakované epidurální či spinální punkce. Pacienty je třeba často monitorovat, zda nejeví známky a příznaky neurologického poškození (například necitlivost nebo slabost dolních končetin, dysfunkce střev nebo močového měchýře). Pokud se zjistí neurologické potíže, je nutno urgentně stanovit diagnózu a zajistit léčbu. Před neuroaxiální intervencí lékař zváží potenciální přínos a riziko u pacientů na antikoagulační terapii i u pacientů, kde hodlá antikoagulační léčbu podat v rámci tromboprofylaxe.
S použitím 20 mg rivaroxabanu v těchto situacích nejsou klinické zkušenosti.
Ke snížení možného rizika krvácení během současného užívání rivaroxabanu při neuroaxiální (spinální nebo epidurální) anestezii nebo spinální punkci se bere v úvahu farmakokinetický profil rivaroxabanu. Zavedení nebo odstranění epidurálního katetru nebo lumbální punkci je nejlépe provést, když je odhadovaný antikoagulační účinek rivaroxabanu nízký (viz bod 5.2). Přesný čas, kdy je u každého pacienta antikoagulační účinek dostatečně nízký, však není znám.
Odstranění epidurálního katetru by mělo být na základě farmakokinetických vlastností nejméně za dobu představující 2x poločas, to je nejméně 18 hodin u mladých pacientů a 26 hodin u starších pacientů po posledním podání rivaroxabanu (viz bod 5.2). Další dávka rivaroxabanu se nepodává dříve než 6 hodin po vyjmutí katetru.
Pokud dojde k traumatické punkci, podávání rivaroxabanu se odloží o 24 hodin.
Doporučení pro dávkování před a po invazivních procedurách a chirurgickém výkonu
Pokud je nutná invazivní procedura nebo chirurgický zákrok, měl by být přípravek Xarelto 20 mg vysazen
minimálně 24 hodin před zákrokem, pokud je to podle posouzení lékaře možné.
Pokud není možné výkon odložit, je třeba posoudit zvýšené riziko krvácení s ohledem na neodkladnost zákroku.
Léčba přípravkem Xarelto má být znovu zahájena po invazivní proceduře nebo chirurgickém zákroku co nejdříve, pokud to situace umožní a pokud je podle úsudku ošetřujícího lékaře dosaženo odpovídající hemostázy (viz bod 5.2).
Starší populace
Se zvyšujícím se věkem se může zvyšovat riziko krvácení (viz bod 5.2).
Informace o pomocných látkách
Xarelto obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a P-gp
Současné podávání rivaroxabanu s ketokonazolem (400 mg jednou denně) nebo ritonavirem (600 mg dvakrát denně) vedlo k 2,6 resp. 2,5násobnému nárůstu střední hodnoty AUC rivaroxabanu a
1,7 resp. 1,6 násobnému nárůstu jeho střední hodnoty Cmax, s významným zesílením farmakodynamických účinků, což může vést ke zvýšenému riziku krvácení. Proto se použití přípravku Xarelto nedoporučuje u pacientů užívajících současně a systémově azolová antimykotika, jako je ketokonazol, itrakonazol, vorikonazol a posakonazol, nebo inhibitory proteáz HIV. Tyto léčivé látky jsou silnými inhibitory obou systémů CYP3A4 a současně P-gp (viz bod 4.4).
Léčivé látky silně inhibující pouze jednu z metabolických cest eliminace rivaroxabanu (buď CYP3A4, nebo P-gp) podle všeho zvyšují plazmatické koncentrace rivaroxabanu méně. Například klaritromycin (500 mg dvakrát denně), který je považován za silného inhibitora CYP3A4 a středně silného inhibitora P-gp, způsobuje 1,5 násobný nárůst středních hodnot AUC rivaroxabanu a 1,4 násobný nárůst Cmax. Tento nárůst není považován za klinicky relevantní. (Pacienti s poruchou funkce ledvin: viz bod 4.4).
Erythromycin (500 mg třikrát denně), který středně silně inhibuje 3A4 a P-gp, způsobuje 1,3 násobný nárůst středních hodnot AUC a Cmax rivaroxabanu. Tento nárůst není považován za klinicky relevantní.
U pacientů s mírnou renální insuficiencí vedlo podávání erythromycinu (500 mg třikrát denně) k 1,8 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu Cmax ve srovnání s pacienty s normální renální funkcí. U pacientů se středně těžkým renálním poškozením vedl erythromycin k 2,0 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu v Cmax ve srovnání s pacienty s normální renální funkcí. Účinek erythromycinu je aditivní k renálnímu poškození (viz bod 4.4).
Flukonazol (400 mg jednou denně), který je považován za středně silný inhibitor CYP3A4, vedl k
1,4 násobnému zvýšení průměrné AUC rivaroxabanu a k 1,3 násobnému zvýšení průměrné Cmax. Toto zvýšení není považováno za klinicky významné. (Pacienti se sníženou funkcí ledvin viz bod 4.4).
Dronedaron by neměl být podáván spolu s rivaroxabanem, vzhledem k omezeným klinickým údajům, které jsou k dispozici.
Antikoagulační přípravky
Po kombinovaném podávání enoxaparinu (40 mg, jednorázová dávka) s rivaroxabanem (10 mg, jednorázová dávka) byl zjištěn aditivní vliv na inhibici faktoru Xa, a to bez dalších účinků na výsledky testů srážení krve (PT, apTT). Enoxaparin neovlivňoval farmakokinetiku rivaroxabanu.
Vzhledem ke zvýšenému riziku krvácení je třeba postupovat opatrně, pokud jsou pacienti současně léčeni jinými antikoagulačními přípravky (viz body 4.3 a 4.4).
NSAID / inhibitory srážení trombocytů
Při současném podávání rivaroxabanu (15 mg) a 500 mg naproxenu nebylo zjištěno klinicky relevantní prodloužení doby krvácení. Některé osoby však mohou mít silnější farmakodynamickou odezvu.
Žádné klinicky významné farmakokinetické ani farmakodynamické interakce nebyly zjištěny při současném podání rivaroxabanu s 500 mg kyseliny acetylsalicylové.
Clopidogrel (úvodní dávka 300 mg, poté udržovací dávka 75 mg) nevykazoval farmakokinetické interakce s rivaroxabanem (15 mg), ale u části populace pacientů došlo k relevantnímu nárůstu doby krvácení, který nekoreloval s agregací trombocytů, ani hladinami P-selektinu nebo receptoru GPIIb/IIIa.
Postupovat opatrně je třeba, pokud jsou pacienti současně léčeni NSAID (včetně kyseliny acetylsalicylové) a inhibitory agregace trombocytů, protože tyto léčivé přípravky obvykle zvyšují riziko krvácení (viz bod 4.4).
Warfarin
Konverze pacientů z antagonisty vitaminu K warfarinu (INR 2,0 až 3,0) na rivaroxaban (20 mg) nebo z rivaroxabanu (20 mg) na warfarin (INR 2,0 až 3,0) vedla ke zvýšení protrombinového času/INR (Neoplastin) více než aditivně (mohou být pozorovány jednotlivé hladiny INR až 12), zatímco účinky na aPTT, inhibici aktivity faktoru Xa a potenciál endogenního trombinu byly aditivní.
Pokud je třeba testovat farmakodynamické účinky rivaroxabanu během fáze konverze, mohou být použity anti Xa aktivita, PiCT a Heptest, protože tyto testy nebyly ovlivněny warfarinem. Čtvrtý den po poslední dávce warfarinu odráží všechny testy (včetně PT, aPTT, inhibice aktivity faktoru Xa a ETP) pouze účinek rivaroxabanu.
Pokud je třeba testovat farmakodynamické účinky warfarinu během fáze převodu, může být použito měření INR při Cmin rivaroxabanu (24 hodin po předchozím užití rivaroxabanu), protože tento test je v tento okamžik minimálně ovlivněný rivaroxabanem.
Mezi warfarinem a rivaroxabanem nebyla pozorována žádná farmakokinetická interakce.
Induktory CYP3A4
Současné podávání rivaroxabanu se silným induktorem CYP3A4 rifampicinem vedlo k přibližně 50% poklesu střední hodnoty AUC rivaroxabanu, s odpovídajícím poklesem farmakodynamického účinku. Současné použití rivaroxabanu s jinými silnými induktory CYP3A4 (například fenytoinem, karbamazepinem, fenobarbitalem nebo třezalkou (Hypericum perforatum)) může také vést ke snížení plazmatických koncentrací rivaroxabanu. Proto je třeba se vyhnou současnému podávání silných induktorů CYP3A4, pokud není pacient pozorně sledován kvůli známkám a příznakům trombózy.
Jiné současně podávané léky
Žádné klinicky významné farmakokinetické nebo farmakodynamické interakce nebyly zjištěny při současném podávání rivaroxabanu s midazolamem (substrát CYP3A4), digoxinem (substrát P-gp), atorvastatinem (substrát CYP3A4 a P-gp) nebo omeprazolem (inhibitor protonové pumpy). Rivaroxaban neinhibuje ani neindukuje významné izoformy CYP jako je CYP3A4.
Laboratorní parametry
Parametry srážení krve (například PT, aPTT, Heptest) j sou ovlivněny podle očekávání na základě mechanismu působení rivaroxabanu (viz bod 5.1).
4.6 Fertilita, těhotenství a kojení
Bezpečnost a účinnost přípravku Xarelto nebyly u těhotných žen stanoveny. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Vzhledem k možné reprodukční toxicitě, známému riziku krvácení a důkazu, že rivaroxaban prochází placentou, je přípravek Xarelto kontraindikován v těhotenství (viz bod 4.3). Ženy ve fertilním věku musí během léčby rivaroxabanem zabránit otěhotnění.
Kojení
Bezpečnost a účinnost přípravku Xarelto nebyly u kojících žen stanoveny. Údaje z experimentů na zvířatech signalizují, že je rivaroxaban vylučován do mléka. Podávání přípravku Xarelto je během kojení kontraindikováno (viz bod 4.3). Je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit léčbu.
Fertilita
Nebyly provedeny žádné specifické studie užívání rivaroxabanu u lidí s cílem vyhodnotit účinky na fertilitu. Ve studii samčí a samičí fertility na potkanech nebyly pozorovány žádné účinky (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xarelto má malý vliv na schopnost řídit a obsluhovat stroje. Byly hlášeny nežádoucí účinky jako synkopa (frekvence výskytu: méně časté) a závrať (frekvence výskytu: časté) (viz bod 4.8). Pacienti, kteří zaznamenali tyto nežádoucí účinky, by neměli řídit vozidla a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostních informací
Bezpečnost rivaroxabanu byla hodnocena v jedenácti studiích fáze III u 32 625 pacientů, kteří byli vystaveni rivaroxabanu (viz tabulka 1).
Tabulka 1: Počet hodnocených pacientů, maximální ^ denní dávka a délka léčby ve studiích fáze II
|
Indikace |
Počet pacientů* |
Maximální denní dávka |
Maximální délka léčby |
|
Prevence žilního tromboembolismu u dospělých pacientů podstupujících elektivní operativní náhradu kyčelního nebo kolenního kloubu |
6 097 |
10 mg |
39 dnů |
|
Prevence žilního tromboembolismu u hospitalizovaných nechirurgických pacientů |
3 997 |
10 mg |
39 dnů |
|
Léčba hluboké žilní trombózy a plicní embolie a prevence jejich recidivy |
4 556 |
Den 1 - 21: 30 mg Den 22 a dále: 20 mg |
21 měsíců |
|
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní |
7 750 |
20 mg |
41 měsíců |
|
Prevence aterotrombotických příhod u pacientů po AKS |
10 225 |
5 mg nebo 10 mg, podávaných společně s ASA nebo s kombinací ASA plus klopidogrel či tiklopidin |
31 měsíců |
*Pacienti exponovaní minimálně jedné dávce rivaroxabanu
Nejčastěji hlášenými nežádoucí účinky u pacientů, kteří dostávali rivaroxaban, bylo krvácení (viz bod 4.4 a níže uvedený „Popis vybraných nežádoucích účinků“). Nejčastěji hlášeným krvácením (> 4 %) byla epistaxe (5,9 %) a gastrointestinální krvácení (4,2 %).
Celkem asi u 67% pacientů exponovaných minimálně jedné dávce rivaroxabanu byl hlášen výskyt nežádoucích příhod. Asi u 22% pacientů se vyskytly nežádoucí příhody, které byly považovány za související s léčbou podle posouzení zkoušejícími. U pacientů léčených přípravkem Xarelto v dávce 10 mg podstupujících chirurgickou náhradu kyčelního nebo kolenního kloubu a u nechirurgických pacientů hospitalizovaných pro jiné onemocnění se krvácivé příhody vyskytly asi u 6,8% a 12,6% pacientů a anémie se objevila asi u 5,9% a 2,1% pacientů. U pacientů léčených přípravkem Xarelto buď v dávce 15 mg dvakrát denně s následným podáváním 20 mg jednou denně pro léčbu hluboké žilní trombózy nebo plicní embolie nebo dávkou 20 mg jednou denně z důvodu prevence recidivující hluboké žilní trombózy a plicní embolie se krvácivé příhody vyskytly asi u 27,8% pacientů a anémie se objevila asi u 2,2% pacientů. U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace bylo krvácení jakéhokoli typu a závažnosti hlášeno s frekvencí 28 na 100 paciento-roků a anémie s frekvencí 2,5 na 100 paciento-roků. U pacientů léčených z důvodu prevence kardiovaskulárního úmrtí a infarktu myokardu po akutním koronárním syndromu (ACS), bylo krvácení jakéhokoli typu a závažnosti hlášeno s frekvencí 22 na 100 paciento-roků. Anémie byla hlášena s frekvencí 1,4 na 100 paciento-roků
Seznam nežádoucích účinků uvedený v tabulce
Výskyt nežádoucích účinků hlášený u přípravku Xarelto je shrnutý v tabulce 2 níže podle orgánové klasifikace (v MedDRA) a podle frekvence výskytu.
Četnosti j sou definovány takto:
velmi časté (> 1/10)
časté (> 1/100 až < 1/10)
méně časté (> 1/1 000 až < 1/100)
vzácné (> 1/10 000 až < 1/1 000)
velmi vzácné ( < 1/10,000)
není známo ( z dostupných údajů nelze určit)
Tabulka 2: Všechny s léčbou související nežádoucí účinky hlášené u pacientů ve studiích fáze III
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (včetně příslušných laboratorních parametrů) |
Trombocytémie (včetně zvýšeného počtu trombocytů)A | ||
|
Poruchy imunitního systému | |||
|
Alergická reakce, alergická dermatitida | |||
|
Poruchy nervového systému | |||
|
Závratě, bolesti hlavy |
Cerebrální a intrakraniální krvácení, synkopa | ||
|
Poruchy oka | |||
|
Oční krvácení (včetně krvácení do spojivek) | |||
|
Srdeční poruchy | |||
|
Cévní poruchy | |||
|
Hypotenze, hematom | |||
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe, hemoptýza | |||
|
Gastrointestinální poruchy | |||
|
Krvácení z dásní, krvácení z gastrointestinálního traktu (včetně rektálního krvácení), gastrointestinální a abdominální bolest, dyspepsie, nausea, A |
Sucho v ústech | ||
|
Poruchy jater a žlučových cest | |||
|
Abnormity jaterní funkce |
Žloutenka | ||
|
Poruchy kůže a podkožní tkáně | |||
|
Pruritus (včetně méně častých případů generalizovaného pruritu), vyrážka, ekchymóza, kožní a podkožní krvácení |
Kopřivka | ||
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |||
|
Bolest v končetináchA |
Hemartróza |
Krvácení do svalů |
Kompartment syndrom sekundárně po krvácení |
|
Poruchy ledvin a močových cest | |||
|
Urogenitální krvácení (včetně hematurie a menorhagieB), poškození ledvin (včetně zvýšení hladin kreatininu a močoviny v krvi)A |
Renální selhání/akutní renální selhání vzniklé sekundárně po krvácení natolik silném, aby způsobilo hypoperfúzi | ||
|
Celkové poruchy a reakce v místě aplikace | |||
|
HorečkaA, periferní edém, pokles celkové síly a energie (včetně únavy a tělesné slabosti) |
Pocit indispozice (včetně malátnosti) |
Lokalizovaný edémA | |
|
Vyšetření | |||
|
Zvýšená hladina transamináz |
Zvýšená hladina bilirubinu, alkalické fosfatázyA, LDHA, lipázyA, amylázyA, GMTA |
Zvýšení konjugovaného bilirubinu (s přidruženým zvýšením ALT nebo bez zvýšení ALT) | |
|
Poranění, otravy a procedurální komplikace | |||
|
Pooperační krvácení (včetně pooperační anémie a krvácení z rány), kontuze, sekrece z ranA |
Cévní pseudoaneurysmaC | ||
A: pozorováno u prevenze žilního tromboembolismu u dospělých pacientů, kteří podstoupili chirurgickou náhradu kyčelního nebo kolenního kloubu
B: pozorováno u léčby hluboké žilní trombózy, plicní embolie a u prevence recidivy jako velmi časté u žen do 55 let
C: pozorováno méně často u prevence aterotrombotických příhod u pacientů po akutním koronárním syndromu (po perkutánní koronární intervenci)
Popis vybraných nežádoucích účinků
Vzhledem k farmakologickému mechanismu působení může být užívání přípravku Xarelto spojeno se zvýšeným rizikem okultního nebo zjevného krvácení z jakékoli tkáně nebo orgánu s možným následkem posthemoragické anémie. Známky, příznaky a závažnost (včetně možného fatálního zakončení) se mohou různit podle místa a stupně nebo rozsahu krvácení a/nebo anémie (viz bod 4.9 Léčba krvácení). V klinických studiích bylo během dlouhodobé léčby rivaroxabanem ve srovnání s léčbou VKA častěji pozorováno slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Proto, kromě adekvátního klinického sledování, pokud je shledáno vhodným, může laboratorní vyšetření hemoglobinu/hematokritu být přínosem pro detekci okultního krvácení. Riziko krvácení bude možná zvýšeno u některých skupin pacientů, například osob s těžkou arteriální hypertenzí neupravenou léčbou a/nebo souběžnou léčbou ovlivňující krevní srážlivost (viz Riziko krvácení v bodu 4.4). Menstruační krvácení může být zesíleno a/nebo prodlouženo. Hemoragické komplikace se mohou projevovat jako celková slabost, bledost, závratě, bolesti hlavy nebo nevysvětlitelné otoky, dušnost a nevysvětlitelný šok. V některých případech byly v důsledku anémie pozorovány příznaky srdeční ischémie, jako je například bolest na hrudníku nebo angina pectoris.
V souvislosti s užíváním přípravku Xarelto byly hlášeny známé sekundární komplikace závažného krvácení, jako je například kompartment syndrom a renální selhání v důsledku hypoperfuze. Možnost krvácení je proto třeba zvážit při posuzování stavu pacientů s jakoukoli antikoagulační léčbou.
Post-marketingové sledování
V postmarketingovém sledování byly v časové souvislosti s užitím přípravku Xarelto hlášeny následující nežádoucí účinky. Frekvenci těchto nežádoucích účinků hlášených po uvedení na trh nelze určit.
Poruchy imunitního systému: angioedém a alergický edém (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Poruchy jater a žlučových cest: cholestáza, hepatitida (včetně hepatocelulárního poškození) (v souhrnných studiích fáze III byly tyto příhody vzácné (>1/10 000 až <1/1000)).
Poruchy krve a lymfatického systému: trombocytopenie (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny vzácné případy předávkování dávkou až 600 mg bez krvácivých komplikací nebo jiných nežádoucích reakcí. Z důvodu omezené absorpce se očekává strop účinku bez dalšího zvýšení průměrné plazmatické hladiny v případě vyšší než terapeutické dávky 50 mg rivaroxabanu nebo dávek vyšších. Specifické antidotum blokující farmakodynamický účinek rivaroxabanu není k dispozici.
Lze zvážit podání aktivního uhlí ke snížení absorpce v případě předávkování rivaroxabanem.
Léčba krvácení
Pokud dojde ke krvácivým komplikacím u pacienta léčeného rivaroxabanem, musí se podání další dávky rivaroxabanu odložit nebo se léčba musí ukončit, dle potřeby. Rivaroxaban má biologický poločas asi 5 až 13 hodin (viz bod 5.2). Léčba by měla být individuální podle závažnosti a lokalizace krvácení. Podle potřeby je třeba použít vhodnou symptomatickou léčbu, jako je mechanická komprese (např. u závažné epistaxe), chirurgická hemostáza se zajištěním kontroly krvácení, náhradou tekutin a zajištěním hemodynamické podpory, krevními deriváty (erytrocyty nebo čerstvá zmrazená plasma, v závislosti na související anémii nebo koagulopatii) nebo trombocyty.
Pokud krvácení nelze kontrolovat výše uvedenými opatřeními, lze zvážit podávání specifické prokoagulační reverzní látky, jako je koncentrát protrombinového komplexu (PCC), aktivovaný koncentrát protrombinového komplexu (APCC) nebo rekombinantní faktor VIIa (r-FVIIa). V současnosti jsou však k dispozici velmi omezené klinické zkušenosti s použitím těchto přípravků u osob užívajících rivaroxaban. Doporučení je též podloženo omezenými neklinickými údaji. Opakované podání rekombinantního faktoru VIIa je třeba zvážit a titrovat v závislosti na zlepšování krvácení. V případě závažného krvácení je třeba konzultovat odborníka na koagulaci, pokud je odborník v místě dostupný (viz bod 5.1).
Protamin sulfát a vitamin K podle všeho nebudou ovlivňovat antikoagulační aktivitu rivaroxabanu. U osob užívajících rivaroxaban jsou omezené zkušenosti s použítím kyseliny tranexamové a neexistují zkušenosti s použitím kyseliny aminokaproové a aprotininu. Neexistují ani vědecké důvody přínosu ani zkušenosti s použitím systémového hemostatika desmopressinu u osob užívajících rivaroxaban. Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: přímé inhibitory faktoru Xa, ATC kód: B01AF01 Mechanismus účinku
Rivaroxaban je vysoce selektivní přímý inhibitor faktoru Xa biologicky dostupný při perorálním podání. Inhibice faktoru Xa blokuje vnitřní a vnější metabolické cesty koagulační kaskády, a inhibuje vznik trombinu i vytváření trombů. Rivaroxaban neinhibuje trombin (aktivovaný faktor II) a nebyly prokázány žádné účinky na trombocyty.
Farmakodynamické účinky
U lidí byla zjištěna inhibice faktoru Xa přímo úměrná dávce. Protrombinový čas (PT) je rivaroxabanem ovlivňován úměrně dávce, objevuje se vysoká korelace s plazmatickými koncentracemi (hodnota R je 0,98), pokud je při testu jako assay použit Neoplastin. Jiné reagenty mohou přinést jiné výsledky. Hodnotu PT je nutno stanovit v sekundách, protože INR (mezinárodní normalizovaný poměr) je kalibrován a validován pouze pro kumariny, a nelze jej využívat pro jiné antikoagulanty.
U pacientů užívajících rivaroxaban v léčbě hluboké žilní trombózy a plicní embolie a k prevenci jejich recidivy se v 5/95 percentilu hodnoty PT (Neoplastin) za 2 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 17 až 32 s pro dávku 15 mg rivaroxabanu dvakrát denně a od 15 do 30 s pro dávku 20 mg rivaroxabanu jednou denně. Nejnižší hodnoty se v 5/95 percentilu pohybovaly od 14 do 24 s pro dávku 15 mg dvakrát denně (8 -16 hodin po požití) a od 13 do 20 s pro dávku 20 mg jednou denně (18 - 30 hodin po požití). U pacientů s nevalvulární fibrilací síní užívajících rivaroxaban v prevenci cévní mozkové příhody a systémové embolizace se v 5/95 percentilu hodnoty PT (Neoplastin) za 1 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 14 až 40 s pro dávku 20 mg rivaroxabanu jednou denně a od 10 do 50 s u pacientů se středně závažným poškozením renálních funkcí léčených dávkou 15 mg jednou denně. Nejnižší hodnoty (16 - 36 hodin po požití) se v 5/95 percentilu pohybovaly od 12 do 26 s pro dávku 20 mg jednou denně a u pacientů se středně závažným poškozením renálních funkcí léčených 15 mg jednou denně se hodnoty pohybovaly od 12 do 26 s.
V klinické farmakologické studii sledující reverzi farmakodynamického účinku rivaroxabanu u zdravých dospělých osob (n=22) byl hodnocen účinek jednotlivé dávky (50 IU/kg) u dvou rozdílných typů PCC, 3-faktorového PCC (faktory II, IX a X) a 4-faktorového PCC (II, VII, IX a X). 3-faktorový PCC redukoval průměrnou hodnotu PT času (protrombinového času), hodnocenou neoplastinem, přibližně o 1,0 sekundy během 30 minut, ve srovnání s přibližně 3,5 sekundami pozorovanými u 4-faktorového PCC. Naproti tomu, 3-faktorový PCC měl větší a rychlejší celkový efekt na zvrácení změny tvorby endogenního trombinu než 4-faktorový PCC (viz bod 4.9).
Aktivovaný parciální tromboplastinový čas (aPTT) a hodnoty analýzy HepTest jsou také prodlouženy úměrně dávce; nedoporučuje se však tyto metody používat k hodnocení farmakodynamických účinků rivaroxabanu. Během léčby rivaroxabanem v běžné klinické praxi není třeba monitorovat parametry koagulace. Pokud však je klinicky indikováno, lze hladiny rivaroxabanu měřit pomocí kalibrovaných kvantitativních testů anti-faktoru Xa (viz bod 5.2).
Klinická účinnost a bezpečnost
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní Klinický program přípravku Xarelto byl navržen tak, aby prokázal účinnost přípravku Xarelto v prevenci cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní.
V pivotní dvojitě zaslepené studii ROCKET AF bylo 14 264 pacientů přiřazeno buď do léčby přípravkem Xarelto 20 mg jednou denně (15 mg jednou denně u pacientů s clearance kreatininu 30 - 49 ml/min) nebo léčby warfarinem titrovaným na cílovou hodnotu INR 2,5 (terapeutické rozmezí 2,0 až 3,0). Střední doba léčby byla 19 měsíců a celková doba léčby byla až 41 měsíců.
34,9% pacientů bylo léčeno kyselinou acetylsalicylovou a 11,4% bylo léčeno pomocí antiarytmik třídy III, včetně amiodaronu.
V porovnání s warfarinem dosáhl přípravek non-inferiority co do primárního kompozitního cílového ukazatele cévní mozkové příhody a systémové embolizace nepostihující CNS. U populace „per protocol“ (dle protokolu) v období sledování „on treatment“ (po dobu léčby), se cévní mozková příhoda nebo systémová embolizace vyskytla u 188 pacientů na rivaroxabanu (1,71% za rok) a u 241 pacientů na warfarinu (2,16% za rok) (HR 0,79; 95% CI, 0,66 - 0,96; P<0,001 pro non-inferioritu). Mezi všemi randomizovanými pacienty analyzovanými podle ITT, se primární cílový parametr vyskytl u 269 pacientů na rivaroxabanu (2,12% za rok) a u 306 pacientů na warfarinu (2,42% za rok) (HR 0,88; 95% CI, 0,74 - 1,03; P<0,001 pro non-inferioritu; P=0,117 pro superioritu). Výsledky sekundárních cílových ukazatelů, v pořadí, jak byly testovány v ITT analýze, jsou ukázány v tabulce 3.
Mezi pacienty na léčbě warfarinem, byly hodnoty INR uvnitř terapeutického rozmezí (2,0 až 3,0) v průměru 55% doby (median 58%; rozsah mezi kvartily byl 43 až 71). Účinek rivaroxabanu se nelišil napříč úrovněmi TTR v centru (čas v cílovém INR rozmezí 2,0 až 3,0) ve stejnoměrně velkých kvartálech (P=0,74 pro interakci). V centrech v nejvyšším kvartilu bylo HR rivaroxaban versus warfarin 0,74 (95% CI, 0,49 -1,12). Četnost incidence pro hlavní bezpečnostní ukazatel (závažné a klinicky významné méně závažné krvácivé příhody) byla pro obě léčebné skupiny podobná (viz tabulka 4).
Tabulka 3: Výsledky účinnosti ze studie fáze III ROCKET AF
|
Populace studie |
ITT analýzy účinnosti u pacientů s nevalvulární fibrilací síní | ||
|
Xarelto |
Warfarin |
Poměr rizik HR | |
|
20 mg jednou denně |
titrovaný na cílovou |
(95% CI) | |
|
(15 mg jednou denně u |
hladinu INR 2,5 |
p-hodnota, test pro | |
|
pacientů se středně |
(terapeutické rozmezí |
superioritu | |
|
Dávkování |
závažnou renální insuficiencí |
2,0 až 3,0) | |
|
Výskyt příhod |
Výskyt příhod | ||
|
(100 paciento-roků) |
(100 paciento-roků) | ||
|
Cévní mozková příhoda a |
269 |
306 |
0,88 (0,74 - 1,03) |
|
systémová embolizace nepostihující CNS |
(2,12) |
(2,42) |
0,117 |
|
Cévní mozková příhoda, |
572 |
609 |
0,94 (0,84 - 1,05) |
|
systémová embolizace nepostihující CNS a vaskulární úmrtí |
(4,51) |
(4,81) |
0,265 |
|
Cévní mozková příhoda, |
659 |
709 |
0,93 (0,83 - 1,03) |
|
systémová embolizace nepostihující CNS, vaskulární úmrtí a infarkt myokardu |
(5,24) |
(5,65) |
0,158 |
|
Cévní mozková příhoda |
253 |
281 |
0,90 (0,76 - 1,07) |
|
(1,99) |
(2,22) |
0,221 | |
|
Systémová embolizace |
20 |
27 |
0,74 (0,42 - 1,32) |
|
nepostihující CNS |
(0,16) |
(0,21) |
0,308 |
|
Infarkt myokardu |
130 |
142 |
0,91 (0,72 - 1,16) |
|
(1,02) |
(1,11) |
0,464 | |
|
Populace studie |
Pacienti s nevalvulární fibrilací sínía) | ||
|
Dávkování |
Xarelto 20 mg jednou denně (15 mg jednou denně u pacientů se středně závažnou renální insuficiencí |
Warfarin titrovaný na cílovou hladinu INR 2,5 (terapeutické rozmezí 2,0 až 3,0) |
Poměr rizik (95% CI) p-hodnota |
|
Výskyt příhod (100 paciento-roků) |
Výskyt příhod (100 paciento-roků) | ||
|
Závažné a méně závažné klinicky významné příhody krvácení |
1 475 (14,91) |
1 449 (14,52) |
1,03 (0,96 - 1,11) 0,442 |
|
Závažné příhody krvácení |
395 (3,60) |
386 (3,45) |
1,04 (0,90 - 1,20) 0,576 |
|
Úmrtí v důsledku krvácení2 |
27 (0,24) |
55 (0,48) |
0,50 (0,31 - 0,79) 0,003 |
|
Krvácení do kritického orgánu2 |
91 (0,82) |
133 (1,18) |
0,69 (0,53 - 0,91) 0,007 |
|
Intrakraniální krvácení2 |
55 (0,49) |
84 (0,74) |
0,67 (0,47 - 0,93) 0,019 |
|
Pokles hemoglobinu2 |
305 (2,77) |
254 (2,26) |
1,22 (1,03 - 1,44) 0,019 |
|
Transfuze 2 nebo více jednotek erytrocytů nebo plné krve2 |
183 (1,65) |
149 (1,32) |
1,25 (1,01 - 1,55) 0,044 |
|
Méně závažné klinicky významné krvácivé příhody |
1 185 (11,80) |
1 151 (11,37) |
1,04 (0,96 - 1,13) 0,345 |
|
Úmrtí z jakékoli příčiny |
208 (1,87) |
250 (2,21) |
0,85 (0,70 - 1,02) 0,073 |
a) „Safety“ populace „on treatment“ (populace, ve které byla hodnocena bezpečnost, po dobu léčby)
Pacienti podstupující kardioverzi
Prospektivní, randomizovaná, otevřená, multicentrická, analytická studie se zaslepeným hodnocením cílů (X-VERT) byla provedena u 1504 pacientů (bez předchozí léčby perorálními antikoagulancii nebo předléčených) s nevalvulární fibrilací síní naplánovaných ke kardioverzi, srovnávající rivaroxaban s adjustovanou dávkou VKA (randomizovaných v poměru 2:1) v prevenci kardiovaskulárních příhod.
Byly sledovány buď kardioverze s provedenou TEE (1-5 dní léčby) nebo konvenční kardioverze (nejméně tři týdny léčby). Primární cíl účinnosti (všechny CMP, transitorní ischemická ataka, systémová embolie mimo CNS, IM a úmrtí z kardiovaskulárních příčin) nastal u 5 (0,5%) pacientů léčených rivaroxabanem (n=978) a u 5 (1,0%) pacientů léčených VKA (n=492; RR% 0,5; CI 0,15-1,73; modifikovaná ITT populace). Hlavní bezpečnostní ukazatel (závažné krvácení) se vyskytl u 6 (0,6%) pacientůléčených rivaroxabanem (n= 988) a u 4 (0,8%) pacientů léčených VKA (n=499), (RR 0,76; 95% CI 0,21-2,67; safety populace). Tato analytická studie ukázala srovnatelnou účinnost a bezpečnost mezi skupinami s rivaroxabanem a VKA v případě kardioverze.
Léčba hluboké žilní trombózy, plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie Klinický program přípravku Xarelto byl navržen tak, aby prokázal účinnost přípravku Xarelto v úvodní a pokračující léčbě hluboké žilní trombózy a plicní embolie a prevenci jejich recidivy.
Více než 4 600 pacientů bylo hodnoceno ve třech randomizovaných kontrolovaných studiích fáze III (Einstein DVT, Einstein PE a Einstein Extension) a poté byla provedena predefinovaná poolovaná analýza studií Einstein DVT a Einstein PE. Celková kombinovaná délka léčby ve všech studiích byla až 21 měsíců.
Ve studii Einstein DVT bylo hodnoceno 3 449 pacientů s akutní hlubokou žilní trombózou v léčbě hluboké žilní trombózy a prevenci recidivující hluboké žilní trombózy a plicní embolie (pacienti, kteří měli symptomatickou plicní embolii, byli z této studie vyřazeni). Délka léčby byla až 12 měsíců v závislosti na klinickém posouzení zkoušejícím.
V úvodní 3-týdenní léčbě akutní hluboké žilní trombózy byl podáván rivaroxaban v dávce 15 mg dvakrát denně. Poté následovalo podávání dávky 20 mg rivaroxabanu jednou denně.
Ve studii Einstein PE bylo hodnoceno 4 832 pacientů s akutní plicní embolií v léčbě plicní embolie a v prevenci recidivující hluboké žilní trombózy a plicní embolie. Délka léčby byla 3, 6 nebo 12 měsíců v závislosti na klinickém posouzení zkoušejícím.
V úvodní léčbě akutní PE bylo podáváno 15 mg rivaroxabanu dvakrát denně 3 týdny. Poté následovalo podávání dávky 20 mg rivaroxabanu jednou denně.
V obou studiích Einstein DVT a Einstein PE zahrnoval srovnávaný léčebný režim enoxaparin podávaný minimálně 5 dnů v kombinaci s antagonisty vitaminu K do dosažení terapeutického rozmezí PT/INR (> 2,0). Léčba pokračovala antagonistou vitaminu K, jehož dávka byla upravena pro udržení hodnot PT/INR v terapeutickém rozmezí 2,0 až 3,0.
Ve studii Einstein Extension bylo hodnoceno 1 197 pacientů s hlubokou žilní trombózou nebo plicní embolií v prevenci recidivující hluboké žilní trombózy a plicní embolie. Trvání léčby bylo dalších 6 nebo 12 měsíců u pacientů, kteří dokončili 6 až 12 měsíců léčby pro žilní tromboembolismus v závislosti na klinickém posouzení zkoušejícím. Přípravek Xarelto 20 mg jednou denně byl srovnáván s placebem.
Všechny studie fáze III využívaly stejné předem definované primární a sekundární parametry účinnosti. Primární parametr účinnosti byl symptomatický recidivující žilní tromboembolismus definovaný jako kompozit recidivující hluboké žilní trombózy nebo fatální či nefatální plicní embolie. Sekundární parametr účinnosti byl definovaný jako kompozit recidivující hluboké žilní trombózy, nefatální plicní embolie a mortality ze všech příčin.
Ve studii Einstein DVT (viz tabulka 5) prokázal rivaroxaban non-inferioritu proti enoxaparinu/antagonistům vitaminu K v primárním parametru účinnosti (p < 0,0001 (test non-inferiority); poměr rizik: 0,680 (0,443 - 1,042), p = 0,076 (test superiority)). Předem definovaný čistý klinický přínos (primární parametr účinnosti plus závažná krvácivá příhoda) byl hlášen s poměrem rizik 0,67 ((95% CI: 0,47-0,95) s nominální hodnotou p = 0,027) ve prospěch rivaroxabanu. Hodnoty INR byly uvnitř terapeutického rozmezí
s průměrem 60,3% pro průměrnou dobu léčby 189 dní a 55,4%, 60,1% a 62,8% doby pro skupiny s plánovanou léčbou 3, 6 a 12 měsíců. Ve skupině enoxaparin/VKA nebyl jasný vztah mezi hladinou TTR v centru (doba v cílovém INR rozmezí 2,0 -3,0) ve stejně velkých tertilech a incidencí recidivující HZT (P=0,932 pro interakci). V centrech v nejvyšším tertilu bylo HR rivaroxaban versus warfarin byl 0,69 (95% CI: 0,35 - 1,35).
Výskyt primárního bezpečnostního ukazatele (závažné nebo klinicky významné méně závažné krvácivé příhody) stejně jako sekundárního bezpečnostního ukazatele (závažné krvácivé příhody) byl podobný v obou léčebných skupinách.
Tabulka 5: Výsledky účinnosti a bezpečnosti ze studie fáze III Einstein DVT (hluboká žilní trombóza)
|
Populace studie |
3 449 pacientů se symptomatickou hlubokou žilní trombózou | |
|
Dávkování a délka léčby |
Xareltoa) |
Enoxaparin/VKAb) |
|
3, 6 nebo 12 měsíců |
3, 6 nebo 12 měsíců | |
|
N = 1 731 |
N = 1 718 | |
|
Symptomatický recidivující žilní |
36 |
51 |
|
tromboembolismu* |
(2,1%) |
(3,0%) |
|
Symptomatická recidivující |
20 |
18 |
|
plicní embolizace |
(1,2%) |
(1,0%) |
|
Symptomatická recidivující |
14 |
28 |
|
hluboká žilní trombóza |
(0,8%) |
(1,6%) |
|
Symptomatická plicní embolie a |
1 |
0 |
|
hluboká žilní trombóza |
(0,1%) | |
|
Fatální plicní embolie/úmrtí, |
4 |
6 |
|
kde plicní embolie nemůže být vyloučena |
(0,2%) |
(0,3%) |
|
Závažné nebo klinicky významné |
139 |
138 |
|
méně závažné krvácení |
(8,1%) |
(8,1%) |
|
Závažné krvácivé příhody |
14 |
20 |
|
(0,8%) |
(1,2%) | |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů vitaminu K
* p < 0,0001 (non-inferiorita k stanovenému poměru rizik 2,0); poměr rizik: 0,680 (0,443 - 1,042), p = 0,076 (superiorita)
Ve studii Einstein PE (viz tabulka 6) prokázal rivaroxaban non-inferioritu proti enoxaparinu/antagonistům vitaminu K v primárním parametru účinnosti (p = 0,0026 (test non-inferiority); poměr rizik: 1,123 (0,749 - 1,684)). Předem definovaný čistý klinický přínos (primární parametr účinnosti plus závažná krvácivá příhoda) byl hlášen s poměrem rizik 0,849 ((95% CI: 0,633-1,139), s nominální hodnotou p = 0,275). Hodnoty INR byly uvnitř terapeutického rozmezí s průměrem 63% pro průměrnou dobu léčby 215 dní a 57%, 62% a 65% doby pro skupiny s plánovanou léčbou 3, 6 a 12 měsíců. Ve skupině enoxaparin/VKA nebyl jasný vztah mezi hladinou TTR v centru (doba v cílovém INR rozmezí 2,0 - 3,0) ve stejně velkých tertilech a incidencí recidivující HZT (P=0,082 pro interakci). V centrech v nejvyšším tertilu bylo HR rivaroxaban versus warfarin 0,642 (95% CI: 0,277 - 1,484).
Výskyt primárního bezpečnostního ukazatele (závažné nebo klinicky významné méně závažné krvácivé příhody) byl lehce nižší ve skupině léčené rivaroxabanem (10,3% (249/2412)) než ve skupině léčené enoxaparinem/antagonisty vitaminu K (11,4% (274/2405)). Výskyt sekundárního bezpečnostního ukazatele (závažné krvácivé příhody) byl nižší ve skupině léčené rivaroxabanem (1.1% (26/2412)) než ve skupině enoxaparin/antagonisté vitaminu K (2.2% (52/2405)) s poměrem rizik 0,493 (95% CI: 0,308 - 0,789).
|
Populace studie |
4 832 pacientů s akutní symptomatickou PE | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Dávkování a doba léčby |
3, 6 nebo 12 měsíců |
3, 6 nebo 12 měsíců |
|
N=2 419 |
N=2 413 | |
|
Symptomatický recidivující žilní |
50 |
44 |
|
tromboembolismus* |
(2,1%) |
(1,8%) |
|
Symptomatická recidivující PE |
23 (1,0%) |
20 (0,8%) |
|
Symptomatická recidivující |
18 |
17 |
|
hluboká žilní trombóza |
(0,7%) |
(0,7%) |
|
Symptomatická recidivující plicní embolie a hluboká žilní trombóza |
0 |
2 (<0,1%) |
|
Fatální plicní embolie/úmrtí, kde plicní embolie nemůže být vyloučena |
11 (0,5%) |
7 (0,3%) |
|
Závažné nebo klinicky významné |
249 |
274 |
|
méně závažné krvácení |
(10,3%) |
(11,4%) |
|
Závažné krvácivé příhody |
26 (1,1%) |
52 (2,2%) |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů
vitaminu K
* p < 0,0026 (non-inferiorita k predefinovanému poměru rizik 2,0); poměr rizik: 1,123 (0,749 - 1,684)
Byla provedena predefinovaná poolovaná analýza výsledku studií Einstein DVT a PE (viz tabulka 7).
Tabulka 7: Výsledky účinnosti a bezpečnosti z poolované analýzy studií fáze III Einstein DVT a Einstein PE
|
Populace studie |
8281 pacientů s akutní symptomatickou HZT nebo PE | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Dávkování a doba léčby |
3, 6 nebo 12 měsíců |
3, 6 nebo12 měsíců |
|
N=4150 |
N=4131 | |
|
Symptomatický recidivující žilní |
86 |
95 |
|
tromboembolismus* |
(2,1%) |
(2,3%) |
|
Symptomatická recidivující |
43 |
38 |
|
(1,0%) |
(0,9%) | |
|
Symptomatická recidivující |
32 |
45 |
|
hluboká žilní trombóza |
(0,8%) |
(1,1%) |
|
Symptomatická recidivující plicní embolie a hluboká žilní trombóza |
1 (<0,1%) |
2 (<0,1%) |
|
Fatální plicní embolie/úmrtí, kde |
15 |
13 |
|
plicní embolie nemůže být vyloučena |
(0,4%) |
(0,3%) |
|
Závažné nebo klinicky významné |
388 |
412 |
|
méně závažné krvácení |
(9,4%) |
(10,0%) |
|
Závažné krvácivé příhody |
40 (1,0%) |
72 (1,7%) |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů vitaminu K
* p < 0,0001 (non-inferiorita k predefinovanému poměru rizik 1,75); poměr rizik: 0,886 (0,661 - 1,186)
Predefinovaný čistý klinický prospěch (výsledek primární účinnosti plus závažné krvácivé příhody) poolované analýzy byl hlášen s poměrem rizik 0,771 ((95% CI: 0,614 - 0,967), nominální hodnota p= 0,0244).
Ve studii Einstein Extension (viz tabulka 8) byl rivaroxaban lepší než placebo v primárních a sekundárních parametrech účinnosti. U primárního bezpečnostního ukazatele (závažné krvácivé příhody) byl nevýznamný numericky vyšší výskyt u pacientů léčených rivaroxabanem v dávce 20 mg jednou denně v srovnání s placebem. Sekundární bezpečnostní ukazatel (závažné nebo klinicky významné méně závažné krvácivé příhody) prokázal vyšší výskyt u pacientů léčených rivaroxabanem 20 mg jednou denně ve srovnání s placebem.
|
Populace studie |
Pokračování léčby u 1197 pacientů, u nichž byla podávána léčba a prevence recidivujícího žilního tromboembolismu | |
|
Dávkování a doba léčby |
Xareltoa) |
Placebo |
|
6 nebo 12 měsíců |
6 nebo 12 měsíců | |
|
N = 602 |
N = 594 | |
|
Symptomatický recidivující žilní |
8 |
42 |
|
tromboembolismus* |
(1,3%) |
(7,1%) |
|
Symptomatická recidivující |
2 |
13 |
|
plicní embolizace |
(0,3%) |
(2,2%) |
|
Symptomatická recidivující |
5 |
31 |
|
hluboká žilní trombóza |
(0,8%) |
(5,2%) |
|
Fatální plicní embolie/úmrtí, |
1 |
1 |
|
kde plicní embolie nemůže být vyloučena |
(0,2%) |
(0,2%) |
|
Závažné krvácivé příhody |
4 |
0 |
|
(0,7%) |
(0,0%) | |
|
Klinicky významné méně závažné |
32 |
7 |
|
krvácení |
(5,4%) |
(1,2%) |
a) Rivaroxaban 20 mg j ednou denně
* p < 0,0001 (superiorita), poměr rizik: 0,185 (0,087 - 0,393)
Kromě studií fáze III programu EINSTEIN byla provedena prospektivní, neintervenční, otevřená kohortová studie (XALIA) s centrálním vyhodnocováním sledovaných ukazatelů zahrnujících recidivující žilní tromboembolismus, závažné krvácení a úmrtí. Bylo zařazeno 5 142 pacientů s akutní hlubokou žilní trombózou za účelem posoudit dlouhodobou bezpečnost rivaroxabanu v porovnání se standardní antikoagulační terapií v klinické praxi. Výskyt závažného krvácení, recidivujícího žilního tromboembolismu a úmrtí ze všech příčin byly v rivaroxabanové větvi 0,7 %, 1,4 % a 0,5 %.
Ve vstupních charakteristikách pacientů byly rozdíly včetně věku, výskytu nádorových onemocnění a ledvinové nedostatečnosti. Přestože byla pro úpravu získaných základních rozdílů použita předem stanovená analýza stratifikovaná dle propensity skóre, mohou reziduální zavádějící faktory tyto výsledky ovlivnit. Upravené poměry rizik srovnávající rivaroxaban a standardní léčbu pro závažné krvácení, recidivující žilní tromboembolismus a mortalitu ze všech příčin byly 0,77 (95% CI: 0,40 - 1,50), 0,91 (95% CI: 0,54 - 1,54) a 0,51 (95% CI: 0,24 - 1,07).
Tato pozorování z klinické praxe jsou v souladu s potvrzeným bezpečnostním profilem v této indikaci. Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xarelto v léčbě tromboembolických příhod u jedné nebo více podskupin pediatrické populace. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xarelto v prevenci tromboembolických příhod u všech podskupin pediatrické populace (informace o použití u pediatrické populace viz bod 4.2.)
5.2 Farmakokinetické vlastnosti
Absorpce
Rivaroxaban je rychle absorbován; maximální koncentrace (Cmax) se objeví 2 - 4 hodiny po užití tablety.
Bez ohledu na stav na lačno nebo po jídle je u dávky 2,5 mg a 10 mg rivaroxabanu tablety perorální absorpce téměř kompletní a perorální biologická dostupnost vysoká (80 - 100 %). Užívání při jídle neovlivňuje při 2,5 mg a 10mg dávce AUC ani Cmax rivaroxabanu.
Pro 20 mg tabletu byla v důsledku sníženého rozsahu absorpce stanovena biologická dostupnost 66 % při stavu nalačno. Když byly užívány 20 mg tablety přípravku Xarelto společně s jídlem, došlo ke zvýšení průměrné AUC o 39 % ve srovnání s užíváním tablety nalačno, což ukazuje na téměř kompletní absorpci a vysokou biologickou dostupnost po perorálním podání. Přípravek Xarelto 15 mg a 20 mg se má užívat s jídlem (viz bod 4.2).
Farmakokinetické vlastnosti rivaroxabanu jsou až do denní dávky 15 mg nalačno přibližně lineární. Po jídle byla u přípravku Xarelto 10 mg, 15 mg a 20 mg prokázána farmakokinetika závislá na dávce. Ve vyšších dávkách je absorbce rivaroxabanu omezena disolucí, dochází ke snížení biologické dostupností a stupeň absorbce se snižuje se zvyšující se dávkou.
Variabilita farmakokinetiky rivaroxabanu je střední, s interindividuální variabilitou v rozmezí od 30% do 40%.
Absorpce rivaroxabanu je závislá na místě jeho uvolnění v gastrointestinálním traktu. Bylo hlášeno 29% a 56% snížení AUC a Cmax ve srovnání s tabletou, pokud byl rivaroxaban v granulátu uvolněn v proximální časti tenkého střeva. Expozice je dále snížena, když je rivaroxaban uvolněn v distální části tenkého střeva nebo ve vzestupné části tračníku. Podání rivaroxabanu distálně od žaludku by se mělo zabránit, jelikož to může vést ke snížení absorpce a související expozice rivaroxabanu.
Biologická dostupnost (AUC and Cmax) byla pro podání 20 mg rivaroxabanu per os ve formě rozdrcené tablety vmíchané do jablečného pyré nebo rozpuštěné ve vodě a pro podání žaludeční sondou následované tekutou stravou, v porovnání s celou tabletou srovnatelná. Vzhledem k předvídatelnému dávce úměrnému farmakokinetickému profilu rivaroxabanu odpovídají výsledky biologické dostupnosti z této studie pravděpodobně nižším dávkám rivaroxabanu.
Distribuce
Vazba na plazmatické proteiny u lidí je vysoká, přibližně 92% - 95%, přičemž hlavní část se váže na sérový albumin. Distribuční objem je střední, Vss činí přibližně 50 litrů.
Biotransformace a eliminace
Z podané dávky rivaroxabanu se přibližně 2/3 metabolicky degradují, z čehož je polovina vylučována ledvinami a druhá polovina stolicí. Zbývající 1/3 podané dávky je vylučována ledvinami přímo jako nezměněná léčivá látka, hlavně prostřednictvím aktivní ledvinové sekrece.
Rivaroxaban je metabolizován prostřednictvím systémů CYP3A4 a CYP2J2 i mechanismy na CYP nezávislými. Hlavními cestami transformace je oxidativní degradace morfolinonové části a hydrolýza amidových vazeb. Na základě in vitro experimentů je zřejmé, že rivaroxaban slouží jako substrát transportních proteinů - P-gp (P-glykoprotein) a Bcrp (breast cancer resistance protein).
Nezměněný rivaroxaban je nejvýznamnější formou přípravku v lidské plazmě; v krevním oběhu nejsou žádné významné nebo aktivní metabolity. Rivaroxaban lze vzhledem ke systémové clearance asi 10 l/h klasifikovat jako látku s nízkou clearance. Po intravenózním podání dávky 1 mg je eliminační poločas asi
4.5 hodiny. Po perorálním podání je eliminace limitována stupněm absorpce. K eliminaci rivaroxabanu
z plazmy dochází s terminálním poločasem 5 až 9 hodin u mladších osob a s terminálním poločasem 11 -13 hodin u starších osob.
Zvláštní skupiny
Pohlaví
Mezi muži a ženami nebyl žádný klinicky relevantní rozdíl ve farmakokinetice a farmakodynamice přípravku.
Starší populace
Starší pacienti vykazovali vyšší plazmatické koncentrace než mladší, s průměrnou hodnotou AUC přibližně
1.5 x vyšší, hlavně vzhledem ke snížené (zdánlivé) celkové a ledvinové clearance. Žádná úprava dávky není nutná.
Různé váhové kategorie
Extrémy v tělesné hmotnosti (< 50 kg nebo > 120 kg) měly pouze malý vliv na plazmatické koncentrace rivaroxabanu (méně než 25%). Žádná úprava není dávky nutná.
Rozdíly mezi etniky
Žádné klinicky relevantní rozdíly mezi etniky nebyly ve farmakokinetice a farmakodynamice rivaroxabanu zjištěny u pacientů z řad bělochů, Afroameričanů, Hispánců, Japonců ani Číňanů.
Jaterní nedostatečnost
Pacienti s cirhózou s mírnou jatemí nedostatečností (Child-Pugh A) vykazovali pouze menší změny ve farmakokinetice rivaroxabanu (v průměru 1,2x nárůst AUC rivaroxabanu), a výsledky byly téměř srovnatelné s kontrolní skupinou zdravých dobrovolníků. U pacientů trpících cirhózou se středně závažnou jaterní nedostatečností (Child-Pugh B) průměrná AUC rivaroxabanu významně stoupla - 2,3x v porovnání se zdravými dobrovolníky. AUC nevázané látky stoupla 2,6x. Tito pacienti měli současně sníženou renální eliminaci rivaroxabanu, podobně jako pacienti se středně závažnou ledvinovou nedostatečností. O farmakokinetice u pacientů s těžkým jaterním poškozením nejsou k dispozici žádné údaje.
Inhibice aktivity faktoru Xa byla u pacientů se středně závažnou jaterní nedostatečností zvýšena ve srovnání se zdravými dobrovolníky 2,6x; prodloužení PT bylo obdobně zvýšeno 2,1x. Pacienti se středně závažnou jaterní nedostatečností byli na rivaroxaban citlivější, a vztah mezi koncentrací a PT měl tak strmější průběh. Přípravek Xarelto je kontraindikován u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child Pugh B a C (viz bod 4.3).
Ledvinová nedostatečnost
Byl zjištěn nárůst expozice rivaroxabanu související s poklesem funkce ledvin, která byla posuzována prostřednictvím hodnot clearance kreatininu. U osob s lehkou (clearance kreatininu 50 - 80 ml/min), střední (clearance kreatininu 30 - 49 ml/min) a těžkou (clearance kreatininu 15 - 29 ml/min) ledvinovou nedostatečností byly plazmatické koncentrace rivaroxabanu (AUC) zvýšeny 1,4, 1,5 resp. 1,6 x.
Odpovídající zesílení farmakodynamických účinků bylo výraznější. U osob s lehkou, střední a těžkou ledvinovou nedostatečností byla celková inhibice faktoru Xa ve srovnání se zdravými dobrovolníky zvýšena 1,5, 1,9 resp. 2,0 x; prodloužení PT bylo obdobně zvýšeno 1,3, 2,2 a 2,4 x. O použití u pacientů s clearance kreatininu < 15 ml/min nejsou žádné údaje.
Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min. U pacientů s clearance kreatininu 15 - 29 ml/min je nutno Xarelto používat s opatrností (viz bod 4.4).
Farmakokinetické údaje u pacientů
U pacientů užívajících rivaroxaban 20 mg jednou denně k léčbě akutní hluboké žilní trombózy (HZT) byl geometrický průměr koncentrace (90% interval přepovědi) 2 - 4 hodiny a přibližně 24 hodin po podání dávky (zhruba představující maximální a minimální koncentrace během dávkovacího intervalu) 215 (22535) a 32 (6-239) pg/l.
Farmakokinetické a farmakodynamické vztahy
Po podání různě velkých dávek (5 - 30 mg dvakrát denně) byl hodnocen farmakokinetický a farmakodynamický (PK/PD) vztah mezi plazmatickou koncentrací rivaroxabanu a několika konečnými cílovými ukazateli PD (inhibice faktoru Xa, PT, aPTT, Heptest). Vztah mezi plazmatickou koncentrací rivaroxabanu a aktivitou faktoru Xa byl nejlépe popsán pomocí modelu Emax. U PT byly údaje lépe vyjádřeny pomocí lineárního ohraničeného modelu. Hodnoty PT se významně lišily v závislosti na použitých reagenciích. Při použití Neoplastinu byl výchozí PT asi 13 sekund a odchylka hodnot přibližně 3 až 4 s/(100 pg/l). Výsledek analýz PK/PD ve studiích fáze II a III byl v souladu s údaji získanými u zdravých jedinců.
Pediatrická populace
Bezpečnost a účinnost nebyly stanoveny u dětí a dospívajících do 18 let.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po jednorázovém podání, fototoxicity, genotoxicity, kancerogenního potenciálu a reprodukční juvenilní toxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studii toxicity při opakovaném podání byly způsobeny hlavně zesílenou farmakologickou aktivitou rivaroxabanu. Při klinicky relevantních úrovních expozice byly u potkanů pozorovány zvýšené plazmatické hladiny IgG a IgA.
U potkanů nebyly pozorovány žádné účinky na fertilitu samců nebo samic. Studie na zvířatech prokázaly reprodukční toxicitu související s farmakologickým mechanismem působení rivaroxabanu (např. hemoragickými komplikacemi). V klinicky relevantních plazmatických koncentracích byla pozorována embryonální a fetální toxicita (post-implantační ztráta, opožděná nebo progredující osifikace, hepatální mnohočetné světle zbarvené skvrny) a zvýšený výskyt běžných malformací a také placentárních změn.
V prenatálních a postnatálních experimentech u potkanů byla zjištěna snížená životaschopnost potomků, a to v dávkách toxických pro matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Sodná sůl kroskarmelózy Monohydrát laktózy Hypromelóza Natrium-lauryl-sulfát Magnesium-stearát.
Potah tablety:
Makrogol 3350
Hypromelóza
Oxid titaničitý (E 171)
Červený oxid železitý (E 172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Blistry z PP/Al fólie, v krabičkách po 10, 14, 28 nebo 98 potahovaných tabletách nebo perforované jednodávkové blistry v krabičkách obsahující 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních obsahujících 100 potahovaných tablet (10 balení po 10 x 1 tabletě).
HDPE lahvičky obsahující 100 potahovaných tablet se šroubovacím PP uzávěrem.
Blistry z fólie Al/PP v pouzdru obsahující 42 potahovaných tablet přípravku Xarelto 15 mg a 7 potahovaných tablet přípravku Xarelto 20 mg (balení pro zahájení léčby).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/472/017-021, EU/1/08/472/024, EU/1/08/472/037, EU/1/08/472/039, EU/1/08/472/040
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. září 2008
Datum posledního prodloužení registrace: 22. května 2013
10. DATUM REVIZE TEXTU {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Balení pro zahájení léčby
'VTento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Xarelto 15 mg potahované tablety Xarelto 20 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna 15mg potahovaná tableta obsahuje rivaroxabanum 15 mg.
Jedna 20mg potahovaná tableta obsahuje rivaroxabanum 20 mg.
Pomocná látka se známým účinkem:
Jedna 15mg potahovaná tableta obsahuje 24,13 mg laktózy (jako monohydrátu), viz bod 4.4. Jedna 20mg potahovaná tableta obsahuje 21,76 mg laktózy (jako monohydrátu), viz bod 4.4.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
15mg potahovaná tableta: červené, kulaté, bikonvexní tablety (o průměru 6 mm, poloměru zakřivení 9 mm) označené logem (kříž) BAYER na jedné straně a číslem „15“ a trojúhelníkem na druhé straně.
20mg potahovaná tableta: hnědo-červené, kulaté, bikonvexní tablety (o průměru 6 mm, poloměru zakřivení 9 mm) označené logem (kříž) BAYER na jedné straně a číslem „20“ a trojúhelníkem na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba hluboké žilní trombózy (HŽT) a plicní embolie (PE) a prevence recidivující hluboké žilní trombózy a plicní embolie u dospělých (hemodynamicky nestabilní pacienti s PE viz bod 4.4).
4.2 Dávkování a způsob podání
Dávkování
Léčba hluboké žilní trombózy, léčba plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie
Doporučená dávka pro úvodní léčbu akutní hluboké žilní trombózy nebo plicní embolie je 15 mg dvakrát denně po dobu prvních tří týdnů a dále 20 mg jednou denně jako udržovací léčba a prevence hluboké žilní trombózy a plicní embolie podle tabulky uvedené níže.
|
Dávkování |
Maximální denní dávka | |
|
1. až 21. den |
15 mg dvakrát denně |
30 mg |
|
22. den a dále |
20 mg jednou denně |
20 mg |
Pro pacienty, kteří budou od 22. dne léčby přecházet z 15 mg dvakrát denně na 20 mg jednou denně (viz bod 6.5) je určeno 4týdenní balení pro zahájení léčby přípravku Xarelto.
Pro pacienty se středně závažnou nebo závažnou ledvinovou nedostatečností, u kterých bylo rozhodnuto, že budou od 22. dne užívat 15 mg jednou denně, jsou dostupná balení přípravku obsahující pouze 15mg potahované tablety (viz pokyny pro dávkování v bodu Speciální populace dále v textu).
Délka léčby by měla být stanovena individuálně po pečlivém zhodnocení přínosu léčby a rizika krvácení (viz bod 4.4). Krátkodobá léčba (nejméně 3 měsíce) by měla být indikována na základě přechodných rizikových faktorů (např. operace prodělaná v nedávné době, úraz, imobilizace) a dlouhodobá léčba by měla být indikována na základě trvalých rizikových faktorů nebo idiopatické HZT nebo PE.
Pokud dojde k vynechání dávky během té fáze léčby, kdy je přípravek podáván v dávce 15 mg dvakrát denně (1. - 21. den), měl by pacient užít přípravek Xarelto co nejdříve, aby se zajistilo dávkování 30 mg přípravku Xarelto denně. V tomto případě mohou být užity dvě 15 mg tablety najednou. Pacient by měl pokračovat s pravidelným užíváním dávky 15 mg dvakrát denně následující den podle doporučení.
Pokud dojde k vynechání dávky během té fáze léčby, kdy je přípravek podáván v jedné denní dávce (22. den a dále), měl by pacient užít přípravek Xarelto co nejdříve a pokračovat s užíváním jednou denně následující den podle doporučení. Dávka by neměla být pro nahrazení vynechané dávky ve stejný den zdvojnásobena.
Převod z antagonistů vitaminu K (VKA) na přípravek Xarelto
U pacientů léčených pro hlubokou žilní trombózu, plicní embolii a pro prevenci recidivy by měly být antagonisté vitaminu K vysazeny a léčba přípravkem Xarelto by měla být zahájena při hladině INR < 2,5.
Při převodu pacientů z antagonistů vitaminu K na přípravek Xarelto, budou po užití přípravku Xarelto hladiny INR falešně zvýšeny. Test INR není pro měření antikoagulační aktivity přípravku Xarelto validní a proto by neměl být používán (viz bod 4.5).
Převod z přípravku Xarelto na antagonisty vitaminu K (VKA)
Během přechodu z přípravku Xarelto na antagonisty vitaminu K existuje možnost neadekvátní antikoagulace. Během jakéhokoli převodu na alternativní antikoagulancia by měla být zajištěna kontinuální adekvátní antikoagulace. Je třeba uvést, že přípravek Xarelto může přispět ke zvýšení INR.
U pacientů, kteří jsou převáděni z přípravku Xarelto na antagonisty vitaminu K by měli být tito antagonisté podáváni současně, dokud není hladina INR > 2,0. Po dobu prvních dvou dnů fáze převodu by mělo být použito standardní úvodní dávkování antagonistů vitaminu K s následným dávkováním těchto antagonistů na základě testování INR. Během doby, kdy pacienti užívají jak přípravek Xarelto tak antagonisty vitaminu K by nemělo být prováděno testování INR dříve než 24 hodin po předchozí dávce, ale před další dávkou přípravku Xarelto. Jakmile je přípravek Xarelto vysazen, může být testování INR spolehlivě provedeno minimálně 24 hodin po poslední dávce (viz body 4.5 a 5.2).
Převod z parenterálních antikoagulancií přípravků na přípravek Xarelto
U pacientů, kteří dostávají parenterální antikoagulancia, přerušte podávání parenterálního antikoagulancia a začněte léčbu přípravkem Xarelto v rozmezí 0 až 2 hodiny před tím, než by mělo dojít k dalšímu plánovanému podání parenterálního přípravku (např. nízkomolekulární hepariny) nebo v době vysazení kontinuálně podávaného parenterálního přípravku (např. intravenózní nefrakciovaný heparin).
Převod z přípravku Xarelto na parenterální antikoagulancia
Podejte první dávku parenterálního antikoagulancia v době, kdy by měla být užita další dávka přípravku Xarelto.
Speciální populace
Ledvinová nedostatečnost
Omezené klinické údaje u nemocných s těžkou renální nedostatečností (clearance kreatininu 15 - 29 ml/min) signalizují, že u této populace pacientů jsou plazmatické koncentrace rivaroxabanu významně zvýšeny. Xarelto je proto u těchto pacientů nutno užívat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.4 a 5.2).
U pacientů se středně závažnou (clearance kreatininu 30 - 49 ml/min) nebo závažnou (clearance kreatininu 15-29 ml/min) renální nedostatečností platí následující doporučení pro dávkování:
- Pro léčbu hluboké žilní trombózy, léčbu plicní embolie a prevenci recidivující hluboké žilní trombózy a plicní embolie: pacienti by měli být léčeni dávkou 15 mg dvakrát denně po dobu prvních tří týdnů. Poté je doporučená dávka 20 mg jednou denně. Snížení dávky z 20 mg jednou denně na 15 mg jednou denně je třeba zvážit, pokud u pacienta riziko krvácení převáží riziko vzniku recidivující HZT a PE. Doporučení pro použití dávky 15 mg je založeno na farmakokinetickém modelu a nebylo v těchto klinických podmínkách studováno (viz body 4.4, 5.1 a 5.2).
Úprava dávky není nutná u pacientů s mírnou renální nedostatečností (clearance kreatininu 50 - 80 ml/min) (viz bod 5.2).
Jaterní nedostatečnost
Xarelto je kontraindikováno u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child Pugh B a C (viz body 4.3 a 5.2).
Starší populace
Dávky bez úprav (viz bod 5.2).
Tělesná hmotnost
Dávky bez úprav (viz bod 5.2).
Pohlaví
Dávky bez úprav (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Xarelto u dětí ve věku 0 až 18 let nebyla ještě stanovena. Nejsou dostupné žádné údaje. Podávání přípravku Xarelto dětem do 18 let se proto nedoporučuje.
Způsob podání Perorální podání.
Tablety se mají užívat s jídlem (viz bod 5.2).
Pacientům, kteří nejsou schopni polykat celé tablety, může být tableta přípravku Xarelto před užitím rozdrcena a smíchána s vodou nebo s jablečným pyré, a poté podána perorálně. Po podání rozdrcené potahované tablety Xarelto 15 mg nebo 20 mg musí být dávka okamžitě následována jídlem.
Rozdrcená tableta přípravku Xarelto může být také podána gastrickou sondou poté, co je potvrzeno správné umístění sondy v žaludku. Umístění sondy v žaludku by mělo být před podáním přípravku Xarelto potvrzeno. Rozdrcená tableta by měla být podána žaludeční sondou v malém množství vody a sonda by poté měla být propláchnuta vodou. Po podání rozdrcené potahované tablety Xarelto 15 mg nebo 20 mg musí být poté dávka okamžitě následována enterální výživou (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku. Aktivní klinicky významné krvácení.
Léze nebo stavy, které jsou považovány za významné riziko závažného krvácení. Mohou mezi ně patřit současné nebo nedávno prodělané ulcerace gastrointestinálního traktu, přítomnost maligních nádorů s vysokým rizikem krvácení, nedávno prodělané poranění mozku nebo míchy, operace mozku, míchy nebo oka v nedávné době, intrakraniální krvácení v nedávné době, jícnové varixy nebo podezření na ně, arteriovenózní malformace, cévní aneurysma nebo významné cévní abnormality v míše nebo mozku.
Souběžná léčba jinými antikoagulačními přípravky, např. nefrakcionovaným heparinem (UFH), nízkomolekulámími hepariny (enoxaparin, dalteparin atd), heparinovými deriváty (fondaparinux atd), orálními antikoagulancii (warfarin, dabigatran etexilát, apixaban, atd.), se nedoporučuje s výjimkou specifické situace, kdy je pacient převáděn z antikoagulační léčby (viz bod 4.2), nebo když je podáván UFH v dávkách nezbytných pro udržení průchodnosti centrálního žilního nebo arteriálního katetru (viz bod 4.5).
Jaterní onemocnění, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně pacientů s cirhózou s klasifikací Child Pugh B a C (viz bod 5.2).
Těhotenství a kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
V průběhu léčby se doporučuje pacienta klinicky sledovat v souladu s praxí běžnou při podávání antikoagulační léčby.
Riziko krvácení
Jako v případě jiných antikoagulancií, u pacientů užívajících přípravek Xarelto mají být pečlivě sledovány známky krvácení. Doporučuje se opatrnost při použití přípravku v případě zvýšeného rizika krvácení. Pokud se objeví závažné krvácení, podávání přípravku Xarelto je třeba přerušit.
V klinických studiích bylo během dlouhodobé léčby rivaroxabanem ve srovnání s léčbou VKA častěji pozorováno slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Proto, kromě adekvátního klinického sledování, pokud je shledáno vhodným, může být přínosem pro detekci okultního krvácení laboratorní vyšetření hemoglobinu/hematokritu.
U několika podskupin pacientů (podrobně uvedených dále) hrozí zvýšené riziko krvácení. Tyto pacienty je třeba pečlivě sledovat, zda se po zahájení léčby neobjeví známky a příznaky krvácivých komplikací a anémie (viz bod 4.8).
Při jakémkoli nevysvětlitelném poklesu hladin hemoglobinu nebo krevního tlaku je třeba hledat místo krvácení.
Přestože léčba rivaroxabanem nevyžaduje rutinní monitorování expozice, hladiny rivaroxabanu měřené kalibrovanou kvantitativní analýzou anti-faktoru Xa mohou být užitečné ve výjimečných situacích, kdy znalost expozice rivaroxabanu může pomoci při klinických rozhodováních, např. při předávkování nebo při urgentních chirurgických zákrocích (viz body 5.1 a 5.2).
Ledvinová nedostatečnost
U pacientů s těžkou ledvinovou nedostatečností (clearance kreatininu < 30 ml/min) mohou být plazmatické hladiny rivaroxabanu významně zvýšeny (1,6 násobek průměru), což může vést ke zvýšenému riziku krvácení. Xarelto je u pacientů s clearance kreatininu 15 - 29 ml/min nutno používat s opatrností. Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min (viz body 4.2 a 5.2).
Xarelto musí být používáno s opatrností u pacientů s renálním poškozením, kteří současně užívají jiné léčivé přípravky, které zvyšují koncentraci rivaroxabanu v plazmě (viz bod 4.5).
Interakce s jinými léčivými přípravky
Použití přípravku Xarelto se nedoporučuje u pacientů současně léčených systémovými azolovými antimykotiky (jako jsou ketokonazol, itrakonazol, vorikonazol a posakonazol) nebo inhibitory proteáz HIV (například ritonavir). Tyto léčivé látky jsou silnými inhibitory současně obou systémů CYP3A4 a P-gp, a proto mohou zvyšovat plazmatické koncentrace rivaroxabanu v klinicky významném rozsahu (v průměru 2,6 násobek), což může vést ke zvýšenému riziku krvácení (viz bod 4.5).
Postupujte opatrně, pokud jsou pacienti současně léčeni léčivými přípravky ovlivňujícími krevní srážlivost, jako jsou například nesteroidní antirevmatika (NSAID), kyselina acetylsalicylová a inhibitory agregace trombocytů. U pacientů s rizikem vředové gastrointestinální choroby lze zvážit vhodnou profylaktickou léčbu (viz bod 4.5).
Jiné rizikové faktory krvácení
Podobně jako v případě jiných antitrombotik, se použití rivaroxabanu nedoporučuje u pacientů se zvýšeným rizikem krvácení, například:
• vrozené nebo získané krvácivé poruchy
• léčbou neupravená těžká arteriální hypertenze
• jiné gastrointestinální onemocnění bez aktivní ulcerace, které může potenciálně vést ke krvácivým komplikacím (např. zánětlivé střevní onemocnění, esofagitida, gastritida a gastroesofageální refluxní choroba)
• cévní retinopatie
• bronchiektázie nebo plicní krvácní v anamnéze
Hemodynamicky nestabilní pacienti s plicní embolií nebo pacienti, kteří vyžadují trombolýzu nebo plicní embolektomii
Přípravek Xarelto se nedoporučuje používat jako alternativní léčbu k nefrakcionovanému heparinu u pacientů s plicní embolií, kteří jsou hemodynamicky nestabilní nebo kteří mohou podstoupit trombolýzu nebo plicní embolektomii, protože bezpečnost a účinnost přípravku Xarelto nebyla pro tyto klinické situace stanovena.
Spinální / epidurální anestezie nebo punkce
Pokud je u pacienta provedena anestezie (spinální či epidurální anestezie) nebo spinální resp. epidurální punkce, u pacientů preventivně léčených antitrombotiky pro prevenci tromboembolických komplikací hrozí riziko vývinu epidurálního či spinálního hematomu, který může vyústit v dlouhodobou nebo trvalou paralýzu. Riziko těchto příhod může dále zvýšit epidurální katetr dlouhodobě zavedený po operaci, nebo současné použití léčivých přípravků ovlivňujících krevní srážlivost. Riziko může také zvýšit provedení traumatické nebo opakované epidurální či spinální punkce. Pacienty je třeba často monitorovat, zda nejeví známky a příznaky neurologického poškození (například necitlivost nebo slabost dolních končetin, dysfunkce střev nebo močového měchýře). Pokud se zjistí neurologické potíže, je nutno urgentně stanovit diagnózu a zajistit léčbu. Před neuroaxiální intervencí lékař zváží potenciální přínos a riziko u pacientů na antikoagulační terapii i u pacientů, kde hodlá antikoagulační léčbu podat v rámci tromboprofylaxe.
S použitím 15 mg nebo 20 mg rivaroxabanu v těchto situacích nejsou klinické zkušenosti.
Ke snížení možného rizika krvácení během současného užívání rivaroxabanu při neuroaxiální (spinální nebo epidurální) anestezii nebo spinální punkci se bere v úvahu farmakokinetický profil rivaroxabanu. Zavedení nebo odstranění epidurálního katetru nebo lumbální punkci je nejlépe provést, když je odhadovaný antikoagulační účinek rivaroxabanu nízký (viz bod 5.2). Přesný čas, kdy je u každého pacienta antikoagulační účinek dostatečně nízký, však není znám.
Odstranění epidurálního katetru by mělo být na základě farmakokinetických vlastností nejméně za dobu představující 2x poločas, to je nejméně 18 hodin u mladých pacientů a 26 hodin u starších pacientů po posledním podání rivaroxabanu (viz bod 5.2).
Další dávka rivaroxabanu se nepodává dříve než 6 hodin po vyjmutí katetru.
Pokud dojde k traumatické punkci, podávání rivaroxabanu se odloží o 24 hodin.
Doporučení pro dávkování před a po invazivních procedurách a chirurgickém výkonu
Pokud je nutná invazivní procedura nebo chirurgický zákrok, měl by být přípravek Xarelto 15 mg / Xarelto
20 mg vysazen minimálně 24 hodin před zákrokem, pokud je to podle posouzení lékaře možné.
Pokud není možné výkon odložit, je třeba posoudit zvýšené riziko krvácení s ohledem na neodkladnost zákroku.
Léčba přípravkem Xarelto má být znovu zahájena po invazivní proceduře nebo chirurgickém zákroku co nejdříve, pokud to situace umožní a pokud je podle úsudku ošetřujícího lékaře dosaženo odpovídající hemostázy (viz bod 5.2).
Starší populace
Se zvyšujícím se věkem se může zvyšovat riziko krvácení (viz bod 5.2).
Informace o pomocných látkách
Xarelto obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a P-gp
Současné podávání rivaroxabanu s ketokonazolem (400 mg jednou denně) nebo ritonavirem (600 mg dvakrát denně) vedlo k 2,6 resp. 2,5 násobnému nárůstu střední hodnoty AUC rivaroxabanu a
1,7 resp. 1,6 násobnému nárůstu jeho střední hodnoty Cmax, s významným zesílením farmakodynamických účinků, což může vést ke zvýšenému riziku krvácení. Proto se použití přípravku Xarelto nedoporučuje u pacientů užívajících současně a systémově azolová antimykotika, jako je ketokonazol, itrakonazol, vorikonazol a posakonazol, nebo inhibitory proteáz HIV. Tyto léčivé látky jsou silnými inhibitory obou systémů CYP3A4 a současně P-gp (viz bod 4.4).
Léčivé látky silně inhibující pouze jednu z metabolických cest eliminace rivaroxabanu (buď CYP3A4, nebo P-gp) podle všeho zvyšují plazmatické koncentrace rivaroxabanu méně. Například klaritromycin (500 mg dvakrát denně), který je považován za silného inhibitora CYP3A4 a středně silného inhibitora P-gp, způsobuje 1,5 násobný nárůst středních hodnot AUC rivaroxabanu a 1,4 násobný nárůst Cmax. Tento nárůst není považován za klinicky relevantní. (Pacienti s poruchou funkce ledvin: viz bod 4.4).
Erythromycin (500 mg třikrát denně), který středně silně inhibuje 3A4 a P-gp, způsobuje 1,3násobný nárůst středních hodnot AUC a Cmax rivaroxabanu. Tento nárůst není považován za klinicky relevantní.
U pacientů s mírnou renální insuficiencí vedlo podávání erythromycinu (500 mg třikrát denně) k 1,8 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu Cmax ve srovnání s pacienty s normální renální funkcí. U pacientů se středně těžkým renálním poškozením vedl erythromycin k 2,0 násobnému nárůstu střední hodnoty AUC rivaroxabanu a 1,6 násobnému nárůstu v Cmax ve srovnání s pacienty s normální renální funkcí. Účinek erythromycinu je aditivní k renálnímu poškození (viz bod 4.4).
Flukonazol (400 mg jednou denně), který je považován za středně silný inhibitor CYP3A4, vedl k
1,4 násobnému zvýšení průměrné AUC rivaroxabanu a k 1,3 násobnému zvýšení průměrné Cmax. Toto zvýšení není považováno za klinicky významné. (Pacienti se sníženou funkcí ledvin: viz bod 4.4).
Dronedaron by neměl být podáván spolu s rivaroxabanem, vzhledem k omezeným klinickým údajům, které jsou k dispozici.
Antikoagulační přípravky
Po kombinovaném podávání enoxaparinu (40 mg, jednorázová dávka) s rivaroxabanem (10 mg, jednorázová dávka) byl zjištěn aditivní vliv na inhibici faktoru Xa, a to bez dalších účinků na výsledky testů srážení krve (PT, apTT). Enoxaparin neovlivňoval farmakokinetiku rivaroxabanu.
Vzhledem ke zvýšenému riziku krvácení je třeba postupovat opatrně, pokud jsou pacienti současně léčeni jinými antikoagulačními přípravky (viz body 4.3 a 4.4).
NSAID / inhibitory srážení trombocytů
Při současném podávání rivaroxabanu (15 mg) a 500 mg naproxenu nebylo zjištěno klinicky relevantní prodloužení doby krvácení. Některé osoby však mohou mít silnější farmakodynamickou odezvu.
Žádné klinicky významné farmakokinetické ani farmakodynamické interakce nebyly zjištěny při současném podání rivaroxabanu s 500 mg kyseliny acetylsalicylové.
Clopidogrel (úvodní dávka 300 mg, poté udržovací dávka 75 mg) nevykazoval farmakokinetické interakce s rivaroxabanem (15 mg), ale u části populace pacientů došlo k relevantnímu nárůstu doby krvácení, který nekoreloval s agregací trombocytů, ani hladinami P-selektinu nebo receptoru GPIIb/IIIa.
Postupovat opatrně je třeba, pokud jsou pacienti současně léčeni NSAID (včetně kyseliny acetylsalicylové) a inhibitory agregace trombocytů, protože tyto léčivé přípravky obvykle zvyšují riziko krvácení (viz bod 4.4).
Warfarin
Konverze pacientů z antagonisty vitaminu K warfarinu (INR 2,0 až 3,0) na rivaroxaban (20 mg) nebo z rivaroxabanu (20 mg) na warfarin (INR 2,0 až 3,0) vedla ke zvýšení protrombinového času/INR (Neoplastin) více než aditivně (mohou být pozorovány jednotlivé hladiny INR až 12), zatímco účinky na aPTT, inhibici aktivity faktoru Xa a potenciál endogenního trombinu byly aditivní.
Pokud je třeba testovat farmakodynamické účinky rivaroxabanu během fáze konverze, mohou být použity anti Xa aktivita, PiCT a Heptest, protože tyto testy nebyly ovlivněny warfarinem. Čtvrtý den po poslední dávce warfarinu odráží všechny testy (včetně PT, aPTT, inhibice aktivity faktoru Xa a ETP) pouze účinek rivaroxabanu.
Pokud je třeba testovat farmakodynamické účinky warfarinu během fáze převodu, může být použito měření INR při Cmin rivaroxabanu (24 hodin po předchozím užití rivaroxabanu), protože tento test je v tento okamžik minimálně ovlivněný rivaroxabanem.
Mezi warfarinem a rivaroxabanem nebyla pozorována žádná farmakokinetická interakce.
Induktory CYP3A4
Současné podávání rivaroxabanu se silným induktorem CYP3A4 rifampicinem vedlo k přibližně 50% poklesu střední hodnoty AUC rivaroxabanu, s odpovídajícím poklesem farmakodynamického účinku. Současné použití rivaroxabanu s jinými silnými induktory CYP3A4 (například fenytoinem, karbamazepinem, fenobarbitalem nebo třezalkou (Hypericum perforatum)) může také vést ke snížení plazmatických koncentrací rivaroxabanu. Proto je třeba se vyhnou současnému podávání silných induktorů CYP3A4, pokud není pacient pozorně sledován kvůli známkám a příznakům trombózy.
Jiné současně podávané léky
Žádné klinicky významné farmakokinetické nebo farmakodynamické interakce nebyly zjištěny při současném podávání rivaroxabanu s midazolamem (substrát CYP3A4), digoxinem (substrát P-gp), atorvastatinem (substrát CYP3A4 a P-gp) nebo omeprazolem (inhibitor protonové pumpy). Rivaroxaban neinhibuje ani neindukuje významné izoformy CYP jako je CYP3A4.
Laboratorní parametry
Parametry srážení krve (například PT, aPTT, Heptest) j sou ovlivněny podle očekávání na základě mechanismu působení rivaroxabanu (viz bod 5.1).
4.6 Fertilita, těhotenství a kojení
Bezpečnost a účinnost přípravku Xarelto nebyly u těhotných žen stanoveny. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Vzhledem k možné reprodukční toxicitě, známému riziku krvácení a důkazu, že rivaroxaban prochází placentou, je přípravek Xarelto kontraindikován v těhotenství (viz bod 4.3). Ženy ve fertilním věku musí během léčby rivaroxabanem zabránit otěhotnění.
Kojení
Bezpečnost a účinnost přípravku Xarelto nebyly u kojících žen stanoveny. Údaje z experimentů na zvířatech signalizují, že je rivaroxaban vylučován do mléka. Podávání přípravku Xarelto je během kojení kontraindikováno (viz bod 4.3). Je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit léčbu.
Fertilita
Nebyly provedeny žádné specifické studie užívání rivaroxabanu u lidí s cílem vyhodnotit účinky na fertilitu. Ve studii samčí a samičí fertility na potkanech nebyly pozorovány žádné účinky (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Xarelto má malý vliv na schopnost řídit a obsluhovat stroje. Byly hlášeny nežádoucí účinky jako synkopa (frekvence výskytu: méně časté) a závrať (frekvence výskytu: časté) (viz bod 4.8). Pacienti, kteří zaznamenali tyto nežádoucí účinky, by neměli řídit vozidla a obsluhovat stroje.
Souhrn bezpečnostních informací
Bezpečnost rivaroxabanu byla hodnocena v jedenácti studiích fáze III u 32 625 pacientů, kteří byli vystaveni rivaroxabanu (viz tabulka 1).
Tabulka 1: Počet hodnocených pacientů, maximální ^ denní dávka a délka léčby ve studiích fáze II
|
Indikace |
Počet pacientů* |
Maximální denní dávka |
Maximální délka léčby |
|
Prevence žilního tromboembolismu u dospělých pacientů podstupujících elektivní operativní náhradu kyčelního nebo kolenního kloubu |
6 097 |
10 mg |
39 dnů |
|
Prevence žilního tromboembolismu u hospitalizovaných nechirurgických pacientů |
3 997 |
10 mg |
39 dnů |
|
Léčba hluboké žilní trombózy a plicní embolie a prevence jejich recidivy |
4 556 |
Den 1-21: 30 mg Den 22 a dále: 20 mg |
21 měsíců |
|
Prevence cévní mozkové příhody a systémové embolizace u pacientů s nevalvulární fibrilací síní |
7 750 |
20 mg |
41 měsíců |
|
Prevence aterotrombotických příhod u pacientů po AKS |
10 225 |
5 mg nebo 10 mg, podávaných společně s ASA nebo s kombinací ASA plus klopidogrel či tiklopidin |
31 měsíců |
*Pacienti exponovaní minimálně jedné dávce rivaroxabanu
Nejčastěji hlášenými nežádoucí účinky u pacientů, kteří dostávali rivaroxaban, bylo krvácení (viz bod 4.4 a níže uvedený „Popis vybraných nežádoucích účinků“). Nejčastěji hlášeným krvácením (>4 %) byla epistaxe (5,9 %) a gastrointestinální krvácení (4,2 %).
Celkem asi u 67% pacientů exponovaných minimálně jedné dávce rivaroxabanu byl hlášen výskyt nežádoucích příhod. Asi u 22% pacientů se vyskytly nežádoucí příhody, které byly považovány za související s léčbou podle posouzení zkoušejícími. U pacientů léčených přípravkem Xarelto v dávce 10 mg podstupujících chirurgickou náhradu kyčelního nebo kolenního kloubu a u nechirurgických pacientů hospitalizovaných pro jiné onemocnění se krvácivé příhody vyskytly asi u 6,8% a 12,6% pacientů a anémie se objevila asi u 5,9% a 2,1% pacientů. U pacientů léčených přípravkem Xarelto buď v dávce 15 mg dvakrát denně s následným podáváním 20 mg jednou denně pro léčbu hluboké žilní trombózy nebo plicní embolie nebo dávkou 20 mg jednou denně z důvodu prevence recidivující hluboké žilní trombózy a plicní embolie se krvácivé příhody vyskytly asi u 27,8% pacientů a anémie se objevila asi u 2,2% pacientů. U pacientů léčených z důvodu prevence cévní mozkové příhody a systémové embolizace bylo krvácení jakéhokoli typu a závažnosti hlášeno s frekvencí 28 na 100 paciento-roků a anémie s frekvencí 2,5 na 100 paciento-roků. U pacientů léčených z důvodu prevence kardiovaskulárního úmrtí a infarktu myokardu po akutním koronárním syndromu (ACS), bylo krvácení jakéhokoli typu a závažnosti hlášeno s frekvencí 22 na 100 paciento-roků. Anémie byla hlášena s frekvencí 1,4 na 100 paciento-roků.
Seznam nežádoucích účinků uvedený v tabulce
Výskyt nežádoucích účinků hlášený u přípravku Xarelto je shrnutý v tabulce 2 níže podle orgánové klasifikace (v MedDRA) a podle frekvence výskytu.
Četnosti j sou definovány takto:
velmi časté (> 1/10)
časté (> 1/100 až < 1/10)
méně časté (> 1/1 000 až < 1/100)
vzácné (> 1/10 000 až < 1/1 000)
velmi vzácné ( < 1/10,000)
není známo (z dostupných údajů nelze určit)
Tabulka 2: Všechny s léčbou související nežádoucí účinky hlášené u pacientů ve studiích fáze III
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (včetně příslušných laboratorních parametrů) |
Trombocytémie (včetně zvýšeného počtu trombocytů)A | ||
|
Poruchy imunitního systému | |||
|
Alergická reakce, alergická dermatitida | |||
|
Poruchy nervového systému | |||
|
Závratě, bolesti hlavy |
Cerebrální a intrakraniální krvácení, synkopa | ||
|
Poruchy oka | |||
|
Oční krvácení (včetně krvácení do spojivek) | |||
|
Srdeční poruchy | |||
|
Cévní poruchy | |||
|
Hypotenze, hematom | |||
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe, hemoptýza | |||
|
Gastrointestinální poruchy | |||
|
Krvácení z dásní, krvácení z gastrointestinálního traktu (včetně rektálního krvácení), gastrointestinální a abdominální bolest, dyspepsie, nausea, A |
Sucho v ústech | ||
|
Poruchy jater a žlučových cest | |||
|
Abnormity jaterní funkce |
Žloutenka | ||
|
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy kůže a podkožní tkáně | |||
|
Pruritus (včetně méně častých případů generalizovaného pruritu), vyrážka, ekchymóza, kožní a podkožní krvácení |
Kopřivka | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |||
|
Bolest v končetináchA |
Hemartróza |
Krvácení do svalů |
Kompartment syndrom sekundárně po krvácení |
|
Poruchy ledvin a močových cest | |||
|
Urogenitální krvácení (včetně hematurie a menorhagieB), poškození ledvin (včetně zvýšení hladin kreatininu a močoviny v krvi)A |
Renální selhání/akutní renální selhání vzniklé sekundárně po krvácení natolik silném, aby způsobilo hypoperfúzi | ||
|
Celkové poruchy a reakce v místě aplikace | |||
|
HorečkaA, periferní edém, pokles celkové síly a energie (včetně únavy a tělesné slabosti) |
Pocit indispozice (včetně malátnosti) |
Lokalizovaný edémA | |
|
Vyšetření | |||
|
Zvýšená hladina transamináz |
Zvýšená hladina bilirubinu, alkalické fosfatázyA, LDHA, lipázyA, amylázyA, GMTA |
Zvýšení konjugovaného bilirubinu (s přidruženým zvýšením ALT nebo bez zvýšení ALT) | |
|
Poranění, otravy a procedurální komplikace | |||
|
Pooperační krvácení (včetně pooperační anémie a krvácení z rány), kontuze, sekrece z ranA |
Cévní pseudoaneurysmaC | ||
A: pozorováno u prevence žilního tromboembolismu u dospělých pacientů, kteří podstoupili chirurgickou náhradu kyčelního nebo kolenního kloubu
B: pozorováno u léčby hluboké žilní trombózy, plicní embolie a u prevence recidivy jako velmi časté u žen do 55 let
C: pozorováno méně často u prevence aterotrombotických příhod u pacientů po akutním koronárním syndromu (po perkutánní koronární intervenci)
Popis vybraných nežádoucích účinků
Vzhledem k farmakologickému mechanismu působení může být užívání přípravku Xarelto spojeno se zvýšeným rizikem okultního nebo zjevného krvácení z jakékoli tkáně nebo orgánu s možným následkem posthemoragické anémie. Známky, příznaky a závažnost (včetně možného fatálního zakončení) se mohou různit podle místa a stupně nebo rozsahu krvácení a/nebo anémie (viz bod 4.9 Léčba krvácení). V klinických studiích bylo během dlouhodobé léčby rivaroxabanem ve srovnání s léčbou VKA častěji pozorováno slizniční krvácení (tj. epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Proto, kromě adekvátního klinického sledování, pokud je shledáno vhodným, může laboratorní vyšetření hemoglobinu/hematokritu být přínosem pro detekci okultního krvácení. Riziko krvácení bude možná zvýšeno u některých skupin pacientů, například osob s těžkou arteriální hypertenzí neupravenou léčbou, a/nebo souběžnou léčbou ovlivňující krevní srážlivost (viz Riziko krvácení v bodu 4.4). Menstruační krvácení může být zesíleno a/nebo prodlouženo. Hemoragické komplikace se mohou projevovat jako celková slabost, bledost, závratě, bolesti hlavy nebo nevysvětlitelné otoky, dušnost a nevysvětlitelný šok. V některých případech byly v důsledku anémie pozorovány příznaky srdeční ischémie, jako je například bolest na hrudníku nebo angina pectoris.
V souvislosti s užíváním přípravku Xarelto byly hlášeny známé sekundární komplikace závažného krvácení, jako je například kompartment syndrom a renální selhání v důsledku hypoperfúze. Možnost krvácení je proto třeba zvážit při posuzování stavu pacientů s jakoukoli antikoagulační léčbou.
Post-marketingové sledování
V postmarketingovém sledování byly v časové souvislosti s užitím přípravku Xarelto hlášeny následující nežádoucí účinky. Frekvenci těchto nežádoucích účinků hlášených po uvedení na trh nelze určit.
Poruchy imunitního systému: angioedém a alergický edém (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Poruchy jater a žlučových cest: cholestáza, hepatitida (včetně hepatocelulárního poškození) (v souhrnných studiích fáze III byly tyto příhody vzácné (>1/10 000 až <1/1000)).
Poruchy krve a lymfatického systému: trombocytopenie (v souhrnných studiích fáze III byly tyto příhody méně časté (>1/1 000 až <1/100)).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly hlášeny vzácné případy předávkování dávkou až 600 mg bez krvácivých komplikací nebo jiných nežádoucích reakcí. Z důvodu omezené absorpce se očekává strop účinku bez dalšího zvýšení průměrné plazmatické hladiny v případě vyšší než terapeutické dávky 50 mg rivaroxabanu nebo dávek vyšších. Specifické antidotum blokující farmakodynamický účinek rivaroxabanu není k dispozici.
Lze zvážit podání aktivního uhlí ke snížení absorpce v případě předávkování rivaroxabanem.
Léčba krvácení
Pokud dojde ke krvácivým komplikacím u pacienta léčeného rivaroxabanem, musí se podání další dávky rivaroxabanu odložit nebo se léčba musí ukončit, dle potřeby. Rivaroxaban má biologický poločas asi 5 až 13 hodin (viz bod 5.2). Léčba by měla být individuální podle závažnosti a lokalizace krvácení. Podle potřeby je třeba použít vhodnou symptomatickou léčbu, jako je mechanická komprese (např. u závažné epistaxe), chirurgická hemostáza se zajištěním kontroly krvácení, náhradou tekutin a zajištěním hemodynamické podpory, krevní deriváty (erytrocyty nebo čerstvá zmrazená plasma, v závislosti na související anémii nebo koagulopatii) nebo trombocyty.
Pokud krvácení nelze kontrolovat výše uvedenými opatřeními, lze zvážit podávání specifické prokoagulační reverzní látky, jako je koncentrát protrombinového komplexu (PCC), aktivovaný koncentrát protrombinového komplexu (APCC) nebo rekombinantní faktor VIIa (r-FVIIa). V současnosti jsou však k dispozici velmi omezené klinické zkušenosti s použitím těchto přípravků u osob užívajících rivaroxaban. Doporučení je též podloženo omezenými neklinickými údaji. Opakované podání rekombinantního faktoru VIIa je třeba zvážit a titrovat v závislosti na zlepšování krvácení. V případě závažného krvácení je třeba konzultovat odborníka na koagulaci, pokud je odborník v místě dostupný (viz bod 5.1).
Protamin sulfát a vitamin K podle všeho nebudou ovlivňovat antikoagulační aktivitu rivaroxabanu. U osob užívajících rivaroxaban jsou omezené zkušenosti s použítím kyseliny tranexamové a neexistují zkušenosti s použitím kyseliny aminokaproové a aprotininu. Neexistují ani vědecké důvody přínosu ani zkušenosti s použitím systémového hemostatika desmopressinu u osob užívajících rivaroxaban. Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: přímé inhibitory faktoru Xa, ATC kód: B01AF01 Mechanismus účinku
Rivaroxaban je vysoce selektivní přímý inhibitor faktoru Xa biologicky dostupný při perorálním podání. Inhibice faktoru Xa blokuje vnitřní a vnější cesty koagulační kaskády, a inhibuje vznik trombinu i vytváření trombů. Rivaroxaban neinhibuje trombin (aktivovaný faktor II) a nebyly prokázány žádné účinky na trombocyty.
Farmakodynamické účinky
U lidí byla zjištěna inhibice faktoru Xa přímo úměrná dávce. Protrombinový čas (PT) je rivaroxabanem ovlivňován úměrně dávce, objevuje se vysoká korelace s plazmatickými koncentracemi (hodnota R je 0,98), pokud je při testu jako assay použit Neoplastin. Jiné reagenty mohou přinést jiné výsledky. Hodnotu PT je nutno stanovit v sekundách, protože INR (mezinárodní normalizovaný poměr) je kalibrován a validován pouze pro kumariny, a nelze jej využívat pro jiné antikoagulanty.
U pacientů užívajících rivaroxaban v léčbě hluboké žilní trombózy a plicní embolie a k prevenci jejich recidivy se v 5/95 percentilu hodnoty PT (Neoplastin) za 2 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 17 až 32 s pro dávku 15 mg rivaroxabanu dvakrát denně a od 15 do 30 s pro dávku 20 mg rivaroxabanu jednou denně. Nejnižší hodnoty se v 5/95 percentilu pohybovaly od 14 do 24 s pro dávku 15 mg dvakrát denně (8 - 16 hodin po požití) a od 13 do 20 s pro dávku 20 mg jednou denně (18 - 30 hodin po požití). U pacientů s nevalvulární fibrilací síní užívajících rivaroxaban v prevenci cévní mozkové příhody a systémové embolizace se v 5/95 percentilu hodnoty PT (Neoplastin) za 1 - 4 hodiny po užití tablety (tedy v době maximálního účinku) pohybovaly v rozsahu 14 až 40 s pro dávku 20 mg rivaroxabanu jednou denně a od 10 do 50 s u pacientů se středně závažným poškozením renálních funkcí léčených dávkou 15 mg jednou denně. Nejnižší hodnoty (16 - 36 hodin po požití) se v 5/95 percentilu pohybovaly od 12 do 26 s pro dávku 20 mg jednou denně a u pacientů se středně závažným poškozením renálních funkcí léčených 15 mg jednou denně se hodnoty pohybovaly od 12 do 26 s.
V klinické farmakologické studii sledující reverzi farmakodynamického účinku rivaroxabanu u zdravých dospělých osob (n=22) byl hodnocen účinek jednotlivé dávky (50 IU/kg) u dvou rozdílných typů PCC, 3-faktorového PCC (faktory II, IX a X) a 4-faktorového PCC (II, VII, IX a X). 3-faktorový PCC redukoval průměrnou hodnotu PT času (protrombinového času), hodnocenou neoplastinem, přibližně o 1,0 sekundy během 30 minut, ve srovnání s přibližně 3,5 sekundami pozorovanými u 4-faktorového PCC. Naproti tomu, 3-faktorový PCC měl větší a rychlejší celkový efekt na reverzní změny generace endogenního trombinu než 4-faktorový PCC (viz bod 4.9).
Aktivovaný parciální tromboplastinový čas (aPTT) a hodnoty analýzy HepTest jsou také prodlouženy úměrně dávce; nedoporučuje se však tyto metody používat k hodnocení farmakodynamických účinků rivaroxabanu. Během léčby rivaroxabanem v běžné klinické praxi není třeba monitorovat parametry koagulace. Pokud však je klinicky indikováno, lze hladiny rivaroxabanu měřit pomocí kalibrovaných kvantitativních testů anti-faktoru Xa (viz bod 5.2).
Klinická účinnost a bezpečnost
Léčba hluboké žilní trombózy, plicní embolie a prevence recidivující hluboké žilní trombózy a plicní embolie Klinický program přípravku Xarelto byl navržen tak, aby prokázal účinnost přípravku Xarelto v úvodní a pokračující léčbě hluboké žilní trombózy a plicní embolie a prevenci jejich recidivy.
Více než 9 400 pacientů bylo hodnoceno ve třech randomizovaných kontrolovaných studiích fáze III (Einstein DVT, Einstein PE a Einstein Extension) a poté byla provedena predefinovaná poolovaná analýza studií Einstein DVT a Einstein PE. Celková kombinovaná délka léčby ve všech studiích byla až 21 měsíců.
Ve studii Einstein DVT bylo hodnoceno 3 449 pacientů s akutní hlubokou žilní trombózou v léčbě hluboké žilní trombózy a prevenci recidivující hluboké žilní trombózy a plicní embolie (pacienti, kteří měli symptomatickou plicní embolii, byli z této studie vyřazeni). Délka léčby byla 3, 6 nebo 12 měsíců v závislosti na klinickém posouzení zkoušejícím.
V úvodní 3-týdenní léčbě akutní hluboké žilní trombózy byl podáván rivaroxaban v dávce 15 mg dvakrát denně. Poté následovalo podávání dávky 20 mg rivaroxabanu jednou denně.
Ve studii Einstein PE bylo hodnoceno 4 832 pacientů s akutní plicní embolií v léčbě plicní embolie a v prevenci recidivující hluboké žilní trombózy a plicní embolie. Délka léčby byla 3, 6 nebo 12 měsíců v závislosti na klinickém posouzení zkoušejícím.
V úvodní léčbě akutní PE bylo podáváno 15 mg rivaroxabanu dvakrát denně 3 týdny. Poté následovalo podávání dávky 20 mg rivaroxabanu jednou denně.
V obou studiích Einstein DVT a Einstein PE zahrnoval srovnávaný léčebný režim enoxaparin podávaný minimálně 5 dnů v kombinaci s antagonisty vitaminu K do dosažení terapeutického rozmezí PT/INR (> 2,0). Léčba pokračovala antagonistou vitaminu K, jehož dávka byla upravena pro udržení hodnot PT/INR v terapeutickém rozmezí 2,0 až 3,0.
Ve studii Einstein Extension bylo hodnoceno 1 197 pacientů s hlubokou žilní trombózou nebo plicní embolií v prevenci recidivující hluboké žilní trombózy a plicní embolie. Trvání léčby bylo dalších 6 nebo 12 měsíců u pacientů, kteří dokončili 6 až 12 měsíců léčby pro žilní tromboembolismus v závislosti na klinickém posouzení zkoušejícím. Přípravek Xarelto 20 mg jednou denně byl srovnáván s placebem.
Všechny studie fáze III využívaly stejné předem definované primární a sekundární parametry účinnosti. Primární parametr účinnosti byl symptomatický recidivující žilní tromboembolismus definovaný jako kompozit recidivující hluboké žilní trombózy nebo fatální či nefatální plicní embolie. Sekundární parametr účinnosti byl definovaný jako kompozit recidivující hluboké žilní trombózy, nefatální plicní embolie a mortality ze všech příčin.
Ve studii Einstein DVT (viz tabulka 3) prokázal rivaroxaban non-inferioritu proti enoxaparinu/antagonistům vitaminu K v primárním parametru účinnosti (p < 0,0001 (test non-inferiority); poměr rizik: 0,680 (0,443 - 1,042), p = 0,076 (test superiority)). Předem definovaný čistý klinický přínos (primární parametr účinnosti plus závažná krvácivá příhoda) byl hlášen s poměrem rizik 0,67 ((95% CI: 0,47- 0,95), s nominální hodnotou p = 0,027) ve prospěch rivaroxabanu. Hodnoty INR byly uvnitř terapeutického rozmezí s průměrem 60,3% pro průměrnou dobu léčby 189 dní a 55,4%, 60,1% a 62,8% doby pro skupiny s plánovanou léčbou 3, 6 a 12 měsíců. Ve skupině enoxaparin/VKA nebyl jasný vztah mezi hladinou TTR v centru (doba v cílovém INR rozmezí 2,0 -3,0) ve stejně velkých tertilech a incidencí recidivující HZT (P=0,932 pro interakci). V centrech v nejvyšším tertilu bylo HR rivaroxaban versus warfarin 0,69 (95% CI: 0,35 - 1,35).
Výskyt primárního bezpečnostního ukazatele (závažné nebo klinicky významné méně závažné krvácivé příhody) stejně jako sekundárního bezpečnostního ukazatele (závažné krvácivé příhody) byl podobný v obou léčebných skupinách.
|
Populace studie |
3 449 pacientů se symptomatickou hlubokou žilní trombózou | |
|
Dávkování a délka léčby |
Xareltoa) |
Enoxaparin/VKAb) |
|
3, 6 nebo 12 měsíců |
3, 6 nebo 12 měsíců | |
|
N = 1 731 |
N = 1 718 | |
|
Symptomatický recidivující žilní |
36 |
51 |
|
tromboembolismus* |
(2,1%) |
(3,0%) |
|
Symptomatická recidivující |
20 |
18 |
|
(1,2%) |
(1,0%) | |
|
Symptomatická recidivující |
14 |
28 |
|
hluboká žilní trombóza |
(0,8%) |
(1,6%) |
|
Symptomatická plicní embolie a |
1 |
0 |
|
hluboká žilní trombóza |
(0,1%) | |
|
Fatální plicní embolie/úmrtí, |
4 |
6 |
|
kde plicní embolie nemůže být vyloučena |
(0,2%) |
(0,3%) |
|
Závažné nebo klinicky významné |
139 |
138 |
|
méně závažné krvácení |
(8,1%) |
(8,1%) |
|
Závažné krvácivé příhody |
14 |
20 |
|
(0,8%) |
(1,2%) | |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou
denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů vitaminu K
* p < 0,0001 (non-inferiorita k stanovenému poměru rizik 2,0); poměr rizik: 0,680 (0,443 - 1,042), p = 0,076 (superiorita)
Ve studii Einstein PE (viz tabulka 4) prokázal rivaroxaban non-inferioritu proti enoxaparinu/antagonistům vitaminu K v primárním parametru účinnosti (p = 0,0026 (test non-inferiority); poměr rizik: 1,123 (0,749 - 1,684)). Předem definovaný čistý klinický přínos (primární parametr účinnosti plus závažná krvácivá příhoda) byl hlášen s poměrem rizik 0,849 ((95% CI: 0,633-1,139), s nominální hodnotou p = 0,275). Hodnoty INR byly uvnitř terapeutického rozmezí s průměrem 63% pro průměrnou dobu léčby 215 dní a 57%, 62% a 65% doby pro skupiny s plánovanou léčbou 3, 6 a 12 měsíců. Ve skupině enoxaparin/VKA nebyl jasný vztah mezi hladinou TTR v centru (doba v cílovém INR rozmezí 2,0 - 3,0) ve stejně velkých tertilech a incidencí recidivující HZT (P=0,082 pro interakci). V centrech v nejvyšším tertilu bylo HR rivaroxaban versus warfarin 0,642 (95% CI: 0,277 - 1,484).
Výskyt primárního bezpečnostního ukazatele (závažné nebo klinicky významné méně závažné krvácivé příhody) byl lehce nižší ve skupině léčené rivaroxabanem (10,3% (249/2412)) než ve skupině léčené enoxaparinem/antagonisty vitaminu K (11,4% (274/2405)). Výskyt sekundárního bezpečnostního ukazatele (závažné krvácivé příhody) byl nižší ve skupině léčené rivaroxabanem (1.1% (26/2412)) než ve skupině enoxaparin/antagonisté vitaminu K (2.2% (52/2405)) s poměrem rizik 0,493 (95% CI: 0,308 - 0,789).
|
Populace studie |
4 832 pacientů s akutní symptomatickou PE | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Dávkování a doba léčby |
3, 6 nebo 12 měsíců |
3, 6 nebo 12 měsíců |
|
N=2 419 |
N=2 413 | |
|
Symptomatický recidivující žilní |
50 |
44 |
|
tromboembolismus* |
(2,1%) |
(1,8%) |
|
Symptomatická recidivující PE |
23 (1,0%) |
20 (0,8%) |
|
Symptomatická recidivující |
18 |
17 |
|
hluboká žilní trombóza |
(0,7%) |
(0,7%) |
|
Symptomatická recidivující plicní embolie a hluboká žilní trombóza |
0 |
2 (<0,1%) |
|
Fatální plicní embolie/úmrtí, kde plicní embolie nemůže být vyloučena |
11 (0,5%) |
7 (0,3%) |
|
Závažné nebo klinicky významné |
249 |
274 |
|
méně závažné krvácení |
(10,3%) |
(11,4%) |
|
Závažné krvácivé příhody |
26 (1,1%) |
52 (2,2%) |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním antagonistů
vitaminu K
* p < 0,0026 (non-inferiorita k predefinovanému poměru rizik 2,0); poměr rizik: 1,123 (0,749 - 1,684)
Byla provedena predefinovaná poolovaná analýza výsledku studií Einstein DVT a PE (viz tabulka 5).
Tabulka 5: Výsledky účinnosti a bezpečnosti z poolované analýzy studií fáze III Einstein DVT a Einstein PE
|
Populace studie |
8281 pacientů s akutní symptomatickou HZT nebo PE | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Dávkování a doba léčby |
3, 6 nebo 12 měsíců |
3, 6 nebo12 měsíců |
|
N=4150 |
N=4131 | |
|
Symptomatický recidivující žilní |
86 |
95 |
|
tromboembolismus* |
(2,1%) |
(2,3%) |
|
Symptomatická recidivující |
43 |
38 |
|
(1,0%) |
(0,9%) | |
|
Symptomatická recidivující |
32 |
45 |
|
hluboká žilní trombóza |
(0,8%) |
(1,1%) |
|
Symptomatická recidivující plicní embolie a hluboká žilní trombóza |
1 (<0,1%) |
2 (<0,1%) |
|
Fatální plicní embolie/úmrtí, kde |
15 |
13 |
|
plicní embolie nemůže být vyloučena |
(0,4%) |
(0,3%) |
|
Závažné nebo klinicky významné |
388 |
412 |
|
méně závažné krvácení |
(9,4%) |
(10,0%) |
|
Závažné krvácivé příhody |
40 (1,0%) |
72 (1,7%) |
a) Rivaroxaban 15 mg dvakrát denně po dobu 3 týdnů s následným podáváním 20 mg jednou
denně
b) Enoxaparin po dobu minimálně 5 dnů se současným a poté následným podáváním
antagonistů vitaminu K
* p < 0.0001 (non-inferiorita k předefinovanému poměru rizik 1,75); poměr rizik: 0,886
(0,661 - 1,186)
Předefinovaný čistý klinický prospěch (výsledek primární účinnosti plus závažné krvácivé příhody) poolované analýzy byl hlášen s poměrem rizik 0,771 ((95% CI: 0,614 - 0,967), nominální hodnota p= 0,0244).
Ve studii Einstein Extension (viz tabulka 6) byl rivaroxaban lepší než placebo v primárních a sekundárních parametrech účinnosti. U primárního bezpečnostního ukazatele (závažné krvácivé příhody) byl nevýznamný numericky vyšší výskyt u pacientů léčených rivaroxabanem v dávce 20 mg jednou denně v srovnání s placebem. Sekundární bezpečnostní ukazatel (závažné nebo klinicky významné méně závažné krvácivé příhody) prokázal vyšší výskyt u pacientů léčených rivaroxabanem 20 mg jednou denně ve srovnání s placebem.
Tabulka 6: Výsledky účinnosti a bezpečnosti ze studie fáze III Einstein Extension
|
Populace studie |
pokračování léčby u 1 197 pacientů, u nichž byla podávána léčba a prevence recidivujícího žilního tromboembolismu | |
|
Dávkování a doba léčby |
Xareltoa) |
Placebo |
|
6 nebo 12 měsíců |
6 nebo 12 měsíců | |
|
N = 602 |
N = 594 | |
|
Symptomatický recidivující žilní |
8 |
42 |
|
tromboembolismus* |
(1,3%) |
(7,1%) |
|
Symptomatická recidivující plicní |
2 |
13 |
|
embolie |
(0,3%) |
(2,2%) |
|
Symptomatická recidivující |
5 |
31 |
|
hluboká žilní trombóza |
(0,8%) |
(5,2%) |
|
Fatální plicní embolie/úmrtí, kde |
1 |
1 |
|
plicní embolie nemůže být vyloučena |
(0,2%) |
(0,2%) |
|
Závažné krvácivé příhody |
4 |
0 |
|
(0,7%) |
(0,0%) | |
|
Klinicky významné méně závažné |
32 |
7 |
|
krvácení |
(5,4%) |
(1,2%) |
a) Rivaroxaban 20 mg j ednou denně
* p < 0,0001 (superiorita), poměr rizik: 0,185 (0,087 - 0,393)
Kromě studií fáze III programu EINSTEIN byla provedena prospektivní, neintervenční, otevřená kohortová studie (XALIA) s centrálním vyhodnocováním sledovaných ukazatelů zahrnujících recidivující žilní tromboembolismus, závažné krvácení a úmrtí. Bylo zařazeno 5 142 pacientů s akutní hlubokou žilní trombózou za účelem posoudit dlouhodobou bezpečnost rivaroxabanu v porovnání se standardní antikoagulační terapií v klinické praxi. Výskyt závažného krvácení, recidivujícího žilního tromboembolismu a úmrtí ze všech příčin byly v rivaroxabanové větvi 0,7 %, 1,4 % a 0,5 %.
Ve vstupních charakteristikách pacientů byly rozdíly včetně věku, výskytu nádorových onemocnění a ledvinové nedostatečnosti. Přestože byla pro úpravu získaných základních rozdílů použita předem stanovená analýza stratifikovaná dle propensity skóre, mohou reziduální zavádějící faktory tyto výsledky ovlivnit. Upravené poměry rizik srovnávající rivaroxaban a standardní léčbu pro závažné krvácení, recidivující žilní tromboembolismus a mortalitu ze všech příčin byly 0,77 (95% CI: 0,40 - 1,50), 0,91 (95% CI: 0,54 - 1,54) a 0,51 (95% CI: 0,24 - 1,07).
Tato pozorování z klinické praxe jsou v souladu s potvrzeným bezpečnostním profilem v této indikaci. Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xarelto v léčbě tromboembolických příhod u jedné nebo více podskupin pediatrické populace. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xarelto v prevenci tromboembolických příhod u všech podskupin pediatrické populace (informace o použití u pediatrické populace viz bod 4.2)
5.2 Farmakokinetické vlastnosti
Absorpce
Rivaroxaban je rychle absorbován; maximální koncentrace (Cmax) se objeví 2 - 4 hodiny po užití tablety.
Bez ohledu na stav na lačno nebo po jídle je u dávky 2,5 mg a 10 mg rivaroxabanu tablety perorální absorpce téměř kompletní a perorální biologická dostupnost vysoká (80 - 100 %). Užívání při jídle neovlivňuje při 2,5 mg a 10 mg dávce AUC ani Cmax rivaroxabanu.
Pro 20 mg tabletu byla v důsledku sníženého rozsahu absorpce stanovena biologická dostupnost 66% při stavu nalačno. Když byly užívány 20 mg tablety přípravku Xarelto společně s jídlem, došlo ke zvýšení průměrné AUC o 39% ve srovnání s užíváním tablety nalačno, což ukazuje na téměř kompletní absorpci a vysokou biologickou dostupnost po perorálním podání. Přípravek Xarelto 15 mg a 20 mg se má užívat s jídlem (viz bod 4.2).
Farmakokinetické vlastnosti rivaroxabanu jsou až do denní dávky 15 mg nalačno přibližně lineární. Po jídle byla u přípravku Xarelto 10 mg, 15 mg a 20 mg prokázána farmakokinetika závislá na dávce. Ve vyšších dávkách je absorbce rivaroxabanu omezena disolucí, dochází ke snížení biologické dostupností a stupeň absorbce se snižuje se zvyšující se dávkou.
Variabilita farmakokinetiky rivaroxabanu je střední, s interindividuální variabilitou v rozmezí od 30% do 40%.
Absorpce rivaroxabanu je závislá na místě jeho uvolnění v gastrointestinálním traktu. Bylo hlášeno 29% a 56% snížení AUC a Cmax ve srovnání s tabletou, pokud byl rivaroxaban v granulátu uvolněn v proximální časti tenkého střeva. Expozice je dále snížena, když je rivaroxaban uvolněn v distální části tenkého střeva nebo ve vzestupné části tračníku. Podání rivaroxabanu distálně od žaludku by se mělo zabránit, jelikož to může vést ke snížení absorpce a související expozice rivaroxabanu.
Biologická dostupnost (AUC and Cmax) byla pro podání 20 mg rivaroxabanu per os ve formě rozdrcené tablety vmíchané do jablečného pyré nebo rozpuštěné ve vodě a pro podání žaludeční sondou následované tekutou stravou, v porovnání s celou tabletou srovnatelná. Vzhledem k předvídatelnému dávce úměrnému farmakokinetickému profilu rivaroxabanu odpovídají výsledky biologické dostupnosti z této studie pravděpodobně nižším dávkám rivaroxabanu.
Distribuce
Vazba na plazmatické proteiny u lidí je vysoká, přibližně 92% - 95%, přičemž hlavní část se váže na sérový albumin. Distribuční objem je střední, Vss činí přibližně 50 litrů.
Biotransformace a eliminace
Z podané dávky rivaroxabanu se přibližně 2/3 metabolicky degradují, z čehož je polovina vylučována ledvinami a druhá polovina stolicí. Zbývající 1/3 podané dávky je vylučována ledvinami přímo jako nezměněná léčivá látka, hlavně prostřednictvím aktivní ledvinové sekrece.
Rivaroxaban je metabolizován prostřednictvím systémů CYP3A4 a CYP2J2 i mechanismy na CYP nezávislými. Hlavními cestami transformace je oxidativní degradace morfolinonové části a hydrolýza amidových vazeb. Na základě in vitro experimentů je zřejmé, že rivaroxaban slouží jako substrát transportních proteinů - P-gp (P-glykoprotein) a Bcrp (breast cancer resistance protein).
Nezměněný rivaroxaban je nejvýznamnější formou přípravku v lidské plazmě; v krevním oběhu nejsou žádné významné nebo aktivní metabolity. Rivaroxaban lze vzhledem ke systémové clearance asi 10 l/h klasifikovat jako látku s nízkou clearance. Po intravenózním podání dávky 1 mg je eliminační poločas asi
4,5 hodiny. Po perorálním podání je eliminace limitována stupněm absorpce. K eliminaci rivaroxabanu z plazmy dochází s terminálním poločasem 5 až 9 hodin u mladších osob a s terminálním poločasem 11 -13 hodin u starších osob.
Zvláštní skupiny
Pohlaví
Mezi muži a ženami nebyl žádný klinicky relevantní rozdíl ve farmakokinetice a farmakodynamice přípravku.
Starší populace
Starší pacienti vykazovali vyšší plazmatické koncentrace než mladší, s průměrnou hodnotou AUC přibližně
1,5 x vyšší, hlavně vzhledem ke snížené (zdánlivé) celkové a ledvinové clearance. Žádná úprava dávky není nutná.
Různé váhové kategorie
Extrémy v tělesné hmotnosti (< 50 kg nebo > 120 kg) měly pouze malý vliv na plazmatické koncentrace rivaroxabanu (méně než 25%). Žádná úprava není dávky nutná.
Rozdíly mezi etniky
Žádné klinicky relevantní rozdíly mezi etniky nebyly ve farmakokinetice a farmakodynamice rivaroxabanu zjištěny u pacientů z řad bělochů, Afroameričanů, Hispánců, Japonců ani Číňanů.
Jaterní nedostatečnost
Pacienti s cirhózou s mírnou jaterní nedostatečností (Child-Pugh A) vykazovali pouze menší změny ve farmakokinetice rivaroxabanu (v průměru 1,2x nárůst AUC rivaroxabanu), a výsledky byly téměř srovnatelné s kontrolní skupinou zdravých dobrovolníků. U pacientů trpících cirhózou se středně závažnou jaterní nedostatečností (Child-Pugh B) průměrná AUC rivaroxabanu významně stoupla - 2,3x v porovnání se zdravými dobrovolníky. AUC nevázané látky stoupla 2,6x. Tito pacienti měli současně sníženou renální eliminaci rivaroxabanu, podobně jako pacienti se středně závažnou ledvinovou nedostatečností. O farmakokinetice u pacientů s těžkým jaterním poškozením nejsou k dispozici žádné údaje.
Inhibice aktivity faktoru Xa byla u pacientů se středně závažnou jaterní nedostatečností zvýšena ve srovnání se zdravými dobrovolníky 2,6x; prodloužení PT bylo obdobně zvýšeno 2,1x. Pacienti se středně závažnou jaterní nedostatečností byli na rivaroxaban citlivější, a vztah mezi koncentrací a PT měl tak strmější průběh. Přípravek Xarelto je kontraindikován u pacientů s jaterním onemocněním, které je spojeno s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s klasifikací Child Pugh B a C (viz bod 4.3).
Ledvinová nedostatečnost
Byl zjištěn nárůst expozice rivaroxabanu související s poklesem funkce ledvin, která byla posuzována prostřednictvím hodnot clearance kreatininu. U osob s lehkou (clearance kreatininu 50 - 80 ml/min), střední (clearance kreatininu 30 - 49 ml/min) a těžkou (clearance kreatininu 15 - 29 ml/min) ledvinovou nedostatečností byly plazmatické koncentrace rivaroxabanu (AUC) zvýšeny 1,4, 1,5 resp. 1,6 x. Odpovídající zesílení farmakodynamických účinků bylo výraznější. U osob s lehkou, střední a těžkou ledvinovou nedostatečností byla celková inhibice faktoru Xa ve srovnání se zdravými dobrovolníky zvýšena 1,5, 1,9 resp. 2,0 x; prodloužení PT bylo obdobně zvýšeno 1,3, 2,2 a 2,4 x. O použití u pacientů s clearance kreatininu < 15 ml/min nejsou žádné údaje.
Vzhledem k vysoké vazbě na plazmatické proteiny se u rivaroxabanu neočekává možnost odstranění dialýzou.
Použití se nedoporučuje u pacientů s clearance kreatininu < 15 ml/min. U pacientů s clearance kreatininu 15 - 29 ml/min je nutno Xarelto používat s opatrností (viz bod 4.4).
Farmakokinetické údaje u pacientů
U pacientů užívajících rivaroxaban 20 mg jednou denně k léčbě akutní hluboké žilní trombózy (HŽT) byl geometrický průměr koncentrace (90% interval přepovědi) 2 - 4 hodiny a přibližně 24 hodin po podání dávky (zhruba představující maximální a minimální koncentrace během dávkovacího intervalu) 215 (22535) a 32 (6-239) ^g/l.
Farmakokinetické a farmakodynamické vztahy
Po podání různě velkých dávek (5 - 30 mg dvakrát denně) byl hodnocen farmakokinetický a farmakodynamický (PK/PD) vztah mezi plazmatickou koncentrací rivaroxabanu a několika konečnými cílovými ukazateli PD (inhibice faktoru Xa, PT, aPTT, Heptest). Vztah mezi plazmatickou koncentrací rivaroxabanu a aktivitou faktoru Xa byl nejlépe popsán pomocí modelu Emax. U PT byly údaje lépe vyjádřeny pomocí lineárního ohraničeného modelu. Hodnoty PT se významně lišily v závislosti na použitých reagenciích. Při použití Neoplastinu byl výchozí PT asi 13 sekund a odchylka hodnot přibližně 3 až 4 s/(100 ^g/l). Výsledek analýz PK/PD ve studiích fáze II a III byl v souladu s údaji získanými u zdravých jedinců.
Pediatrická populace
Bezpečnost a účinnost nebyly stanoveny u dětí a dospívajících do 18 let.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po jednorázovém podání, fototoxicity, genotoxicity, kancerogenního potenciálu a juvenilní toxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studii toxicity při opakovaném podání byly způsobeny hlavně zesílenou farmakologickou aktivitou rivaroxabanu. Při klinicky relevantních úrovních expozice byly u potkanů pozorovány zvýšené plazmatické hladiny IgG a IgA.
U potkanů nebyly pozorovány žádné účinky na fertilitu samců nebo samic. Studie na zvířatech prokázaly reprodukční toxicitu související s farmakologickým mechanismem působení rivaroxabanu (např. hemoragickými komplikacemi). V klinicky relevantních plazmatických koncentracích byla pozorována embryonální a fetální toxicita (post-implantační ztráta, opožděná nebo progredující osifikace, hepatální mnohočetné světle zbarvené skvrny) a zvýšený výskyt běžných malformací, a také placentárních změn.
V prenatálních a postnatálních experimentech u potkanů byla zjištěna snížená životaschopnost potomků, a to v dávkách toxických pro matky.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulóza Sodná sůl kroskarmelózy Monohydrát laktózy Hypromelóza Natrium-lauryl-sulfát Magnesium-stearát
Potah tablety:
Makrogol 3350
Hypromelóza
Oxid titaničitý (E171)
Červený oxid železitý (E172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Balení pro zahájení léčby pro první 4 týdny léčby:
Blistry z fólie Al/PP v pouzdru obsahujícím 49 potahovaných tablet: 42 potahovaných tablet přípravku Xarelto 15 mg a 7 potahovaných tablet přípravku Xarelto 20 mg.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/472/040
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. září 2008
Datum posledního prodloužení registrace: 22. května 2013
10. DATUM REVIZE TEXTU {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců odpovědných za propouštění šarží
Bayer Pharma AG 51368 Leverkusen Německo
Bayer HealthCare Manufacturing Srl.
Via delle Groane, 126 20024 Garbagnate Milanese Itálie
V tištěné příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Před uvedením na trh držitel rozhodnutí o registraci poskytne všem lékařům, u kterých se očekává preskripce/podávání přípravku Xarelto, edukační balíček.
Tento edukační balíček je určený k zvýšení povědomí o potenciálním riziku krvácení během léčby přípravkem Xarelto a poskytuje návod, jak toto riziko zvládnout.
Edukační balíček pro lékaře musí obsahovat:
• Souhrn údajů o přípravku
• Doporučení pro předepisujícího lékaře
• Informační karty pro pacienty (text je obsažen v Příloze III)
Před distribucí edukačního balíčku se držitel rozhodnutí o registraci dohodne o obsahu a formě Doporučení pro předepisujícího lékaře a komunikačního plánu s příslušnou národní kompetentní autoritou daného členského státu.
Doporučení pro předepisujícího lékaře musí obsahovat následující klíčová bezpečnostní sdělení:
• Detailní informace o populacích s potencionálně vyšším rizikem krvácení
• Doporučení ke snížení dávky u rizikových populací
• Návod na změnu léčby na rivaroxaban nebo z rivaroxabanu
• Nutnost užívat 15 mg a 20 mg tablety s jídlem
• Postup při předávkování
• Používání koagulačních testů a jejich interpretace
• Všichni pacienti musí být poučeni o následujícím:
> Příznaky a proj evy krvácení a kdy j e nutno vyhledat odbornou pomoc.
> Význam dodržování správného režimu léčby
> Nutnost užívat 15 mg a 20 mg tablety s jídlem
> Nutnost trvale nosit u sebe Informační kartu,která je součástí každého balení
> Nutnost informovat lékaře o užívání přípravku Xarelto, pokud nastane potřeba nějakého chirurgického nebo invazivního výkonu.
Držitel rozhodnutí o registraci také v každém balení poskytne bezpečnostní Informační kartu pro pacienta, jejíž text je obsažen v Příloze III.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Poregistrační studijní program týkající se bezpečnosti rivaroxabanu v sekundární prevenci akutního koronárního syndromu mimo prostředí klinické studie, zvláště s ohledem na incidenci, závažnost, léčbu a výsledek krvácivých příhod v celé populaci a zejména u pacientů se zvýšeným rizikem krvácení. |
• Interim analýzy budou předkládány každý rok počínaje 4.čtvrtletím 2015 až do ukončení studijního programu. • Kumulativní interim zpráva bude předložena do 4.čtvrtletí 2017. • Závěrečná zpráva o studii bude předložena do 4.čtvrtletí 2020. |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA PRO 2,5 MG_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 2,5 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 2,5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
14 potahovaných tablet 28 potahovaných tablet 56 potahovaných tablet 60 potahovaných tablet 98 potahovaných tablet 168 potahovaných tablet 196 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta 30 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/025
EU/1/08/472/026
EU/1/08/472/027
EU/1/08/472/028
EU/1/08/472/029
EU/1/08/472/030
EU/1/08/472/031
EU/1/08/472/032
EU/1/08/472/033
EU/1/08/472/035
14 potahovaných tablet 28 potahovaných tablet 56 potahovaných tablet 60 potahovaných tablet 98 potahovaných tablet 168 potahovaných tablet 196 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta 30 potahovaných tablet
(PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr) (PP/Al blistr)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 2,5 mg
VNĚJŠÍ KRABIČKA PRO MULTIBALENÍ (VČETNĚ BLUE BOXU) PRO 2,5 MG 1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 2,5 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 2,5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Multibalení 100 (10 balení po 10 x1) potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/034 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 2,5 mg
JEDNOTLIVÁ KRABIČKA V MULTIBALENÍ (BEZ BLUE BOXU) PRO 2,5 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 2,5 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 2,5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10 x1 potahovaná tableta.
Balení je součástí multibalení, jednotlivý prodej není možný.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/034 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 2,5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 2,5 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
BAYER (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 2,5 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 2,5 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Po
Út
St
Čt
Pá
So
Ne
symbol slunce symbol měsíce
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA PRO 10 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 10 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
5 potahovaných tablet 10 potahovaných tablet 30 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/001
EU/1/08/472/002
EU/1/08/472/003
EU/1/08/472/004
EU/1/08/472/005
EU/1/08/472/006
EU/1/08/472/007
EU/1/08/472/008
EU/1/08/472/009
EU/1/08/472/010
5 potahovaných tablet 10 potahovaných tablet 30 potahovaných tablet 100 x 1 potahovaná tableta 5 potahovaných tablet 10 potahovaných tablet 30 potahovaných tablet 100 x 1 potahovaná tableta 10 x 1 potahovaná tableta 10 x 1 potahovaná tableta
blistry z fólie PVC/PVDC/Al blistry z fólie PVC/PVDC/Al blistry z fólie PVC/PVDC/Al blistry z fólie PVC/PVDC/Al blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie PVC/PVDC/Al blistry z fólie Al/PPl
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 10 mg
VNĚJŠÍ KRABIČKA PRO MULTIBALENÍ (VČETNĚ BLUE BOXU) PRO 10 MG 1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 10 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Multibalení 100 (10 balení po 10 x1) potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/022 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 10 mg
JEDNOTLIVÁ KRABIČKA V MULTIBALENÍ (BEZ BLUE BOXU) PRO 10 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 10 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10 x1 potahovaná tableta.
Balení je součástí multibalení, jednotlivý prodej není možný.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/022 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 10 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR PRO 10 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 10 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
BAYER (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA PRO JEDNOTLIVÉ BALENÍ PRO 15 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 15 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
10 potahovaných tablet 14 potahovaných tablet 28 potahovaných tablet 42 potahovaných tablet 98 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/011
EU/1/08/472/012
EU/1/08/472/013
EU/1/08/472/014
EU/1/08/472/015
EU/1/08/472/016
EU/1/08/472/038
14 potahovaných tablet 28 potahovaných tablet 42 potahovaných tablet 98 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta 10 potahovaných tablet
blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 15 mg
VNĚJŠÍ KRABIČKA PRO MULTIBALENÍ (VČETNĚ BLUE BOXU) PRO 15 MG 1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 15 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Multibalení 100 (10 balení po 10 x1) potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/023 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 15 mg
JEDNOTLIVÁ KRABIČKA V MULTIBALENÍ (BEZ BLUE BOXU) PRO 15 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 15 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10 x1 potahovaná tableta.
Balení je součástí multibalení, jednotlivý prodej není možný.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/023 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 15 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
BAYER (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Po
Út
St
Čt
Pá
So
Ne
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 15 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
100 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/036 100 potahovaných tablet (HDPE lahvička)
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 15 mg (uvádí se pouze na krabičce, neuvádí se na nálepce na lahvičku)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
10 potahovaných tablet 14 potahovaných tablet 28 potahovaných tablet 98 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/017
EU/1/08/472/018
EU/1/08/472/019
EU/1/08/472/020
EU/1/08/472/021
EU/1/08/472/039
14 potahovaných tablet 28 potahovaných tablet 98 potahovaných tablet 10 x 1 potahovaná tableta 100 x 1 potahovaná tableta 10 potahovaných tablet
blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl blistry z fólie Al/PPl
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 20 mg
VNĚJŠÍ KRABIČKA PRO MULTIBALENÍ (VČETNĚ BLUE BOXU) PRO 20 MG 1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Multibalení 100 (10 balení po 10 x1) potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/024 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 20 mg
JEDNOTLIVÁ KRABIČKA V MULTIBALENÍ (BEZ BLUE BOXU) PRO 20 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10 x1 potahovaná tableta.
Balení je součástí multibalení, jednotlivý prodej není možný.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/024 100 potahovaných tablet (10 x 10 x 1) (multibalení) (blistry z fólie Al/PP)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 20 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
BAYER (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Po
Út
St
Čt
Pá
So
Ne
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM A VNITŘNÍM OBALU KRABIČKA A NÁLEPKA NA HDPE LAHVIČKU PRO 20 MG
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 20 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rivaroxabanum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
100 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/037 100 potahovaných tablet (HDPE lahvička)
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 20 mg (uvádí se pouze na krabičce, neuvádí se na nálepce na lahvičku)
VNĚJŠÍ KRABIČKA PRO BALENÍ PRO ZAHÁJENÍ LÉČBY (42 POTAHOVANÝCH TABLET 15 MG A 7 POTAHOVANÝCH TABLET 20 MG) (VČETNĚ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg Xarelto 20 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Červená potahovaná tableta pro týdny 1, 2 a 3 obsahuje rivaroxabanum 15 mg. Hnědo-červená potahovaná tableta pro týden 4 obsahuje rivaroxabanum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Jedno balení se 49 potahovanými tabletami obsahuje:
42 potahovaných tablet obsahujících 15 mg rivaroxabanu. 7 potahovaných tablet obsahujících 20 mg rivaroxabanu.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Balení pro zahájení léčby
Toto balení pro zahájení léčby je určeno pouze pro první 4 týdny léčby.
DÁVKOVÁNÍ
Den 1 až 21: jedna 15mg tableta dvakrát denně (jedna 15mg tableta ráno a jedna večer) současně s jídlem. Ode dne 22: jedna 20mg tableta jednou denně (užívaná ve stejnou denní dobu) současně s jídlem.
Den 1 až 21: 15 mg 1 tableta dvakrát denně (jedna 15mg tableta ráno a jedna večer) současně s jídlem. Ode dne 22: 20 mg 1 tableta jednou denně (užívaná ve stejnou denní dobu) současně s jídlem.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO
DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/040 42 potahovaných tablet obsahujících rivaroxabanum 15 mg a
7 potahovaných tablet obsahujících rivaroxabanum 20 mg (balení pro zahájení léčby)
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
xarelto 15 mg xarelto 20 mg
POUZDRO PRO BALENÍ PRO ZAHÁJENÍ LÉČBY (42 POTAHOVANÝCH TABLET 15 MG A 7 POTAHOVANÝCH TABLET 20 MG) (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg Xarelto 20 mg potahované tablety rivaroxabanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Červená potahovaná tableta pro týdny 1, 2 a 3 obsahuje rivaroxabanum 15 mg. Hnědo-červená potahovaná tableta pro týden 4 obsahuje rivaroxabanum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu. Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Jedno balení se 49 potahovanými tabletami obsahuje:
42 potahovaných tablet obsahujících 15 mg rivaroxabanu. 7 potahovaných tablet obsahujících 20 mg rivaroxabanu.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Balení pro zahájení léčby
Toto balení pro zahájení léčby je určeno pouze pro první 4 týdny léčby.
Den 1 až 21: 15 mg 1 tableta dvakrát denně (jedna 15mg tableta ráno a jedna večer) současně s jídlem. Ode dne 22: 20 mg 1 tableta jednou denně (užívaná ve stejnou denní dobu) současně s jídlem.
DÁVKOVÁNÍ a DÁVKOVACÍ SCHÉMA
Den 1 až 21: jedna 15mg tableta dvakrát denně (jedna 15mg tableta ráno a jedna večer).
Ode dne 22: jedna 20mg tableta jednou denně (užívaná ve stejnou denní dobu).
Úvod léčby Xarelto 15 mg dvakrát denně první 3 týdny
Pokračování léčby Xarelto 20 mg jednou denně 4. týden
Navštivte svého lékaře k zajištění pokračování léčby.
Užívá se s jídlem.
Xarelto 15 mg
Začátek léčby 15 mg
dvakrát denně Datum zahájení
TÝDEN 1, TÝDEN 2, TÝDEN 3
DEN 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
symbol slunce symbol měsíce
Změna dávky Xarelto 20 mg 20 mg
jednou denně
užívat ve stejnou denní dobu Datum změny dávky TÝDEN 4
DEN 22 DEN 23 DEN 24 DEN 25 DEN 26 DEN 27 DEN 28
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG 13342 Berlin Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/472/040 42 potahovaných tablet obsahujících rivaroxabanum 15 mg a
7 potahovaných tablet obsahujících rivaroxabanum 20 mg (balení pro zahájení léčby)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
BLISTR PRO BALENÍ PRO ZAHÁJENÍ LÉČBY V POUZDRU (42 POTAHOVANÝCH TABLET 15 MG A 7 POTAHOVANÝCH TABLET 20 MG)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Xarelto 15 mg tablety Xarelto 20 mg tablety rivaroxabanum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Bayer (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Informační karta pro pacienta
Bayer (logo)
Xarelto 2,5 mg Xarelto 15 mg Xarelto 20 mg
♦ Tuto kartu noste vždy při sobě.
♦ Kartu ukažte každému lékaři nebo zubnímu lékaři ještě před ošetřením.
Užívám antikoagulační léčbu přípravkem Xarelto (rivaroxaban)
Jméno:
Adresa:
Datum narození:
Krevní skupina:
Hmotnost:
Jiná medikace/onemocnění:
V naléhavém případě prosím informujte:
Jméno lékaře:
Telefonní číslo lékaře:
Razítko lékaře:
Rovněž informujte:
Jméno:
Telefonní číslo:
Příbuzenský vztah:
Informace pro ošetřujícího lékaře:
♦ Hodnoty INR by neměly být používány, neboť nejsou spolehlivým ukazatelem antikoagulační aktivity přípravku Xarelto.
Co musím vědět o přípravku Xarelto?
♦ Přípravek Xarelto ovlivňuje srážlivost krve a brání tak tvorbě nebezpečných krevních sraženin.
♦ Přípravek Xarelto se musí vždy užívat přesně podle pokynů Vašeho lékaře. Aby byla zajištěna optimální prevence tvorby krevních sraženin, nikdy nevynechávejte žádnou dávku.
♦ Bez předchozí konzultace se svým lékařem nikdy nepřestávejte užívat přípravek Xarelto, neboť by se mohlo zvýšit riziko tvorby krevních sraženin ve Vašem těle.
♦ Než začnete užívat přípravek Xarelto, informujte svého lékaře o všech jiných lécích, které užíváte, užíval(a) jste v nedávné době, nebo které se chystáte užívat.
♦ Informujte svého lékaře o tom, že užíváte přípravek Xarelto před každým chirurgickým nebo jiným invazivním zákrokem.
Kdy svého lékaře žádat o radu?
Jestliže užíváte přípravek snižující srážlivost krve, jako je Xarelto, je důležité, abyste si byl(a) vědom(a) jeho možných nežádoucích účinků. Nejčastěji se vyskytujícím nežádoucím účinkem je krvácení. Jestliže víte, že Vám hrozí riziko krvácení, bez porady s lékařem nezačínejte přípravek Xarelto užívat. Svého ošetřujícího lékaře ihned informujte, jestliže se u Vás objeví příznaky nebo známky krvácení, jako například:
♦ bolest
♦ otok nebo nepříjemný pocit
♦ bolest hlavy, závrať nebo slabost
♦ neobvyklé modřiny, krvácení z nosu, krvácení z dásní, rány, které dlouho krvácejí
♦ menstruační nebo vaginální krvácení silnější než obvykle
♦ krev v moči, která může být růžově nebo hnědě zbarvená, červená nebo černá stolice
♦ vykašlávání krve nebo zvracení krve nebo zvratky, které vypadají jako kávová sedlina
Jak se přípravek Xarelto užívá?
♦ Aby byla zajištěna optimální ochrana, přípravek Xarelto
- 2,5 mg se může užívat s jídlem nebo nezávisle na jídle
- 15 mg se musí užívat s jídlem
- 20 mg se musí užívat s jídlem.
Xarelto 2,5 mg potahované tablety
rivaroxabanum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Xarelto a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
3. Jak se přípravek Xarelto užívá
4. Možné nežádoucí účinky
5. Jak přípravek Xarelto uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xarelto a k čemu se používá
Byl Vám předepsán přípravek Xarelto, protože Vám byl zjištěn akutní koronární syndrom (skupina onemocnění, která zahrnuje infarkt myokardu a nestabilní anginu pectoris, které se projevují silnou bolestí na hrudi). Zároveň jste měl(a) zvýšené určité krevní testy, které ukazují na poškození srdce.
Přípravek Xarelto u dospělých snižuje riziko dalšího infarktu myokardu nebo snižuje riziko úmrtí na onemocnění srdce nebo cév.
Xarelto obsahuje léčivou látku rivaroxaban a patří do skupiny léků nazývaných antitrombotika Účinkuje tak, že blokuje faktor krevní srážlivosti (faktor Xa), čímž snižuje sklon k tvorbě krevních sraženin.
Xarelto nebudete užívat jako jediný lék. Váš lékař Vám k němu přidá:
• kyselinu acetylsalicylovou (známou také jako aspirin) nebo
• kyselinu acetylsalicylovou a klopidogrel nebo tiklopidin.
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat Neužívejte přípravek Xarelto
- jestliže jste alergický(á) na rivaroxaban nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže silně krvácíte
- jestliže máte onemocnění nebo postižení některého orgánu, které zvyšují riziko závažného krvácení (například vřed žaludku, poranění nebo krvácení v mozku, nedávno prodělanou operaci mozku nebo očí)
- jestliže užíváte léky, které brání srážení krve (např. warfarin, dabigatran, apixaban nebo heparin),
s výjimkou situací provázených změnou antikoagulační léčby nebo při podávání heparinu zavedenou sondou (katetrem nebo kanylou) v žíle nebo tepně, aby byla zachována její průchodnost
- jestliže máte akutní koronární syndrom a dříve jste měl(a) krvácení nebo krevní sraženinu v mozku (cévní mozkovou příhodu)
- jestliže máte onemocnění jater, které vede ke zvýšenému riziku krvácení
- jestliže jste těhotná nebo kojíte
Xarelto neužívejte a informujte lékaře, pokud se Vás týká cokoli z výše uvedeného.
Upozornění a opatření
Před užitím přípravku Xarelto se poraďte se svým lékařem nebo lékarníkem.
Přípravek Xarelto se nemá užívat v kombinaci s určitými dalšími léky, které snižují srážení krve, jako např. prasugrel nebo tikagrelor, jinými než aspirin a klopidogrel/tiklopidin.
Zvláštní opatrnosti při použití přípravku Xarelto je zapotřebí
- pokud máte zvýšené riziko krvácení, které se může vyskytnout v situacích, jako například:
■ těžké onemocnění ledvin, protože funkce ledvin může ovlivnit množství léku ve Vašem těle
■ jestliže užíváte další léky, které brání srážení krve (např. warfarin, dabigatran, apixaban nebo heparin), při změně protisrážlivé léčby nebo jestliže dostáváte heparin žilní nebo tepennou kanylou/katetrem z důvodu zachování její průchodnosti (viz bod „Další léčivé přípravky a přípravek Xarelto“)
■ krvácivé poruchy
■ velmi vysoký krevní tlak, neupravený léčbou
■ onemocnění žaludku nebo střeva, která mohou mít za následek krvácení, např. zánět střev nebo žaludku nebo zánět jícnu, způsobený např. refluxní chorobou (onemocnění, při kterém se žaludeční kyselina dostává nahoru do jícnu)
■ problém s cévami na očním pozadí (retinopatie)
■ onemocnění plic, při kterém j sou průdušky rozšířené a vyplněné hnisem (bronchiektázie), nebo předchozí výskyt krvácení z plic
■ je vám více než 75 let
■ vážíte 60 kg nebo méně
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře ještě předtím, než začnete Xarelto užívat. Lékař rozhodne, zda máte být léčen(a) tímto přípravkem a zda máte být pečlivě sledován(a).
Pokud musíte jít na operaci:
- je velmi důležité, abyste před operací a po ní užíval(a) Xarelto přesně v časech stanovených lékařem.
- Pokud při operaci bude použit katetr nebo injekce do páteřního kanálu (například při epidurální nebo spinální anestezii nebo k tlumení bolesti):
■ je velmi důležité užívat Xarelto před injekcí a po injekci nebo odstranění katetru přesně tak, jak Vám lékař řekl
■ okamžitě informujte svého lékaře, pokud zaznamenáte po anestezii necitlivost nebo slabost dolních končetin nebo střevní potíže anebo potíže s močovým měchýřem, protože je třeba okamžitá léčba.
Děti a dospívající
Přípravek Xarelto se nedoporučuje jedincům ve věku do 18 let. O použití tohoto přípravku u dětí a dospívajících není k dispozici dostatek informací.
Další léčivé přípravky a přípravek Xarelto
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste užíval(a) v nedávné době, nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
- Jestliže užíváte:
■ některé léky k léčbě plísňových infekcí (například ketokonazol, itrakonazol, vorikonazol, posakonazol), s výjimkou léků aplikovaných pouze na kůži
■ některé antivirové léky k léčbě infekce virem HIV / AIDS (například ritonavir)
■ jiné léky k omezení tvorby krevních sraženin (například enoxaparin, klopidogrel nebo antagonisté vitaminu K, například warfarin a acenokumarol)
■ protizánětlivé léky a léky proti bolesti (například naproxen nebo kyselina acetylsalicylová)
■ dronedaron, lék k léčbě poruch srdečního rytmu
Jestliže užíváte některý z výše uvedených léků, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít ke zvýšení účinku přípravku Xarelto. Váš lékař rozhodne, zda máte být léčen(a) tímto přípravkem a zda máte být pečlivěji sledován(a).
Pokud se Váš lékař domnívá, že u Vás existuje zvýšené riziko vzniku vředů žaludku nebo střeva, může u Vás rovněž použít preventivní protivředovou léčbu.
- Jestliže užíváte:
■ některé léky k léčbě epilepsie (fenytoin, karbamazepin, fenobarbital)
■ třezalku tečkovanou (Hypericum perforatum), rostlinný přípravek k léčbě deprese
■ rifampicin, antibiotikum
Jestliže užíváte některý z výše uvedených léků, informujte lékaře před zahájením užívání přípravku Xarelto, protože může dojít k zeslabení účinku přípravku Xarelto. Váš lékař rozhodne, zda máte být léčen(a) přípravkem Xarelto a zda máte být pečlivěji sledován(a).
Těhotenství a kojení
Xarelto neužívejte, jestliže jste těhotná nebo kojíte. Pokud byste mohla otěhotnět, používejte během léčby přípravkem Xarelto spolehlivou antikoncepci. Pokud během léčby tímto přípravkem otěhotníte, ihned informujte lékaře. Ten pak rozhodne o další léčbě.
Řízení dopravních prostředků a obsluha strojů
Přípravek Xarelto může způsobovat závratě (častý nežádoucí účinek) nebo mdloby (méně častý nežádoucí účinek) (viz bod 4 „Možné nežádoucí účinky“). Pokud zaznamenáte tyto příznaky, nesmíte řídit vozidla a obsluhovat stroje.
Přípravek Xarelto obsahuje laktózu
Pokud Vám lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se Xarelto užívá
Vždy užívejte tento léčivý přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik přípravku užívat
Doporučená dávka přípravku je jedna 2,5mg tableta dvakrát denně. Přípravek Xarelto užívejte každý den přibližně ve stejnou denní dobu (například jednu tabletu ráno a jednu večer). Tento přípravek lze užívat při jídle nebo nezávisle na jídle.
Pokud máte obtíže polknout celou tabletu, poraďte se s lékařem o dalších možnostech, jak užívat přípravek Xarelto. Tableta může být rozdrcena a smíchána s vodou nebo jablečným pyré, bezprostředně před tím, než ji užijete.
Je-li to nutné, lékař Vám také může podat rozdrcenou tabletu přípravku Xarelto žaludeční sondou.
Xarelto nebudete užívat jako jediný lék. Váš lékař Vám k němu přidá:
• kyselinu acetylsalicylovou (známou také jako aspirin) nebo
• kyselinu acetylsalicylovou a klopidogrel nebo tiklopidin.
Váš lékař Vám sdělí, jakou dávku těchto přípravků budete užívat (obvykle 75 až 100 mg kyseliny acetylsalicylové denně nebo denní dávku 75 až 100 mg kyseliny acetylsalicylové plus denní dávku 75 mg klopidogrelu nebo standardní denní dávku tiklopidinu).
Kdy začít užívat Xarelto
Léčbu přípravkem Xarelto je třeba zahájit co nejdříve po stabilizaci akutního koronárního syndromu, nejdříve za 24 hodin po přijetí do nemocnice a v době, kdy by normálně byla ukončena parenterální (injekční) antikoagulační léčba.
Lékař rozhodne, jak dlouho musíte v léčbě pokračovat.
Jestliže jste užil(a) více přípravku Xarelto, než jste měl(a)
Pokud jste užil(a) příliš mnoho tablet, kontaktujte ihned svého lékaře. Nadměrné množství přípravku Xarelto zvyšuje riziko krvácení.
Jestliže jste zapomněl(a) užít přípravek Xarelto
Pokud jednu dávku vynecháte, nezdvojujte následující dávku, abyste ji nahradil(a). Pokud jednu dávku vynecháte, užijte následující dávku v obvyklou dobu.
Jestliže jste přestal(a) užívat přípravek Xarelto
Užívejte přípravek Xarelto pravidelně tak dlouho, dokud Vám jej Váš lékař bude předepisovat.
Užívání přípravku Xarelto nepřerušujte bez předchozí konzultace s lékařem. Jestliže tento přípravek přestanete užívat, může se zvýšit riziko, že dostanete další infarkt myokardu nebo cévní mozkovou příhodu nebo se může zvýšit riziko, že zemřete na onemocnění související s Vaším srdcem nebo cévami.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i přípravek Xarelto nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Stejně jako jiné podobné léky (antitrombotika), může i Xarelto způsobit krvácení, které může být potenciálně životu nebezpečné. Nadměrné krvácení může vést k náhlému poklesu krevního tlaku (šok).
V některých případech toto krvácení nemusí být zjevné.
Možné nežádoucí účinky, které mohou být známkou krvácení:
Ihned informujte lékaře, jestliže se u Vás projeví některý z následujících nežádoucích účinků:
- dlouhotrvající nebo rozsáhlé krvácení
- výjimečná slabost, únava, bledost, závratě, bolest hlavy, otok z neznámých příčin, dušnost, bolest na hrudníku nebo angina pectoris, protože to mohou být známky krvácení.
Lékař vás možná bude chtít pečlivě sledovat, nebo změní způsob léčby.
Časté (mohou postihovat až 1 osobu z 10):
- krvácení v žaludku nebo střevech, z močopohlavního traktu (včetně výskytu krve v moči a silného menstruačního krvácení), krvácení z nosu a z dásní
- krvácení do oka (včetně krvácení do očního bělma)
- krvácení do tkáně nebo tělní dutiny (modřiny, podlitiny)
- vykašlávání krve
- krvácení z kůže nebo do kůže
- krvácení po operaci
- vytékání krve nebo tekutiny z operační rány
- otoky končetin
- bolest končetin
- horečka
- snížení počtu červených krvinek, což může způsobit bledost kůže a slabost nebo dušnost
- bolest břicha, poruchy trávení, nevolnost nebo zvracení, zácpa, průjem
- nízký krevní tlak (příznaky mohou být pocity závratě nebo mdloby při vstávání)
- pokles celkové síly a energie (slabost, únava), bolest hlavy, závratě
- vyrážka, svědění kůže
- porucha funkce ledvin (může se prokázat na základě testů prováděných Vaším lékařem)
- zvýšení hodnot některých jaterních enzymů v krevních testech
Méně časté (mohou postihovat až 1 osobu ze 100):
- krvácení do mozku nebo lebeční dutiny
- krvácení do kloubu, které vede k bolesti a otoku kloubu
- omdlévání
- celkový pocit nemoci
- pocit sucha v ústech
- zrychlený srdeční tep
- alergické reakce, včetně alergických kožních reakcí
- kopřivka
- porucha funkce ledvin nebo jater (může se prokázat na základě testů prováděných Vaším lékařem)
- krevní testy mohou ukázat zvýšení bilirubinu, některých pankreatických nebo jaterních enzymů nebo počtu krevních destiček
Vzácné (mohou postihovat až 1 osobu z 1 000):
- krvácení do svalů
- místní otok
- žloutnutí kůže a očí (žloutenka)
- nahromadění krve (hematom) v tříslech jako komplikace srdečního výkonu, při kterém je katetr zaveden do tepny na dolní končetině (pseudoaneuryzma)
Není známo (frekvenci z dostupných údajů nelze určit)
- zvýšený tlak uvnitř svalů na nohách nebo pažích vzniklý po krvácení, který vede k bolesti, otoku, poruše citlivosti, necitlivosti nebo obrně (kompartment syndrom po krvácení)
- selhání ledvin po těžkém krvácení
Následující nežádoucí účinky byly hlášeny po schválení registrace tohoto léčivého přípravku:
- angioedém a alergický otok (otékání tváře, rtů, úst, jazyka nebo hrdla)
- cholestáza (snížení toku žluči), hepatitida včetně hepatocelulárního poškození (zánět jater včetně poškození jater)
- trombocytopenie (snížené množství krevních destiček, což jsou buňky, které pomáhají srážení krve) Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Xarelto uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a každém blistru za „EXP". Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Xarelto obsahuje
- Léčivou látkou je rivaroxabanum. Jedna tableta obsahuje rivaroxabanum 2,5 mg.
- Pomocnými látkami jsou:
Jádro tablety: mikrokrystalická celulóza, sodná sůl kroskarmelózy, monohydrát laktózy, hypromelóza, natrium-lauryl-sulfát, magnesium-stearát.
Potah tablety: makrogol 3350, hypromelóza, oxid titaničitý (E171), žlutý oxid železitý (E172).
Jak přípravek Xarelto vypadá a co obsahuje toto balení
Xarelto 2,5 mg potahované tablety jsou světle žluté, kulaté, bikonvexní a označené logem (BAYER kříž) na jedné straně a číslem „2,5“ a trojúhelníkem na druhé straně.
Dodávají se v blistrech balených do krabiček, a to po 14, 28, 30, 56, 60, 98, 168 nebo 196 potahovaných tabletách nebo v jednodávkových blistrech balených do krabiček obsahujících 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních skládajících se z 10 krabiček, každá obsahující 10 x 1 potahovanou tabletu.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Bayer HealthCare Manufacturing Srl.
Via delle Groane, 126 20024 Garbagnate Milanese Itálie
Belgie / Belgique / Belgien
Lietuva
UAB Bayer
Tel: +370-5-233 68 68
Luxembourg / Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel: +36-1-487 4100
Malta
Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +4723 13 05 00 Osterreich
Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska
Bayer Sp. z o.o.
Tel: +48-22-572 35 00
Portugal
Bayer Portugal, Lda.
Tel: +351-21-416 42 00
Románia
SC Bayer SRL
Tel: +40-(0)21-528 59 00
Slovenija
Bayer d. o. o.
Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o.
Tel: +421-(0)2-59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358-(0)20-78521
Sverige
Bayer AB
Tel: +46-(0)8-580 223 00 United Kingdom
Bayer plc
Tel: +44-(0)1635-563000
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
BtnrapHH
Eaňep EtnrapHn EOOfl Ten: +359-(0)2-81 401 01 Česká republika Bayer s.r.o.
Tel: +420-266 101 111
Danmark
Bayer A/S
Tlf: +45-45 235 000
Deutschland
Bayer Vital GmbH
Tel: +49-(0)214-30 513 48
Eesti
Bayer OU
Tel: +372-655 85 65 EXláSa
Bayer ET7ág ABEE Tn^: +30-210-618 75 00 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: + 385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353-(0)1-2999 313
Ísland
Icepharma hf.
Sími: +354-540 80 00 Italia
Bayer S.p.A.
Tel: +39-02-3978 1 Kúnpoq
NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer
Tel: +371-67 84 55 63
Tato příbalová informace byla naposledy revidována: {MM.RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Xarelto 10 mg potahované tablety
rivaroxabanum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Xarelto a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
3. Jak se Xarelto užívá
4. Možné nežádoucí účinky
5. Jak přípravek Xarelto uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xarelto a k čemu se používá
Xarelto obsahuje léčivou látku rivaroxaban a používá se u dospělých k zabránění vzniku krevních sraženin v žilách po operativní náhradě kyčelního nebo kolenního kloubu. Lékař Vám tento lék předepsal, protože po operaci máte zvýšené riziko tvorby krevních sraženin.
Xarelto patří do skupiny léků nazývaných antitrombotika. Účinkuje tak, že blokuje faktor krevní srážlivosti (faktor Xa), čímž snižuje i tvorbu krevních sraženin.
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
Neužívejte přípravek Xarelto
- jestliže jste alergický(á) na rivaroxaban nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže silně krvácíte
- jestliže máte onemocnění nebo postižení některého orgánu, které zvyšují riziko závažného krvácení (například vřed žaludku, poranění nebo krvácení v mozku, nedávno prodělanou operaci mozku nebo očí)
- jestliže užíváte léky, které brání srážení krve, například warfarin, dabigatran, apixaban nebo heparin), s výjimkou situací provázených změnou antikoagulační léčby nebo při podávání heparinu zavedeným katetrem nebo kanylou v žíle nebo tepně, aby byla zachována jejich průchodnost
- jestliže máte onemocnění jater, které vede ke zvýšenému riziku krvácení
- jestliže jste těhotná nebo kojíte
Xarelto neužívejte a informujte lékaře, pokud máte některou z výše uvedených komplikací.
Upozornění a opatření
Před užitím přípravku Xarelto se poraďte se svým lékařem nebo lékárníkem.
Zvláštní opatrnosti při použití přípravku Xarelto je zapotřebí
- pokud máte zvýšené riziko krvácení, které se může vyskytnout v situacích, jako například:
■ středně závažné nebo těžké onemocnění ledvin, protože funkce ledvin může ovlivnit množství léku ve Vašem těle
■ Jestliže užíváte jiné léky, které brání srážení krve (např. warfarin, dabigatran, apixaban nebo heparin), při změně antikoagulační léčby nebo při podávání heparinu zavedeným katetrem nebo kanylou v žíle nebo tepně, aby byla zachována jejich průchodnost (viz bod „Další léčivé přípravky a přípravek Xarelto”)
■ krvácivé poruchy
■ velmi vysoký krevní tlak, neupravený léčbou
■ onemocnění žaludku nebo střeva, která mohou mít za následek krvácení, např. zánět střev nebo žaludku nebo zánět jícnu, způsobený např. refluxní chorobou (onemocnění, při kterém se žaludeční kyselina dostává nahoru do jícnu)
■ problém s cévami na očním pozadí (retinopatie)
■ onemocnění plic, při kterém jsou průdušky rozšířené a vyplněné hnisem (bronchiektázie), nebo předchozí výskyt krvácení z plic
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře ještě předtím, než začnete Xarelto
užívat. Lékař rozhodne, zda máte být léčen(a) tímto léčivým přípravkem a zda máte být pečlivě sledován(a).
Pokud vám byl při operaci zaveden katetr do páteře nebo jste do ní dostali injekci (například k epidurální či spinální anestezii nebo ke snížení bolesti):
■ je velmi důležité užívat Xarelto přesně v časech stanovených lékařem.
■ informujte ihned svého lékaře, pokud po ukončení anestezie zjistíte necitlivost nebo slabost dolních končetin nebo potíže se střevy nebo močovým měchýřem, protože potřebujete okamžitou lékařskou péči.
Děti a dospívající
Přípravek Xarelto se nedoporučuje jedincům ve věku do 18 let. O použití tohoto přípravku u dětí a dospívajících není k dispozici dostatek informací.
Další léčivé přípravky a přípravek Xarelto
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste užíval(a) v nedávné době, nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
- Jestliže užíváte:
■ některé léky proti plísňovým infekcím (například ketokonazol, itrakonazol, vorikonazol, posakonazol) - s výjimkou léků aplikovaných pouze na kůži
■ některé antivirové léky proti HIV a AIDS (například ritonavir)
■ jiné léky k omezení tvorby krevních sraženin (například enoxaparin, klopidogrel nebo antagonisté vitamínu K, například warfarin a acenokumarol)
■ protizánětlivé léky a léky proti bolesti (například naproxen nebo kyselina acetylsalicylová)
■ dronedaron, lék k léčbě poruch srdečního rytmu
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít ke zvýšení účinku přípravku Xarelto.
Váš lékař rozhodne, zda máte být léčen(a) tímto léčivým přípravkem a zda máte být pečlivě sledován(a). Pokud se Váš lékař domnívá, že u Vás existuje zvýšené riziko vzniku vředů žaludku nebo střeva, může u Vás rovněž použít preventivní protivředovou léčbu.
- Jestliže užíváte:
■ některé léky na léčbu epilepsie (fenytoin, karbamazepin, fenobarbital),
■ Třezalku (hypericum perforatum), rostlinný přípravek na depresi,
■ rifampicin, antibiotikum.
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít k zeslabení účinku přípravku Xarelto.
Váš lékař rozhodne, zda máte být léčen(a) přípravkem Xarelto a zda máte být pečlivě sledován(a).
Těhotenství a kojení
Xarelto neužívejte, jestliže jste těhotná nebo kojíte. Pokud byste mohla otěhotnět, používejte během léčby přípravkem Xarelto spolehlivou antikoncepci. Pokud během léčby tímto léčivým přípravkem otěhotníte, ihned informujte lékaře. Ten pak rozhodne o další léčbě.
Řízení dopravních prostředků a obsluha strojů
Přípravek Xarelto může způsobovat závratě (častý nežádoucí účinek) nebo mdloby (méně častý nežádoucí účinek) (viz bod 4 „Možné nežádoucí účinky“). Pokud zaznamenáte tyto příznaky, nesmíte řídit vozidla a obsluhovat stroje.
Přípravek Xarelto obsahuje laktózu.
Pokud vám váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se Xarelto užívá
Vždy užívejte tento léčivý přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik přípravku užívat
Doporučená dávka přípravku je j edna tableta (10 mg) denně.
Tabletu pokud možno zapíjejte vodou.
Xarelto lze užívat při jídle nebo nezávisle na jídle.
Pokud máte obtíže polknout celou tabletu, poraďte se s lékařem o dalších možnostech, jak užívat přípravek Xarelto. Tableta může být rozdrcena a smíchána s vodou nebo jablečným pyré, bezprostředně před tím, než ji užijete.
Je-li to nutné, lékař Vám také může podat rozdrcenou tabletu přípravku Xarelto žaludeční sondou.
Jak se Xarelto užívá
První tabletu užijte 6 - 10 hodin po operaci.
Poté užívejte jednu tabletu denně, dokud vám lékař neřekne, abyste léčbu ukončil(a).
Tablety užívejte ve stejnou denní dobu - snáze si na užívání vzpomenete.
Pokud jste absolvoval(a) rozsáhlou operaci kyčle, budete pravděpodobně tablety užívat 5 týdnů.
Pokud j ste absolvoval(a) rozsáhlou operaci kolena, budete pravděpodobně tablety užívat 2 týdny.
Jestliže jste užil(a) více přípravku Xarelto, než jste měl(a)
Pokud jste užil(a) příliš mnoho tablet, kontaktujte ihned svého lékaře. Nadměrné množství přípravku Xarelto zvyšuje riziko krvácení.
Jestliže jste zapomněl(a) užít přípravek Xarelto
Pokud jednu dávku vynecháte, užijte ji, jakmile si vzpomenete. Další tabletu užijte následující den, a poté pokračujte v užívání tablet jednou denně jako dřív.
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou tabletu.
Jestliže jste přestal(a) užívat přípravek Xarelto
Užívání přípravku nepřerušujte bez předchozí konzultace s lékařem, protože brání vzniku závažných komplikací.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i přípravek Xarelto nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Stejně jako jiné podobné léky (antitrombotika), může i Xarelto způsobit krvácení, které může být potenciálně životu nebezpečné. Nadměrné krvácení může vést k náhlému poklesu krevního tlaku (šok).
V některých případech toto krvácení nemusí být zřejmé.
Možné nežádoucí účinky, které mohou být známkou krvácení:
Ihned informujte lékaře, jestliže se u Vás projeví některý z následujících nežádoucích účinků:
- dlouhotrvající nebo rozsáhlé krvácení
- výjimečná slabost, únava, bledost, závratě, bolesti hlavy, otok z neznámých příčin, dušnost, bolesti na hrudníku nebo angina pectoris, protože to mohou být známky krvácení.
Lékař vás možná bude chtít pečlivě sledovat, nebo změní způsob léčby.
Seznam možných nežádoucích účinků:
Časté (mohou postihovat až 1 osobu z 10):
- krvácení v žaludku nebo střevech, z močopohlavního traktu (včetně výskytu krve v moči a silného menstruačního krvácení), krvácení z nosu a z dásní
- krvácení do oka (včetně krvácení do očního bělma)
- krvácení do tkáně nebo tělesné dutiny (modřiny, podlitiny)
- vykašlávání krve
- krvácení z kůže nebo do kůže- krvácení po operaci
- vytékání krve nebo tekutiny z operační rány
- otoky končetin
- bolest končetin
- horečka
- snížení počtu červených krvinek, což může způsobit bledost kůže a slabost nebo dušnost
- bolesti žaludku, poruchy trávení, pocit nevolnosti nebo nevolnost, zácpa, průjem
- nízký krevní tlak (příznaky mohou být pocity závratě nebo mdloby při vstávání)
- pokles celkové síly a energie (slabost, únava), bolesti hlavy, závratě
- vyrážka, svědění kůže
- porucha funkce ledvin (může se prokázat na základě testů prováděných Vaším lékařem)
- zvýšení hodnot některých jaterních enzymů v krevních testech
Méně časté (mohou postihovat až 1 osobu ze 100):
- krvácení do mozku nebo lebeční dutiny
- krvácení do kloubu, které vede k bolesti a otoku kloubu
- mdloby
- obecně se necítit dobře
- pocit sucha v ústech
- zrychlený srdeční tep
- alergické reakce, včetně alergických kožních reakcí
- kopřivka
- poškozená funkce jater (může se prokázat na základě testů prováděných vaším lékařem)
- krevní testy mohou ukázat zvýšení bilirubinu, některých pankreatických nebo jatemích enzymů nebo počtu krevních destiček
Vzácné (mohou postihovat až 1 osobu z 1 000):
- krvácení do svalů
- místní otok
- žloutnutí kůže a očí (žloutenka)
- nahromadění krve (hematom) v tříslech jako komplikace srdečního výkonu, při kterém je katetr zaveden do tepny na dolní končetině (pseudoaneuryzma)
Není známo (frekvenci z dostupných údajů nelze určit)
- zvýšený tlak uvnitř svalů na nohách nebo pažích vzniklý po krvácení, který vede k bolesti, otoku, poruše citlivosti, necitlivosti nebo obrně (kompartment syndrom po krvácení)
- selhání ledvin po těžkém krvácení
Následující nežádoucí účinky byly hlášeny po schválení registrace tohoto léčivého přípravku:
- angioedém a alergický otok (otékání tváře, rtů, úst, jazyka nebo hrdla)
- cholestáza (snížení toku žluči), hepatitida včetně hepatocelulárního poškození (zánět jater včetně poškození jater)
- trombocytopenie (snížené množství krevních destiček, což jsou buňky, které pomáhají srážení krve) Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Xarelto uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a každém blistru za „EXP". Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Xarelto obsahuje
- Léčivou látkou je rivaroxabanum. Jedna tableta obsahuje rivaroxabanum 10 mg.
- Pomocnými látkami jsou:
Jádro tablety: mikrokrystalická celulóza, sodná sůl kroskarmelózy, monohydrát laktózy, hypromelóza, natrium-lauryl-sulfát, magnesium-stearát.
Potah tablety: makrogol 3350, hypromelóza, oxid titaničitý (E171), červený oxid železitý (E172).
Jak přípravek Xarelto vypadá a co obsahuje toto balení
Xarelto 10 mg potahované tablety jsou světle červené, kulaté, bikonvexní a označené logem (kříž) BAYER na jedné straně a číslem „10“ a trojúhelníkem na druhé straně. Dodávají se v blistrech balených do krabiček, a to po 5, 10 nebo 30 potahovaných tabletách nebo v jednodávkových blistrech balených do krabiček
obsahujících 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních skládajících se z 10 krabiček, každá obsahující 10 x 1 potahovanou tabletu.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Bayer HealthCare Manufacturing Srl. Via delle Groane, 126 20024 Garbagnate Milanese Itálie
Belgie / Belgique / Belgien
Lietuva
UAB Bayer
Tel: +370-5-233 68 68
Luxembourg / Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel: +36-1-487 4100
Malta
Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +4723 13 05 00 Osterreich
Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska
Bayer Sp. z o.o.
Tel: +48-22-572 35 00
Portugal
Bayer Portugal, Lda.
Tel: +351-21-416 42 00
Románia
SC Bayer SRL
Tel: +40-(0)21-528 59 00
Slovenija
Bayer d. o. o.
Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o.
Tel: +421-(0)2-59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358-(0)20-78521
Sverige
Bayer AB
Tel: +46-(0)8-580 223 00 United Kingdom
Bayer plc
Tel: +44-(0)1635-563000
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
BtnrapHH
Eaňep EtnrapHn EOOfl Ten: +359-(0)2-81 401 01 Česká republika Bayer s.r.o.
Tel: +420-266 101 111
Danmark
Bayer A/S
Tlf: +45-45 235 000
Deutschland
Bayer Vital GmbH
Tel: +49-(0)214-30 513 48
Eesti
Bayer OU
Tel: +372-655 85 65 EXláSa
Bayer ET7ág ABEE Tn^: +30-210-618 75 00 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: + 385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353-(0)1-2999 313
Ísland
Icepharma hf.
Sími: +354-540 80 00 Italia
Bayer S.p.A.
Tel: +39-02-3978 1 Kúnpoq
NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer
Tel: +371-67 84 55 63
Tato příbalová informace byla naposledy revidována: {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Xarelto 15 mg potahované tablety Xarelto 20 mg potahované tablety
rivaroxabanum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Xarelto a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
3. Jak se Xarelto užívá
4. Možné nežádoucí účinky
5. Jak přípravek Xarelto uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xarelto a k čemu se používá
Xarelto obsahuje léčivou látku rivaroxaban a používá se u dospělých k:
- zabránění vzniku krevních sraženin v mozku (cévní mozková příhoda) a v dalších krevních cévách v těle, pokud máte typ arytmie (nepravidelného srdečního rytmu) označovaný jako nevalvulární fibrilace síní.
- léčbě krevních sraženin v žilách dolních končetin (hluboká žilní trombóza) a v krevních cévách plic (plicní embolie) a k prevenci vzniku opakovaných krevních sraženin v krevních cévách dolních končetin a/nebo plic.
Xarelto patří do skupiny léků nazývaných antitrombotika. Účinkuje tak, že blokuje faktor krevní srážlivosti
(faktor Xa), čímž snižuje i tvorbu krevních sraženin.
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
Neužívejte přípravek Xarelto
- jestliže jste alergický(á) na rivaroxaban nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže silně krvácíte
- jestliže máte onemocnění nebo postižení některého orgánu, které zvyšuje riziko závažného krvácení (např. žaludeční vřed, poranění nebo krvácení v mozku, nedávný chirurgický výkon na mozku nebo očích)
- jestliže užíváte léky zabraňující srážení krve (například warfarin, dabigatran, apixaban nebo heparin), s výjimkou změny antikoagulační léčby nebo pokud dostáváte heparin přes žilní nebo tepenný katetr
(hadičku) k udržení jeho průchodnosti
- jestliže máte onemocnění jater, které vede ke zvýšenému riziku krvácení
- j estliže j ste těhotná nebo koj íte
Xarelto neužívejte a informujte lékaře, pokud máte některou z výše uvedených komplikací.
Upozornění a opatření
Před užitím přípravku Xarelto se poraďte se svým lékařem nebo lékarníkem.
Zvláštní opatrnosti při použití přípravku Xarelto je zapotřebí
- pokud máte zvýšené riziko krvácení, které se může vyskytnout v situacích, jako například:
■ závážné onemocnění ledvin, protože funkce ledvin může ovlivnit množství léku ve Vašem těle
■ pokud užíváte jiné léky bránící srážení krve (například warfarin, dabigatran, apixaban nebo heparin), při změně antikoagulační léčby nebo pokud dostáváte heparin přes žilní nebo tepenný katetr (hadičku) k udržení jeho průchodnosti (viz bod „Další léčivé přípravky a přípravek Xarelto“)
■ krvácivé poruchy
■ velmi vysoký krevní tlak, neupravený léčbou
■ onemocnění žaludku nebo střeva, která mohou mít za následek krvácení, např. zánět střev nebo žaludku nebo zánět jícnu, způsobený např. refluxní chorobou (onemocnění, při kterém se žaludeční kyselina dostává nahoru do jícnu)
■ problém s cévami na očním pozadí (retinopatie)
■ onemocnění plic, při kterém jsou průdušky rozšířené a vyplněné hnisem (bronchiektázie) nebo předchozí výskyt krvácení z plic
- pokud máte umělou srdeční chlopeň
- pokud lékař rozhodne, že je Váš krevní tlak nestabilní nebo je plánována jiná léčba nebo chirurgický
zákrok k odstranění krevní sraženiny z Vašich plic
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře ještě předtím než začnete Xarelto užívat. Lékař rozhodne, zda máte být léčen(a) tímto léčivým přípravkem a zda máte být pečlivě sledován(a).
Pokud musíte podstoupit operaci:
- je velmi důležité, abyste užíval(a) přípravek Xarelto před a po operaci přesně v době, kdy Vám to řekl Váš lékař.
- Pokud při operaci bude použit katetr nebo injekce do páteřního kanálu (například při epidurální nebo spinální anestezii nebo k tlumení bolesti):
■ je velmi důležité užívat Xarelto před injekcí a po injekci nebo odstranění katetru přesně tak, jak Vám lékař řekl
■ okamžitě informujte svého lékaře, pokud zaznamenáte po anestezii necitlivost nebo slabost dolních končetin nebo střevní potíže anebo potíže s močovým měchýřem, protože je třeba okamžitá léčba.
Děti a dospívající
Přípravek Xarelto se nedoporučuje jedincům ve věku do 18 let. O použití tohoto přípravku u dětí a dospívajících není k dispozici dostatek informací.
Další léčivé přípravky a přípravek Xarelto
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Jestliže užíváte:
■ některé léky proti plísňovým infekcím (například ketokonazol, itrakonazol, vorikonazol, posakonazol), s výjimkou léků aplikovaných pouze na kůži
■ některé antivirové léky proti HIV a AIDS (například ritonavir)
■ jiné léky k omezení tvorby krevních sraženin (například enoxaparin, klopidogrel nebo antagonisté vitaminu K, jako je warfarin a acenokumarol)
■ protizánětlivé léky a léky proti bolesti (například naproxen nebo kyselina acetylsalicylová)
■ dronedaron, lék k léčbě poruch srdečního rytmu
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít ke zvýšení jeho účinku.
Váš lékař rozhodne, zda máte být léčen(a) tímto léčivým přípravkem a zda máte být pečlivě sledován(a).
Pokud se Váš lékař domnívá, že u Vás existuje zvýšené riziko vzniku vředů žaludku nebo střeva, může rovněž použít preventivní protivředovou léčbu.
Jestliže užíváte:
■ některé léky na léčbu epilepsie (fenytoin, karbamazepin, fenobarbital),
■ třezalku (Hypericum perforatum), rostlinný přípravek na depresi,
■ rifampicin, antibiotikum.
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít k zeslabení účinku přípravku Xarelto.
Lékař rozhodne, zda máte být léčen(a) přípravkem Xarelto a zda máte být pečlivě sledován(a).
Těhotenství a kojení
Xarelto neužívejte, jestliže jste těhotná nebo kojíte. Pokud byste mohla otěhotnět, používejte během léčby přípravkem Xarelto spolehlivou antikoncepci. Pokud během léčby tímto léčivým přípravkem otěhotníte, ihned informujte lékaře. Ten pak rozhodne o další léčbě.
Řízení dopravních prostředků a obsluha strojů
Přípravek Xarelto může způsobovat závratě (častý nežádoucí účinek) nebo mdloby (méně častý nežádoucí účinek) (viz bod 4 „Možné nežádoucí účinky“). Pokud zaznamenáte tyto příznaky, nesmíte řídit vozidla a obsluhovat stroje.
Přípravek Xarelto obsahuje laktózu
Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se Xarelto užívá
Vždy užívejte tento léčivý přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik přípravku užívat
- K prevenci krevních sraženin v mozku (cévní mozková příhoda) a dalších krevních cévách v těle doporučená dávka je jedna 20 mg tableta jednou denně.
Pokud máte onemocnění ledvin, může být dávka snížena na jednu 15 mg tabletu jednou denně.
- K léčbě krevních sraženin v žilách dolních končetin a k léčbě krevních sraženin v cévách plic a k prevenci opakovaného výskytu krevních sraženin
doporučená dávka je jedna 15 mg tableta dvakrát denně po dobu prvních 3 týdnů. Poté je doporučená dávka jedna 20 mg tableta jednou denně.
Pokud máte onemocnění ledvin, může se lékař rozhodnout po třech týdnech snížit dávku na jednu 15 mg tabletu jednou denně, jestliže je riziko krvácení vyšší než riziko vzniku další sraženiny.
Tabletu/tablety pokud možno zapíjejte vodou.
Přípravek Xarelto užívejte s jídlem.
Pokud máte obtíže polknout celou tabletu, poraďte se s lékařem o dalších možnostech, jak užívat přípravek Xarelto. Tableta může být rozdrcena a smíchána s vodou nebo jablečným pyré, bezprostředně před tím, než ji užijete. Poté by ihned mělo následovat požití jídla.
Je-li to nutné, lékař Vám také může podat rozdrcenou tabletu přípravku Xarelto žaludeční sondou.
Kdy se Xarelto užívá
Užívejte tabletu/tablety denně, dokud Vám lékař neřekne, abyste léčbu ukončil(a).
Tabletu/tablety užívejte ve stejnou denní dobu, což umožní, že si snáze na užívání vzpomenete.
Váš lékař se rozhodne, jak dlouho bude léčba trvat.
Prevence krevních sraženin v mozku (mrtvice) a v ostatních cévách těla:
Pokud srdeční akce srdce musí být převedena na normální hodnoty postupem zvaným kardioverze, užívejte Xarelto v časových intervalech podle pokynů svého lékaře.
Jestliže jste užil(a) více přípravku Xarelto, než jste měl(a)
Pokud jste užil(a) příliš mnoho tablet, kontaktujte ihned svého lékaře. Nadměrné množství přípravku Xarelto zvyšuje riziko krvácení.
Jestliže jste zapomněl(a) užít přípravek Xarelto
- Pokud užíváte jednu 20 mg nebo jednu 15 mg tabletu jednou denně a zapomněl(a) jste užít dávku, užijte ji co nejdříve si vzpomenete. Neužívejte více než jednu tabletu denně, abyste nahradil(a) zapomenutou dávku. Další tabletu užijte následující den, a poté pokračujte v užívání tablet jednou denně.
- Pokud užíváte jednu 15 mg tabletu dvakrát denně a vynechal(a) jste dávku, užijte ji co nejdříve si vzpomenete. Neužívejte více než dvě 15 mg tablety během jednoho dne. Jestliže zapomenete užít jednu dávku, můžete užít dvě 15 mg tablety najednou, aby bylo dosaženo celkového množství dvou tablet (30 mg) v jednom dni. Následující den pokračujte v užívání jedné 15 mg tablety dvakrát denně.
Jestliže jste přestal(a) užívat přípravek Xarelto
Užívání přípravku nepřerušujte bez předchozí konzultace s lékařem, protože léčí a zabraňuje vzniku závažných komplikací.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i přípravek Xarelto nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Stejně jako jiné podobné léky (antitrombotika), může i Xarelto způsobit krvácení, které může být potenciálně životu nebezpečné. Nadměrné krvácení může vést k náhlému poklesu krevního tlaku (šok).
V některých případech toto krvácení nemusí být zřejmé.
Možné nežádoucí účinky, které mohou být známkou krvácení:
Ihned informujte lékaře, jestliže se u Vás projeví některý z následujících nežádoucích účinků:
- dlouhotrvající nebo rozsáhlé krvácení
- výjimečná slabost, únava, bledost, závratě, bolesti hlavy, otok z neznámých příčin, dušnost, bolesti na hrudníku nebo angina pectoris, což mohou být známky krvácení.
Váš lékař Vás možná bude chtít pečlivě sledovat, nebo změní způsob léčby.
Souhrnný seznam možných nežádoucích účinků:
Časté (mohou postihovat až 1 osobu z 10):
- krvácení v žaludku nebo střevech, krvácení v močovém a pohlavním ústrojí (včetně krve v moči a silného menstruačního krvácení), krvácení z nosu, krvácení z dásní
- krvácení do oka (včetně krvácení do očního bělma)
- krvácení do tkání nebo tělesných dutin (podlitiny, modřiny)
- vykašlávání krve
- krvácení do kůže a pod kůži
- krvácení po operaci
- vytékání krve nebo tekutiny z operační rány
- otok dolních končetin
- bolest dolních končetin
- horečka
- snížení počtu červených krvinek, což může způsobit bledost kůže a slabost nebo dušnost
- bolesti žaludku, poruchy trávení, pocit nevolnosti, zácpa, průjem
- nízký krevní tlak (příznaky mohou zahrnovat závratě nebo mdloby při vstávání)
- pokles celkové síly a energie (slabost, únava), bolesti hlavy, závratě, mdloby
- vyrážka, svědění kůže
- porucha funkce ledvin (může se zjistit z testů, které lékař provede)
- krevní testy mohou ukázat zvýšení některých jaterních testů
Méně časté (mohou postihovat až 1 osobu ze 100):
- krvácení do mozku nebo lebeční dutiny
- krvácení do kloubu vedoucí k bolesti a otoku
- mdloby
- obecně se necítit dobře
- sucho v ústech
- zrychlený srdeční tep
- alergické reakce, včetně alergických kožních reakcí
- kopřivka
- porucha funkce jater (může být zjištěna při vyšetření prováděném lékařem)
- vyšetření krve může prokázat zvýšení hladiny bilirubinu, některých enzymů slinivky břišní nebo jater nebo počtu krevních destiček
Vzácné (mohou postihovat až 1 osobu z 1000):
- krvácení do svalů
- lokalizovaný otok
- zežloutnutí kůže a očí (žloutenka)
- výron krve (hematom) v tříslech jako komplikace srdečního výkonu, při kterém je katetr zaveden do tepny na dolní končetině (pseudoaneuryzma)
Není známo (frekvenci z dostupných údajů nelze určit)
- zvýšený tlak uvnitř svalů na nohách nebo pažích vzniklý po krvácení, který vede k bolesti, otoku, poruše citlivosti, necitlivosti nebo obrně (kompartment syndrom po krvácení)
- selhání ledvin po těžkém krvácení
Následující nežádoucí účinky byly hlášeny po schválení registrace tohoto léčivého přípravku:
- angioedém a alergický otok (otékání tváře, rtů, úst, jazyka nebo hrdla)
- cholestáza (snížení toku žluči), hepatitida včetně hepatocelulárního poškození (zánět jater včetně poškození jater)
- trombocytopenie (snížené množství krevních destiček, což jsou buňky, které pomáhají srážení krve) Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xarelto uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a každém blistru za „EXP". Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Xarelto obsahuje
- Léčivou látkou je rivaroxabanum. Jedna tableta obsahuje rivaroxabanum 15 mg nebo 20 mg.
- Pomocnými látkami jsou:
Jádro tablety: mikrokrystalická celulóza, sodná sůl kroskarmelózy, monohydrát laktózy, hypromelóza, natrium-lauryl-sulfát, magnesium-stearát.
Potah tablety: makrogol 3350, hypromelóza, oxid titaničitý (E 171), červený oxid železitý (E 172). Jak Xarelto vypadá a co obsahuje toto balení
Xarelto 15 mg potahované tablety jsou červené, kulaté, bikonvexní a označené logem (kříž) BAYER na jedné straně a číslem „15“ a trojúhelníkem na druhé straně. Dodávají se v blistrech balených do krabiček, a to po 10, 14, 28, 42 nebo 98 potahovaných tabletách nebo v jednodávkových blistrech, balených do krabiček obsahujících 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních skládajících se z 10 krabiček, každá obsahující 10 x 1 potahovanou tabletu nebo v lahvičkách obsahujících 100 potahovaných tablet.
Xarelto 20 mg potahované tablety jsou hnědo-červené, kulaté, bikonvexní a označené logem (kříž) BAYER na jedné straně a číslem „20“ a trojúhelníkem na druhé straně.
Dodávají se v blistrech balených do krabiček, a to po 10, 14, 28 nebo 98 potahovaných tabletách nebo v jednodávkových blistrech, balených do krabiček obsahujících 10 x 1 nebo 100 x 1 potahovanou tabletu nebo v multibaleních skládajících se z 10 krabiček, každá obsahující 10 x 1 potahovanou tabletu nebo v lahvičkách obsahujících 100 potahovaných tablet.
Na trhu nemusí být všechny velikosti balení.
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Bayer HealthCare Manufacturing Srl. Via delle Groane, 126 20024 Garbagnate Milanese Itálie
Belgie / Belgique / Belgien
Lietuva
UAB Bayer
Tel: +370-5-233 68 68
Luxembourg / Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel: +36-1-487 4100
Malta
Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +4723 13 05 00 Osterreich
Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska
Bayer Sp. z o.o.
Tel: +48-22-572 35 00
Portugal
Bayer Portugal, Lda.
Tel: +351-21-416 42 00
Románia
SC Bayer SRL
Tel: +40-(0)21-528 59 00
Slovenija
Bayer d. o. o.
Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o.
Tel: +421-(0)2-59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358-(0)20-78521
Sverige
Bayer AB
Tel: +46-(0)8-580 223 00 United Kingdom
Bayer plc
Tel: +44-(0)1635-563000
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
BtnrapHH
Eaňep EtnrapHn EOOfl Ten: +359-(0)2-81 401 01 Česká republika Bayer s.r.o.
Tel: +420-266 101 111
Danmark
Bayer A/S
Tlf: +45-45 235 000
Deutschland
Bayer Vital GmbH
Tel: +49-(0)214-30 513 48
Eesti
Bayer OU
Tel: +372-655 85 65 EXláSa
Bayer ET7ág ABEE Tn^: +30-210-618 75 00 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: + 385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353-(0)1-2999 313
Ísland
Icepharma hf.
Sími: +354-540 80 00 Italia
Bayer S.p.A.
Tel: +39-02-3978 1 Kúnpoq
NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer
Tel: +371-67 84 55 63
Tato příbalová informace byla naposledy revidována: {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Xarelto 15 mg potahované tablety Xarelto 20 mg potahované tablety
Balení pro zahájení léčby
rivaroxabanum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Xarelto a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
3. Jak se Xarelto užívá
4. Možné nežádoucí účinky
5. Jak přípravek Xarelto uchovávat
6. Obsah balení a další informace
1. Co je přípravek Xarelto a k čemu se používá
Xarelto obsahuje léčivou látku rivaroxaban a používá se u dospělých k:
- léčbě krevních sraženin v žilách dolních končetin (hluboká žilní trombóza) a v krevních cévách plic (plicní embolie) a k prevenci vzniku opakovaných krevních sraženin v krevních cévách dolních končetin a/nebo plic.
Xarelto patří do skupiny léků nazývaných antitrombotika. Účinkuje tak, že blokuje faktor krevní srážlivosti (faktor Xa), čímž snižuje i tvorbu krevních sraženin.
2. Čemu musíte věnovat pozornost, než začnete Xarelto užívat
Neužívejte přípravek Xarelto
- jestliže jste alergický(á) na rivaroxaban nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže silně krvácíte
- jestliže máte onemocnění nebo postižení některého orgánu, které zvyšuje riziko závažného krvácení (např. žaludeční vřed, poranění nebo krvácení v mozku, nedávný chirurgický výkon na mozku nebo očích)
- jestliže užíváte léky zabraňující srážení krve (například warfarin, dabigatran, apixaban nebo heparin), s výjimkou změny antikoagulační léčby nebo pokud dostáváte heparin přes žilní nebo tepenný katetr
(hadičku) k udržení jeho průchodnosti
- jestliže máte onemocnění jater, které vede ke zvýšenému riziku krvácení
- j estliže j ste těhotná nebo koj íte
Xarelto neužívejte a informujte lékaře, pokud máte některou z výše uvedených komplikací.
Upozornění a opatření
Před užitím přípravku Xarelto se poraďte se svým lékařem nebo lékarníkem.
Zvláštní opatrnosti při použití přípravku Xarelto je zapotřebí
- pokud máte zvýšené riziko krvácení, které se může vyskytnout v situacích, jako například:
■ závážné onemocnění ledvin, protože funkce ledvin může ovlivnit množství léku ve Vašem těle
■ pokud užíváte jiné léky bránící srážení krve (například warfarin, dabigatran, apixaban nebo heparin), při změně antikoagulační léčby nebo pokud dostáváte heparin přes žilní nebo tepenný katetr (hadičku) k udržení jeho průchodnosti (viz bod „Další léčivé přípravky a přípravek Xarelto“)
■ krvácivé poruchy
■ velmi vysoký krevní tlak, neupravený léčbou
■ onemocnění žaludku nebo střeva, která mohou mít za následek krvácení, např. zánět střev nebo žaludku nebo zánět jícnu, způsobený např. refluxní chorobou (onemocnění, při kterém se žaludeční kyselina dostává nahoru do jícnu)
■ problém s cévami na očním pozadí (retinopatie)
■ onemocnění plic, při kterém jsou průdušky rozšířené a vyplněné hnisem (bronchiektázie) nebo předchozí výskyt krvácení z plic
- pokud máte umělou srdeční chlopeň
- pokud lékař rozhodne, že je Váš krevní tlak nestabilní nebo je plánována jiná léčba nebo chirurgický zákrok k odstranění krevní sraženiny z Vašich plic
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře ještě předtím než začnete Xarelto užívat. Lékař rozhodne, zda máte být léčen(a) tímto léčivým přípravkem a zda máte být pečlivě sledován(a).
Pokud musíte podstoupit operaci:
- je velmi důležité, abyste užíval(a) přípravek Xarelto před a po operaci přesně v době, kdy Vám to řekl Váš lékař.
- Pokud při operaci bude použit katetr nebo injekce do páteřního kanálu (například při epidurální nebo spinální anestezii nebo k tlumení bolesti):
■ je velmi důležité užívat Xarelto před injekcí a po injekci nebo odstranění katetru přesně tak, jak Vám lékař řekl
■ okamžitě informujte svého lékaře, pokud zaznamenáte po anestezii necitlivost nebo slabost dolních končetin nebo střevní potíže anebo potíže s močovým měchýřem, protože je třeba okamžitá léčba.
Děti a dospívající
Přípravek Xarelto se nedoporučuje jedincům ve věku do 18 let. O použití tohoto přípravku u dětí a dospívajících není k dispozici dostatek informací.
Další léčivé přípravky a přípravek Xarelto
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Jestliže užíváte:
■ některé léky proti plísňovým infekcím (například ketokonazol, itrakonazol, vorikonazol, posakonazol), s výjimkou léků aplikovaných pouze na kůži
■ některé antivirové léky proti HIV a AIDS (například ritonavir)
■ jiné léky k omezení tvorby krevních sraženin (například enoxaparin, klopidogrel nebo antagonisté vitaminu K, jako je warfarin a acenokumarol)
■ protizánětlivé léky a léky proti bolesti (například naproxen nebo kyselina acetylsalicylová)
■ dronedaron, lék k léčbě poruch srdečního rytmu
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít ke zvýšení jeho účinku.
Váš lékař rozhodne, zda máte být léčen(a) tímto léčivým přípravkem a zda máte být pečlivě sledován(a).
Pokud se Váš lékař domnívá, že u Vás existuje zvýšené riziko vzniku vředů žaludku nebo střeva, může rovněž použít preventivní protivředovou léčbu.
Jestliže užíváte:
■ některé léky na léčbu epilepsie (fenytoin, karbamazepin, fenobarbital),
■ třezalku (Hypericum perforatum), rostlinný přípravek na depresi,
■ rifampicin, antibiotikum.
Pokud se Vás cokoli z výše uvedeného týká, informujte svého lékaře před zahájením užívání přípravku Xarelto, protože může dojít k zeslabení účinku přípravku Xarelto.
Lékař rozhodne, zda máte být léčen(a) přípravkem Xarelto a zda máte být pečlivě sledován(a).
Těhotenství a kojení
Xarelto neužívejte, jestliže jste těhotná nebo kojíte. Pokud byste mohla otěhotnět, používejte během léčby přípravkem Xarelto spolehlivou antikoncepci. Pokud během léčby tímto léčivým přípravkem otěhotníte, ihned informujte lékaře. Ten pak rozhodne o další léčbě.
Řízení dopravních prostředků a obsluha strojů
Přípravek Xarelto může způsobovat závratě (častý nežádoucí účinek) nebo mdloby (méně častý nežádoucí účinek) (viz bod 4 „Možné nežádoucí účinky“). Pokud zaznamenáte tyto příznaky, nesmíte řídit vozidla a obsluhovat stroje.
Přípravek Xarelto obsahuje laktózu
Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se Xarelto užívá
Vždy užívejte tento léčivý přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik přípravku užívat
Doporučená dávka je jedna 15mg tableta dvakrát denně po dobu prvních 3 týdnů. Poté je doporučená dávka jedna 20mg tableta jednou denně.
Toto balení pro zahájení léčby přípravku Xarelto 15 mg a 20 mg je určeno pouze pro první 4 týdny léčby.
Po dokončení užívání tohoto úvodního balení bude léčba dále pokračovat užíváním přípravku Xarelto 20 mg jednou denně tak, jak určil Váš lékař.
Pokud máte onemocnění ledvin, může se lékař rozhodnout po třech týdnech snížit dávku na jednu 15 mg tabletu jednou denně, jestliže je riziko krvácení vyšší než riziko vzniku další sraženiny.
Tabletu/tablety pokud možno zapíjejte vodou. Přípravek Xarelto užívejte s jídlem.
Pokud máte obtíže polknout celou tabletu, poraďte se s lékařem o dalších možnostech, jak užívat přípravek Xarelto. Tableta může být rozdrcena a smíchána s vodou nebo jablečným pyré, bezprostředně před tím, než ji užijete. Poté by ihned mělo následovat požití jídla.
Je-li to nutné, lékař Vám také může podat rozdrcenou tabletu přípravku Xarelto žaludeční sondou.
Kdy se Xarelto užívá
Užívejte tabletu/tablety denně, dokud Vám lékař neřekne, abyste léčbu ukončil(a).
Tabletu/tablety užívejte ve stejnou denní dobu, což umožní, že si snáze na užívání vzpomenete.
Váš lékař se rozhodne, jak dlouho bude léčba trvat.
Jestliže jste užil(a) více přípravku Xarelto, než jste měl(a)
Pokud jste užil(a) příliš mnoho tablet, kontaktujte ihned svého lékaře. Nadměrné množství přípravku Xarelto zvyšuje riziko krvácení.
Jestliže jste zapomněl(a) užít přípravek Xarelto
- Pokud užíváte jednu 15mg tabletu dvakrát denně a vynechal(a) jste dávku, užijte ji co nejdříve si vzpomenete. Neužívejte více než dvě 15mg tablety během jednoho dne. Jestliže zapomenete užít jednu dávku, můžete užít dvě 15mg tablety najednou, aby bylo dosaženo celkového množství dvou tablet (30 mg) v jednom dni. Následující den pokračujte v užívání jedné 15mg tablety dvakrát denně.
- Pokud užíváte jednu 20mg tabletu jednou denně a zapomněl(a) jste užít dávku, užijte ji co nejdříve si vzpomenete. Neužívejte více než jednu tabletu denně, abyste nahradil(a) zapomenutou dávku. Další tabletu užijte následující den, a poté pokračujte v užívání tablet jednou denně.
Jestliže jste přestal(a) užívat přípravek Xarelto
Užívání přípravku nepřerušujte bez předchozí konzultace s lékařem, protože léčí a zabraňuje vzniku závažných komplikací.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i přípravek Xarelto nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Stejně jako jiné podobné léky (antitrombotika), může i Xarelto způsobit krvácení, které může být potenciálně životu nebezpečné. Nadměrné krvácení může vést k náhlému poklesu krevního tlaku (šok).
V některých případech toto krvácení nemusí být zřejmé.
Možné nežádoucí účinky, které mohou být známkou krvácení:
Ihned informujte lékaře, jestliže se u Vás projeví některý z následujících nežádoucích účinků:
- dlouhotrvající nebo rozsáhlé krvácení
- výjimečná slabost, únava, bledost, závratě, bolesti hlavy, otok z neznámých příčin, dušnost, bolesti na hrudníku nebo angina pectoris, což mohou být známky krvácení.
Váš lékař Vás možná bude chtít pečlivě sledovat, nebo změní způsob léčby.
Souhrnný seznam možných nežádoucích účinků:
Časté (mohou postihovat až 1 osobu z 10):
- krvácení v žaludku nebo střevech, krvácení v močovém a pohlavním ústrojí (včetně krve v moči a silného menstruačního krvácení), krvácení z nosu, krvácení z dásní
- krvácení do oka (včetně krvácení do očního bělma)
- krvácení do tkání nebo tělesných dutin (podlitiny, modřiny)
- vykašlávání krve
- krvácení do kůže a pod kůži
- krvácení po operaci
- vytékání krve nebo tekutiny z operační rány
- otok dolních končetin
- bolest dolních končetin
- horečka
- snížení počtu červených krvinek, což může způsobit bledost kůže a slabost nebo dušnost
- bolesti žaludku, poruchy trávení, pocit nevolnosti, zácpa, průjem
- nízký krevní tlak (příznaky mohou zahrnovat závratě nebo mdloby při vstávání)
- pokles celkové síly a energie (slabost, únava), bolesti hlavy, závratě, mdloby
- vyrážka, svědění kůže
- porucha funkce ledvin (může se zjistit z testů, které lékař provede)
- krevní testy mohou ukázat zvýšení některých jaterních testů
Méně časté (mohou postihovat až 1 osobu ze 100):
- krvácení do mozku nebo lebeční dutiny
- krvácení do kloubu vedoucí k bolesti a otoku
- mdloby
- obecně se necítit dobře
- sucho v ústech
- zrychlený srdeční tep
- alergické reakce, včetně alergických kožních reakcí
- kopřivka
- porucha funkce jater (může být zjištěna při vyšetření prováděném lékařem)
- vyšetření krve může prokázat zvýšení hladiny bilirubinu, některých enzymů slinivky břišní nebo jater nebo počtu krevních destiček
Vzácné (mohou postihovat až 1 osobu z 1000):
- krvácení do svalů
- lokalizovaný otok
- zežloutnutí kůže a očí (žloutenka)
- výron krve (hematom) v tříslech jako komplikace srdečního výkonu, při kterém je katetr zaveden do tepny na dolní končetině (pseudoaneuryzma)
Není známo (frekvenci z dostupných údajů nelze určit)
- zvýšený tlak uvnitř svalů na nohách nebo pažích vzniklý po krvácení, který vede k bolesti, otoku, poruše citlivosti, necitlivosti nebo obrně (kompartment syndrom po krvácení)
- selhání ledvin po těžkém krvácení
Následující nežádoucí účinky byly hlášeny po schválení registrace tohoto léčivého přípravku:
- angioedém a alergický otok (otékání tváře, rtů, úst, jazyka nebo hrdla)
- cholestáza (snížení toku žluči), hepatitida včetně hepatocelulárního poškození (zánět jater včetně poškození jater)
- trombocytopenie (snížené množství krevních destiček, což jsou buňky, které pomáhají srážení krve). Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Xarelto uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a každém blistru za „EXP". Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Xarelto obsahuje
- Léčivou látkou je rivaroxabanum. Jedna tableta obsahuje rivaroxabanum 15 mg nebo 20 mg v daném pořadí.
- Pomocnými látkami jsou:
Jádro tablety: mikrokrystalická celulóza, sodná sůl kroskarmelózy, monohydrát laktózy, hypromelóza, natrium-lauryl-sulfát, magnesium-stearát.
Potah tablety: makrogol 3350, hypromelóza, oxid titaničitý (E 171), červený oxid železitý (E 172). Jak Xarelto vypadá a co obsahuje toto balení
Xarelto 15 mg potahované tablety jsou červené, kulaté, bikonvexní a označené logem (kříž) BAYER na jedné straně a číslem „15“ a trojúhelníkem na druhé straně.
Xarelto 20 mg potahované tablety jsou hnědo-červené, kulaté, bikonvexní a označené logem (kříž) BAYER na jedné straně a číslem „20“ a trojúhelníkem na druhé straně.
4 týdenní balení pro zahájení léčby: jedno balení se 49 potahovanými tabletami pro první 4 týdny léčby obsahuje:
42 potahovaných tablet obsahujících 15 mg rivaroxabanu a 7 potahovaných tablet obsahujících 20 mg rivaroxabanu v pouzdru.
Držitel rozhodnutí o registraci
Bayer Pharma AG 13342 Berlin Německo
Výrobce
Bayer Pharma AG 51368 Leverkusen Německo
Bayer HealthCare Manufacturing Srl. Via delle Groane, 126 20024 Garbagnate Milanese Itálie
Belgie / Belgique / Belgien
Lietuva
UAB Bayer
Tel: +370-5-233 68 68
Luxembourg / Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária KFT
Tel: +36-1-487 4100
Malta
Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf: +4723 13 05 00 Osterreich
Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska
Bayer Sp. z o.o.
Tel: +48-22-572 35 00
Portugal
Bayer Portugal, Lda.
Tel: +351-21-416 42 00
Románia
SC Bayer SRL
Tel: +40-(0)21-528 59 00
Slovenija
Bayer d. o. o.
Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o.
Tel: +421-(0)2-59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358-(0)20-78521
Sverige
Bayer AB
Tel: +46-(0)8-580 223 00 United Kingdom
Bayer plc
Tel: +44-(0)1635-563000
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
BtnrapHH
Eaňep EtnrapHn EOOfl Ten: +359-(0)2-81 401 01 Česká republika Bayer s.r.o.
Tel: +420-266 101 111
Danmark
Bayer A/S
Tlf: +45-45 235 000
Deutschland
Bayer Vital GmbH
Tel: +49-(0)214-30 513 48
Eesti
Bayer OU
Tel: +372-655 85 65 EXláSa
Bayer ET7ág ABEE Tn^: +30-210-618 75 00 Espaňa
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare
Tél (N° vert): +33-(0)800 87 54 54
Hrvatska
Bayer d.o.o.
Tel: + 385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353-(0)1-2999 313
Ísland
Icepharma hf.
Sími: +354-540 80 00 Italia
Bayer S.p.A.
Tel: +39-02-3978 1 Kúnpoq
NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer
Tel: +371-67 84 55 63
Tato příbalová informace byla naposledy revidována: {MM.RRRR}
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
184
Nominálně významné
Kromě studie fáze III ROCKET AF byla provedena prospektivní, jednoramenná, poregistrační, neintervenční, otevřená kohortová studie (XANTUS) s centrálním vyhodnocováním sledovaných ukazatelů zahrnujících tromboembolické příhody a závažné krvácení u 6 785 pacientů s nevalvulární fibrilací síní v prevenci cévní mozkové příhody a systémové embolie nepostihující CNS v klinické praxi. Průměrné CHADS2 a HAS-BLED skóre bylo v obou případech 2,0 ve studii XANTUS v porovnání s průměrným CHADS2 a HAS-BLED skóre 3,5 a 2,8 ve studii ROCKET AF. Výskyt závažného krvácení činil 2,1 na100 pacientoroků. Fatální krvácení bylo hlášeno v 0,2 případech na 100 pacientoroků a intrakraniální krvácení v 0,4 případech na 100 pacientoroků. Cévní mozková příhoda nebo systémová embolie byla zaznamenána v 0,8 případech na 100 pacientoroků. Tato pozorování z klinické praxe jsou v souladu s potvrzeným bezpečnostním profilem v této indikaci.
Nominálně významné
Kromě studie fáze III ROCKET AF byla provedena prospektivní, jednoramenná, poregistrační, neintervenční, otevřená kohortová studie (XANTUS) s centrálním vyhodnocováním sledovaných ukazatelů zahrnujících tromboembolické příhody a závažné krvácení u 6 785 pacientů s nevalvulární fibrilací síní v prevenci cévní mozkové příhody a systémové embolie nepostihující CNS v klinické praxi. Průměrné CHADS2 a HAS-BLED skóre bylo v obou případech 2,0 ve studii XANTUS v porovnání s průměrným CHADS2 a HAS-BLED skóre 3,5 a 2,8 ve studii ROCKET AF. Výskyt závažného krvácení činil 2,1 na100 pacientoroků. Fatální krvácení bylo hlášeno v 0,2 případech na 100 pacientoroků a intrakraniální krvácení v 0,4 případech na 100 pacientoroků. Cévní mozková příhoda nebo systémová embolie byla zaznamenána v 0,8 případech na 100 pacientoroků. Tato pozorování z klinické praxe jsou v souladu s potvrzeným bezpečnostním profilem v této indikaci.