Xalkori 200 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
XALKORI 200 mg tvrdé tobolky XALKORI 250 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
XALKORI 200 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje crizotinibum 200 mg.
XALKORI 250 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje crizotinibum 250 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
XALKORI 200 mg tvrdé tobolky
Tvrdé želatinové tobolky, bílé a růžové barvy, neprůhledné, s potiskem „Pfizer“ na víčku a označením „CRZ 200“ na těle tobolky.
XALKORI 250 mg tvrdé tobolky
Tvrdé želatinové tobolky, růžové barvy, neprůhledné, s potiskem „Pfizer“ na víčku a označením „CRZ 250“ na těle tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek XALKORI je indikován jako léčba první linie dospělých pacientů s pokročilým nemalobuněčným karcinomem plic (NSCLC) pozitivním na kinázu anaplastického lymfomu (ALK).
Přípravek XALKORI je indikován jako léčba dospělých pacientů s již dříve léčeným pokročilým nemalobuněčným karcinomem plic (NSCLC) pozitivním na kinázu anaplastického lymfomu (ALK).
Přípravek XALKORI je indikován jako léčba dospělých pacientů s ROS1-pozitivním pokročilým nemalobuněčným karcinomem plic (NSCLC) .
4.2 Dávkování a způsob podání
Léčba přípravkem XALKORI má být zahájena a vedena pod dohledem lékaře se zkušenostmi s podáváním protinádorových léčivých přípravků.
Testování ALK a ROS1
K výběru pacientů pro léčbu přípravkem XALKORI je nezbytná přesná a ověřená analýza ALK nebo ROS1 (viz bod 5.1 informace o testech používaných v klinických studiích).
Pozitivitu NSCLC na ALK nebo ROS1 je třeba stanovit před zahájením léčby krizotinibem. Hodnocení by měly provádět laboratoře s prokázanou zkušeností v používání specifické technologie (viz bod 4.4).
Dávkování
Doporučená dávka přípravku XALKORI je 250 mg dvakrát denně (500 mg denně) užívaná bez přerušení.
Pokud pacient vynechá dávku, měl by si ji vzít co nejdříve, jakmile si to uvědomí, pokud to není méně než 6 hodin do další dávky, v takovém případě by si pacient vynechanou dávku brát neměl. Pacienti nemají užívat 2 dávky najednou jako náhradu za vynechanou dávku.
Úprava dávky
Na základě individuální bezpečnosti a snášenlivosti může být nezbytné přerušení dávkování a/nebo snížení dávky. U 1722 pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC léčených krizotinibem v klinických studiích byly neutropenie, zvýšené transaminázy, zvracení a nauzea nejčastějšími nežádoucími účinky (> 3 %) spojenými s přerušením léčby. Nejčastějšími nežádoucími účinky (> 3 %) spojenými se snížením dávky, byly zvýšené transaminázy a neutropenie. Je-li snížení dávky nezbytné, pak by měla být dávka přípravku XALKORI snížena na 200 mg dvakrát denně. Je-li nezbytné další snížení dávky, pak by měla být provedena úprava na 250 mg jednou denně na základě individuální bezpečnosti a snášenlivosti. Pokyny pro snižování dávek u hematologické a nehematologické toxicity jsou uvedeny v tabulkách 1 a 2.
Tabulka 1. Úprava dávky přípravku XALKORI - hematologická toxicitaa b
|
Stupeň dle CTCAEc |
Léčba přípravkem XALKORI |
|
Stupeň 3 |
Pozastavit, dokud nedojde ke zmírnění na stupeň < 2, poté pokračovat ve stejném dávkování |
|
Stupeň 4 |
Pozastavit, dokud nedojde ke zmírnění na stupeň < 2, poté pokračovat v dávkování 200 mg dvakrát denněd |
a. S výjimkou lymfopenie (není-li spojena s klinickými příhodami, např. oportunními infekcemi).
b. U pacientů, u kterých dojde k rozvoji neutropenie a leukopenie, viz body 4.4 a 4.8.
c. Obecná terminologická kritéria NCI definující nežádoucí účinky.
d. V případě rekurence pozastavit, dokud nedojde ke zmírnění na stupeň < 2, poté pokračovat
v dávkování 250 mg jednou denně. Trvale vysadit v případě dalšího objevení příznaků stupně 4.
Tabulka 2. Úprava dávky přípravku XALKORI - nehematologická toxicita
|
Stupeň dle CTCAEa |
Léčba přípravkem XALKORI |
|
Stupeň 3 nebo 4 zvýšení alaninaminotransferázy (ALT) nebo aspartátaminotransferázy (AST) se stupněm < 1 celkového bilirubinu |
Pozastavit, dokud nedojde ke zmírnění na stupeň < 1 nebo na výchozí stav, poté pokračovat s dávkou 250 mg jednou denně a zvýšit dávku na 200 mg dvakrát denně, jestliže je klinicky tolerovánab |
|
Stupeň dle CTCAEa |
Léčba přípravkem XALKORI |
|
Stupeň 2, 3 nebo 4 zvýšení ALT nebo AST se současným stupněm 2, 3 nebo 4 celkového zvýšení bilirubinu (v nepřítomnosti cholestázy nebo hemolýzy) |
Trvale vysadit |
|
Jakýkoli stupeň intersticiálního plicního onemocnění (ILD) / pneumonitidy |
Pozastavit v případě podezření na ILD/pneumonitidu a trvale vysadit, je-li diagnostikována ILD/pneumonitida související s léčbouc |
|
Stupeň 3 prodloužení QTc |
Pozastavit, dokud nedojde ke zmírnění na stupeň < 1, provést kontrolu a popřípadě upravit ionty, poté pokračovat v 200 mg dvakrát denněb |
|
Stupeň 4 prodloužení QTc |
Trvale vysadit |
|
Stupeň 2,3 bradykardiec,d Symptomatická, může být závažná nebo ze zdravotního hlediska významná, indikovaná lékařská intervence |
Pozastavit, dokud nedojde ke zmírnění na stupeň < 1 nebo dosažení srdečního rytmu 60 a více Vyhodnotit souběžně podávané léčivé přípravky, o nichž je známo, že přispívají k bradykardii, jakož i hypertenziva Jestliže je zjištěno a ukončeno souběžné podávání léčivého přípravku, který přispívá k bradykardii, nebo je dávka tohoto léku upravena, pokračujte v podávání s předchozí dávkou, jakmile dojde ke zmírnění na stupeň < 1 nebo k dosažení srdečního rytmu 60 a více. Jestliže není zjištěn žádný souběžně podávaný léčivý přípravek, který přispívá k bradykardii, nebo není léčba takovými léčivými přípravky ukončena nebo není upraveno jejich dávkování, pokračujte se sníženým dávkováním, jakmile dojde ke zmírnění na stupeň < 1 nebo k dosažení srdečního rytmu 60 a více. |
|
Stupeň 4 bradykardiec,d,e Život ohrožující následky, indikovaná naléhavá intervence |
Trvale vysadit, pokud není zjištěn žádný souběžně podávaný léčivý přípravek, který přispívá k bradykardii Jestliže je zjištěno a ukončeno souběžné podávání léčivého přípravku, který přispívá k bradykardii, nebo je dávka tohoto léčivého přípravku upravena, pokračujte s 250 mg jednou denně, jakmile dojde ke zmírnění na stupeň < 1 nebo k dosažení srdečního rytmu 60 a více, často monitorujte. |
|
Stupeň 4 poruchy zraku (ztráta zraku) |
Přerušit během vyšetřování těžké ztráty zraku |
a. Obecná terminologická kritéria NCI definující nežádoucí účinky
b. Trvale vysadit v případě dalšího objevení příznaků stupně >3. Viz body 4.4 a 4.8.
c. Viz body 4.4 a 4.8
d. Srdeční rytmus nižší než 60 tepů za minutu.
e. Trvale vysadit z důvodu rekurence.
Porucha funkce jater
Krizotinib nebyl studován u pacientů s poruchou funkce jater. Z prováděných klinických studií byli vyřazeni pacienti s AST nebo ALT > 2,5 x ULN nebo z důvodu základního maligního onemocnění, pacienti s >5,0x ULN nebo s celkovým bilirubinem >1,5x ULN. Léčba krizotinibem má být u pacientů s lehkou a středně těžkou poruchou funkce jater použita s opatrností. Krizotinib nesmí být podáván pacientům s těžkou poruchou jater (viz body 4.3, 4.4 a 4.8).
Porucha funkce ledvin
U pacientů s lehkou (60 < clearance kreatininu [CLcr] < 90 ml/min) nebo středně těžkou (30 < CLcr < 60 ml/min) poruchou funkce ledvin není doporučena úprava zahajovací dávky, neboť populační farmakokinetická analýza neukázala u těchto pacientů žádné klinicky významné změny expozice krizotinibu v ustáleném stavu. Plazmatické koncentrace krizotinibu mohou být u pacientů s těžkou poruchou funkce ledvin (CLcr <30 ml/min) zvýšeny. Zahajovací dávka krizotinibu má být upravena na 250 mg užívaných perorálně jednou denně u pacientů s těžkou poruchou funkce ledvin nevyžadující peritoneální dialýzu nebo hemodialýzu. Dávka může být zvýšena na 200 mg dvakrát denně na základě individuální bezpečnosti a snášenlivosti po alespoň 4 týdnech léčby (viz body 4.4 a 5.2).
Starší pacienti
Není nutná žádná úprava zahajovací dávky (viz body 5.1 a 5.2)
Pediatrická populace
Bezpečnost a účinnost krizotinibu u pediatrických pacientů nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Tobolky se mají spolknout celé, nejlépe zapít sklenicí vody. Nesmí se drtit, rozpouštět nebo otvírat. Mohou se užívat s jídlem nebo bez jídla. Grapefruit nebo grapefruitová šťáva mohou zvýšit plazmatickou koncentraci krizotinibu, proto je třeba se jim vyhnout. Třezalka tečkovaná může snížit plazmatickou koncentraci krizotinibu, a je třeba se jí vyhnout (viz bod 4.5).
4.3 Kontraindikace
Hypersensitivita na krizotinib nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Těžká porucha funkce jater (viz body 4.2, 4.4 a 4.8).
4.4 Zvláštní upozornění a opatření pro použití
Hodnocení stavu ALK a ROS1
Při vyhodnocování stavu ALK nebo ROS1 u pacienta je důležité zvolit dobře validovanou a robustní metodiku, aby byly vyloučeny falešně negativní nebo falešně pozitivní výsledky.
Hepatotoxicita
U pacientů léčených krizotinibem v klinických studiích byla hlášena lékem indukovaná hepatotoxicita (včetně případů s fatálním průběhem), (viz bod 4.8). Krizotinib se nesmí podávat pacientům s těžkou poruchou funkce jater (včetně pacientů s celkovým bilirubinem > 3* ULN bez ohledu na ALT/AST), (viz body 4.2, 4.3, a 4.8). Testy funkce jater včetně stanovení ALT, AST a celkového bilirubinu mají být prováděny jednou týdně během prvních 2 měsíců léčby a poté jednou měsíčně a dle klinické potřeby, přičemž v případě zvýšení stupně 2, 3 nebo 4 by se testování mělo opakovat častěji. Pacienti, u nichž se rozvine zvýšení transamináz, viz bod 4.2.
Intersticiální plicní onemocnění/pneumonitida
U pacientů léčených krizotinibem se může vyskytnout těžké, život ohrožující nebo fatální intersticiální plicní onemocnění (ILD) / pneumonitida. Pacienti s plicními příznaky ukazujícími na ILD/pneumonitidu mají být sledováni. Při podezření na ILD/pneumonitidu je nutné léčbu krizotinibem pozastavit. Lékem indukované ILD/pneumonitida je třeba zvážit při diferenciální diagnostice u pacientů s onemocněními podobajícími se ILD, například: pneumonitidou, radiační pneumonitidou, hypersenzitivní pneumonitidou, intersticiální pneumonitidou, plicní fibrózou, syndromem akutní respirační tísně (ARDS), alveolitidou, plicní infiltrací, pneumonií, plicním edémem, chronickým obstrukčním plicním onemocněním, pleurálním výpotkem, aspirační pneumonií, bronchitidou, obliterující bronchiolitidou a bronchiektáziemi. Další potenciální příčiny ILD/pneumonitidy mají být vyloučeny. Krizotinib má být trvale vysazen u pacientů s diagnózou ILD/pneumonitidy související s léčbou (viz body 4.2 a 4.8).
Prodloužení QT intervalu
V klinických studiích bylo u pacientů léčených krizotinibem pozorováno prodloužení QTc intervalu (viz body 4.8 a 5.2), které může vést ke zvýšenému riziku ventrikulární tachykardie (např. Torsades de Pointes) nebo náhlé smrti. Před zahájením terapie krizotinibem je třeba zvážit přínosy a potenciální rizika u pacientů s preexistující bradykardií, kteří mají prodloužení QTc intervalu v anamnéze nebo mají k prodloužení QTc intervalu predispozice a kteří užívají antiarytmika nebo jiné léčivé přípravky, o nichž je známo, že prodlužují QT interval, a u pacientů s významným preexistujícím srdečním onemocněním a/nebo porušenou rovnováhou elektrolytů. U těchto pacientů má být krizotinib podáván s opatrností a je nutné provádět pravidelné elektrokardiografické vyšetření (EKG) a vyšetření iontů a renální funkce. Při podávání krizotinibu je třeba provést vyšetření EKG a elektrolytů (např. vápníku, hořčíku, draslíku) v době co nejblíže před první dávkou a doporučuje se pravidelně monitorovat EKG a ionty především na začátku léčby v případě zvracení, průjmu, dehydratace nebo poruchy funkce ledvin. Elektrolyty dle potřeby upravte. Jestliže se QTc zvýší o 60 ms nebo více oproti počáteční hodnotě, ale QTc je <500 ms, je třeba léčbu krizotinibem pozastavit a konzultovat s kardiologem. Při zvýšení QTc na 500 ms nebo více se musí léčba okamžitě konzultovat s kardiologem. Pacienti, u nichž se rozvine prodloužení QTc, viz body 4.2, 4.8 a 5.2.
Bradykardie
Bradykardie z jakýchkoli příčin byla v klinických studiích hlášena u 13 % pacientů léčených krizotinibem. U pacientů užívajících krizotinob se může objevit symptomatická bradykardie (např. synkopa, závrať, hypotenze). Celkový účinek krizotinibu na snížení srdečního rytmu se může rozvinout až po několika týdnech od zahájení léčby. Vzhledem ke zvýšenému riziku symptomatické bradykardie se v maximální možné míře vyvarujte podávání krizotinibu v kombinaci s jinými léky vyvolávajícími bradykardii (např. beta-blokátory, blokátory kalciových kanálů jinými než dihydropyridiny, jako jsou verapamil a diltiazem, klonidin, digoxin). Pravidelně monitorujte srdeční rytmus a krevní tlak. Úprava dávky není nutná v případě asymptomatické bradykardie. Pokyny pro léčbu pacientů, u nichž se rozvine symptomatická bradykardie, najdete v bodech Úprava dávky a Nežádoucí účinky (viz body 4.2 a 4.8).
Srdeční selhání
V klinických studiích s krizotinibem a během sledování po uvedení na trh bylo jako nežádoucí účinek hlášeno těžké, život ohrožující nebo fatální srdeční selhání (viz bod 4.8).
Jak pacienti s preexistujícími srdečními poruchami, tak pacienti bez nich, kteří užívají krizotinib, mají být sledováni na přítomnost známek a příznaků srdečního selhání (dyspnoe, edém, rychlý přírůstek hmotnosti způsobený retencí tekutin). Při zjištění takových příznaků je nutno zvážit přerušení podávání, snížení dávky nebo vysazení.
Neutropenie a leukopenie
V klinických studiích s krizotinibem u pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC byla velmi často (12 %) hlášena neutropenie stupně 3 nebo 4. Často (3 %) byla hlášena leukopenie stupně 3 nebo 4 (viz bod 4.8). U méně než 0,5 % pacientů v klinických studiích s krizotinibem byla zaznamenána febrilní neutropenie. Kompletní krevní obraz včetně diferenciálního počtu leukocytů má být sledován dle klinické potřeby, s častějším opakováním testování v případě, že jsou pozorovány abnormality stupně 3 nebo 4, nebo pokud se objeví horečka či infekce (viz bod 4.2).
Gastrointestinální perforace
V klinických studiích s krizotinibem byly hlášeny případy gastrointestinální perforace. V rámci užívání krizotinibu po jeho uvedení na trh byly hlášeny fatální případy gastrointestinální perforace (viz bod 4.8).
U pacientů s rizikem gastrointestinální perforace (např. divertikulitidou v anamnéze, metastázami do trávicího traktu, souběžným užíváním léčivých přípravků se známým rizikem gastrointestinální perforace) je třeba krizotinib používat s opatrností.
Podávání krizotinibu je nutné přerušit, pokud u pacienta dojde ke gastrointestinální perforaci. Pacienty je třeba poučit o prvních příznacích takového stavu a instruovat je, aby se ihned obrátili na lékaře v případě, že se u nich příznaky gastrointestinální perforace projeví.
Účinky na ledviny
U pacientů účastnících se klinických studií s krizotinibem bylo pozorováno zvýšení kreatininu v krvi a snížení clearance kreatininu. U pacientů léčených krizotinibem v rámci klinických studií a také po uvedení přípravku na trh byly hlášeny případy renálního selhání a akutního renálního selhání. Zjištěny byly rovněž případy s fatálním průběhem, případy vyžadující hemodialýzu a případy hyperkalemie 4. stupně. Na počátku a poté během léčby krizotinibem se doporučuje sledovat u pacientů renální funkce. Zvláštní pozornost je třeba věnovat pacientům s rizikovými faktory poruchy funkce ledvin a pacientům s poruchou funkce ledvin v anamnéze (viz bod 4.8).
Porucha funkce ledvin
U pacientů, kteří mají těžkou poruchu funkce ledvin nevyžadující peritoneální dialýzu ani hemodialýzu, má být dávka krizotinibu upravena (viz body 4.2 a 5.2).
Účinky na. zrak
V klinických studiích s krizotinibem byl hlášen u pacientů s ALK-pozitivním nebo ROSl-pozitivním NSCLC (N=1722) defekt zrakového pole stupně 4 se ztrátou zraku u 4 (0,2 %) pacientů. Jako možné příčiny ztráty zraku byly uváděny atrofie a porucha optického nervu.
U pacientů s novým vznikem těžké ztráty zraku (nejlépe korigovaná zraková ostrost menší než 6/60 na jednom nebo obou očích) je třeba léčbu krizotinibem přerušit (viz bod 4.2). Je třeba provést oftalmologické vyšetření sestávající z nejlépe korigované zrakové ostrosti, fotografií sítnice, vyšetření zrakového pole, optické koherentní tomografie (OCT) a dalších vyšetření odpovídajících novému výskytu těžké ztráty zraku. Neexistuje dostatek informací k charakterizování rizika opětovného zahájení podávání krizotinibu u pacientů s těžkou ztrátou zraku. Při rozhodování o obnovení podávání krizotinibu je třeba zvážit možný přínos pro pacienta.
Pokud poruchy zraku přetrvávají nebo se zhorší jejich závažnost, doporučuje se oftalmologické vyšetření, (viz bod 4.8).
Lékové interakce
Je třeba se vyhnout současnému podávání krizotinibu se silnými inhibitory CYP3A nebo se silnými a středně silnými induktory CYP3A (viz bod 4.5).
Je třeba se vyhnout současnému podávání krizotinibu se substráty CYP3A4 s úzkým terapeutickým indexem (viz bod 4.5). Vyvarujte se podávání krizotinibu v kombinaci s jinými léky vyvolávajícími bradykardii, léčivými přípravky, o nichž je známo, že prodlužují QT interval, a/nebo antiarytmiky (viz bod 4.4 Prodloužení QT intervalu, Bradykardie a bod 4.5).
Histologický typ jiný než adenokarcinom
U pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC s jiným histologickým typem než adenokarcinom, včetně dlaždicobuněčného karcinomu (SCC), jsou dostupné omezené informace (viz bod 5.1).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakokinetické interakce
Látky, které mohou zvyšovatplazmatickou koncentraci krizotinibu
Současné podávání krizotinibu se silnými inhibitory CYP3A může zvyšovat plazmatické koncentrace krizotinibu. Současné podání jednotlivé dávky 150 mg krizotinibu perorálně při podávání ketokonazolu (200 mg dvakrát denně), silného inhibitoru CYP3A, vedlo ke zvýšení systémové expozice krizotinibu, s hodnotami plochy pod křivkou plazmatické koncentrace proti času od nuly do nekonečna (AUCinf) a hodnotami maximální pozorované plazmatické koncentrace (Cmax) krizotinibu, které byly přibližně 3,2násobek respektive 1,4násobek hodnot, které byly zaznamenány, pokud byl krizotinib podáván samotný.
Je třeba se proto vyhnout současnému užívání silných inhibitorů CYP3A (některé inhibitory proteáz jako atazanavir, indinavir, nelfinavir, ritonavir, sachinavir a některá azolová antimykoticka jako itrakonazol, ketokonazol a vorikonazol, některé makrolidy jako klarithromycin, telithromycin a troleandomycinu). Grapefruit nebo grapefruitová šťáva mohou rovněž zvýšit plazmatické koncentrace krizotinibu a je nutné se jim vyhnout (viz body 4.2 a 4.4). Navíc, účinek inhibitorů CYP3A na rovnovážnou expozici krizotinibu nebyl stanoven.
Látky, které mohou snižovat plazmatickou koncentraci krizotinibu
Současné podání opakovaných dávek krizotinibu (250 mg dvakrát denně) s opakovanými dávkamu rifampicinu (600 mg jednou denně), silným induktorem CYP3A4 vedlo ke snížení rovnovážných hodnot AUCtau a Cmax na 84 % respektive 79 %, v porovnání s hodnotami při podávání krizotinibu samotného. Je třeba se vyhnout současnému podávání se silnými induktory CYP3A, včetně, ovšem nikoliv pouze, karbamazepinu, fenobarbitalu, fenytoinu, rifampicinu a třezalky tečkované (viz bod 4.4).
Účinek středně silného induktoru, včetně ovšem nikoliv pouze, efavirenzu nebo rifabutinu nebyl jasně stanoven a proto je třeba se jeho kombinaci s krizotinibem vyhnout (viz bod 4.4).
Současné podávání s léčivými přípravky, které zvyšují pH v žaludku
Rozpustnost krizotinibu ve vodě závisí na pH, přičemž nízké (kyselé) pH vede k vyšší rozpustnosti. Podání jednorázové dávky 250 mg krizotinibu po léčbě esomeprazolem 40 mg jednou denně po dobu 5 dnů vedlo k přibližně 10% snížení celkové expozice krizotinibu (AUCf a nevedlo k žádné změně maximální expozice (Cmax); rozsah změny celkové expozice krizotinibu nebyl klinicky významný. Proto není nutná úprava zahajovací dávky, když se krizotinib podává současně s látkami, které zvyšují pH v žaludku (např. inhibitory protonové pumpy, H2-blokátory nebo antacida).
Látky, jejichž plazmatické koncentrace mohou být ovlivněny krizotinibem
Po 28denním podávání krizotinibu v dávce 250 mg dvakrát denně pacientům s karcinomem, hodnota AUC perorálně podaného midazolamu byla 3,7násobek hodnoty zaznamenané při podávání midazolamu samotného. To naznačuje, že krizotinib je středně silným inhibitorem CYP3A. Je proto třeba se vyhnout současnému užívání krizotinibu a substrátů CYP3A4 s úzkým terapeutickým indexem, včetně, ovšem nikoliv pouze alfentanilu, cisapridu, cyklosporinu, derivátů ergotaminu, fentanylu, pimozidu, chinidinu, sirolimusu, a takrolimusu (viz bod 4.4). Pokud je tato kombinace nutná, má být prováděno pečlivé klinické sledování.
In vitro studie ukázaly, že krizotinib je inhibitor CYP2B6, a může proto zvyšovat plazmatické koncentrace současně podávaných léčiv, která jsou metabolizována CYP2B6 (např. bupropion, efavirenz).
In vitro studie na lidských hepatocytech ukázaly, že krizotinib může indukovat pregnanový X receptor (PXR) a enzymy regulované konstitutivním androstanovým receptorem (CAR), (např. CYP3A4, CYP2B6, CYP2C8, CYP2C9, UGT1A1). Když byl však krizotinib současně podáván s midazolamem jako zkušebním substrátem CYP3A4, nebyla pozorována žádná indukce in vivo. Opatrnost je proto nutná při podávání krizotinibu v kombinaci s léčivými přípravky, které jsou přednostně metabolizovány těmito enzymy. Účinnost současně podávaných perorálních kontraceptiv může být snížena.
In vitro studie ukázaly, že krizotinib je slabým inhibitorem uridin difosfát glukuronosyltransferázy (UGT)1A1 a UGT2B7. Proto může mít krizotinib potenciál ke zvyšování plazmatické koncentrace současně podávaných léků, které jsou metabolizovány převážně prostřednictvím UGT1A1 (např. raltegravir, irinotekan) nebo UGT2B7 (morfin, naloxon).
Na základě in vitro studie se předpokládá, že krizotinib inhibuje intestinální P-gp (P-glykoprotein). Podávání krizotinibu s léčivými přípravky, které jsou substráty P-glykoproteinu (např. digoxin, dabigatran, kolchicin, pravastatin) může zvýšit jejich terapeutický účinek a nežádoucí účinky. Pečlivý klinický dohled se doporučuje, pokud je krizotinib podáván s těmito léčivými přípravky.
Krizotinib je inhibitorem OCT1 a OCT2 in vitro. Proto může mít krizotinib potenciál k zvyšování plazmatické koncentrace současně podávaných léků, které jsou substráty OCT1 nebo OCT2 (např. metformin, prokainamid).
Farmakodynamické interakce
V klinických studiích s krizotinibem bylo pozorováno prodloužení QT intervalu. Z toho důvodu je nutné pečlivě zvážit současné podání krizotinibu s léčivými přípravky, o kterých je známo, že mohou prodloužit QT interval nebo s léčivými přípravky, které mohou indukovat torsades de pointes (např. třída IA [chinidin, disopyramid] nebo třída III [např. amiodaron, sotalol, dofetilid, ibutilid], methadon, cisaprid, moxifloxacin, antipsychotika, atd.). Monitorování QT intervalu je nutné v případě kombinace těchto léčivých přípravků (viz body 4.2 a 4.4).
Bradykardie byla hlášena během klinických studií, proto je nutná opatrnost při podávání krizotinibu vzhledem k riziku extrémní bradykardie při podání v kombinaci s jinými léky vyvolávajícími bradykardii (např. non-dihydropyridinové blokátory kalciového kanálu jako jsou verapamil a diltiazem, beta-blokátory, klonidin, guanfacin, digoxin, meflochin, anticholinesterázy, pilokarpin),
(viz body 4.2 a 4.4).
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Ženy ve fertilním věku je třeba upozornit, aby se v době užívání přípravku XALKORI vyvarovaly otěhotnění.
V průběhu léčby a po dobu alespoň 90 dnů po ukončení léčby mají být používány vhodné antikoncepční metody (viz bod 4.5).
Přípravek XALKORI může způsobit poškození plodu, pokud je podáván těhotným ženám. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Neexistují žádné údaje ohledně těhotných žen užívajících krizotinib. Tento léčivý přípravek lze v těhotenství použít pouze tehdy, když klinický stav matky vyžaduje léčbu. Těhotné ženy, pacientky, které otěhotní v době užívání krizotinibu, nebo těhotné partnerky léčených pacientů mají být informovány o možném riziku pro plod.
Kojení
Není známo, zda se krizotinib a jeho metabolity vylučují do lidského mateřského mléka. Vzhledem k možnému riziku pro dítě matky mají být poučeny, aby při léčbě přípravkem XALKORI nekojily (viz bod 5.3).
Fertilita
Na základě neklinických bezpečnostních zjištění může léčba přípravkem XALKORI snížit mužskou i ženskou fertilitu (viz bod 5.3). Informace ohledně uchování fertility mají být poskytnuty před zahájením léčby mužům i ženám.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Je třeba dbát zvýšené opatrnosti při řízení nebo obsluze strojů, protože u pacientů se mohou vyskytnout během užívání přípravku XALKORI příznaky bradykardie (např. synkopy, závratě, hypotenze), poruchy vidění nebo únava (viz body 4.2, 4.4 a 4.8).
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Níže uvedená data reflektují vystavení vlivu přípravku XALKORI u 1669 pacientů s ALK-pozitivním pokročilým NSCLC, kteří se zúčastnili 2 randomizovaných studií fáze III (studie 1007 a 1014) a 2 jednoramenných studií (studie 1001 a 1005), a u 53 pacientů s ROS1-pozitivním pokročilým NSCLC, kteří se zúčastnili jednoramenné studie 1001, celkem tedy u 1722 pacientů (viz bod 5.1). Tito pacienti dostávali zahajovací dávku 250 mg perorálně dvakrát denně bez přerušení. Ve studii 1014 činil medián doby trvání hodnocené léčby 47 týdnů u pacientů v rameni s krizotinibem (N=171); medián doby trvání léčby u pacientů, kteří přešli z ramene s chemoterapií na léčbu krizotinibem (N=109), byl 23 týdnů. Ve studii 1007 činil medián doby trvání hodnocené léčby 48 týdnů u pacientů v rameni s krizotinibem (N=172). U pacientů s ALK-pozitivním pokročilým NSCLC byl ve studii 1001 (N=154) medián doby trvání léčby 57 týdnů a ve studii 1005 (N=1063) činil 45 týdnů. U pacientů s ROS1-pozitivním pokročilým NSCLC ve studii 1001 (N=53) byl medián doby trvání léčby 101 týdnů.
Nejzávažnějšími nežádoucími účinky u 1722 pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC byly hepatotoxicita, ILD/pneumonitida, neutropenie a prodloužení QT intervalu (viz bod 4.4). Nejčastějšími nežádoucími účinky (> 25 %) u pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC byly poruchy zraku, nauzea, průjem, zvracení, otoky, zácpa, zvýšené transaminázy, únava, snížená chuť k jídlu, závrať a neuropatie.
Seznam nežádoucích účinků v tabulkovém formátu
Tabulka 3 uvádí nežádoucí účinky hlášené u 1722 pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC, kteří byli léčeni krizotinibem ve 2 randomizovaných studiích fáze III (studie 1007 a 1014) a 2 jednoramenných klinických studiích (1001 a 1005), (viz bod 5.1).
Nejčastějšími nežádoucími účinky (> 3 %, četnost nežádoucích účinků z jakýchkoli příčin) spojenými s přerušením léčby, byly neutropenie (11 %), zvýšené transaminázy (7 %), zvracení (5 %) a nauzea (4 %). Nejčastějšími nežádoucími účinky (> 3 %, četnost nežádoucích účinků z jakýchkoli příčin) spojenými se snížením dávky, byly zvýšené transaminázy (4 %) a neutropenie (3 %). Nežádoucí účinky z jakýchkoli příčin spojené s trvalým vysazením léčby, se vyskytly u 302 (18 %) pacientů, přičemž nejčastějšími (> 1 %) byly intersticiální plicní onemocnění (1 %) a zvýšené transaminázy (1 %).
Nežádoucí účinky uvedené v tabulce 3 jsou seřazeny podle tříd orgánových systémů a frekvencí, které jsou definovány jako: velmi časté (>1/10), časté (>1/100 až < 1/10), méně časté (>1/1000 až < 1/100), vzácné (>1/10 000 až < 1/1000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Tabulka 3. Nežádoucí účinky |
ílášené v klinických studiích s krizotinibem (N=1722) | ||
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Poruchy krve a lymfatického systému |
Neutropeniea (22 %) Anemieb (15 %) Leukopeniec (15 %) | ||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu (30 %) |
Hypofosfatemi e (6 %) | |
|
Poruchy nervového systému |
Neuropatied (25 %) Dysgeusie (21 %) | ||
|
Poruchy oka |
Poruchy zrakue (63 %) | ||
|
Srdeční poruchy |
Závratěf (26 %) Bradykardieg (13 %) |
Srdeční selháníh (1 %) Prodloužení QT na EKG (4%) Synkopa(3 %) | |
|
Respirační, hrudní a mediastinální poruchy |
Intersticiální plicní onemocněnť (3 %) | ||
|
Gastrointestinální poruchy |
Zvracení (51 %) Průjem (54 %) Nauzea (57 %) Zácpa (43 %) Bolest břichaJ (21 %) |
Dyspepsie (8 %) Ezofagitidak (2%) |
Gastrointestináln í perforacel (< 1 %) |
|
Poruchy jater a žlučových cest |
Zvýšení transamináz m (32 %) |
Zvýšení hladiny alkalické fosfatázy v krvi (7%) |
Selhání jater (< 1 %) |
|
Poruchy kůže a podkožní tkáně |
Vyrážka (13 %) | ||
|
Poruchy ledvin a močových cest |
Ledvinová cysta11 (3 %) Zvýšený kreatinin v krvio (8 %) |
Akutní renální selhání (< 1 %) Renální selhání (< 1 %) | |
|
Celkové poruchy a reakce v místě aplikace |
Otokyp (47 %) Únava (30 %) | ||
|
Vyšetření |
Testosteron v krvi sníženýq (2%) |
Termíny označující příhody, které odpovídají stejnému zdravotnímu stavu, byly pro potřeby tabulky 3
seskupeny a hlášeny v rámci téhož nežádoucího účinku. Termíny skutečně hlášené v rámci studie až
do termínu uzavření údajů a náležící k příslušným nežádoucím účinkům jsou uvedeny v závorkách
níže.
a. Neutropenie (febrilní neutropenie, neutropenie, snížený počet neutrofilů).
b. Anemie (anemie, snížený hemoglobin, hypochromní anemie).
c. Leukopenie (leukopenie, počet leukocytů snížený).
d. Neuropatie (pálivý pocit, dysestezie, mravenčení, porucha chůze, hyperestezie, hypestezie, hypotonie, motorická dysfunkce, svalová atrofie, svalová slabost, neuralgie, neuritida, periferní neuropatie, neurotoxicita, parestézie, periferní motorická neuropatie, periferní senzomotorická neuropatie, periferní senzorická neuropatie, obrna nervus peroneus, polyneuropatie, senzitivní poškození, pálení kůže).
e. Poruchy vidění (diplopie, halo vidění, fotofobie, fotopsie, rozmazané vidění, snížená zraková ostrost, zraková percepce jasu, poruchy zraku, vizuální perseverace, sklivcové vločky).
f. Závrať (porucha rovnováhy, závrať, posturální závrať, presynkopa).
g. Bradykardie (bradykardie,srdeční frekvence snížená, sinusová bradykardie).
h. Srdeční selhání (srdeční selhání, městnavé srdeční selhání, ejekční frakce snížená, selhání levé komory, plicní edém). Napříč klinickými studiemi (n=1722) mělo 19 (1,1 %) pacientů léčených krizotinibem nějaký stupeň srdečního selhání, 8 (0,5 %) pacientů mělo stupeň 3 nebo 4 a u 3 pacientů (0,2 %) mělo smrtelný průběh.
i. Intersticiální plicní onemocnění (syndrom akutní respirační tísně, alveolitida, intersticiální plicní onemocnění, pneumonitida).
j. Bolest břicha (břišní diskomfort, bolest břicha, bolest dolní poloviny břicha, bolest horní poloviny břicha, břišní citlivost).
k. Ezofagitida (ezofagitida, jícnový vřed).
l. Gastrointestinální perforace (gastrointestinální perforace, intestinální perforace, perforace tračníku).
m. Zvýšené transaminázy (zvýšení alaninaminotransferázy, zvýšení aspartátaminotransferázy, zvýšení gamaglutamyltransferázy, jaterní enzymy zvýšené, abnormální jaterní funkce, funkční jaterní test abnormální, zvýšené transaminázy).
n. Renální cysta (renální absces, renální cysta, krvácení do renální cysty, infekce ledvinové cysty).
o. Otoky (edém obličeje, generalizovaný edém, lokální zduření, lokální edém, otok, periferní edém, periorbitální edém).
p. Testosteron v krvi snížený (testosteron v krvi snížený, hypogonadismus, sekundární hypogonadismus).
Popis vybraných nežádoucích účinků
Hepatotoxicita
Došlo k výskytu lékem indukované hepatotoxicity s fatálním průběhem u 0,1 % ze 1722 pacientů léčených krizotinibem v klinických studiích. Současná zvýšení ALT a/nebo AST > 3* ULN a celkového bilirubinu > 2* ULN bez významných zvýšení alkalické fosfatázy_(< 2* ULN) byla pozorována u méně než 1 % pacientů léčených krizotinibem.
Zvýšení ALT nebo AST na stupeň 3 nebo 4 byla pozorována u 187 (11 %) a 95 (6 %) pacientů v tomto pořadí. U sedmnácti (1 %) pacientů bylo nutné trvalé vysazení léčby v souvislosti se zvýšenými transaminázami, což naznačuje, že tyto příhody byly obecně zvládnutelné úpravami dávkování popsanými v tabulce 2 (viz bod 4.2). V randomizované studii 1014 fáze III byla pozorována zvýšení ALT nebo AST na stupeň 3 nebo 4 u 15% a 8% pacientů užívajících krizotinib oproti 2 % a 1 % pacientů léčených chemoterapií. V randomizované studii 1007 fáze III byla pozorována zvýšení ALT nebo AST na stupeň 3 nebo 4 u18% a 9% pacientů užívajících krizotinib a 5 % a < 1 % pacientů léčených chemoterapií.
Zvýšení transamináz se obvykle objevilo během prvních 2 měsíců léčby. Napříč studiemi s krizotinibem u pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC činil medián doby do nástupu zvýšení transamináz stupně 1 nebo 2 23 dní. Medián doby do nástupu zvýšení transamináz stupně 3 nebo 4 činil 43 dní.
Zvýšení transamináz na stupeň 3 a 4 bylo obecně reversibilní po přerušení léčby. Napříč studiemi s krizotinibem u pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC (N=1722) se vyskytla snížení dávky spojená se zvýšením transamináz u 76 (4 %) pacientů. U sedmnácti (1 %) pacientů bylo nutné trvalé vysazení léčby.
Krizotinib se nesmí podávat pacientům se těžkou poruchou funkce jater (viz body 4.2, 4.3, a 4.4).
U pacientů má být sledována hepatotoxicita a kontrolována podle doporučení v bodech 4.2 a 4.4.
Gastrointestinální účinky
Nauzea (57 %), průjem (54 %), zvracení (51 %) a zácpa (43 %) byly nejčastěji uváděnými gastrointestinálními příhodami z jakýchkoli příčin. Většina těchto příhod byla lehká až středně těžká. Medián doby do nástupu nauzey a zvracení byl 3 dny a četnost výskytu těchto příhod klesla po 3 týdnech léčby. Podpůrná léčby by měla zahrnovat podávání antiemetik. Medián doby do nástupu průjmu byl 13 dnů a do nástupu zácpy 17 dnů. Podpůrná léčba u průjmu a zácpy by měla zahrnovat podávání standardních léků proti průjmům, resp. projímadel.
V klinických studiích s krizotinibem byly hlášeny případy gastrointestinální perforace. V rámci užívání krizotinibu po jeho uvedení na trh byly hlášeny fatální případy gastrointestinální perforace (viz bod 4.4).
Prodloužení QT intervalu
Napříč studiemi u pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC byl QTcF (QT upravené metodou Fridericia) > 500 ms zaznamenán u 34 (2,1 %) z 1619 pacientů s alespoň
I provedeným vyšetřením EKG po vstupu do studie a maximální zvýšení oproti výchozí hodnotě u QTcF > 60 ms bylo pozorováno u 79 (5,0 %) z 1585 pacientů s výchozím vyšetřením EKG a alespoň 1 provedeným vyšetřením EKG po vstupu do studie. Prodloužení QT na EKG stupně 3 a 4 z jakýchkoli příčin bylo hlášeno u 27 (1,6 %) z 1722 pacientů (viz body 4.2, 4.4, 4.5 a 5.2).
V jednoramenné dílčí studii EKG (viz bod 5.2), v níž bylo použito zaslepené ruční měření EKG, mělo
II pacientů (21 %) zvýšení QTcF oproti výchozí hodnotě > 30 až < 60 ms a 1 pacient (2 %) měl zvýšení QTcF oproti výchozí hodnotě > 60 ms. Žádní pacienti neměli maximální QTcF > 480 ms. Analýza centrální tendence ukazuje, že největší průměrná změna oproti výchozím hodnotám QTcF byla 12,3 ms (95% CI 5,1-19,5 ms, střední hodnota nejmenších čtverců [LS] z analýzy rozptylu [ANOVA]) a došlo k ní 6 hodin po podání dávky 1. dne 2. cyklu. Všechny horní hranice 90% CI pro průměrnou změnu LS oproti výchozím hodnotám QTcF ve všech časových bodech 1. dne 2. cyklu byly < 20 ms.
Prodloužení QT může vést k arytmii a je rizikovým faktorem náhlého úmrtí. Prodloužení QT se může klinicky manifestovat jako bradykardie, závrať, a synkopa. Nerovnováha elektrolytů, dehydratace a bradykardie mohou dále zvyšovat riziko prodloužení QTc, a proto se u pacientů s gastrointestinální toxicitou doporučuje periodický monitoring EKG a hladin elektrolytů (viz bod 4.4).
Bradykardie
Ve studiích s krizotinibem u pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC, se bradykardie z jakýchkoli příčin vyskytla u 219 (13 %) ze 1722 pacientů léčených krizotinibem. Většina příhod byla co do závažnosti mírná. Celkem 259 (16 %) z 1666 pacientů s alespoň 1 posouzením životních funkcí po vstupu do studie mělo tepovou frekvenci < 50 tepů za minutu.
Souběžné podávání léčivých přípravků spojených s bradykardií je nutné pečlivě zhodnotit. Pacienty, u nichž se rozvine symptomatická bradykardie, je třeba léčit podle doporučení uvedených v bodech Úprava dávky a Upozornění a opatření (viz body 4.2, 4.4 a 4.5).
Intersticiální plicní onemocnění/pneumonitida
U pacientů léčených krizotinibem se může vyskytovat těžké, život ohrožující nebo fatální intersticiální plicní onemocnění (ILD) / pneumonitida. Napříč studiemi u pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC (N=1722) mělo 50 (3 %) pacientů léčených krizotinibem některý stupeň ILD z jakýchkoli příčin, včetně 18 (1 %) pacientů se stupněm 3 nebo 4 a u 8 (< 1 %) pacientů s fatálním průběhem. Podle analýzy pacientů s ALK-pozitivním NSCLC (N=1669) nezávislého hodnotícího výboru (IRC) mělo 20 (1,2 %) pacientů intersticiální plicní onemocnění (ILD) / pneumonitidu, včetně 10 (<1 %) pacientů s fatálním průběhem. Tyto případy se obecně vyskytly do 3 měsíců od zahájení léčby. Pacienti s plicními příznaky ukazujícími na ILD/pneumonitidu mají být sledováni. Další potenciální příčiny ILD/pneumonitidy mají být vyloučeny (viz body 4.2 a 4.8).
Účinky na zrak
V klinických studiích s krizotinibem byl hlášen u pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC (N=1722) defekt zrakového pole stupně 4 se ztrátou zraku u 4 (0,2 %) pacientů. Jako možné příčiny ztráty zraku byly uváděny atrofie a porucha optického nervu (viz bod 4.4).
Poruchy zraku všech stupňů z jakýchkoli příčin, nejčastěji poškození zraku, fotopsie, rozmazané vidění a sklivcové vločky, byly zaznamenány u 1084 (63 %) ze 1722 pacientů léčených krizotinibem. Z 1084 pacientů s poruchou zraku mělo 95 % pacientů příhody, jejichž závažnost byla mírná. U sedmi (0,4 %) pacientů byla dočasně přerušena léčba a u 2 (0,1%) pacientů byla snížena dávka v souvislosti s poruchou zraku. U žádného z 1722 pacientů léčených krizotinibem nedošlo k trvalému ukončení léčby v souvislosti s poruchou zraku.
Podle údajů z dotazníků pro vyhodnocení zrakových symptomů (VSAQ-ALK) hlásili pacienti léčení krizotinibem ve studii 1007 a studii 1014 vyšší incidenci poruch zraku než pacienti léčení chemoterapií. Poruchy zraku obecně začaly nastupovat během prvního týdne podávání léku. Většina pacientů v rameni léčené krizotinibem v randomizovaných studiích 1007 a 1014 fáze III (> 50 %) hlásila poruchy zraku, které se podle údajů z dotazníku VSAQ-ALK vyskytovaly s četností 4 až 7 dnů za týden, trvaly až 1 minutu a měly mírný nebo žádný vliv na denní činnosti (skóre 0 až 3 z maximálního skóre 10).
Byla provedena oftalmologická dílčí studie využívající přesně daná oční vyšetření v určených časových bodech u 54 pacientů s NSCLC, kteří užívali krizotinib v dávce 250 mg dvakrát denně. U třiceti osmi (70,4 %) z 54 pacientů došlo k nežádoucí příhodě, která se z jakýchkoli příčin vyskytla během léčby, řadící se do třídy orgánových systémů Poruchy oka, přičemž u 30 pacientů bylo provedeno oční vyšetření. Z těchto 30 pacientů byla hlášena oční abnormalita jakéhokoli typu u 14 (36,8 %) pacientů a u 16 (42,1 %) pacientů nebyl pozorován žádný oční nález. Nejčastější nálezy se týkaly biomikroskopie štěrbinovou lampou (21,1 %), fundoskopie (15,8 %) a zrakové ostrosti (13,2 %). U mnoha pacientů byly zaznamenány preexistující oční abnormality a souběžná onemocnění, jež mohly přispět k očním nálezům, a nebylo možné přesvědčivě prokázat kauzální vztah ke krizotinibu. Nevyskytly se žádné nálezy v souvislosti s vyšetřením počtu buněk v komorové vodě a záblesku v přední komoře oční. Zdá se, že žádná ze zrakových poruch spojených s užíváním krizotinibu nesouvisí se změnami nejlepší korigované zrakové ostrosti, sklivce, sítnice či zrakového nervu.
U pacientů s novým vznikem těžké ztráty zraku stupně 4je třeba léčbu krizotinibem přerušit a je třeba provést oční vyšetření. Pokud poruchy zraku přetrvávají nebo se zhorší jejich závažnost, jedoporučeno oftalmologické vyšetření (viz body 4.2 a 4.4).
Účinky na nervový systém
Neuropatie z jakýchkoli příčin, jak je definována v tabulce 3, byla zaznamenána u 435 (25 %) z 1722 pacientů léčených krizotinibem. V těchto studiích byly také velmi často uváděny poruchy chuti, které byly primárně 1. stupně závažnosti.
Renální cysty
Komplexní renální cysty z jakýchkoli příčin byly zaznamenány u 52 (3 %) z 1722 pacientů léčených krizotinbibem. U některých pacientů byla pozorována lokální cystická invaze mimo oblast ledvin.
U pacientů, u nichž se rozvinou renální cysty, je třeba zvážit pravidelné sledování pomocí zobrazovacích technik a analýzy moči.
Neutropenie a leukopenie
Napříč studiemi u pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC (N=1722) byla neutropenie 3. nebo 4. stupně pozorována u 212(12%) pacientů léčených krizotinibem. Medián doby do nástupu jakéhokoli stupně neutropenie byl 89 dní. Neutropenie byla spojena se snížením dávky nebo trvalým ukončením léčby u 3 % a < 1 % pacientů. V klinických studiích s krizotinibem byla febrilní neutropenie zaznamenána u méně než 0,5 % pacientů.
Napříč studiemi u pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC (N=1722) byla leukopenie 3. nebo 4. stupně pozorována u 48 (3 %) pacientů léčených krizotinibem. Medián doby do nástupu jakéhokoli stupně leukopenie byl 85 dní.
Leukopenie byla spojena se snížením dávky u < 0,5 % pacientů a u žádných pacientů nebyla léčba krizotinibem trvale ukončena v souvislosti s leukopenií.
V klinických studiích krizotinibu u pacientů s ALK-pozitivním nebo ROS1-pozitivním pokročilým NSCLC byla pozorována snížení leukocytů a neutrofilů na stupeň 3 nebo 4 s frekvencí 4 %, resp.
13 %.
Kompletní krevní obraz včetně diferenciálního počtu leukocytů má být sledován dle klinické potřeby, s častějším opakováním testování v případě, že jsou pozorovány abnormality stupně 3 nebo 4, nebo pokud se objeví horečka či infekce. Pacienti, u nichž se rozvinou hematologické laboratorní abnormality, viz bod 4.2.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Léčba předávkování tímto léčivým přípravkem spočívá v obecných podpůrných opatřeních. Proti přípravku XALKORI neexistuje žádné antidotum.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, inhibitory proteinkináz, kód ATC: L01XE16. Mechanismus účinku
Krizotinib je selektivním nízkomolekulárním inhibitorem receptorové tyrozinkinázy ALK (RTK) a jejích onkogenních variant (tj. případ ALK fúze a vybrané mutace ALK). Krizotinib je také inhibitorem receptoru pro růstový faktor pro hepatocyty (HGFR, c-Met) RTK, ROS1 (c-ros) a Recepteur d’Origine Nantais (RON) RTK. Krizotinib prokázal v závislosti na koncentraci inhibici kinázové aktivity ALK, ROS1 a c-Met v biochemických testech a v buněčných testech inhiboval fosforylaci a modulované fenotypy závislé na kináze. Krizotinib prokázal silnou a selektivní aktivitu inhibice růstu a vyvolával apoptózu u nádorových buněk vykazující fúze ALK (včetně echinodermového s mikrotubuly asociovaného proteinu 4 [EML4]-ALK a nukleofosminu [NPM]-ALK), fúze ROS1 nebo vykazující amplifikaci míst uložení genu ALK nebo MET. Krizotinib prokázal protinádorový účinek včetně podstatné cytoredukční protinádorové aktivity u myší s nádorovými xenoimplantáty, které exprimovaly fúzní proteiny ALK. Protinádorový účinek krizotinibu byl závislý na dávce a souvisel s farmakodynamickou inhibicí fosforylace ALK fúzních proteinů (včetně EML4-ALK a NPM-ALK), u nádorů in vivo. Krizotinib také prokázal značnou protinádorovou aktivitu v xenograftových studiích u myší, v nichž byly nádory vytvořeny pomocí panelu buněčných linií NIH-3T3 navržených tak, aby exprimovaly hlavní fúze ROS1 identifikované v nádorech u člověka. Protinádorový účinek krizotinibu byl závislý na dávce a demonstroval korelaci s inhibicí fosforylace ROS1 in vivo.
Klinické studie
Dosud neléčený ALK-pozitivní pokročilý NSCLC - randomizovaná studie 1014 fáze III Účinnost a bezpečnost krizotinibu při léčbě pacientů s ALK-pozitivním metastazujícím NSCLC, kteří nebyli dříve léčeni systémovou léčbou pro pokročilé onemocnění, byla prokázána v celosvětové, randomizované, otevřené studii 1014.
Úplná analyzovaná populace zahrnovala 343 pacientů s ALK-pozitivním pokročilým NSCLC indetifikovaným pomocí fluorescenční in situ hybridizace (FISH) před randomizací: 172 pacientů bylo randomizováno do ramene s krizotinibem a 171 pacientů bylo randomizováno do ramene s chemoterapií (pemetrexed + karboplatina nebo cisplatina; až 6 cyklů léčby). Demografické údaje a charakteristiky onemocnění celkové studované populace byly 62 % žen, medián věku 53 let, výkonnost podle skupiny Eastern Cooperative Oncology Group (ECOG) na počátku 0 nebo 1 (95 %), 51 % bílé a 46 % asijské rasy, 4 % představovali aktivní kuřáci, 32 % bývalí kuřáci a 64 % nikdy nekouřilo. Charakteristiky onemocnění celkové studované populace byly metastazující onemocnění u 98 % pacientů, u 92 % pacientů byly nádory histologicky klasifikovány jako adenokarcinomy a 27 % pacientů mělo mozkové metastázy.
Dle uvážení zkoušejícího mohli pacienti v léčbě krizotinibem pokračovat i po době progrese onemocnění podle kritérií hodnocení odpovědi solidního nádoru na léčbu (RECIST), jestliže byl u pacienta ještě pozorován klinický přínos léčby. Šedesát pět z 89 (73 %) pacientů léčených krizotinibem a 11 ze 132 (8,3 %) pacientů léčených chemoterapií pokračovalo v léčbě po dobu nejméně 3 týdnů po objektivní progresi onemocnění. Pacienti randomizovaní do ramene s chemoterapií mohli přejít na léčbu krizotinibem při progresi onemocnění podle kritérií RECIST, potvrzené na základě nezávislého radiologického vyhodnocení (IRR). Jedno sto dvacet (70 %) pacientů ve skupině s chemoterapií bylo následně léčeno krizotinibem.
Krizotinib v porovnání s chemoterapií významně prodloužil primární cíl studie, přežití bez progrese (PFS), dle hodnocení na základě IRR. Přínos krizotinibu z hlediska PFS byl konzistentní napříč podskupinami pacientů podle výchozích charakteristik, například podle věku, pohlaví, rasy, kouření, doby od diagnózy, stavu výkonnosti podle ECOG a přítomnosti mozkových metastáz. Údaje o účinnosti z randomizované studie 1014 fáze 3 jsou shrnuty v tabulce 4 a Kaplan-Meierovy křivky PFS a celkového přežití (OS) jsou znázorněny na obrázku 1 resp. 2. Údaje o celkovém přežití (OS) nebyly v době analýzy PFS konečné.
Tabulka 4. Výsledky účinnosti z randomizované studie 1014 fáze III (úplná analyzovaná
populace) u pacientů s dosud neléčeným ALK-pozitivním pokročilým NSCLC
|
Parametr léčebné odpovědi |
Krizotinib N=172 |
Chemoterapie N=171 |
|
Přežití bez progrese (na základě IRR) | ||
|
Počet s příhodou, n (%) |
100 (58 %) |
137 (80 %) |
|
Medián PFS v měsících (95% CI) |
10,9 (8,3, 13,9) |
7,0a (6,8, 8,2) |
|
HR(95% CI)b |
0,45 (0,35, 0,60) | |
|
Hodnota pc |
<0,0001 | |
|
Celkové přežitíd | ||
|
Počet úmrtí, n (%) |
44 (26 %) |
46 (27 %) |
|
Medián OS v měsících (95% CI) |
NR |
NR |
|
HR (95% CI)b |
0,82 (0,54, 1,26) | |
|
Hodnota pc |
0,] |
804 |
|
Pravděpodobnost přežití v délce 12 měsíců,d % (95% CI) |
83,5 (76,7, 88,5) |
78,6 (71,3, 84,2) |
|
Pravděpodobnost přežití v délce 18 měsíců,d % (95% CI) |
68,6 (59,5, 76,1) |
67,3 (58,1, 74,9) |
|
Výskyt objektivní léčebné odpovědi (na základě IRR) | ||
|
Výskyt objektivní léčebné odpovědi % (95% CI) |
74% (67, 81) |
45 %e (37, 53) |
|
Hodnota pf |
<0,0001 | |
|
Trvání léčebné odpovědi | ||
|
Mediáng, měsíce (95% CI) |
11,3 (8,1, 13,8) |
5,3 (4,1, 5,8) |
Zkratky: CI = interval spolehlivosti; HR = poměr rizik; IRR = nezávislé radiologické vyhodnocení; N = celkový počet pacientů; NR = nedosaženo; PFS = přežití bez progrese; OS = celkové přežití.
a. Medián PFS byl 6,9 měsíců (95% CI: 6,6, 8,3) u pemetrexedu/cisplatiny (HR=0,49; hodnota p < 0,0001 pro krizotinib v porovnání s pemetrexedem/cisplatinou) a 7,0 měsíců (95% CI: 5,9, 8,3)
u pemetrexedu/karboplatiny (HR=0,45; hodnota p < 0,0001 pro krizotinib v porovnání s pemetrexedem/karboplatinou).
b. Založeno na stratifikované Coxově analýze proporcionálních rizik.
c. Založeno na stratifikovaném log-rank testu (1stranném).
d. Analýza OS nebyla korigována s ohledem na potenciálně zkreslující účinky křížení.
e. ORR byl 43 % (95% CI: 37, 58) u pemetrexedu/cisplatiny (hodnota p < 0,0001 v porovnání s krizotinibem) a 44 % (95% CI: 32, 55) u pemetrexedu/karboplatiny (hodnota p < 0,0001 v porovnání s krizotinibem).
f. Založeno na stratifikovaném Cochran-Mantel-Haenszelově testu (2stranném).
g. Hodnoceno za použití Kaplan-Meierovy metody.
Obrázek 1. Kaplan-Meierovy křivky přežití bez progrese (na základě IRR) podle léčebných skupin v randomizované studii 1014 fáze III (úplná analyzovaná populace) u pacientů s dosud neléčeným ALK-pozitivním pokročilým NSCLC
100
XALKORI (N = 172)
Medián 10.9 mesicu
171)
Chemoterapie (N
Medián 7.0 mesicu
Poměr rizik = 0.45
p < 0,0001
_
+-1
Doba (mesice)
Počet rizikových subjektů
XALKORI 172 Chemoterapie 171
Obrázek 2. Kaplan-Meierovy křivky OS podle léčebných skupin v randomizované studii 1014 fáze III (úplná analyzovaná populace) u pacientů s dosud neléčeným ALK-pozitivním pokročilým NSCLC
100
Si 80
XALKORI (N
Medián nebyl dosazen
Chemoterap
Medián nebyl dosazen
Poměr rizik 95% Cl 0,54, 1,26
0.1804
15 20
Doba (měsíce)
Počet rizikových subjektů
XALKORI 172 Chemoterapie 171
U pacientů s dříve léčenými mozkovými metastázami při vstupu do studie byl medián intrakraniálního času do progrese (IC-TTP) 15,7 měsíců ve skupině krizotinibu (N=39) a 12,5 měsíců ve skupině s chemoterapií (N=40), (HR=0,45 [95% CI: 0,19, 1,07]; 1stranná hodnotap = 0,0315). U pacientů bez mozkových metastáz při vstupu do studie nebylo mediánu IC-TTP dosaženo ani ve skupině krizotinibu (N=132), ani ve skupině chemoterapie (N=131), (HR=0,69 [95% CI: 0,33, 1,45]; 1stranná hodnota p = 0,1617).
Údaje o příznacích a celkové kvalitě života (QOL) hlášené pacientem byly shromažďovány pomocí dotazníku EORTC QLQ-C30 a jeho modulu pro karcinom plic (EORTC QLQ-LC13). Celkem 166 pacientů ve skupině krizotinibu a 163 pacientů ve skupině chemoterapie vyplnilo při vstupu do studie a alespoň při 1 návštěvě po vstupu do studie dotazníky EORTC QLQ-C30 a LC13 . Ve skupině s krizotinibem bylo pozorováno významně větší zlepšení celkové QOL v porovnání se skupinou s chemoterapií (celkový rozdíl ve změně od výchozích skóre 13,8; hodnota p < 0,0001).
Doba do zhoršení (TTD) byla předem určena jako první výskyt > 10bodového zvýšení skóre od výchozího stavu u příznaků bolesti na hrudi, kašle nebo dyspnoe, na základě hodnocení pomocí dotazníku EORTC QLQ-LC13.
Krizotinib vedl v porovnání s chemoterapií k významnému prodloužení TTD příznaků (medián
2,1 měsíců oproti 0,5 měsícům; HR=0,59; 95% CI: 0,45, 0,77; log-rank s Hochbergovou korekcí 2stranná hodnota p = 0,0005).
Dříve léčený ALK-pozitivnípokročilý NSCLC - randomizovaná studie 1007fáze III Účinnost a bezpečnost krizotinibu při léčbě pacientů s ALK-pozitivním metastazujícím NSCLC, kteří byli dříve léčeni systémovou léčbou pro pokročilé onemocnění, byla prokázána v celosvětové, randomizované, otevřené studii 1007.
Úplná analyzovaná populace zahrnovala 347 pacientů s ALK-pozitivním pokročilým NSCLC identifikovaným pomocí FISH před randomizací. Jedno sto sedmdesát tři (173) pacientů bylo randomizováno do ramene s krizotinibem a 174 pacientů bylo randomizováno do ramene chemoterapie (buď pemetrexed nebo docetaxel). Demografické údaje a charakteristiky onemocnění celkové studované populace byly 56 % žen, medián věku 50 let, výkonnost podle ECOG na počátku 0 (39 %) nebo 1 (52 %), 52 % bílé a 45 % asijské rasy, 4 % představovali aktivní kuřáci, 33 % bývalí kuřáci a 63 % nikdy nekouřilo, metastazující onemocnění u 93 % pacientů a u 93 % pacientů byly nádory histologicky klasifikovány jako adenokarcinomy.
Dle uvážení zkoušejícího mohli pacienti v přiřazené léčbě pokračovat i po době progrese onemocnění podle kritérií RECIST, které bylo zhodnoceno na základě IRR, jestliže byl u pacienta pozorován klinický přínos léčby. Padesát osm z 84 (69 %) pacientů léčených krizotinibem a 17 ze 119 (14 %) pacientů léčených chemoterapií pokračovalo v léčbě po dobu nejméně 3 týdnů po objektivní progresi onemocnění. Pacienti randomizovaní do ramene s chemoterapií mohli přejít na léčbu krizotinibem při progresi onemocnění podle kritérií RECIST, potvrzené na základě IRR. Celkem 112 (64 %) pacientů bylo následně léčeno krizotinibem.
Krizotinib v porovnání s chemoterapií významně prodloužil PFS, primární cíl studie, dle hodnocení na základě IRR. Přínos krizotinibu z hlediska PFS byl konzistentní napříč podskupinami pacientů podle vstupních parametrů, například podle věku, pohlaví, rasy, kouření, doby od diagnózy, stavu výkonnosti ECOG, přítomnosti mozkových metastáz a předchozí léčby inhibitory tyrozinkinázy EGFR.
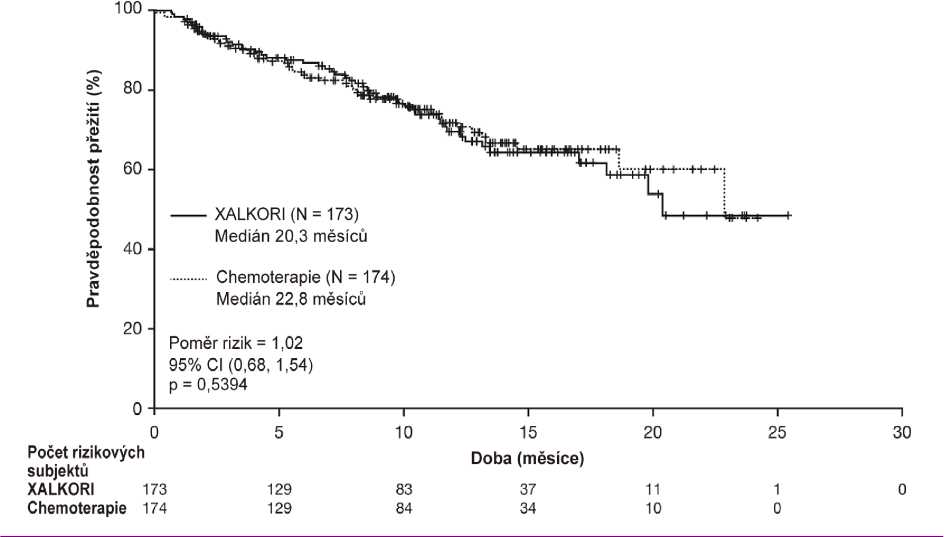
Údaje ze studie 1007 týkající se účinnosti jsou shrnuty v tabulce 5 a Kaplan-Meierovy křivky parametru PFS a OS jsou znázorněny na obrázku 3 resp. 4. Údaje o celkovém přežití (OS) nebyly v době analýzy PFS konečné.
Tabulka 5. Výsledky účinnosti z randomizované studie 1007 fáze III (úplná analyzovaná
populace) u pacientů s dříve léčeným ALK-pozitivním pokročilým NSCLC
|
Parametr léčebné odpovědi |
Krizotinib N=173 |
Chemoterapie N=174 |
|
Přežití bez progrese (na základě IRR) | ||
|
Počet s příhodou, n (%) |
100 (58 %) |
127 (73 %) |
|
Typ příhody, n (%) | ||
|
Progresivní onemocnění |
84 (49 %) |
119 (68 %) |
|
Smrt bez objektivní progrese |
16 (9 %) |
8 (5 %) |
|
Medián PFS v měsících (95% CI) |
7,7 (6,0, 8,8) |
3,0a (2,6, 4,3) |
|
HR(95% CI)b |
0,49 (0,37, 0,64) | |
|
Hodnota pc |
<0,0001 | |
|
Celkové přežitíd | ||
|
Počet úmrtí, n (%) |
49 (28 %) |
47 (27 %) |
|
Medián OS v měsících (95% CI) |
20,3 (18,1, NR) |
22,8 (18,6, NR) |
|
HR (95% CI)b |
1,02 (0,68, 1,54) | |
|
Hodnota pc |
0,5394 | |
|
Pravděpodobnost přežití v délce 6 měsíců,e % (95% CI) |
86,8 (80,4, 91,2) |
83,8 (77,0, 88,7) |
|
Pravděpodobnost přežití v délce 1 roku,e % (95% CI) |
69,5 (60,6, 76,8) |
71,8 (63,3, 78,7) |
|
Výskyt objektivní léčebné odpovědi (na základě IRR) | ||
|
Výskyt objektivní léčebné odpovědi % (95% CI) |
65 % (58, 72) |
20 %f (14, 26) |
|
Hodnota pg |
<0,0001 | |
|
Trvání léčebné odpovědi | ||
|
Mediáne, měsíce (95% CI) |
7,4 (6,1, 9,7) |
5,6 (3,4, 8,3) |
Zkratky: CI = interval spolehlivosti; HR = poměr rizik; IRR = nezávislé radiologické vyhodnocení; N = celkový počet pacientů; NR = nedosaženo; PFS = přežití bez progrese; OS = celkové přežití. a. Mediány doby PFS byly 4,2 měsíce (95% CI: 2,8, 5,7) u pemetrexedu (HR=0,59; hodnota p=0,0004 pro
krizotinib v porovnání s pemetrexedem) a 2,6 měsíce (95% CI: 1,6, 4,0) u docetaxelu (HR=0,30; hodnota p < 0,0001 pro krizotinib v porovnání s docetaxelem).
b. Založeno na stratifikované Coxově analýze proporcionálních rizik.
c. Založeno na stratifikovaném log-rank testu (1stranném).
d. Interim analýza OS provedená při dosažení 40 % všech příhod potřebných pro závěrečnou analýzu. Analýza OS nebyla korigována s ohledem na potenciálně zkreslující účinky křížení.
e. Hodnoceno za použití Kaplan-Meierovy metody.
f. ORR byly 29 % (95% CI: 21, 39) u pemetrexedu (hodnota p < 0,0001 v porovnání s krizotinibem) a 7 % (95% CI: 2, 16) u docetaxelu (hodnota p < 0,0001 v porovnání s krizotinibem).
g. Založeno na stratifikovaném Cochran-Mantel-Haenszelově testu (2stranném).
Obrázek 3. Kaplan-Meierovy křivky přežití bez progrese (na základě IRR) podle léčebných skupin v randomizované studii 1007 fáze III (úplná analyzovaná populace) u pacientů s dříve léčeným ALK-pozitivním pokročilým NSCLC
100
80
60
40-
20-
Poměr rizik = 0,49
95% Cl 0,37, 0,64)
p < 0,0001
XALKORI (N =173)
Medián 7,7 měsíce
Chemoterapie (N =174)
Medián 3,0 měsíce
Počet rizikových
subjektu
XALKORI
173
Chemoterapie
174
Obrázek 4. Kaplan-Meierovy křivky celkového přežití podle léčebných skupin v
randomizované studii 1007 fáze III (úplná analyzovaná populace) u pacientů s dříve léčeným ALK-pozitivním pokročilým NSCLC
Padesát dva (52) pacientů léčených krizotinibem a 57 pacientů léčených chemoterapií s dříve léčenými nebo neléčenými asymptomatickými mozkovými metastázami bylo zařazeno do randomizované studie 1007 fáze III. Míra kontroly intrakraniálního onemocnění (IC-DCR) ve 12. týdnu činila 65 % u pacientů léčených krizotinibem a 46 % u pacientů na chemoterapii.
Údaje o příznacích a celkové kvalitě života (QOL) hlášené pacientem byly shromažďovány pomocí dotazníku EORTC QLQ-C30 a jeho modulu pro karcinom plic (EORTC QLQ-LC13) při vstupu do studie (den 1 cyklus 1) a v den 1 každého následného cyklu léčby. Celkem 162 pacientů ve skupině krizotinibu a 151 pacientů ve skupině chemoterapie vyplnilo při vstupu do studie a nejméně při 1 návštěvě po vstupu do studie dotazníky EORTC QLQ-C30 a LC-13.
Krizotinib vedl v porovnání s chemoterapií k významnému prodloužení doby do zhoršení (medián 4,5 měsíce versus 1,4 měsíce) příznaků u pacientů, kteří hlásili příznaky bolesti na hrudi, dyspnoe nebo kašle (HR 0,504; 95% CI: 0,37, 0,66; log-rank s Hochbergovou korekcí 2stranná hodnota p < 0,0001).
Krizotinib v porovnání s chemoterapií prokázal významně větší zlepšení oproti výchozímu stavu u alopecie (2. až 15. cyklus; hodnota p < 0,05), kašle (2. až 20. cyklus; hodnota p < 0,0001), dyspnoe (2. až 20. cyklus; hodnota p < 0,0001), hemoptýzy (2. až 20. cyklus; hodnota p < 0,05), bolesti v paži nebo rameni (2. až 20. cyklus; hodnota p < 0,0001), bolesti na hrudi (2. až 20. cyklus; hodnota p < 0,0001) a bolesti v jiných částech těla (2. až 20. cyklus; hodnota p < 0,05). Krizotinib v porovnání s chemoterapií vedl k významně menšímu zhoršení oproti výchozímu stavu u periferní neuropatie (6. až 20. cyklus; hodnota p < 0,05), dysfagie (5. až 11. cyklus; hodnota p < 0,05) a bolesti v ústech (2. až 20. cyklus; hodnota p < 0,05).
Krizotinib vedl k celkovému zlepšení celkové kvality života, přičemž ve skupině krizotinibu bylo pozorováno významně větší zlepšení oproti výchozímu stavu než ve skupině chemoterapie (2. až 20. cyklus; hodnota p < 0,05).
Jednoramenné studie u pacientů s ALK-pozitivním pokročilým NSCLC
Použití samotného krizotinibu (v monoterapii) při léčbě ALK-pozitivního pokročilého NSCLC bylo hodnoceno ve 2 mezinárodních, jednoramenných studiích (studie 1001 a 1005). Z pacientů zařazených do těchto studií níže uvedení pacienti dříve podstoupili systémovou léčbu lokálně pokročilého nebo metastazujícího onemocnění. Primárním cílovým parametrem účinnosti v obou studiích byl výskyt objektivní léčebné odpovědi (ORR) podle kritérií RECIST.
K termínu uzavření údajů pro analýzu PFS a ORR bylo do studie 1001 zařazeno celkem 149 pacientů s ALK-pozitivním pokročilým NSCLC, včetně 125 pacientů s dříve léčeným ALK-pozitivním pokročilým NSCLC k termínu uzavření údajů pro analýzu PFS a ORR. Demografické údaje a charakteristiky onemocnění byly 50 % žen, medián věku 51 let, výkonnost ECOG na počátku 0 (32 %) nebo 1 (55 %), 61 % bílé a 30 % asijské rasy, méně než 1 % představovali aktivní kuřáci,
27 % bývalí kuřáci, 72 % nikdy nekouřilo, 94 % metastazujících onemocnění a 98 % nádorů bylo histologicky klasifikováno jako adenokarcinom. Medián doby trvání léčby byl 42 týdnů.
K termínu uzavření údajů pro analýzu PFS a ORR bylo ve studii 1005 léčeno krizotinibem celkem 934 pacientů s ALK-pozitivním pokročilým NSCLC. Demografické údaje a charakteristiky onemocnění byly 57 % žen, medián věku 53 let, výkonnost ECOG na počátku 0/1 (82 %) nebo 2/3 (18 %), 52 % bílé a 44 % asijské rasy, 4 % představovali aktivní kuřáci, 30 % bývalí kuřáci, 66 % nikdy nekouřilo, 92 % metastazujících onemocnění a 94 % nádorů bylo histologicky klasifikováno jako adenokarcinom. Medián doby trvání léčby u těchto pacientů byl 23 týdnů. Dle uvážení zkoušejícího mohli pacienti v léčbě pokračovat i po době progrese onemocnění podle kritérií RECIST. Sedmdesát sedm ze 106 pacientů (73 %) pokračovalo v léčbě krizotinibem po dobu nejméně 3 týdnů po objektivní progresi onemocnění.
Tabulka 6. Výsledky účinnosti u ALK-pozitivních pokročilých NSCLC ze studií 1001 a 1005
|
Parametr účinnosti |
Studie 1001 N=125a |
Studie 1005 N=765a |
|
Objektivní míra odpovědib [% (95% CI)1 |
60 (51, 69) |
48 (44, 51) |
|
Doba do odpovědi [medián (rozsah)], týdny |
7,9 (2,1, 39,6) |
6,1 (3, 49) |
|
Doba trvání odpovědic [medián (95% CI)1, týdny |
48,1 (35,7, 64,1) |
47,3 (36, 54) |
|
Čas do progresec [medián (95% CI)1, měsíce |
9,2 (7,3, 12,7) |
7,8 (6,9, 9,5)d |
|
N=154e |
N=905e | |
|
Počet úmrtí, n (%) |
83 (54 %) |
504 (56 %) |
|
Celkové přežití0 [medián (95% CI)1, měsíce |
28,9 (21,1, 40,1) |
21,5 (19,3, 23,6) |
Zkratky: CI = interval spolehlivosti; N = celkový počet pacientů.
a. Dle dat uzavření údajů 1. června 2011 (studie 1001) a 15. února 2012 (studie 1005).
b. Hodnotitelná odpověď nebyla u tří pacientů ve studii 1001 a u 42 pacientů ve studii 1005.
c. Hodnoceno za použití Kaplan-Meierovy metody.
d. Údaje PFS ze studie 1005 zahrnovaly 807 pacientů v populaci analýzy bezpečnosti, kteří byli identifikováni pomocí FISH analýzy (datum uzavření údajů 15. února 2012)
e. Dle data uzavření údajů 30. listopadu 2013.
ROSl-pozitivní pokročilý NSCLC
Použití samotného krizotinibu (v monoterapii) při léčbě ROS1-pozitivního pokročilého NSCLC bylo hodnoceno v multicentrické, mezinárodní, jednoramenné studii 1001. K datu uzavření údajů bylo do studie zařazeno celkem 53 pacientů s ROS1-pozitivním pokročilým NSCLC, včetně 46 pacientů, kteří dříve podstoupili léčbu ROS1-pozitivního pokročilého NSCLC, a omezeného počtu pacientů (N=7) bez předchozí systémové léčby. Primárním cílovým parametrem účinnosti byl výskyt objektivní léčebné odpovědi (ORR) podle kritérií RECIST. Sekundární cílové parametry zahrnovaly TTR, DR, PFS a OS. Pacienti dostávali krizotinib 250 mg perorálně dvakrát denně.
Demografické údaje byly 57 % žen, medián věku 55 let; výkonnost ECOG na počátku 0 nebo 1 (98 %) nebo 2 (2 %), 57 % bílé a 40 % asijské rasy; 25 % bývalí kuřáci a 75 % nikdy nekouřilo. Charakteristiky onemocnění byly 91 % metastazující, 96 % nádorů bylo histologicky klasifikováno jako adenokarcinom a 13 % bez předchozí systémové léčby metastazujícího onemocnění.
Ve studii 1001 museli pacienti před vstupem do klinické studie mít pokročilý ROS1-pozitivní NSCLC. U většiny pacientů byl ROS1-pozitivní NSCLC identifikován pomocí fluorescenční in situ hybridizace (FISH). Medián doby trvání léčby byl 101 týdnů. Pro ORR 70 % (95 % CI: 56 %, 82 %) bylo dosaženo 5 úplných odpovědí a 32 částečných odpovědí. Mediánu DR nebylo dosaženo (95% CI: 15,2 měsíců, NR). Během prvních 8 týdnů léčby bylo dosaženo padesáti jednoho procenta objektivních odpovědí nádorů. Medián PFS byl k datu uzavření údajů 19,3 měsíců (95% CI:
14,8 NR). Celkové údaje o přežití nebyly k datu uzavření údajů k dispozici.
Tabulka 7. Výsledky účinnosti u ROSl-pozitivních pokročilých NSCLC ze studie 1001
|
Parametr účinnosti |
Studie 1001 N=53a |
|
Objektivní míra odpovědi [% (95% CI)] |
70 (56, 82) |
|
Doba do odpovědi [medián (rozsah)], týdny |
8 (4, 32) |
|
Doba trvání odpovědib [medián (95% CI)], týdny |
NR (15,2, NR) |
|
Čas do progreseb [medián (95% CI)], měsíce |
19,3 (14,8, NR) |
Zkratky: CI = interval spolehlivosti; N = počet pacientů; NR = nedosaženo.
a. Dle dat uzavření údajů 30. listopadu 2014.
b. Hodnoceno za použití Kaplan-Meierovy metody.
Histologie jiná než adenokarcinom
Dvacet jedna pacientů s dosud neléčeným a 12 pacientů s dříve léčeným ALK-pozitivním NSCLC jiným než adenokarcinom bylo zařazeno do randomizovaných studií 1014 a 1007 fáze III. Podskupiny v těchto studiích byly pro vyvození spolehlivých závěrů příliš malé. Je třeba poznamenat, že do skupiny krizotinibu ve studii 1007 nebyli randomizováni žádní pacienti s histologickým nálezem SCC a do studie 1014 nebyli zařazeni žádní pacienti se SCC kvůli režimu založenému na pemetrexedu, který se používal jako srovnávací přípravek.
Informace jsou dostupné pouze ze 45 hodnotitelných odpovědí pacientů s dříve léčeným NSCLC jiným než adenokarcinom (včetně 22 pacientů se SCC) ve studii 1005. Částečné odpovědi byly zaznamenány u 20 ze 45 pacientů s NSCLC jiným než adenokarcinom, přičemž ORR byla 44 %, a u 9 z 22 pacientů se SCC NSCLC, přičemž ORR byla 41 %, což bylo v obou případech méně než ORR hlášená ve studii 1005 (ORR 54 %) pro všechny pacienty.
Opětovná léčba krizotinibem
Nejsou k dispozici žádné údaje o účinnosti týkající se opětovné léčby krizotinibem u pacientů, kterým byl krizotinib podáván v předchozích liniích léčby.
Starší pacienti
Ze 171 pacientů s ALK-pozitivním NSCLC léčených krizotinibem v randomizované studii 1014 fáze III bylo 22 (13 %) ve věku 65 let a starší a ze 109 pacientů s ALK-pozitivním NSCLC léčených krizotinibem, kteří přešli ze skupiny chemoterapie, bylo 26 (24 %) ve věku 65 let a starší. Ze 172 pacientů s ALK-pozitivním NSCLC léčených krizotinibem v randomizované studii 1007 fáze III bylo 27 (16 %) ve věku 65 let a starší. Ze 154 pacientů s ALK-pozitivním NSCLC v jednoramenné studii 1001 bylo 22 (14 %) ve věku 65 let a starších. Z 1063 pacientů s ALK-pozitivním NSCLC v jednoramenné studii 1005 bylo 173 (16 %) ve věku 65 let a starších. U pacientů s ALK-pozitivním NSCLC byla četnost nežádoucích účinků obecně podobná u pacientů ve věku < 65 let a pacientů ve věku > 65 let s výjimkou otoků a zácpy, které byly hlášeny s větší frekvencí (>15% rozdíl) ve studii 1014 u pacientů ve věku > 65 let léčených krizotinibem. Žádný pacient ve věku > 85 let nebyl zařazen do skupiny krizotinibu v randomizovaných studiích 1007 a 1014 fáze III a v jednoramenné studii 1005. V jednoramenné studii 1001 byl jeden pacient s ALK-pozitivním NSCLC ze 154 pacientů ve věku > 85 let (viz také bod 4.2 a 5.2). Z 53 pacientů s ROS1-pozitivním NSCLC v jednoramenné studii 1001 bylo 15 (28 %) ve věku 65 let a starší. Ve studii 1001 nebyl žádný pacient s ROS1-pozitivním NSCLC ve věku > 85 let.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem XALKORI u všech podskupin pediatrické populace s NSCLC (informace o použití u dětí viz bod 4.2).
Podmíněné schválení
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech. Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání jedné dávky na lačno se krizotinib vstřebává s mediánem doby do dosažení maximální koncentrace 4 až 6 hodin. Při dávkování dvakrát denně bylo dosaženo ustáleného stavu během 15 dní. Absolutní biologická dostupnost krizotinibu byla stanovena na 43 % po podání jedné perorální dávky 250 mg.
Tučné jídlo snížilo AUCinf a Cmax krizotinibu přibližně o 14 %, když byla zdravým dobrovolníkům podána jednotlivá dávka 250 mg. Krizotinib lze podávat s jídlem nebo bez něj (viz bod 4.2).
Distribuce
Geometrický průměrný objem distribuce (Vss) krizotinibu činil 1772 l po nitrožilním podání dávky 50 mg, což poukazuje na extenzivní distribuci do tkání z plazmy.
Vazba krizotinibu na lidské plazmatické bílkoviny in vitro je 91 % a je nezávislá na koncentraci léčivého přípravku. In vitro studie naznačují, že krizotinib je substrátem pro P-glykoprotein (P-gp).
Biotransformace
In vitro studie prokázaly, že CYP3A4/5 byly hlavními enzymy, které se podílely se na metabolické clearanci krizotinibu. Primárními metabolickými drahami u člověka byla oxidace piperidinového kruhu na krizotinib laktam a O-dealkylace s následnou konjugací O-dealkylovaných metabolitů fáze 2.
In vitro studie na lidských jaterních mikrozomech prokázaly, že krizotinib je časově závislým inhibitorem CYP2B6 a CYP3A (viz bod 4.5). In vitro studie prokázaly, že v důsledku krizotinibem zprostředkované inhibice metabolismu léčivých přípravků, které jsou substráty CYP1A2, CYP2C8, CYP2C9, CYP2C19 nebo CYP2D6, jsou klinické lékové interakce nepravděpodobné.
In vitro studie ukázaly, že krizotinib je slabým inhibitorem UGT1A1 a UGT2B7 (viz bod 4.5).
Avšak in vitro studie naznačují, že výskyt klinických lékových interakcí v důsledku inhibice metabolismu léků, které jsou substráty UGT1A1, UGT1A4, UGT1A6, UGT1A9 či UGT2B7, zprostředkované krizotinibem, je nepravděpodobný.
In vitro studie lidských hepatocytů naznačily, že klinické lékové interakce v důsledku krizotinibem zprostředkované indukce metabolismu léčiv, která jsou substráty CYP1A2, jsou nepravděpodobné.
Eliminace
Po jednorázových dávkách krizotinibu byl u pacientů zřejmý terminální poločas krizotinibu v plazmě 42 hodin.
Po podání jedné perorální dávky 250 mg radioaktivně značeného krizotinibu u zdravých jedinců bylo ve stolici nalezeno 63 % podané dávky a v moči 22 %. Nezměněný krizotinib reprezentoval přibližně 53 % podané dávky ve stolici a 2,3 % v moči.
Současné podávání s léčivými přípravky, které jsou substráty transportérů
Krizotinib je inhibitorem P-glykoproteinu (P-gp) in vitro. Proto může mít krizotinib potenciál k zvyšování plazmatické koncentrace současně podávaných léků, které jsou substráty P-gp (viz bod 4.5).
Krizotinib je inhibitorem OCT1 a OCT2 in vitro. Proto může mít krizotinib potenciál k zvyšování plazmatické koncentrace současně podávaných léků, které jsou substráty OCT1 nebo OCT2 (viz bod 4.5).
In vitro neinhiboval krizotinib transportní proteiny jaterního vychytávání polypeptidy transportující organické anionty (OATP)1B1 nebo OATP1B3 ani transportní proteiny renálního vychytávání transportéry organických aniontů (OAT) 1 či OAT3 u člověka při klinicky relevantních koncentracích. Proto jsou klinické lékové interakce v důsledku krizotinibem zprostředkované inhibice jaterního či renálního vychytávání léčivých přípravků, které jsou substráty těchto transportérů, nepravděpodobné.
Účinek na jiné transportní proteiny
Krizotinib není in vitro inhibitorem BSEP při klinicky relevantních koncentracích.
Farmakokinetika u zvláštních skupin pacientů Jaterní insuficience
Protože je krizotinib do značné míry metabolizován v játrech, porucha funkce jater pravděpodobně zvýší plazmatické koncentrace krizotinibu. Krizotinib však nebyl studován u pacientů s poruchou funkce jater. Z klinických studií, které byly prováděny, byli vyloučeni pacienti s ALT nebo AST > 2,5 x ULN, nebo pokud z důvodu základního maligního onemocnění bylo >5,0 x ULN nebo s celkovým bilirubinem > 1,5 x ULN (viz bod 4.2). Populační farmakokinetická analýza používající údaje z těchto studií ukázala, že výchozí hladina celkového bilirubinu či AST neměla klinicky významný vliv na farmakokinetiku krizotinibu.
Renální insuficience
Pacienti s lehkou (60 < CLcr < 90 ml/min) a středně těžkou (30 < CLcr < 60 ml/min) poruchou funkce ledvin byli zařazeni do jednoramenných studií 1001 a 1005. Byl hodnocen účinek funkce ledvin měřené jako výchozí clearance kreatininu (CLcr) na pozorované minimální koncentrace krizotinibu v ustáleném stavu (Ctrough, ss). Ve studii 1001 byl upravený geometrický průměr plazmatické koncentrace Ctrough, ss u pacientů s lehkou (N=35) a středně těžkou (N=8) poruchou funkce ledvin o
5,1 % resp. 11 % vyšší než u pacientů s normální funkcí ledvin. Ve studii 1005 byl upravený geometrický průměr koncentrace Ctrough, ss krizotinibu ve skupinách s lehkou (N=191) a středně těžkou (N=65) poruchou funkce ledvin o 9,1 % resp. 15 % vyšší než u pacientů s normální funkcí ledvin. Kromě toho populační farmakokinetická analýza používající údaje ze studií 1001, 1005 a 1007 ukázala, že CLcr neměla klinicky významný účinek na farmakokinetiku krizotinibu. Vzhledem k malému rozsahu zvýšení expozice krizotinibu (5 %-15 %) se u pacientů s lehkou nebo středně těžkou poruchou funkce ledvin nedoporučuje žádná úprava zahajovací dávky.
U subjektů se těžkou poruchou funkce ledvin (CLcr <30 ml/min) nevyžadující peritoneální dialýzu nebo hemodialýzu došlo po jedné dávce 250 mg ke zvýšení AUC a Cmax krizotinibu o 79 % respektive34 %, v porovnání se subjekty s normální funkcí ledvin. Pokud se krizotinib podává pacientům se těžkou poruchou funkce ledvin nevyžadující peritoneální dialýzu nebo hemodialýzu doporučuje se úprava dávky (viz body 4.2 a 4.4).
Věk
Na základě populační farmakokinetické analýzy údajů ze studií 1001, 1005 a 1007 nemá věk žádný vliv na farmakokinetiku krizotinibu (viz body 4.2 a 5.1).
Tělesná hmotnost a pohlaví
Na základě populační farmakokinetické analýzy údajů ze studií 1001, 1005 a 1007 nedošlo k žádnému klinicky významnému vlivu tělesné hmotnosti nebo pohlaví na farmakokinetiku krizotinibu.
Etnické skupiny
Na základě populační farmakokinetické analýzy údajů ze studií 1001, 1005 a 1007 byl predikovaný AUC v rovnovážném stavu (95% CI) u asijských pacientů (N=523) o 23 %-37 % vyšší než v případě neasijských pacientů (N=691).
Ve studiích u pacientů s ALK-pozitivním pokročilým NSCLC (N=1669) byly následující nežádoucí účinky hlášeny s absolutním rozdílem > 10 % u asijských pacientů (N=753) než u neasijských pacientů (N=916): zvýšené transaminázy, snížená chuť k jídlu, neutropenie a leukopenie. Žádné nežádoucí účinky nebyly hlášeny s absolutním rozdílem > 15 %.
Starší pacienti
U této podskupiny pacientů jsou k dispozici pouze omezené údaje (viz body 4.2 a 5.1). Na základě populační farmakokinetické analýzy údajů ze studií 1001, 1005 a 1007 nemá věk žádný vliv na farmakokinetiku krizotinibu.
Elektrofyziologie srdce
Potenciál krizotinibu, pokud jde o prodloužení QT intervalu, byl hodnocen u pacientů s ALK-pozitivním nebo ROS1-pozitivním NSCLC, kterým byl podáván krizotinib v dávce 250 mg dvakrát denně. Pro účely vyhodnocení účinku krizotinibu na QT intervaly bylo třikrát provedeno EKG, po podání jedné dávky a v ustáleném stavu. U třiceti čtyř z 1619 pacientů (2,1 %) s alespoň 1 provedeným vyšetřením EKG po vstupu do studie byly zjištěny QTcF > 500 ms a u 79 z 1585 pacientů (5,0 %) s výchozím vyšetřením EKG a alespoň 1 provedeným vyšetřením EKG po vstupu do studie bylo zaznamenáno zvýšení výchozí hodnoty QTcF > 60 ms na základě automaticky odečítaného hodnocení EKG (viz bod 4.4).
Dílčí studie EKG, v níž bylo použito zaslepené ruční měření EKG, byla provedena na 52 pacientech s ALK pozitivním NSCLC, kteří užívali krizotinib 250 mg dvakrát denně. Jedenáct pacientů (21 %) mělo zvýšení QTcF oproti výchozí hodnotě > 30 až < 60 ms a jeden pacient (2%)měl zvýšení QTcF oproti výchozí hodnotě > 60 ms. Žádní pacienti neměli maximální QTcF > 480 ms. Analýza centrální tendence ukazuje, že všechny horní hranice 90% CI pro průměrnou změnu LS oproti výchozím hodnotám QTcF ve všech časových bodech 1. dne 2. cyklu byly < 20 ms.
Farmakokinetická/farmakodynamická analýza naznačila vztah mezi koncentrací krizotinibu v plazmě a QTc. Kromě toho bylo zjištěno, že pokles srdečního rytmu souvisel se zvyšující se plazmatickou koncentrací krizotinibu (viz bod 4.4), s maximálním průměrným snížením 17,8 tepů za minutu po 8 hodinách 1. dne 2. cyklu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ve studiích toxicity s opakovanou dávkou prováděných u potkanů a psů trvajících až 3 měsíce byly primárně zjištěnými cílovými orgány nežádoucího působení gastrointestinální trakt (zvracení, změněné výkaly, zácpa), hematopoetický systém (hypocelularita kostní dřeně), kardiovaskulární systém (blokáda smíšených iontových kanálů, snížení srdečního rytmu a krevního tlaku, zvýšené intervaly LVEDP, QRS a PR a snížení srdeční kontraktility), nebo reprodukčního systému (degenerace pachytenních spermatocytů, nekróza jednotlivých buněk ovariálních folikulů).
Bez zjistitelných nežádoucích účinků (NOAEL) pro tyto nálezy byly jak subterapeutické tak až 2,6násobky klinické expozice u člověka na základě hodnot AUC. Mezi další nálezy patřil účinek na játra (zvýšení jaterních transamináz) a retinální funkce, a potenciál pro fosfolipidózu v mnoha orgánech bez korelační toxicity.
Krizotinib nebyl mutagenní v in vitro analýze bakteriální reverzní mutace (Ames). Krizotinib byl aneugenní v in vitro mikronukleus testu na buňkách ovárií čínských křečků a v in vitro testu aberace chromozomů lidských lymfocytů. Malá zvýšení v testu strukturální chromozomální aberace při cytotoxických koncentracích byly pozorovány u lidských lymfocytů. Bez zjistitelných nežádoucích účinků (NOAEL) pro aneugenicitu byl přibližně 1,8násobek klinické expozice u člověka na základě hodnot AUC.
Studie kancerogenity s krizotinibem nebyly prováděny.
Pro účely vyhodnocení vlivu na plodnost nebyly prováděny žádné konkrétní studie krizotinibu na zvířatech, nicméně na základě výsledků studií toxicity opakovaných dávek u potkanů se má za to, že krizotinib má potenciál ovlivnit reprodukční funkce a fertilitu u člověka. Nálezy pozorované v samčím pohlavním ústrojí zahrnovaly degeneraci pachytenních spermatocytů u potkanů, kterým bylo podáváno > 50 mg/kg/den po dobu 28 dnů (přibližně 1,1násobek lidské klinické expozice podle AUC). Nálezy pozorované v samičím pohlavním ústrojí zahrnovaly nekrózu jednotlivých buněk ovariálních folikulů u potkanů, kterým bylo podáváno 500 mg/kg/den po dobu 3 dnů.
U březích samic potkanů a králíků nebyly prokázány teratogenní účinky krizotinibu.
U potkanů se zvýšily postimplantační ztráty při dávce >50 mg/kg/den (přibližně 0,4násobek doporučené dávky u člověka podle AUC) a za nežádoucí účinek u potkanů při podávání 200mg/kg/den a u králíků při 60 mg/kg/den (přibližně 1,2 násobek klinické expozice u člověka podle AUC) byla považována snížená tělesná hmotnost plodu.
U mláďat potkanů byla pozorována snížená kostní tvorba u rostoucích dlouhých kostí při dávce 150 mg/kg/den jednou denně po dobu 28 dnů (přibližně 3,3krát vyšší než klinická expozice u člověka podle AUC). U mláďat nebyly hodnoceny jiné potenciální toxicity týkající se pediatrických pacientů.
Výsledky in vitro studie fototoxicity prokázaly, že krizotinib může mít fototoxický potenciál.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
koloidní bezvodý oxid křemičitý mikrokrystalická celulosa hydrogenfosforečnan vápenatý sodná sůl karboxymethylškrobu (Typ A) magnesium-stearát.
Obal tobolky želatina
oxid titaničitý (E171) červený oxid železitý (E172).
Potisk
šelak
propylenglykol hydroxid draselný černý oxid železitý (E172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení XALKORI 200 mg tvrdé tobolky
HDPE lahvičky s polypropylenovým uzávěrem, obsahují 60 tvrdých tobolek.
PVC blistr s fólií obsahující 10 tvrdých tobolek.
Krabička obsahuje 60 tvrdých tobolek XALKORI 250 mg tvrdé tobolky
HDPE lahvičky s polypropylenovým uzávěrem, obsahují 60 tvrdých tobolek.
PVC blistr s fólií obsahující 10 tvrdých tobolek.
Krabička obsahuje 60 tvrdých tobolek Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
XALKORI 200 mg tvrdé tobolky
EU/1/12/793/001
EU/1/12/793/002
XALKORI 250 mg tvrdé tobolky
EU/1/12/793/003
EU/1/12/793/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 23. října 2012
Datum posledního prodloužení registrace: 29. července 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Pfizer Manufacturing Deutschland GmbH Mooswaldallee 1 79090 Freiburg Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci se musí dohodnout s příslušnými národními regulačními orgány ve všech členských státech ještě před uvedením přípravku na trh na formátu a obsahu edukačního materiálu. Konečné znění edukačního materiálu má být v souladu se schválenou informací o přípravku.
Držitel rozhodnutí o registraci zajistí, aby všichni lékaři, u nichž se očekává, že budou
přípravek Xalkori předepisovat, byli seznámeni s edukačním materiálem obsahujícím
následující položky:
1. Souhrn údajů o přípravku a příbalovou informaci
2. Edukační materiál pro lékaře
3. Brožuru pro pacienta včetně kartičky pro pacienta (text schválený CHMP)
Edukační materiál pro lékaře má obsahovat následující klíčové položky:
• XALKORI prodlužuje QTc interval, což může vést ke zvýšenému riziku ventrikulární tachykardie (např. Torsade de Pointes) nebo náhlé smrti.
• Riziko prodloužení QTc intervalu může být zvýšené u pacientů, kteří současně užívají antiarytmika, a u pacientů s významným preexistujícím srdečním onemocněním, bradykardií nebo porušenou rovnováhou elektrolytů (např. sekundárně u průjmu nebo zvracení).
• XALKORI má být podáván s opatrností pacientům:
a. kteří mají v anamnéze nebo mají predispozici k prodloužení QTc intervalu.
b. kteří užívají léky, o nichž je známo, že prodlužují QT interval.
• Při podávání přípravku XALKORI těmto pacientům je vhodné zvážit pravidelné monitorování EKG a elektrolytů.
• Pacienti, u nichž se rozvine prodloužení QTc intervalu stupně 3, mají pozastavit léčbu přípravkem XALKORI, dokud nedojde ke zmírnění na stupeň < 1, poté pokračovat v dávkování 200 mg dvakrát denně.
• Pacienti, u nichž se rozvine prodloužení QTc intervalu stupně 4, mají přípravek XALKORI trvale vysadit.
• Přípravek XALKORI může vyvolat poruchy zraku včetně defektu zrakového pole stupně 4 se ztrátou zraku. Nejčastěji pozorovanými poruchami zraku však byly diplopie, fotopsie, rozmazaného vidění, poruch zraku a sklivcových plovoucích zákalků a většinou byly lehké závažnosti.
• U pacientů s poruchami zraku má být zváženo oftalmologické vyšetření, pokud poruchy zraku přetrvávají nebo se zhorší jejich závažnost. V případě pacientů s novým vznikem těžké ztráty zraku (nejlépe korigovaná zraková ostrost menší než 6/60 na jednom nebo obou očích), je nutné léčbu přípravkem XALKORI přerušit a je třeba provést příslušná vyšetření.
• Přípravek XALKORI může způsobit hepatotoxicitu, symptomatickou bradykardii (např. synkopu, závrať, hypotenzi), intersticiální plicní onemocnění/pneumonitidu, neutropenii a leukopenii, renální cystu, otok, neuropatii a reprodukční toxicitu. Doporučení, jak tato rizika zmírnit pomocí vhodného monitorování a řízení, budou poskytnuta.
• Je třeba se vyhnout současnému užívání přípravku XALKORI se silnými inhibitory/induktory CYP3A4a substráty CYP3A4 s úzkým terapeutickým indexem.
• Přípravek XALKORI může způsobit gastrointestinální perforaci. Z toho důvodu je třeba krizotinib používat s opatrností u pacientů s rizikem rozvojem gastrointestinální perforace; a u pacientů, u kterých dojde k tomuto nežádoucímu účinku, přerušit podávání krizotinibu.
Pacienty je třeba poučit o prvních známkách gastrointestinální perforace a instruovat je, aby se ihned obrátili na lékaře v případě, že se příznaky objeví.
• Přípravek XALKORI může vyvolat těžké, život ohrožující nebo fatální srdeční selhání. Z toho důvodu mají být pacienti s preexistujícími srdečními poruchami i pacienti bez nich, kteří užívají krizotinib, sledováni na přítomnost známek a příznaků srdečního selhání. Při zjištění takových příznaků je nutno zvážit přerušení podávání, snížení dávky nebo vysazení.
• Je třeba informovat pacienty o rizicích souvisejících s podáváním přípravku XALKORI a informovat je, jakých příznaků a známek si všímat a co mají v případě jejich výskytu udělat.
• Význam a použití kartičky pro pacienta.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Držitel rozhodnutí o registraci byl požádán o aktualizaci stavu OS ze studie A8081007 a poskytnutí konečných údajů do 9 měsíců od dosažení požadovaných 238 příhod OS. Zpráva z klinické studie má obsahovat také podrobnou bezpečnostní analýzu. |
30.června 2016 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
XALKORI 200 mg tvrdé tobolky Crizotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje crizotinibum200 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/793/002
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
XALKORI 200 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
XALKORI 200 mg tvrdé tobolky Crizotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje crizotinibum200 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/793/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
XALKORI 200 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
XALKORI 200 mg tvrdé tobolky Crizotinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Ltd (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
XALKORI 250 mg tvrdé tobolky Crizotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje crizotinibum250 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/793/004
13. ČÍSLO ŠARŽE
c.s.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
XALKORI 250 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
XALKORI 250 mg tvrdé tobolky Crizotinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje crizotinibum250 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/793/003
13. ČÍSLO ŠARŽE
c.s.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
XALKORI 250 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
XALKORI 250 mg tvrdé tobolky Crizotinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Ltd (logo)
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
XALKORI 200 mg tvrdé tobolky XALKORI 250 mg tvrdé tobolky
Crizotinibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek XALKORI a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek XALKORI užívat
3. Jak se přípravek XALKORI užívá
4. Možné nežádoucí účinky
5. Jak přípravek XALKORI uchovávat
6. Obsah balení a další informace
1. Co je přípravek XALKORI a k čemu se používá
Přípravek XALKORI je protinádorový lék obsahující léčivou látku krizotinib. Používá se k léčbě dospělých s plicním karcinomem nazývaným nemalobuněčný karcinom plic (NSCLC), u kterého je přítomno specifické přeskupení nebo defekt v genu nazvaném kináza anaplastického lymfomu (ALK) nebo genu nazvaného ROS1.
Přípravek XALKORI Vám může být předepsán k počáteční léčbě, pokud je Vaše onemocnění plicní karcinom v pokročilé fázi.
Přípravek XALKORI Vám může být předepsán, pokud je Vaše onemocnění v pokročilé fázi a předchozí léčba Vám nepomohla k zastavení nemoci.
Přípravek XALKORI může zpomalit nebo zastavit růst plicního karcinomu. To může pomoci zmenšit nádory.
Pokud máte jakékoliv dotazy ohledně toho, jak léčivý přípravek účinkuje, nebo proč Vám byl tento lék předepsán, zeptejte se svého lékaře.
- jestliže jste alergický(á) na krizotinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6 „Co přípravek XALKORI obsahuje“).
- jestliže máte těžké onemocnění jater.
Upozornění a opatření
Před užitím přípravku XALKORI se poraďte se svým lékařem.
• Jestliže máte lehké nebo středně těžké onemocnění jater.
• Jestliže máte nebo jste měl(a) jakékoli jiné plicní problémy. Některé plicní problémy se mohou zhoršit během léčby přípravkem XALKORI, protože přípravek XALKORI může vyvolat během léčby zápal plic. Příznaky jsou podobné příznakům plicního karcinomu. Sdělte svému lékaři okamžitě, pokud máte jakékoli nové příznaky nebo pokud se zhorší stávající příznaky, včetně obtíží s dýcháním, dušnosti nebo kašle s hlenem nebo bez hlenu nebo horečky.
• Jestliže Vám bylo na základě elektrokardiografického vyšetření (EKG) sděleno, že máte abnormalitu srdečního rytmu, známou jako prodloužení QT intervalu.
• Jestliže máte snížený srdeční rytmus.
• Jestliže jste někdy měl(a) žaludeční nebo střevní problémy, jako je proděravění (perforace) žaludku nebo střeva, trpíte onemocněním, které způsobuje nitrobřišní zánět (divertikulitida), nebo máte nádorové onemocnění, které se rozšířilo uvnitř břicha (metastázy).
• Jestliže máte poruchy zraku (vidění záblesků světla, rozmazané vidění a dvojité vidění).
• Jestliže máte těžkou poruchu funkce ledvin.
• Jestliže jste současně léčen(a) některým z léků uvedených v bodě Další léčivé přípravky a XALKORI.
Okamžitě se obraťte na svého lékaře, pokud se u Vás po užití přípravku XALKORI projeví následující obtíže:
• silná bolest žaludku nebo břicha, horečka, zimnice, dušnost, rychlý puls, částečná nebo úplná ztráta zraku (na jednom nebo obou očích) nebo změny ve způsobu vyprazdňování stolice.
Většina dostupných informací je k dispozici od pacientů se specifickým ALK-pozitivním NSCLC histologickým typem (adenokarcinom) a pro jiné histologické typy jsou údaje omezené.
Děti a dospívající
Léčba dětí a dospívajících tímto přípravkem se nedoporučuje. Tato indikace nezahrnuje děti a dospívající.
Další léčivé přípravky a přípravek XALKORI
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně rostlinných přípravků a léků dostupných bez lékařského předpisu.
Zejména následující přípravky mohou zvýšit riziko nežádoucích účinků přípravku XALKORI:
• Klarithromycin, telithromycin, troleandomycin, antibiotika používaná k léčbě bakteriálních infekcí.
• Ketokonazol, itrakonazol, vorikonazol, používané k léčbě plísňových infekcí.
• Atazanavir, indinavir, nelfinavir, ritonavir, sachinavir, používané k léčbě HIV infekcí/AIDS.
• Fenytoin, karbamazepin nebo fenobarbital, anti-epileptika používaná k léčbě epileptických záchvatů a křečí.
• Rifabutin, rifampicin, používané k léčbě tuberkulózy.
• Třezalka tečkovaná (Hypericumperforatum), rostlinný přípravek k léčbě.
Přípravek XALKORI může zvýšit nežádoucí účinky spojené s následujícími léky:
• Alfentanil, a jiné rychle účinkující opioidní analgetika jako je fentanyl (léky proti bolesti používané při chirurgických zákrocích).
• Chinidin, digoxin, disopyramid, amiodaron, sotalol, dofetilid, ibutilid, verapamil, diltiazem používané při problémech se srdečním rytmem.
• Přípravky používané k léčbě vysokého krevního tlaku, tzv. beta-blokátory, například atenolol, propranolol, labetolol.
• Pimozid, používaný k léčbě duševních chorob.
• Metformin, používaný k léčbě cukrovky.
• Prokainamid, používaný k léčbě nepravidelného srdečního rytmu.
• Cisaprid, používaný k léčbě žaludečních problémů.
• Cyklosporin, sirolimus a takrolimus používané u pacientů po transplantaci.
• Ergotaminové alkaloidy (např. ergotamin, dihydroergotamin), používané k léčbě migrény.
• Dabigatran, antikoagulancium používané ke zpomalení srážení krve.
• Kolchicin, používaný k léčbě dny.
• Pravastatin, používaný ke snížení hladin cholesterolu.
• Klonidin, guanfacin, používané k léčbě vysokého tlaku krve.
• Meflochin, používaný k prevenci malárie.
• Pilokarpin, používaný k léčbě zeleného zákalu (glaukomu), (závažné oční onemocnění).
• Anticholinesterázy, používané k obnově funkce svalů.
• Antipsychotika, používaná k léčbě duševních chorob.
• Moxifloxacin, používaný k léčbě bakteriálních infekcí.
• Methadon, používaný k léčbě bolesti a k léčbě závislosti na opioidních analgeticích.
• Bupropion, používaný k léčbě deprese a k odvykání kouření.
• Efavirenz, raltegravir, používané při infekci HIV.
• Irinotekan, lék užívaný při chemoterapii k léčbě nádoru tlustého střeva a konečníku.
• Morfin, používaný k léčbě akutní a nádorové bolesti.
• Naloxon, používaný k léčbě závislosti a k odvykání drog.
Těchto léků je třeba se vyvarovat během léčby přípravkem XALKORI.
Perorální kontraceptiva
Perorální kontraceptiva (tablety proti početí užívané ústy) mohou být neúčinná, pokud jsou užívána během léčby přípravkem XALKORI.
Přípravek XALKORI s jídlem a pitím
Přípravek XALKORI můžete užívat s jídlem nebo bez jídla, ovšem během léčby přípravkem XALKORI byste se měl(a) vyhnout pití grapefruitové šťávy nebo jedení grapefruitu, protože může změnit množství přípravku XALKORI ve Vašem těle.
Těhotenství a kojení
Sdělte svému lékaři nebo lékárníkovi, že jste těhotná, můžete otěhotnět nebo kojíte dříve, než začnete tento přípravek užívat.
Doporučuje se, aby se během léčby přípravkem XALKORI vyhnuli ženy otěhotnění a muži početí dítěte, protože tento přípravek by mohl dítě poškodit. Pokud je jakákoliv pravděpodobnost, že osoba užívající tento přípravek může otěhotnět nebo počít dítě, je nutné během léčby a nejméně 90 dnů po ukončení léčby používat spolehlivou antikoncepci, protože perorální kontraceptiva mohou být při užívání přípravku XALKORI neúčinná.
Během léčby přípravkem XALKORI nekojte. Přípravek XALKORI by mohl poškodit kojené dítě.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Měl(a) byste být opatrný(á), když řídíte nebo obsluhujete stroje, neboť jako pacient užívající přípravek XALKORI můžete zaznamenat poruchy vidění, závratě a únavu.
3. Jak se přípravek XALKORI užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
• Doporučená dávka je jedna tobolka o síle 250 mg užívaná perorálně (ústy) dvakrát denně (celkové množství 500 mg).
• Užívejte jednu tobolku ráno a jednu tobolku večer.
• Užívejte tobolky každý den ve stejnou dobu.
• Tobolky můžete užívat s jídlem nebo bez jídla, vždy se vyhněte grapefruitu.
• Tobolky polykejte celé. Tobolky nedrťte, nerozpouštějte a ani je neotvírejte.
Je-li to nezbytné, Váš lékař se může rozhodnout snížit dávku na 200 mg užívanou perorálně dvakrát denně (celkové množství 400 mg) a je-li nezbytné další snížení dávky, může snížit dávku až na 250 mg jednou denně perorálně.
Jestliže jste užil(a) více přípravku XALKORI, než jste měl(a)
Pokud náhodně užijete příliš mnoho tobolek, informujte o tom ihned svého lékaře nebo lékárníka. Budete možná potřebovat lékařskou péči.
Jestliže jste zapomněl(a) užít přípravek XALKORI
Jak postupovat v případě, že si zapomenete vzít tobolku, závisí na tom, jak dlouho zbývá do příští dávky.
• Pokud je příští dávka za 6 hodin nebo více, vezměte si vynechanou tobolku co nejdříve, jakmile si to uvědomíte. Příští tobolku si vezměte v obvyklou dobu.
• Pokud je příští dávka za méně než 6 hodin, neberte vynechanou tobolku. Příští tobolku si vezměte v obvyklou dobu.
Při příští návštěvě informujte lékaře o vynechané dávce.
Nezdvojnásobujte následující dávku (dvě tobolky najednou), abyste nahradil(a) vynechanou tobolku.
Pokud po užití dávky přípravku XALKORI zvracíte, dávku nenahrazujte a příští dávku si vezměte v obvyklou dobu.
Je důležité užívat přípravek XALKORI každý den tak dlouho, jak Vám předepsal lékař. Pokud nejste schopen(na) užívat přípravek tak, jak Vám lékař předepsal, nebo máte pocit, že ho již nepotřebujete, obraťte se ihned na svého lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Některé nežádoucí účinky by mohly být závažné. Musíte okamžitě informovat svého lékaře, pokud se u Vás objeví kterýkoliv z následujících závažných nežádoucích účinků (viz rovněž bod 2 „Čemu musíte věnovat pozornost, než začnete přípravek XALKORI užívat“):
• Selhání jater
Informujte svého lékaře okamžitě, pokud se cítíte více unaven(a) než obvykle, kůže a bělmo očí se Vám zbarví do žluta, máte tmavou moč nebo hnědě zbarvenou (barva čaje), objeví se pocit na zvracení, zvracení nebo ztráta chuti k jídlu, bolest v pravé horní oblasti břicha, objeví se svědění nebo se Vám tvoří modřiny snáze než obvykle. Lékař by Vám měl provést krevní testy kvůli kontrole funkce jater, a pokud jsou výsledky abnormální, může rozhodnout snížit dávku přípravku XALKORI nebo ukončit Vaši léčbu.
• Zápal plic
Informujte svého lékaře okamžitě, pokud zaznamenáte obtíže s dýcháním, zejména spojené s kašlem a horečkou.
• Snížení počtu bílých krvinek (včetně neutrofilů)
Informujte svého lékaře okamžitě, pokud máte horečku nebo infekci. Lékař Vám udělá krevní testy, a pokud nebudou v pořádku, může se rozhodnout snížit dávku přípravku XALKORI.
• Točení hlavy, mdloby nebo nepříjemný pocit na hrudi
Informujte svého lékaře okamžitě, pokud zaznamenáte tyto příznaky, které mohou být známkou změn v elektrické aktivitě srdce (pozorovatelné na elektrokardiogramu) nebo abnormálního rytmu srdce. Váš lékař Vám natočí elektrokardiogram (EKG), aby zkontroloval, že nenastaly žádné problémy s Vaším srdcem během léčby přípravkem XALKORI.
• Částečná nebo úplná ztráta zraku na jednom nebo obou očích
Okamžitě informujte svého lékaře, pokud zaznamenáte jakoukoli ztrátu zraku, např. potíže s viděním na jedno nebo obě oči. Lékař může rozhodnout o zastavení léčby přípravkem XALKORI a odeslat Vás k očnímu lékaři.
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 pacientů)
• Poruchy vidění (vidění záblesků světla, rozmazané vidění a dvojité vidění, často začínající brzy po zahájení léčby přípravkem XALKORI).
• Žaludeční nevolnost včetně zvracení, průjmu, pocitu na zvracení.
• Edém (přebytek tekutiny v tělesné tkáni způsobující otok rukou a nohou).
• Zácpa.
• Abnormality v jaterních testech.
• Snížená chuť k jídlu.
• Únava.
• Závrať.
• Neuropatie (pocity necitlivosti nebo mravenčení v kloubech nebo končetinách).
• Změna chuti.
• Bolest břicha.
• Snížení počtu červených krvinek (anemie).
• Kožní vyrážka.
• Snížení tepové frekvence.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 pacientů)
• Zažívací potíže.
• Zvýšená hladina enzymu alkalická fosfatáza v krvi (ukazatel špatné funkce nebo poranění orgánů, zejména jater, slinivky břišní, kostí, štítné žlázy nebo žlučníku)
• Hypofosfatemie (nízké hladiny fosfátů v krvi, které mohou způsobovat zmatenost nebo svalovou slabost).
• Uzavřený váček s tekutinou v ledvinách (ledvinné cysty).
• Mdloby.
• Zánět jícnu.
• Snížení hladiny testosteronu, mužského pohlavního hormonu.
• Srdeční selhání.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 pacientů)
• Proděravění (perforace) stěny žaludku nebo střeva.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek XALKORI uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvičce nebo fólii blistru a krabičce za „EXP“ nebo „Použitelné do“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
• Nepoužívejte tento přípravek, pokud si všimnete, že obal je poškozený nebo vykazuje známky porušení.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek XALKORI obsahuje
- Léčivou látkou je crizotinibum.
XALKORI 200 mg: jedna tobolka obsahuje crizotinibum 200 mg XALKORI 250 mg: jedna tobolka obsahuje crizotinibum 250 mg
- Dalšími složkami jsou:
Obsah tobolky: koloidní bezvodý oxid křemičitý, mikrokrystalická celulosa, hydrogenforsforečnan vápenatý, sodná sůl karboxymethylškrobu (Typ A), magnesium-stearát. Obal tobolky: želatina, oxid titaničitý (E171) a červený oxid železitý (E172).
Potiskový inkoust: šelak, propylenglykol, hydroxid draselný a černý oxid železitý (E172).
Jak přípravek XALKORI vypadá a co obsahuje toto balení
Přípravek XALKORI 200 mg je dodáván jako tvrdé želatinové tobolky s růžovým víčkem a bílým tělem, s černým potiskem “Pfizer” na víčku a “CRZ 200” na těle tobolky.
Přípravek XALKORI 250 mg je dodáván jako tvrdé želatinové tobolky s růžovým víčkem a tělem, s černým potiskem “Pfizer” na víčku a “CRZ 250” na těle tobolky.
Je dostupný v blistrech v balení 60 tvrdých tobolek a v plastových lahvičkách obsahujících 60 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ Velká Británie
Výrobce
Pfizer Manufacturing Deutschland GmbH Betriebsstatte Freiburg Mooswaldallee 1 79090 Freiburg Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgique/ Belgie /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11 |
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje Tel. + 370 52 51 4000 |
|
BtnrapHH n$aň3ep HroKceMÓypr CAPH, KnoH Etnrapna Ten.: +359 2 970 4333 |
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11 |
|
Česká republika Pfizer PFE, spol. s r.o. Tel.: +420-283-004-111 |
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00 |
|
Danmark Pfizer ApS Tlf: +45 44 20 11 00 |
Malta V.J. Salomone Pharma Ltd. Tel. +356 21220174 |
|
Deutschland Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000 |
Nederland Pfizer BV Tel: +31 (0)10 406 43 01 |
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel.: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 52 61 00 |
|
EXláSa Pfizer EAAág A.E. TpL: +30 210 6785 800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
Espaňa Pfizer S.L. Tél: +34 91 490 99 00 |
Polska Pfizer Polska Sp.z.o.o Tel.:+48 22 335 61 00 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: +385 1 3908 777 |
Románia Pfizer Romania S.R.L. Tel: +40 (0) 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: + 386 (0)1 52 11 400 |
|
Ísland Icepharma hf. Sími: +354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.: + 421 2 3355 5500 |
Italia
Suomi/Finland
Pfizer Oy
Puh./Tel: +358 (0)9 43 00 40 Sverige
Pfizer Innovations AB Tel: +46 (0)8 550-52000
Pfizer S.r.l.
Tel: +39 06 33 18 21
Kúrcpog
Pfizer EAAág A.E. (Cyprus Branch) T^: +357 22 817690
Latvija United Kingdom
Pfizer Luxembourg SARL filiale Latvija Pfizer Limited
Tel.: + 371 670 35 775 Tel: +44 (0) 1304 616161
Tato příbalová informace byla naposledy revidována MM/RRRR
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
56