Wilate 500
Sp.zn.sukls198779/2013 a k sp.zn.sukls198780/2013 a k sp.zn.sukls106471/2013
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Wilate 500, 500 IU VWF/500 IU FVIII, prášek a rozpouštědlo pro injekční roztok Wilate 1000, 1000 IU VWF/1000 IU FVIII, prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Přípravek Wilate je vyroben z plazmy lidských dárců a je dodáván jako prášek a rozpouštědlo pro injekční roztok obsahující nominálně 500 IU/1000 IU Factor von Willebrand humanus (VWF) a Factor VIII coagulationis humanus v jedné injekční lahvičce.
Po rozpuštění v 5 ml/10 ml vody na injekci s 0,1% polysorbátu 80 přípravek obsahuje přibližně 100 IU/ml lidského von Willebrandova faktoru.
Specifická aktivita Wilate je > 67 IU VWF:RCo/mg proteinu.
Síla VWF (IU) je měřena podle aktivity ristocetin kofaktoru (VWF:RCo) porovnáním s Mezinárodním standardem pro koncentráty von Willebrandova faktoru (WHO).
Po rozpuštění v 5 ml/10 ml vody na injekci s 0,1% polysorbátu 80 přípravek obsahuje přibližně 100 IU/ml lidského koagulačního faktoru VIII.
Síla FVIII (IU) je určena použitím chromogenního testu dle Evropského lékopisu. Specifická aktivita Wilate je > 67 IU FVIII:C/mg proteinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Lyofilizovaný bílý nebo světle žlutý prášek, nebo drobivá hmota.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Von Willebrandova choroba (VWD)
Prevence a léčba krvácivých stavů nebo krvácení při operacích u von Willebrandovy choroby, jestliže léčba samotným desmopresinem (DDAVP) je neúčinná nebo kontraindikována.
Hemofilie A
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
4.2 Dávkování a způsob podání
Léčba by měla být pod dozorem lékaře, který má zkušenosti s léčbou koagulačních poruch. Přípravek je určen k jednorázovému použití a měl by být podán celý obsah lahvičky. Pokud jakékoliv množství zbyde, mělo by být zlikvidováno v souladu s místními předpisy.
Von Willebrandova choroba (VWD)
Poměr mezi VWF:RCo a FVIII:C je 1:1. Obecně, podání 1 IU/kg tělesné hmotnosti VWF:RCo a FVIII:C zvýší plazmatickou hladinu o 1,5 - 2 % normální aktivity u každého z těchto proteinů. Většinou je k dosažení adekvátní hemostázy nutné podání 20 - 50 IU Wilate/kg tělesné hmotnosti. Toto množství zvýší VWF:RCo a FVIII:C u pacientů přibližně o 30 - 100 %.
Může být zapotřebí počáteční dávka Wilate 50 - 80 IU/kg tělesné hmotnosti, obzvláště u pacientů s typem 3 VWD, kde je k udržení adekvátních plazmatických hladin zapotřebí vyšší dávky, než u jiných typů VWD.
Pediatrická populace
Nejsou k dispozici dostatečné údaje pro doporučení použití přípravku Wilate u dětí mladších 6 let.
Prevence krvácení v případech chirurgického zákroku nebo vážného traumatu:
K prevenci krvácení v případě chirurgického zákroku by Wilate měl být podán 1-2 hodiny před chirurgickým zákrokem. Mělo by být dosaženo hladiny > 60 IU/dl (> 60%) VWF:RCo a > 40 IU/dl (> 40%) FVIII:C.
Odpovídající dávka by měla být znovu podávána každých 12 - 24 hodin léčby. Dávka a délka trvání léčby závisí na klinickém stavu pacienta, na typu a závažnosti krvácení a na hladinách obou faktorů VWF:RCo a FVIII:C.
U pacientů léčených přípravky VWF s obsahem FVIII by měla být monitorována hladina FVIII:C, protože její trvalé nadměrné zvýšení může zvyšovat riziko trombotické příhody, zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory. V případě, že jsou zjištěny nadměrné hladiny FVIIFC, je třeba zvážit snížení dávek a/nebo prodloužení intervalů mezi jednotlivými dávkami nebo použití VWF přípravku s nižším obsahem FVIII.
Profylaxe:
Pro dlouhodobou profylaxi krvácení u pacientů s VWD by mělo být podáváno 20 - 40 IU/kg tělesné hmotnosti 2 nebo 3 krát týdně. V některých případech, zejména u pacientů s gastrointestinálním krvácením, mohou být nutné vyšší dávky.
Hemofilie A
Dávka a délka trvání substituční léčby závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Množství podaných jednotek FVIII je vyjádřeno v Mezinárodních jednotkách (IU), které jsou vztaženy k současnému standardu WHO pro přípravky s FVIII. Aktivita FVIII v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v IU (vzhledem k Mezinárodnímu standardu pro FVIII v plazmě).
Jedna IU aktivity faktoru VIII se rovná množství faktoru VIII v 1 ml normální lidské plazmy.
Léčba dle potřeby:
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU FVIII:C/kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o1,5 - 2 % normální aktivity. Potřebná dávka se určuje podle následujícího vzorce:
Požadované množství IU = tělesná hmotnost (kg) x žádaný vzestup FVIII (%) (IU/dl) x 0,5 IU/kg
Dávka a četnost podávání by měly být vždy vztaženy ke klinické účinnosti v individuálním případě. V případě následujících krvácivých příhod by v daném období neměla poklesnout aktivita faktoru VIII:C pod danou hodnotu plazmatické aktivity (v % normálu nebo IU/dl) Následující tabulka může být použita jako vodítko pro stanovení dávky při krvácivých příhodách a chirurgických výkonech:
Léčebné schéma pro krvácivé příhody a chirurgické operace
|
Stupeň krvácení/ Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Četnost dávek (hodiny)/ délka trvání(dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo krvácení do dutiny ústní |
20 - 40 |
Opakovat každých 12 - 24 hodin. Nejméně 1 den, dokud nedojde k zástavě krvácení, které se projeví ukončením bolestí, nebo ke zhojení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom |
30 - 60 |
Opakovat infuzi každých 12 -24 hodin po dobu 3 - 4 dnů nebo déle, dokud nedojde k zastavení bolesti a akutní nemohoucnosti. |
|
Život ohrožující krvácení |
60 - 100 |
Opakovat infuze každých 8 - 24 hodin dokud nepomine ohrožení života. |
|
Chirurgické zákroky | ||
|
Menší chirurgický zákrok včetně vytržení zubu |
30 - 60 |
Každých 24 hodin, nejméně 1 den, dokud nedojde ke zhojení. |
|
Velké chirurgické výkony |
80 - 100 (před a po operaci) |
Opakovat infuzi každých 8 - 24 hodin až do adekvátního zhojení rány, pak pokračovat v léčbě nejméně dalších 7 dní k udržení aktivity faktoru VIII na 30 - 60 % (IU/dl) |
Profylaxe:
Na dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A by měla být podávaná dávka Wilate 20 - 40 IU na kg tělesné hmotnosti v intervalech 2 - 3 dnů. V některých případech, zvláště u mladších pacientů, může být nutné podávat přípravek v kratších intervalech nebo ve vyšších dávkách.
Kontinuální infuze:
Před chirurgickým zákrokem by měla být provedena farmakokinetická analýza, aby mohlo být odhadnuto clearance. Počátečná rychlost infuze je možné vypočítat následovně:
Rychlost infuze (IU/kg/h) = clearance (ml/kg/h) x požadovaná ustálená hladina (IU/ml)
Po počátečních 24 hodinách kontinuální infuze by mělo být clearance počítáno každý den znovu za použití rovnice s naměřenou ustálenou hladinou a známou rychlostí infuze.
V průběhu léčby se doporučuje patřičné sledování hladin FVIII:C, aby bylo možné upravovat dávku, která má být podána a frekvenci opakovaných infuzí. Zvláště v případě větších
chirurgických zákroků je nepostradatelné přesné monitorování substituční léčby pomocí koagulační analýzy (FVIII:C). Odpověď na podání faktoru FVIII se může u jednotlivých pacientů lišit, mohou dosahovat různých hodnot recovery a různé mohou být i poločasy.
Dosud neléčení pacienti
Aktuálně dostupné údaje jsou uvedeny v bodě 4.8
Pediatrická populace
Neexistují dostatečné údaje pro doporučení užití Wilate u dětí s hemofilií A mladších 6 let.
Způsob podání
Intravenózní podání
Rychlost injekce nebo infuze by neměla překročit 2 - 3 ml za minutu.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Stejně jako u infuzí jiných intravenózních přípravků obsahujících deriváty plazmatických bílkovin jsou možné reakce z přecitlivělosti alergického typu. Pacienti musí být důkladně monitorováni a musí u nich být pečlivě sledovány jakékoliv příznaky alergické reakce během infuze.
Pacienti by měli být informováni o všech časných příznacích reakcí z přecitlivělosti, včetně vyrážky, generalizované kopřivky, pocitu tíhy na prsou, sípotu, hypotenze a anafylaxe. Obj eví-li se alergické příznaky, pacienti by měli okamžitě přerušit podávání přípravku a kontaktovat svého lékaře.
V případě šoku má být léčba vedena podle současných standardních lékařských postupů pro léčbu šoku.
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické infekční markery a zařazení účinných výrobních kroků, při nichž jsou viry inaktivovány nebo odstraněny. Přesto nemůže být při podávání léků vyráběných z lidské krve nebo plazmy zcela vyloučena možnost přenosu infekce. To platí i pro jakékoli neznámé nebo vznikající viry a jiné patogeny.
Přijatá opatření jsou považována za účinná u obalených virů, jako například virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV), a u neobaleného viru hepatitidy A. Omezený účinek mají tato opatření u neobalených virů, jako je parvovirus B19.
Infekce parvovirem B19 může být velmi závažná u těhotných žen (fetální infekce) a u imunodeficientních jedinců nebo jedinců se zvýšenou erytropoézou (například hemolytická anemie).
Při každé aplikaci přípravku Wilate se důrazně doporučuje zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
U pacientů, kteří pravidelně nebo opakovaně dostávají přípravky s VWF/FVIII vyrobeným z lidské plazmy, je třeba zvážit vhodné očkování (hepatitida A a B).
Von Willebrandova choroba (VWD)
Při použití VWF přípravku obsahujícího FVIII by si ošetřující lékař měl být vědom toho, že kontinuální léčba může mít za následek silný vzestup FVIII:C. U pacientů, kteří dostávají VWF přípravky obsahující FVIII by měly být sledovány hladiny FVIIFC v plazmě, aby se zamezilo nadměrným hladinám FVIII:C, které mohou zvýšit riziko trombotických příhod.
Existuje riziko výskytu trombotických příhod při podávání VWF přípravků obsahující FVIII, zvláště u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Z tohoto důvodu musí být rizikoví pacienti monitorováni na výskyt časných příznaků trombózy. Měla by být zahájena profylaxe proti žilní tromboembolii dle aktuálně platných doporuční.
U pacientů s VWD, zvláště 3. typu, se mohou vyvinout neutralizující protilátky (inhibitory) proti VWF. Pokud nedojde k očekávanému vzestupu aktivity VWF:RCo v plazmě, nebo pokud není krvácení zvládnuto odpovídající dávkou, je třeba provést vyšetření na přítomnost inhibitorů VWF. U pacientů s vysokými hladinami inhibitoru může být léčba neúčinná a je třeba zvážit jiné léčebné možnosti. Léčba takovýchto pacientů by měla být vedena lékaři, kteří mají zkušenost s péčí o pacienty s hemostatickými poruchami.
Hemofilie A Hypersenzitivita
Hypersenzitivní reakce alergického typu jsou u přípravku Wilate možné. Tento přípravek obsahuje stopová množství lidských proteinů jiných než FVIII. Objeví-li se příznaky hypersenzitivity, mají být pacienti informováni, aby neprodleně přerušili užívání léčivého přípravku a vyhledali svého ošetřujícího lékaře. Pacienti mají být informováni o časných známkách hypersenzitivních reakcí, včetně vyrážky, celkové kopřivky, tlaku na hrudi, sípání, hypotenze a anafylaxe.
V případě šoku je třeba zahájit standardní lékařské postupy pro léčbu šoku.
Inhibitory
Vznik neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby u pacientů s hemofilií A. Tyto inhibitory jsou obyčejně IgG imunoglobuliny působící proti koagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (BU) v jednom ml plazmy za pomocí modifikovaného testu. Riziko vzniku inhibitorů má vztah k podání FVIII, toto riziko je nejvyšší během prvních 20 dnů podávání. Vzácně mohou inhibitory vzniknout po prvních 100 dnech podávání.
Případy opakovaného výskytu inhibitoru (nízký titr) byly pozorovány po přechodu z jednoho přípravku FVIII na jiný u dříve léčených pacientů s více než 100 dny podávání a to u pacientů s anamnézou dřívějšího výskytu inhibitoru. Z tohoto důvodu se doporučuje monitorovat pečlivě všechny pacienty na výskyt inhibitoru po každé změně přípravku.
Obecně platí, že všichni pacienti léčení koagulačními přípravky FVIII mají být pečlivě sledováni s ohledem na rozvoj inhibitorů vhodnými klinickými a laboratorními testy. Pokud nejsou dosaženy očekávané hladiny aktivity faktoru FVIII v plazmě nebo pokud není krvácení zastaveno příslušnou dávkou, měla by být provedena zkouška za účelem stanovení přítomnosti inhibitoru faktoru FVIII. U pacientů s vysokou hladinou inhibitoru může být léčba faktorem FVIII neúčinná a je třeba uvažovat o jiných možnostech léčby. Léčbu takových pacientů má řídit lékař se zkušenostmi s ošetřováním nemocných trpících hemofilií a s inhibitory FVIII.
Tento léčivý přípravek obsahuje až 2,55 mmol (nebo 58,7 mg) sodíku v jedné injekční lahvičce 500 IU VWF a FVIII a až 5,1 mmol (nebo 117,3 mg) sodíku v jedné injekční lahvičce 1000 IU VWF a FVIII. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou známy žádné interakce s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S VWF/FVIII nebyly provedeny reprodukční studie na zvířatech.
Von Willebrandova choroba (VWD)
Zkušenosti s léčbou žen během těhotenství nebo kojení nejsou k dispozici.
Wilate by se měl u žen s deficitem VWF během těhotenství nebo kojení používat pouze tehdy, je-li to nezbytně nutné a mělo by se vzít v úvahu, že porod u těchto pacientek zvyšuje riziko krvácivých příhod.
Hemofilie A
Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou k dispozici zkušenosti s léčbou během těhotenství nebo kojení. Proto by se Wilate měl během těhotenství nebo kojení používat pouze tehdy, je-li to nezbytně nutné.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Wilate nemá žádný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Hypersenzitivita nebo alergická reakce (která může zahrnovat angioedém, pálení a píchání v místě aplikace infuze, zimnici, zarudnutí kůže, generalizovanou kopřivku, bolesti hlavy, vyrážku, hypotenzi, apatii, nevolnost, neklid, tachykardii, pocit tíhy na prsou, mravenčení, zvracení, sípot) byly pozorovány vzácně a v některých případech mohou vyústit k vážné anafylaxi (včetně šoku). Ve vzácných případech byla pozorována horečka.
Von Willebrandova choroba (VWD)
U pacientů s VWD, zvláště typu 3, se mohou velmi vzácně vyvinout neutralizující protilátky (inhibitory) proti VWF. Jestliže se tyto inhibitory objeví, tento stav se projeví jako neadekvátní klinická odpověď organismu. Tyto protilátky se objevují spolu s anafylaktickými reakcemi. Z tohoto důvodu by u pacientů se sklonem k anafylaktické reakci měla být vyšetřena přítomnost inhibitoru.
Ve všech těchto případech se doporučuje kontaktovat specializované hemofilické centrum.
Dosud nebyly hlášeny žádné případy výskytu inhibitoru proti von Willebrandovu faktoru z klinických studií nebo postmarketingových zkušeností s přípravkem Wilate.
Existuje riziko výskytu trombotických příhod, zvláště u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Měla by být zahájena profylaxe proti žilní tromboembolii dle aktuálně platných doporuční.
U pacientů, kteří dostávají VWF přípravky obsahující FVIII, se při trvale zvýšené hladině FVIII:C v plazmě může zvýšit riziko trombotických příhod.
Hemofilie A
U pacientů s hemofilií A mohou vznikat neutralizující protilátky (inhibitory) proti FVIII. Jestliže se tyto inhibitory objeví, tento stav se projeví jako nedostatečná klinická odpověď organismu. V takovém případě se doporučuje spojit se se specializovaným centrem pro léčbu hemofilie.
Zkušenost s podáváním Wilate u dříve neléčených pacientů (PUPs) je omezená. V klinické studii zahrnující 24 PUPs léčených Wilate s minimálně 50 dny podávání byli zachyceni jen tři pacienti s trvalým klinickým projevem inhibitoru nad 5 BU/ml. U tří pacientů se vyvinul nízký titr, přechodný výskyt inhibitoru bez klinického projevu a dva pacienti měli nízký titr inhibitoru při jednom měření, což se již více neopakovalo.
Viz také bod 4.2. U dříve léčených pacientů nebyl pozorován žádný výskyt inhibitorů. Informace o bezpečnosti z hlediska přenosných agens viz bod 4.4.
Tabulkový seznam nežádoucích účinků
Níže uvedená tabulka je vytvořena podle tříd orgánových systémů a preferovaných pojmů databáze MedDRA .
Frekvence výskytu se hodnotí podle následujícího pravidla: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Méně časté |
Vzácné |
Velmi vzácné |
|
Poruchy imunitního systému |
hypersenzitivní reakce |
anafylaktický šok | |
|
Celkové poruchy a reakce v místě aplikace | |||
|
Vyšetření |
Inhibitory FVIII |
Inhibitory VWF |
Popis vybraných nežádoucích účinků
Popis vybraných nežádoucích účinků naleznete v bodě 4.4
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: http://www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování lidským VWF nebo FVIII. Tromboembolické příhody se mohou vyskytnout v případě většího předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemorhagika: krevní koagulační faktory, kombinace Von Willebrandova faktoru a koagulačního faktoru VIII
ATC kód: B02BD06
Von Willebrandova choroba (VWD)
VWF (z koncentrátu) je běžnou složkou lidské plazmy a chová se stejně jako endogenní VWF.
Podání VWF umožňuje korekci hemostatických abnormalit projevující se u pacientů, kteří trpí deficitem VWF (VWD) ve dvou úrovních:
- VWF obnovuje adhezi destiček k cévnímu subendotelu v místě cévního poškození (tím, že se váže k cévnímu subendotelu a k membráně krevních destiček), čímž zajišťuje primární hemostázu projevující se zkrácením doby krvácení. Tento účinek se projeví okamžitě a je ve velké míře závislý na úrovni polymerace proteinu.
- VWF způsobuje zpožděnou korekci asociovaného deficitu FVIII. Při intravenózním podání se VWF váže na endogenní FVIII (který je produkován normálně pacientem) a stabilizací tohoto faktoru zamezuje jeho rychlé degradaci.
Z tohoto důvodu, podání čistého VWF (přípravek obsahující VWF s nízkou hladinou FVIII) obnovuje hladinu FVIII:C na normál, a to jako sekundární účinek po první infuzi.
Podáním přípravku VWF obsahujícího FVIII se obnovuje hladina FVIII:C k normálu bezprostředně po první infuzi.
Kromě role ochranného proteinu faktoru VIII, zprostředkovává von Willebrandův faktor adhezi destiček na místě cévního poranění a hraje hlavní roli v agregaci destiček.
Hemofilie A
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktor VIII a von Willebrandův faktor) s různými fyziologickými funkcemi. Po podání pacientovi s hemofilií se FVIII naváže na VWF v krevním oběhu pacienta.
Aktivovaný faktor VIII (FVIIIa) se chová jako kofaktor pro aktivovaný faktor IX (IXa) urychlující konverzi faktoru X na aktivovaný faktor X (FXa). FXa přeměňuje protrombin na trombin. Trombin pak přeměňuje fibrinogen na fibrin a vytvoří se koagulum.
Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti, jejíž příčinou je snížená hladina FVIII:C a jejímž výsledkem je zvýšené krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánní nebo jako výsledek poranění nebo chirurgického traumatu. Při substituční léčbě se plasmatická hladina faktoru VIII zvýší, tím dojde k přechodné úpravě deficitu faktoru a tím také k úpravě sklonu ke krvácení.
5.2 Farmakokinetické vlastnosti
Von Willebrandova choroba (VWD)
VWF (z koncentrátu) je běžnou složkou lidské plazmy a působí stejně jako endogenní VWF.
Na základě metaanalýzy tří farmakokinetických studií zahrnujících 24 hodnotitelných pacientů se všemi typy VWD byly zjištěny následující údaje.
|
Všechny typy VWD |
VWD typ 1 |
VWD typ 2 |
VWD typ 3 | |||||||||||||||||
|
Parametr |
N |
Střed |
Standa |
Min |
Ma |
N |
Středn |
Standa |
Min |
Ma |
N |
Střed |
Standa |
Min |
Ma |
N |
Stře |
Standa |
Min |
Max. |
|
ní |
rdní |
x. |
í |
rdní |
x. |
ní |
rdní |
x. |
dní |
rdní | ||||||||||
|
hodn |
odchyl |
hodnot |
odchyl |
hodn |
odchyl |
hod |
odchyl | |||||||||||||
|
ota |
ka |
a |
ka |
ota |
ka |
nota |
ka | |||||||||||||
|
Recover |
24 |
1,56 |
0,48 |
0,90 |
2,93 |
2 |
1,19 |
0,07 |
1,14 |
1,24 |
5 |
1,83 |
0,86 |
0,98 |
2,93 |
17 |
1,52 |
0,32 |
0,90 |
2,24 |
|
y (%/IU/k g) | ||||||||||||||||||||
|
AUC (0- |
23 |
1981 |
960 |
593 |
483 |
2 |
2062 |
510 |
170 |
242 |
5 |
2971 |
1383 |
151 |
483 |
16 |
166 |
622 |
593 |
2606 |
|
ro) |
1 |
1 |
3 |
1 |
1 |
2 | ||||||||||||||
|
(h*%) | ||||||||||||||||||||
|
T % (h) |
24 |
23,3 |
12,6 |
7,4 |
58,4 |
2 |
39,7 |
18,3 |
26,7 |
52,7 |
5 |
34,9 |
16 |
17,5 |
58,4 |
17 |
18 |
6,2 |
7,4 |
30,5 |
|
MRT (h) |
24 |
33,1 |
19 |
10,1 |
89,7 |
2 |
53,6 |
25,9 |
35,3 |
71,9 |
5 |
53,5 |
24,6 |
27,8 |
89,7 |
17 |
24,7 |
8,5 |
10,1 |
37,7 |
|
Clearanc e ml/h/kg |
24 |
3,29 |
1,67 |
0,91 |
7,41 |
2 |
2,66 |
0,85 |
2,06 |
3,27 |
5 |
1,95 |
1,02 |
0,91 |
3,31 |
17 |
3,76 |
1,69 |
1,83 |
7,41 |
Vysvětlivky: AUC = plocha pod křivkou, MRT = střední doba zdržení Hemofilie A
FVIII (z koncentrátu) je běžnou složkou lidské plazmy a působí jako endogenní faktor VIII. Po podání přípravku průměrně dvě třetiny až tři čtvrtiny faktoru VIII zůstávají v krevním řečišti. Hladina aktivity FVIII:C v plazmě by měla dosáhnout 80 - 120 % její předpokládané aktivity.
Aktivita FVIII:C se snižuje dvoufázovým exponenciálním rozpadem. V počáteční fázi distribuce mezi intravaskulárními prostory a ostatními částmi (tělní tekutiny) probíhá s poločasem rozpadu v plazmě od 3 do 6 hodin. V následující pomalejší fázi se poločas pohybuje v rozmezí 8-18 hodin s průměrem 15 hodin. Toto odpovídá skutečnému biologickému poločasu.
V klinické studii s 12 pacienty (chromogenní assay, dvojité měření) byly zjištěny následující hodnoty:
|
Parametr |
Základní měření |
Měření po 6 měsících | ||
|
Střední hodnota |
SD |
Střední hodnota |
SD | |
|
Recovery %/IU/kg |
FVIII:C 2,27 |
1,20 |
FVIII:C 2,26 |
1,19 |
|
AUC AUCnorm. |
FVIII:C 31,3 |
7,31 |
FVIII:C 33,8 |
10,9 |
|
% * h/IU/kg | ||||
|
Poločas (h) |
FVIII:C 11,2 |
2,85 |
FVIII:C 11,8 |
3,37 |
|
MRT (h) |
FVIII:C 15,3 |
3,5 |
FVIII:C 16,3 |
4,6 |
|
Clearance ml/h/kg |
FVIII:C 3,37 |
0,86 |
FVIII:C 3,24 |
1,04 |
Vysvětlivky: AUC = plocha pod křivkou, MRT = střední doba zdržení, SD = standardní odchylka
5.3 Předklinické údaje vztahující se k bezpečnosti
VWF a FVIII obsažené ve Wilate jsou běžnou složkou lidské plazmy a působí jako endogenní VWF/FVIII.
Konvenční testování bezpečnosti těchto látek na laboratorních zvířatech nepřidávají k existujícím klinickým zkušenostem využitelné informace a z tohoto důvodu nejsou požadována.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek: Chlorid sodný, glycin, sacharóza, citronan sodný a chlorid vápenatý Rozpouštědlo: Voda na injekci s 0,1% polysorbátu 80
6.2 Inkompatibility
Vzhledem k tomu, že nejsou k dispozici studie kompatibility, nesmí se přípravek Wilate mísit sjinými léčivými přípravky nebo podávat souběžně s jiným intravenózním přípravkem v jednom infuzním setu.
Měla by být použita pouze přiložená injekční/infuzní souprava, protože v důsledku adsorpce FVIII/VWF na vnitřní povrchy některých jiných infuzních zařízení může dojít k selhání léčby.
6.3 Doba použitelnosti
3 roky
Stabilita po rekonstituci před použitím byla prokázána na dobu 4 hodin při pokojové teplotě (max. + 25 °C). Nicméně, aby se zabránilo mikrobiální kontaminaci, roztok má být použit okamžitě.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte prášek a rozpouštědlo v chladničce (2 - 8 °C). Uchovávejte v krabičce, aby byl přípravek chráněn před světlem. Chraňte před mrazem.
Přípravek může být uchováván po dobu 2 měsíců při pokojové teplotě (max. +25°C). V takovém případě je doba použitelnosti 2 měsíce od chvíle, kdy byl přípravek poprvé vyjmut z chladničky. Novou dobu použitelnosti musí pacient vyznačit na krabičce. Rekonstituovaný roztok by měl být použit během jednoho podání. Jakékoli zbylé množství roztoku musí být zlikvidováno.
Informace o podmínkách pro uchovávání léčivého přípravku po rekonstituci jsou uvedeny v bodě 6.3
6.5 Druh obalu a obsah balení
Velikosti balení:
Wilate 500, 500 IU VWF a 500 IU FVIII 1 balení obsahuje:
1 lahvičku s práškem, sklo typu I, uzavřenou zátkou (brombutylová pryž) a opatřenou víčkem.
1 lahvičku s rozpouštědlem (5 ml vody na injekci s 0,1% polysorbátu 80), sklo typu I, uzavřenou zátkou (chlorbutylová pryž) a opatřenou víčkem.
1 krabičku s příslušenstvím (1 jednorázová injekční stříkačka, 1 přepouštěcí set [ Mix2Vial],
1 infuzní set), 2 alkoholové tampony
Wilate 1000, 1000 IU VWF a 1000 IU FVIII
1 balení obsahuje:
1 lahvičku s práškem, sklo typu I, uzavřenou zátkou (brombutylová pryž) a opatřenou víčkem.
1 lahvičku s rozpouštědlem (10 ml vody na injekci s 0,1% polysorbátu 80), sklo typu I, uzavřenou zátkou (chlorbutylová pryž) a opatřenou víčkem.
1 krabičku s příslušenstvím - zdravotnickým prostředkem (1 jednorázová injekční stříkačka,
1 přepouštěcí set [Mix2Vial], 1 infuzní set), 2 alkoholové tampony
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
• Přečtěte si prosím pozorně všechny pokyny a postupujte pečlivě podle nich!
• Nepoužívejte Wilate po uplynutí doby použitelnosti vyznačené na obalu.
• Během níže uvedeného postupu musí být zachována sterilita!
• Roztok ve stříkačce by měl být čirý nebo slabě perlově lesklý. Nepoužívejte zakalené roztoky nebo roztoky s usazeninami.
• Použijte připravený roztok okamžitě, aby se předešlo mikrobiální kontaminaci.
• Používejte pouze přiložený injekční set. Použití jiného injekčního/infuzního zařízení může způsobit další rizika a selhání léčby.
Návod na přípravu roztoku:
1. Nepoužívejte přípravek ihned po vyjmutí z chladničky. Nechte rozpouštědlo i prášek v uzavřených lahvičkách dosáhnout pokojové teploty.
2. Odstraňte víčko z obou lahviček a očistěte pryžové zátky jedním z přiložených alkoholových tamponů.
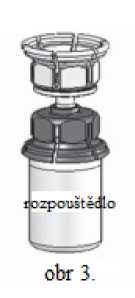
3. Zařízení Mix2Vial je znázorněno na obr. 1. Položte lahvičku s rozpouštědlem na rovný povrch a pevně ji uchopte. Vezměte Mix2Vial a otočte jej horní stranou dolů. Nasaďte Mix2Vial jeho modrým koncem na horní část lahvičky s rozpouštědlem a silně jej zatlačte dolů, dokud nezaklapne (obr. 2 + 3).
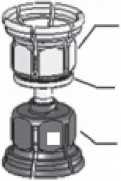
prášek adaptér (transparentní)
integrovaný fittr
rozpouštědlo adaptér (módný
obr L

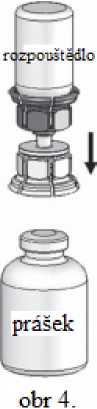
4. Položte lahvičku s práškem na pevný povrch a pevně ji uchopte.
Vezměte lahvičku s rozpouštědlem s připojeným Mix2Vial a otočte
ji dnem vzhůru. Nasaďte ji průhledným koncem na horní část
lahvičky s práškem a silně zatlačte dolů, dokud nezaklapne (obr. 4). Rozpouštědlo přeteče samo do lahvičky s práškem
5. Zlehka otáčejte se spojenými lahvičkami, dokud se přípravek nerozpustí.
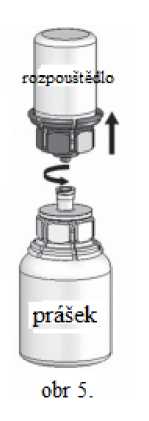
Rozpouštění je dokončeno při pokojové teplotě během méně než 10 minut.
Během přípravy se může objevit jemné napěnění. Rozšroubujte Mix2Vial na dvě části (obr. 5). Napěnění zmizí.
Odstraňte prázdnou lahvičku od rozpouštědla spolu s modrou částí Mix2Vial.
Návod na injekci:
Z preventivních důvodů by vám měl být měřen puls před a během injekce. Pokud se vyskytne výrazné zvýšení vašeho pulsu, zpomalte rychlost injekce nebo podávání na krátký čas přerušte.
1. Nasaďte injekční stříkačku na průhlednou část Mix2Vial. Otočte lahvičku dnem vzhůru a nasajte roztok do stříkačky (obr. 6). Roztok ve stříkačce by měl být čirý nebo slabě perlově lesklý. Jakmile je roztok natažen do stříkačky, uchopte pevně píst stříkačky (držte ji směrem dolů) a oddělte ji od Mix2Vial (obr. 7). Odstraňte Mix2Vial spolu s prázdnou lahvičkou.
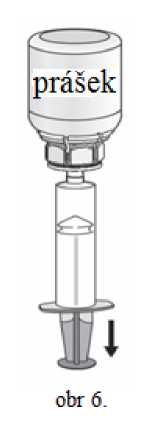
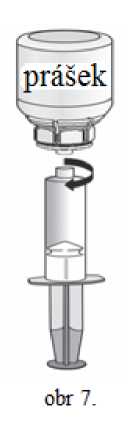
2. Vydesinfikujte vybrané místo aplikace injekce přiloženým alkoholovým tamponem.
3. Nasaďte přiloženou injekční jehlu na stříkačku.
4. Zaveďte injekční jehlu do zvolené žíly. Pokud jste použil(a) škrtidlo pro snadnější viditelnost
žíly, mělo by být uvolněno před zahájením injekce Wilate.
Do stříkačky se nesmí dostat žádná krev, aby nedošlo k riziku tvorby fibrinových sraženin.
5. Vstřikujte roztok pomalu do žíly, ne rychleji než 2 - 3 ml za minutu.
Pokud užíváte více než jednu lahvičku Wilate pro jednu léčbu, můžete použít znovu stejnou injekční jehlu a stříkačku. Zařízení Mix2Vial je však vždy pouze pro jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Octapharma (IP) Limited The Zenith Building 26 Spring Gardens Manchester M2 1AB Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
Wilate 500: 75/001/12-C Wilate 1000: 75/002/12-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE:
Datum první registrace: 11.01.2012
Datum posledního prodloužení registrace: 10.7.2014
10. DATUM REVIZE TEXTU:
15.10.2014
14