Wakix 4,5 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Informace o tom, jak hlásit nežádoucí účinky, naleznete v bodě 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Wakix 4,5 mg potahované tablety Wakix 18 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Wakix 4,5 mg potahované tablety
Každá tableta obsahuje pitolisanti hydrochloridum 5 mg, což odpovídá pitolisantum 4,45 mg. Wakix 18 mg potahované tablety
Každá tableta obsahuje pitolisanti hydrochloridum 20 mg, což odpovídá pitolisantum 17,8 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
Wakix 4,5 mg potahované tablety
Bílé, kulaté, bikonvexní potahované tablety o průměru 3,7 mm označené číslem „5“ na jedné straně. Wakix 18 mg potahované tablety
Bílé, kulaté, bikonvexní potahované tablety o průměru 7,5 mm označené číslem „20“ na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Wakix je indikován u dospělých k léčbě narkolepsie s kataplexií nebo bez kataplexie. (viz také bod 5.1).
4.2 Dávkování a způsob podání
Léčbu by měl zahájit lékař se zkušenostmi v léčbě poruch spánku.
Dávkování
Přípravek Wakix by měl být užíván v nejnižší účinné dávce v závislosti na reakci a toleranci pacienta za použití vzestupné titrace a bez překročení dávky 36 mg/den:
- 1. týden: počáteční dávka 9 mg (dvě 4,5 mg tablety) denně.
- 2. týden: dávku je možné zvýšit na 18 mg (jedna 18 mg tableta) denně, nebo snížit na 4,5 mg (jedna 4,5 mg tableta) denně.
- 3. týden: dávku je možné zvýšit na 36 mg (dvě 18 mg tablety) denně.
Dávku je možné kdykoliv snížit (až na 4,5 mg denně) nebo zvýšit (až na 36 mg denně) na základě posudku lékaře a reakce pacienta.
Celková denní dávka by měla být podávána v jedné dávce ráno při snídani.
Udržení účinnosti
Vzhledem k tomu, že údaje o dlouhodobé účinnosti jsou omezené (viz bod 5.1), je nutné, aby pokračující účinnost léčby pravidelně posuzoval lékař.
Zvláštní populace
Starší osoby
U starších osob jsou dostupné pouze omezené údaje. Dávku je tedy nutné upravit v závislosti na stavu ledvin a jater.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin má být maximální denní dávka 18 mg.
Porucha funkce jater
U pacientů se středně těžkou poruchou funkce jater (Child Pugh B) je možné dva týdny po zahájení léčby zvýšit denní dávku, která nesmí překročit maximální dávku 18 mg (viz bod 5.2).
Pitolisant je kontraindikován u pacientů s těžkou poruchou funkce jater (Child Pugh C) (viz bod 4.3). U pacientů s mírnou poruchou funkce jater není nutná úprava dávkování.
Pediatrická populace
Bezpečnost a účinnost pitolisantu u dětí ve věku 0 až 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Perorální podání.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Těžká porucha funkce jater (Child Pugh C).
Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Psychiatrické poruchy
Při podávání látky pitolisant pacientům, kteří mají v anamnéze psychiatrické poruchy jako je silná úzkost či deprese s rizikem sebevražedných sklonů, je nutné dbát opatrnosti.
Porucha funkce jater nebo ledvin
Je nutné dbát opatrnosti při podávání látky pitolisant pacientům s poruchou funkce ledvin nebo se středně těžkou poruchou funkce jater (Child Pugh B) a upravit dávkovací režim dle pokynů v bodu 4.2.
Gastrointestinální poruchy
U látky pitolisant byly hlášeny reakce gastrických poruch. Je tedy nutné dbát opatrnosti při podávání této látky pacientům s gastrickými obtížemi způsobenými kyselostí (viz bod 4.8) nebo při podávání současně s látkami dráždícími žaludek, jako jsou kortikosteroidy nebo NSAID.
Poruchy výživy
Při podávání látky pitolisant pacientům s těžkými případy obezity či anorexie je třeba dbát opatrnosti (viz bod 4.8). V případě výrazné změny tělesné hmotnosti musí lékař léčbu znovu zvážit.
Srdeční choroby
Během dvou dedikovaných studií QT způsobily dávky nad hranicí léčebného množství pitolisantu (3-6krát víc než je terapeutická dávka, tj. 108-216 mg) mírné až střední prodloužení intervalu QTc (1013 ms). V klinických studiích nebyl při použití terapeutických dávek pitolisantu identifikován žádný konkrétní srdeční bezpečnostní signál. Nicméně, je nutné pozorně sledovat pacienty se srdeční chorobou užívající další přípravky prodlužující QT nebo zvyšující riziko výskytu poruchy repolarizace, pacienty současně užívající léčivé přípravky, které výrazně zvyšují Cmax a AUC poměr pitolisantu (viz bod 4.5) a pacienty s těžkou poruchou funkce ledvin nebo středně těžkou poruchou funkce jater (viz body 4.4 a 4.5).
Při podávání vysokých dávek byly u zvířecích modelů hlášeny křeče (viz bod 5.3). Během klinických studií bylo hlášeno jedno zhoršení epilepsie u epileptického pacienta. U pacientů s těžkou epilepsií je nutné dbát opatrnosti.
Ženy v plodném věku
Ženy v plodném věku musí v průběhu léčby a nejméně 21 dní po ukončení léčby používat účinnou antikoncepci (na základě poločasu rozpadu pitolisantu/metabolitů). Pitolisant může snižovat účinnost hormonální antikoncepce. Z tohoto důvodu by pacientky užívající hormonální antikoncepci měly používat některou z alternativních metod účinné antikoncepce (viz body 4.5 a 4.6).
Lékové interakce
Je třeba vyhnout se kombinaci pitolisantu se substráty CYP3A4 při nízkém terapeutickém indexu (viz bod 4.5).
Opětovné zhoršení syndromů
V průběhu klinických studií nebylo hlášené žádné opětovné zhoršení syndromů. Nicméně, ukončení léčby by mělo být monitorováno.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Antidepresiva
Tri nebo tetracyklická antidepresiva (např. imipramin, clomipramin, mirtazapin) mohou snižovat účinnost pitolisantu, protože působí jako antagonisté histaminového H1 receptoru a mohou rušit účinek endogenního histaminu uvolňovaného v mozku při léčbě.
Antihistaminika
Antihistaminika (antagonisté receptoru H1) prostupující přes hematoencefalickou bariéru (např. feniramin maleát, chlorfeniramin, difenydramin, promethazin, mepyramin) mohou snižovat účinnost pitolisantu.
Přípravky prodlužující QT nebo přípravky zvyšující riziko poruchy repolarizace Kombinace s pitolisantem by měla probíhat za důkladného monitorování (viz bod 4.4). Farmakokinetické interakce
- Induktory enzymů
Současné podávání pitolisantu a rifampicinu v několika dávkách výrazně snižuje průměrnou Cmax a poměr AUC o zhruba 39 %, resp. 50 %. Při současném podávání pitolisantu se silnými induktory CYP3A4 (např. rifampicin, fenobarbital, karbamazepin, fenytoin) je tedy nutné dbát zvýšené pozornosti. V případě současného užívání třezalky tečkované (Hypericum Perforatum) a pitolisantu je nutné dbát zvýšené pozornosti vzhledem k silnému indukčnímu účinku třezalky na CYP3A4. Při kombinaci obou léčivých látek je nutné klinické pozorování a následně úprava dávkování v průběhu současného podávání a jeden týden po podávání induktoru.
- Inhibitory CYP2D6
Současné podávání pitolisantu a paroxetinu výrazně zvyšuje průměrnou Cmax pitolisantu a poměr AUCo-72h o zhruba 47 %, resp. 105 %. Vzhledem k dvojnásobnému zvýšení expozice pitolisantu je nutné v případě současného podávání s inhibitory CYP2D6 (např. paroxetin, fluoxetin, venlafaxin, duloxetin, bupropion, chinidin, terbinafin, cinakalcet) dbát zvýšené opatrnosti. V průběhu současného podávání je možné zvážit úpravu dávkování.
Léčivé přípravky, které mohou ovlivnit metabolismus pitolisantu
- Substráty CYP3A4 a CYP2B6
Na základě in vitro údajů může pitolisant a jeho hlavní metabolity indukovat CYP3A4 a CYP2B6 při terapeutických koncentracích a při extrapolaci CYP2C, UGT a P-gp. Nejsou dostupné žádné klinické údaje o rozsahu této interakce. Je tedy třeba vyhnout se kombinaci pitolisantu se substráty CYP3A4 při nízkém terapeutickém indexu (např. imunosupresiva, docetaxel, inhibitory kinázy, cisaprid, pimozid, halofantrin) (viz bod 4.4). U jiných substrátů CYP3A4, CYP2B6 (např. efavirenz, bupropion), CYP2C (např. repaglinid, fenytoin, warfarin), P-gp (např. dabigatran, digoxin) a UGT (např. morfin, paracetamol, irinotekan) je nutné dbát opatrnosti společně s klinickým monitorováním jejich účinnosti.
U perorálně podávaných antikoncepčních přípravků je nutné se vyhnout kombinaci s pitolisantem a použít jinou, spolehlivou antikoncepční metodu.
- Substráty OCT1
Pitolisant vykazuje více než 50% inhibici OCT1 (organický kationtový transportér 1) při 1,33 ^M, extrapolovaná IC50 pitolisantu je 0,795 ^M.
I v případě, že klinická relevance tohoto účinku nebude stanovena, se doporučuje dbát zvýšené opatrnosti při podávání pitolisantu se substrátem OCT1 (např. metformin (biguanidy)) (viz bod 5.2).
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy v plodném věku
Ženy v plodném věku musí v průběhu léčby a nejméně 21 dní po ukončení léčby používat účinnou antikoncepci (na základě poločasu rozpadu pitolisantu/metabolitů). Pitolisant/metabolity mohou snižovat účinnost hormonální antikoncepce. Z tohoto důvodu by pacientky užívající hormonální antikoncepci měly používat některou z alternativních metod účinné antikoncepce (viz bod 4.5).
Nejsou dostupné žádné údaje nebo jsou dostupné pouze omezené údaje o používání pitolisantu u těhotných žen. Studie u zvířat prokázaly reprodukční toxicitu včetně teratogenicity. U potkanů prostoupil pitolisant/metabolity bariérou placenty (viz bod 5.3).
Pitolisant by během těhotenství neměl být užíván, pokud potenciální přínos nepřevažuje nad potenciálním rizikem pro plod.
Kojení
U zvířat bylo zaznamenáno vylučování pitolisantu/metabolitů do mateřského mléka. Kojení je tedy v průběhu léčby pitolisantem kontraindikováno (viz bod 4.3).
Fertilita
Studie u zvířat ukázaly účinky na parametry spermatu bez závažného dopadu na reprodukční výkonnost samců a na snížení procenta živých plodů u léčených samic (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pitolisant má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
Pacienti s abnormální úrovní ospalosti, kteří užívají pitolisant, by měli být upozorněni na to, že jejich úroveň bdělosti se nemusí vrátit do normálu. Pacienti s nadměrnou spavostí během dne včetně těch, kteří užívají pitolisant, by měli pravidelně absolvovat opětovné vyhodnocení úrovně spavosti a v případě potřeby by jim mělo být doporučeno, aby se vyhýbali řízení nebo jakýmkoli jiným potenciálně nebezpečným činnostem.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastěji hlášené nežádoucí účinky (ADR) při užívání pitolisantu: nespavost (8,4 %), bolest hlavy (7,7 %), nauzea (4,8 %), úzkost (2,1 %), podrážděnost (1,8 %), točení hlavy (1,4 %), deprese (1,3 %), třes (1,2 %), poruchy spánku (1,1 %), únava (1,1 %), zvracení (1,0 %), závratě (1,0 %), dyspepsie (1,0 %), nárůst tělesné hmotnosti (0,9 %), bolest v horní části břicha (0,9 %). Mezi nejzávažnější nežádoucí účinky patří abnormální úbytek tělesné hmotnosti (0,09 %) a spontánní potrat (0,09 %).
Tabulkový seznam nežádoucích účinků
Následující nežádoucí účinky byly hlášeny při užívání pitolisantu v průběhu klinických studií s účastí více než 1094 pacientů s narkolepsií a jinými indikacemi a jsou uvedeny níže dle terminologie MedDRA podle třídy orgánových systémů a četnosti. Četnosti se definují jako: velmi časté (>1/10), časté (>1/100 to <1/10), méně časté (> 1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000). V rámci každé skupiny četnosti jsou nežádoucí účinky uvedeny podle závažnosti v sestupném pořadí:
|
Časté |
Méně časté |
Vzácné | |
|
Infekce a infestace |
Pocení | ||
|
Poruchy metabolismu a příjmu potravy |
Snížená chuť k jídlu Zvýšená chuť k jídlu Zadržování tekutin |
Anorexie Hyperfagie Poruchy chuti k jídlu |
|
Psychiatrické poruchy |
Nespavost Úzkost Podrážděnost Deprese Poruchy spánku |
Agitovanost Halucinace Vizuální a sluchové halucinace Afektivní labilita Abnormální sny Dyssomnie Noční probouzení Obtížné noční usínání Časné ranní probouzení Nervozita Napětí Apatie Noční můry Neklid Záchvat paniky Snížení libida Zvýšení libida |
Abnormální chování Zmatený stav Depresivní nálada Podrážděnost Obsesivní myšlenky Dysforie Hypnopompická halucinace Depresivní symptom Hypnagogická halucinace Mentální porucha |
|
Poruchy nervové soustavy |
Bolest hlavy Točení hlavy Třes |
Dyskineze Porucha rovnováhy Kataplexie Narušení pozornosti Dystonie Fluktuace během dne („on-off phenomenon“) Hypersomnie Migréna Psychomotorická hyperaktivita Syndrom neklidných nohou Somnolence Epilepsie Bradykineze Parestezie |
Ztráta vědomí Tenzní bolest hlavy Porucha paměti Nízká kvalita spánku |
|
Oční poruchy |
Snížená ostrost vidění Blefarospasmus | ||
|
Poruchy sluchu a vnitřního ucha |
Závratě |
Tinnitus | |
|
Srdeční choroby |
Extrasystoly Bradykardie | ||
|
Vaskulární poruchy |
Hypertenze Hypotenze Návaly horka | ||
|
Poruchy dýchání, hrudníku a mezihrudí |
Zívání | ||
|
Gastrointestinální poruchy |
Sucho v ústech Bolest břicha Průjem Mírná bolest břicha Bolest horní části břicha Zácpa Gastroezofageální reflux |
Distenze břicha Dysfagie Plynatost Odynofagie Enterokolitida |
|
Gastritida Gastrointestinální bolest Hyperacidita Parestezie úst Bolest žaludku | |||
|
Poruchy kůže a podkožní tkáně |
Erytém Hyperhidróza |
Toxická kožní erupce Fotosenzitivita | |
|
Poruchy muskuloskeletální a pojivové tkáně |
Bolesti kloubů Bolest zad Svalová ztuhlost Svalová slabost Bolest pohybového ústrojí Bolesti svalů Bolest v končetinách |
Bolest krku Muskuloskeletální bolest hrudníku | |
|
Poruchy ledvin a močového ústrojí |
Polakisurie | ||
|
Potíže v těhotenství, šestinedělí a perinatálním období |
Spontánní potrat | ||
|
Poruchy rozmnožovací soustavy a prsou |
Metroragie | ||
|
Obecné poruchy a podmínky spojené s místem podání přípravku |
Únava |
Slabost Bolest hrudníku Abnormální pocity Malátnost Edém Periferní edém |
Bolest Noční pocení Pocit útlaku |
|
Šetření |
Nárůst tělesné hmotnosti Úbytek tělesné hmotnosti Zvýšení jaterních enzymů |
Zvýšená hladina kreatinfosfokinázy | |
|
Prodloužení QT na elektrokardiogramu Zvýšení tepové frekvence Zvýšená hladina gama-glutamyltransferázy |
Abnormální celkový fyzický stav Abnormalita repolarizace na elektrokardiogramu Inverze vlny T na elektrokardiogramu |
Popis vybraných nežádoucích účinků
V průběhu klinických studií byly hlášeny epizody bolesti hlavy a nespavosti (7,7 % až 8,4 %). Většina z těchto nežádoucích účinků byla mírné nebo středně těžké intenzity. Pokud symptomy přetrvávají, je nutné zvážit sníženou denní dávku nebo přerušení léčby.
Žaludeční potíže
Žaludeční potíže způsobené hyperaciditou byly hlášeny v průběhu klinických studií u 3,5 % pacientů užívajících pitolisant. Tyto účinky byly většinou mírné až středně těžké intenzity. Pokud symptomy přetrvají, je možné zahájit nápravnou léčbu inhibitorem protonové pumpy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku č. V.
4.9 Předávkování
Symptomy
Symptomy předávkování přípravkem Wakix mohou zahrnovat bolest hlavy, nespavost, podrážděnost, nauzeu a bolest břicha.
Řešení
V případě předávkování je doporučeným postupem hospitalizace a sledování životních funkcí. Nebylo stanoveno žádné jednoznačné antidotum.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva nervového systému, kód ATC: N07XX11. Mechanismus účinku
Pitolisant je silný, perorálně aktivní antagonista/inverzní agonista histaminového H3 receptoru, který prostřednictvím zablokování histaminových autoreceptorů zvyšuje aktivitu mozkových histaminergních neuronů, které představují zásadní stimulační systém s rozsáhlým napojením na celý mozek. Pitolisant také moduluje různé systémy neurotransmiterů, zvyšuje uvolňování acetylcholinu, noradrenalinu a dopaminu v mozku. Nicméně, zvýšené uvolňování dopaminu ve striatální oblasti včetně nucleus accumbens nebylo v případě pitolisantu zaznamenáno.
Farmakodynamické účinky
U narkoleptických pacientů s kataplexií nebo bez kataplexie zlepšuje pitolisant úroveň trvání bdělosti a denní pozornosti při hodnocení objektivními prostředky schopnosti udržet bdělost (např. test udržení bdělosti (MWT - Maintenance of Wakefulness Test) a pozornosti (test úkolů na neustálou pozornost (SART - Sustained Attention to Response Task)).
Klinická účinnost a bezpečnost
Narkolepsie (s kataplexií nebo bez kataplexie) je chronické onemocnění. Účinnost pitolisantu až do 36 mg jedenkrát denně při léčbě narkolepsie s kataplexií nebo bez kataplexie byla stanovena ve dvou hlavních, multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích v paralelních skupinách o délce 8 týdnů (Harmony I a Harmony CTP). Harmony Ibis, studie podobné koncepce, byla omezena na 18 mg jedenkrát denně. K dnešnímu datu je k dispozici pouze omezené množství údajů z otevřené studie o dlouhodobé účinnosti přípravku Wakix u této indikace.
Pivotní studie (Harmony 1), dvojitě zaslepená, randomizovaná, oproti placebu a modafinilu (400 mg/den), v paralelních skupinách s flexibilní adaptací dávky zahrnovala 94 pacientů (31 pacientů pitolisant, 30 placebo a 33 modafinil). Dávkování bylo zahájeno na 9 mg jedenkrát denně a bylo zvyšováno v závislosti na reakci a toleranci na 18 mg nebo 36 mg jedenkrát denně v týdenním intervalu. Většina pacientů (60 %) dosáhla dávkování 36 mg jedenkrát denně. Jako primární kritérium účinnosti při hodnocení účinku pitolisantu na nadměrnou denní spavost (EDS - Excessive Daxtime Sleepiness) bylo použito skóre na Epworthské škále spavosti (ESS - Epworth Sleepiness Scale). Výsledky při podávání pitolisantu byly výrazně lepší v porovnání s výsledky v placebo skupině (průměrný rozdíl: -3,33; 95 % CI [-5,83 až -0,83]; p < 0,05), ale nebyly výrazně odlišné od výsledků ve skupině užívající modafinil (průměrný rozdíl: 0,12; 95% CI [-2,5 až 2,7]). Budivý účinek těchto dvou léčivých látek dosahoval podobných hodnot (Obrázek 1).
Obrázek 1: Změny skóre na Epworthské škále spavosti (ESS) (průměr ± SEM) od počátku po 8. týden ve studii Harmony 1
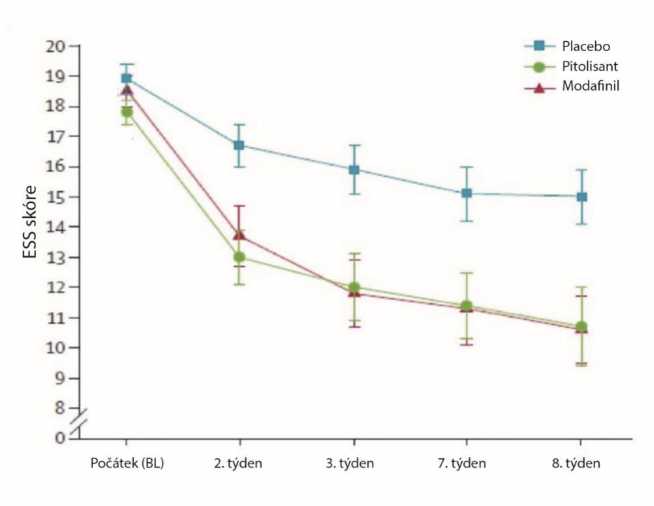
Účinek na Epworthskou škálu byl podpořen ve dvou laboratorních zkouškách bdělosti a pozornosti (Test udržení bdělosti (MWT) (p=0,044) a test úkolů na neustálou pozornost (SART) (p=0,053, téměř významný)).
Četnost atak kataplexie u pacientů vykazujících tento symptom výrazně klesla (p=0,034) při léčbě pitolisantem (-65 %) v porovnání s placebem (-10 %). Denní četnost kataplexie (geometrický průměr) byla 0,52 při zahájení léčby a 0,18 při poslední návštěvě při léčbě pitolisantem a 0,43 při zahájení léčby a 0,39 při poslední návštěvě v případě léčby placebem, s poměrem četností rR=0,38 [0,16; 0,93] (p=0,034).
Druhá pivotní studie (Harmony Ibis) zahrnovala 165 pacientů (67 pacientů léčených pitolisantem, 33 placebem a 65 modafinilem). Design studie byl podobný studii Harmony I s výjimkou toho, že maximální dávka pitolisantu dosažená 75 % pacientů byla 18 mg jedenkrát denně namísto 36 mg v Harmonii I. Jelikož zásadní nerovnováha vedla k porovnávání výsledků se shlukováním pracovišť nebo bez něj, nejkonzervativnější přístup vykazoval nepodstatné snížení skóre ESS s pitolisantem v porovnání s placebem (pitolisant-placebo=-1,94 s p=0,065). Výsledky četnosti kataplexie při 18 mg denně nebyly konzistentní s výsledky první pivotní studie (36 mg denně).
Zlepšení ve dvou objektivních testech bdělosti a pozornosti, MWT a SART, bylo u pitolisantu významné oproti placebu (p=0,009 a p=0,002) a nepodstatné oproti modafinilu (p=0,713 a p=0,294).
Harmony CTP, podpůrná, dvojitě zaslepená, randomizovaná studie v paralelních skupinách, pitolisant oproti placebu, byla navržena ke stanovení účinnosti pitolisantu u pacientů s vysokou četností kataplexie při narkolepsii. Primárním cílovým bodem pro účinnost byla změna v průměrném počtu atak kataplexie za týden mezi 2 týdny od počátku studie a 4 týdny doby stabilní léčby na konci studie. Studie se zúčastnilo 105 narkoleptických pacientů s vysokou počáteční četností výskytu kataplexie za týden (54 pacientům byl podáván pitolisant a 51 placebo). Dávkování bylo zahájeno na 4,5 mg jedenkrát denně a bylo zvyšováno v závislosti na reakci a toleranci na 9 mg, 18 mg nebo 36 mg jedenkrát denně v týdenním intervalu. Většina pacientů (65 %) dosáhla dávkování 36 mg jedenkrát denně.
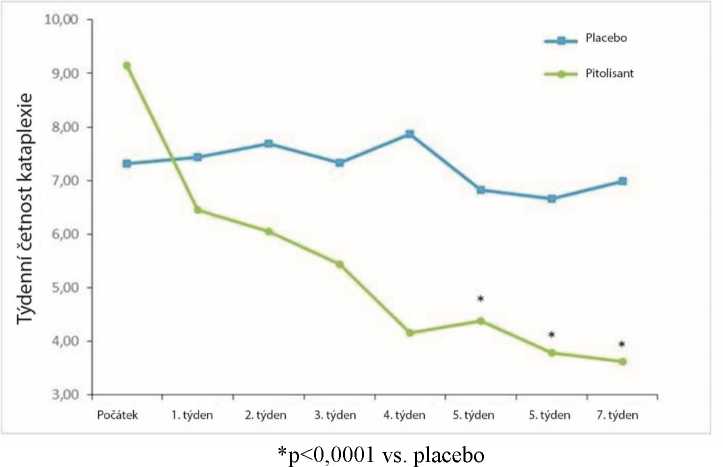
Co se týče primárního cílového bodu účinnosti, týdenní četnosti kataplektických epizod (WRC -Weekly Rate of Cataplexy episodes), výsledky při podávání pitolisantu byly výrazně lepší v porovnání s placebo skupinou (p < 0,0001), s progresivním 64% snížením od počátku studie do konce léčby (Obrázek 2). Při zahájení léčby byl geometrický průměr WRC 7,31 (medián=6.5 [4,5; 12]) a 9,15 (medián=8,5 [5,5; 15,5]) ve skupině s placebem, resp. skupině s pitolisantem. V průběhu stabilní doby (do konce léčby) geometrický průměr WRC klesl na 6,79 (medián=6 [3; 15]) a 3,28 (medián=3 [1,3; 6]) v placebo skupině, resp. ve skupině užívající pitolisant u pacientů, kteří zažili nejméně jednu epizodu kataplexie. Pozorovaná WRC ve skupině užívající pitolisant byla zhruba poloviční oproti placebo skupině: velikost účinku pitolisantu v porovnání s placebem byla shrnuta poměrem četností rR(Pt/Pb), rR=0,512; 95 %CI [0,435 až 0,603]; p < 0,0001). Velikost účinku pitolisantu v porovnání s placebem na základě modelu pro WRC na základě BOCF se středem stanoveným jako fixní účinek byla 0,581, 95 %CI [0,493 až 0,686]; p<0,0001.
Obrázek 2: Změny týdenní četnosti kataplektických epizod (geometrický průměr) od počátku studie do 7. týdnu ve studii Harmony CTP
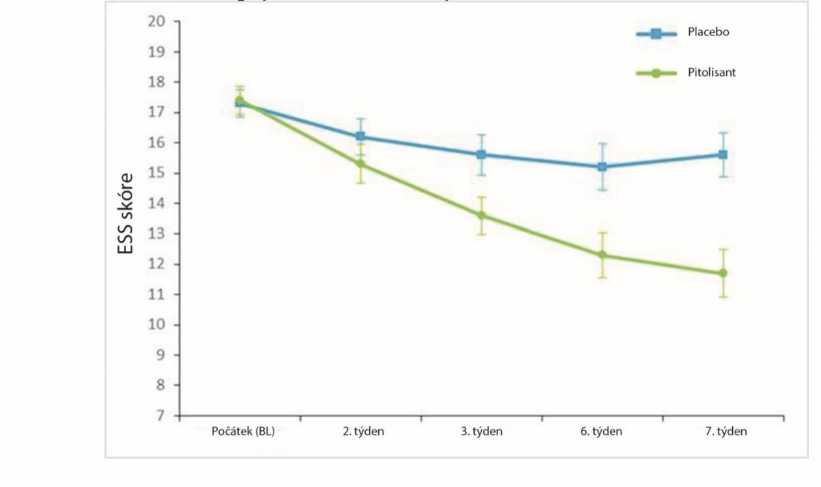
Účinek pitolisantu na EDS byl v této populaci také hodnocen pomocí skóre na ESS. Ve skupině užívající pitolisant skóre na ESS výrazně kleslo mezi počátkem studie a koncem léčby v porovnání s placebem při pozorované průměrné změně -1,9 ± 4,3 a -5,4 ± 4,3 (průměr ± směrodatná odchylka) u placeba, resp. pitolisantu, (p<0,0001) (Obrázek 3). Tento účinek na EDS byl potvrzen výsledky testu udržení bdělosti (MWT). Geometrický průměr poměrů (MWTKonec/MWTpočátek) byl 1,8 (95 % CI 1,19; 2,71, p=0,005). Hodnota MWT ve skupině užívající pitolisant byla o 80 % vyšší než u placebo skupiny.
Obrázek 3: Změny skóre na Epworthské škále spavosti (ESS) (průměr ± SEM) od počátku po 7. týden ve studii Harmony CTP
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Wakix u jedné nebo více podskupin pediatrické populace s narkolepsií s kataplexií nebo bez kataplexie (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Expozice pitolisantu u zdravých dobrovolníků byla hodnocena v rámci studií s účastí více než 200 subjektů, které dostávaly dávky pitolisantu v jediném podání až do 216 mg po dobu až 28 dnů.
Absorpce
Pitolisant se dobře a rychle vstřebává a maximální koncentrace v plazmě dosahuje zhruba tři hodiny po podání.
Distribuce
Pitolisant vykazuje vysokou míru vazby na bílkoviny v séru (>90 %) se zhruba stejnou distribucí mezi červenými krvinkami a plazmou.
Biotransformace
Metabolismus pitolisantu u člověka není úplně charakterizován. Dostupné údaje ukazují, že hlavní nekonjugované metabolity jsou hydroxylované deriváty v několika pozicích. 5-aminovalerová kyselina je hlavním neaktivním metabolitem I. fáze a nachází se v moči a v séru. Ke tvorbě dochází za působení CYP3A4 a CYP2D6. Bylo identifikováno několik konjugovaných metabolitů. Nejvýznamnějším z nich (neaktivní) je konjugát glycinu kyselinového metabolitu O-dealkylovaného desaturovaného pitolisantu a glukuronid metabolitu ketonu monohydroxy desaturovaného pitolisantu.
U jaterních mikrozomů pitolisant nezpůsobuje výraznou inhibici činnosti cytochromů CYP1A2, CYP2C9, CYP2C19, CYP2C8, CYP2B6, CYP2E1 nebo CYP3A4 a uridin difosfát-glukuronosyl transferázy izoforem UGT1A1, UGT1A4, UGT1A6, UGT1A9 a UGT2B7 až do koncentrace 13,3^M, což je úroveň výrazně vyšší než v případě terapeutických dávek. Pitolisant je inhibitor CYP2D6 se střední účinností (IC50 = 2,6 ^M).
Pitolisant indukuje CYP3A4, CYP1A2 a CYP2B6 in vitro. Předpokládají se klinicky relevantní interakce se substráty CYP3A4 a CYP2B6 a při extrapolaci substráty UGT, CYP2C a P-gp (viz bod 4.5).
Studie in vitro naznačují, že pitolisant není substrátem ani inhibitorem lidského P-glykoproteinu a proteinu BCRP (breast cancer resistance protein). Pitolisant není substrát OATP1B1, OATP1B3. Při testované koncentraci pitolisant není silným inhibitorem OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1 nebo MATE2K. Pitolisant vykazuje více než 50% inhibici OCT1 (organický kationtový transportér 1) při 1,33 ^M, extrapolovaná IC50 pitolisantu je 0,795 ^M (viz bod 4.5).
Eliminace
Pitolisant má poločas rozpadu v plazmě 10-12 hodin. Po opakovaném podávání je ustáleného stavu dosaženo po 5-6 dnech podávání, což vede ke zvýšenému množství v séru kolem 100 %. Interindividuální variabilita je poměrně vysoká, u některých dobrovolníků s vysokým profilem (bez potíží s tolerancí).
Eliminace probíhá převážně prostřednictvím moči (zhruba 63 %) přes inaktivní nekonjugovaný metabolit (BP2.951) a glycin konjugovaný metabolit. 25 % dávky je vylučováno ve vydechovaném vzduchu a malý zlomek (<3 %) byl nalezen ve stolici, přičemž množství pitolisantu nebo BP2.951 bylo zanedbatelné.
Linearita/nelinearita
Při zdvojnásobení dávky pitolisantu z 27 na 54 mg se AUC0-<» zvýšila zhruba o 2,3.
Zvláštní populace
Starší osoby
U pacientů ve věku 68 až 80 let se farmakokinetika pitolisantu nelišila od mladších pacientů (18 až 45 let). Nad 80 let kinetika vykazovala mírné odlišnosti bez klinické relevance. U starších osob jsou dostupné pouze omezené údaje. Dávku je tedy nutné upravit v závislosti na stavu ledvin a jater (viz body 4.2 a 4.4).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin (fáze 2 až 4 podle mezinárodní klasifikace chronického onemocnění ledvin, tj. rozsah clearance kreatininu 15 až 89 ml/min), Cmax a AUC měly tendenci 2,5 násobného zvyšování bez dopadu na poločas rozpadu (viz bod 4.2).
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater (Child Pugh A) nebyly zaznamenány žádné výrazné změny ve farmakokinetice v porovnání s normální zdravou populací dobrovolníků. U pacientů se středně těžkou poruchou funkce jater (Child Pugh B) došlo ke 2,4násobnému zvýšení AUC a zdvojnásobení poločasu rozpadu (viz bod 4.2). Farmakokinetika pitolisantu po opakovaném podávání u pacientů s poruchou funkce jater zatím nebyla hodnocena.
Rasa
Vliv rasy na metabolismus pitolisantu nebyl hodnocen.
5.3 Předklinické údaje vztahující se k bezpečnosti
Po 1 měsíci u myší, 6 měsících u potkanů a 9 měsících u opic byla úroveň bez výskytu nežádoucích účinků (NOAEL) 75, resp. 30 a 12 mg/kg/den, p.o., což znamená terapeutický index 9, resp. 1 a 0,4 v porovnání s expozicí přípravku v terapeutických dávkách u lidí. U potkanů se při Tmax vyskytly přechodné reverzibilní křečové epizody, které lze připsat převážně metabolitu, který se hojně vyskytuje u tohoto druhu, ale nikoli u člověka. U opic byly při nejvyšších dávkách zaznamenány přechodné klinické znaky spojené s CNS jako zvracení, třes a křeče. U nejvyšších dávek nebyly zaznamenány žádné histopatologické změny u opic. U potkanů byly u nejvyšších dávek zaznamenány omezené histopatologické změny v některých orgánech (játra, duodenum, brzlík, nadledviny a plíce).
Pitolisant nebyl genotoxický, ani karcinogenní.
Teratogenní účinek pitolisantu byl pozorován u mateřsky toxických dávek (terapeutický index teratogenity <1 u potkanů a králíků). U vysokých dávek pitolisant indukoval abnormality morfologie spermií a snižoval motilitu bez závažných dopadů na indexy fertility u potkaních samců a snižoval procento živých zárodků a zvyšoval postimplantační ztrátu i potkaních samic (terapeutický index 1). Důsledkem bylo zpoždění postnatálního vývoje (terapeutický index 1).
U zvířat pitolisant/metabolity prostoupily bariérou placenty.
Studie toxicity u potkaních mláďat odhalily, že podávání pitolisantu ve vysokých dávkách vyvolalo mortalitu související s dávkou a křečové epizody, které lze připsat metabolitu vyskytujícímu se hojně u potkanů, ale nikoli u člověka.
Pitolisant blokoval hERG kanál s IC50 překračující terapeutické koncentrace a indukoval mírné prodloužení QTc u psů.
V preklinických studiích byly provedeny studie závislosti a náchylnosti ke zneužívání léčiva u myší, opic a potkanů. Nebylo však možné dosáhnout definitivního závěru u studií tolerance, závislosti a samopodání.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulóza Krospovidon typ A Mastek
Magnesium-stearát
Koloidní bezvodý oxid křemičitý
Potahová vrstva tablety
Polyvinylalkohol Oxid titaničitý (E171) Makrogol 3350 Mastek
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
Wakix 4,5 mg tablety 12 měsíců
Wakix 18 mg tablety 30 měsíců
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádná zvláštní opatření pro uchovávání.
6.5 Druh obalu a obsah balení
Lahvička z polyetylenu s vysokou hustotou (HDPE) s polypropylenovým uzávěrem garantujícím neporušenost obalu, dětskou pojistkou a desikantem (silikagel).
Lahvička s 30 potahovanými tabletami.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Bioprojet Pharma 9, rue Rameau 75002 Paris Francie
Tel: +33 (0)1 47 03 66 33 Fax: +33 (0)1 47 03 66 30 e-mail: contact@bioprojet.com
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1068/001
EU/1/15/1068/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců odpovědných za propouštění šarží
Rottendorf
ZI N°2 de Prouvy
Rouvignies
1 rue de Nungesser
59121 Prouvy
Francie
Patheon
40 Boulevard de Champaret 38300 Bourgoin-Jallieu Francie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c, odst. 7 Směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se data odevzdání PSUR a aktualizace RMP shodují, lze je podat současně.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Neintervenční poregistrační studie bezpečnosti (PASS): Multicentrická, neintervenční, poregistrační studie bezpečnosti pro dokumentaci užívání léčivého přípravku Wakix a shromažďování informací o bezpečnosti přípravku Wakix při užívání v běžné lékařské praxi. |
Závěrečná zpráva: 3Q 2023 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Wakix 4,5 mg potahované tablety Pitolisantum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta Wakix 4,5 mg obsahuje pitolisanti hydrochloridum 5 mg, což odpovídá pitolisantum 4,45 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bioprojet Pharma 9, rue Rameau 75002 Paris Francie
12. REGISTRAČNÍ ČÍSLO
EU/1/15/1068/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Wakix 4,5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Wakix 4,5 mg potahované tablety
Pitolisantum
Perorální podání.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
c.s.
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
30 tablet
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Wakix 18 mg potahované tablety Pitolisant
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Každá potahovaná tableta Wakix 18 mg obsahuje pitolisanti hydrochloridum 20 mg, což odpovídá pitolisantum 17,8 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Bioprojet Pharma 9, rue Rameau 75002 Paris Francie
12. REGISTRAČNÍ ČÍSLO
EU/1/15/1068/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Wakix 18 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Wakix 18 mg potahované tablety Pitolisant
K perorálnímu podání.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
c.s.
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
30 tablet
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro pacienta
Wakix 4,5 mg potahované tablety Wakix 18 mg potahované tablety
Pitolisantum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Wakix a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Wakix užívat
3. Jak se přípravek Wakix užívá
4. Možné nežádoucí účinky
5. Jak přípravek Wakix uchovávat
6. Obsah balení a další informace
1. Co je přípravek Wakix a k čemu se používá
Wakix obsahuje léčivou látku pitolisant. Wakix je léčivý přípravek, který se používá k léčbě dospělých pacientů trpících narkolepsií s kataplexií nebo bez kataplexie.
Narkolepsie je porucha, která způsobuje nadměrnou denní spavost a tendenci náhle usínat v nevhodných situacích (spánkové ataky). Kataplexie je nástup náhlé svalové slabosti nebo paralýzy bez ztráty vědomí v reakci na náhlý emocionální podnět jako je vztek, strach, radost, smích nebo překvapení.
Léčivá látka pitolisant se váže na receptory mozkových buněk, které jsou zapojeny do stimulace bdělosti. Tím pomáhá potlačovat denní spavost a kataplexii a podporuje bdělost.
2. Čemu musíte věnovat pozornost, než začnete přípravek Wakix užívat
Neužívejte přípravek Wakix:
- jestliže jste alergický(á) na pitolisant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte vážné obtíže s játry, protože pitolisant se za běžných okolností rozkládá v játrech a u pacientů s výrazně sníženou funkcí jater může dojít k hromadění nadměrného množství léčivé látky.
- jestliže kojíte.
Upozornění a opatření
Před užitím přípravku Wakix se poraďte se svým lékařem, jestliže se Vás týká kterákoli z níže uvedených situací:
- Trpěl(a) jste úzkostí nebo depresí s myšlenkami na sebevraždu.
- Trpíte problémy s játry nebo ledvinami. V takovém případě může být nutné upravit dávkování.
- Máte žaludeční vřed nebo užíváte léky, které mohou dráždit váš žaludek, jako j sou protizánětlivé léky, protože při užívání přípravku Wakix byly hlášeny žaludeční reakce.
- Trpíte obezitou nebo anorexií. Při užívání přípravku Wakix může dojít ke změně Vaší tělesné hmotnosti (zvýšení nebo snížení).
- Trpíte problémy se srdcem. Bude nutné, aby Váš lékař prováděl při užívání přípravku Wakix pravidelné kontroly.
- Máte vážnou formu epilepsie.
Pokud se Vás týká kterákoli situace z výše uvedených, poraďte se se svým lékařem nebo lékárníkem před užitím přípravku Wakix.
Děti a dospívající
Děti a dospívající by neměli užívat přípravek Wakix.
Další léčivé přípravky a přípravek Wakix
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Přípravek Wakix může ovlivňovat účinek jiných léků a stejně tak jiné léčivé přípravky mohou ovlivňovat účinek přípravku Wakix. Váš lékař možná bude muset upravit dávkování přípravku.
Zvláštní opatrnosti je nutné dbát zejména pokud užíváte přípravek Wakix společně s některými antidepresivy (např. imipramin, clomipramin a mirtazapin) a některými léky pro léčbu alergických stavů (antihistaminika, např. feniramin maleát, chlorfeniramin, difenydramin, promethazin, mepyramin).
Informujte svého lékaře nebo lékárníka, pokud užíváte některý z těchto léků: rifampicin (antibiotikum), fenytoin, karbamazepin a fenobarbital (převážně užívané proti záchvatům), chinidin, digoxin (používá se k léčbě abnormálního srdečního rytmu), paroxetin, fluoxetin, venlafaxin, duloxetin (antidepresiva), třezalka tečkovaná (Hypericum perforatum) bylinná léčba deprese, bupropion (antidepresiva pro snadnější odvykání kouření), cinakalcet (léčba poruch příštítných tělísek), terbinafin (používá se k léčbě plísňových infekcí), metformin, repaglinid (používá se k léčbě diabetu), docetaxel, irinotekan (používá se k léčbě rakoviny), cisaprid (používá se k léčbě žaludečního refluxu), pimozid (používá se k léčbě některých duševních poruch), halofantrin (léčba malárie), efavirenz (antivirotikum pro léčbu HIV), morfin, paracetamol (léčba bolesti), dabigatran (používá se k léčbě žilních obtíží), warfarin (používá se k léčbě srdečních chorob).
Wakix může snižovat účinnost hormonální antikoncepce, je tedy nutné používat alternativní metodu účinné antikoncepce (viz bod „Těhotenství “).
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Wakix by neměl být užíván během těhotenství, pokud tak neurčí lékař. Není k dispozici dostatek informací pro určení, zda je s užíváním přípravku Wakix během těhotenství spojeno nějaké konkrétní riziko. Pokud jste žena, musíte v průběhu léčby přípravkem Wakix a nejméně 21 dní po ukončení léčby užívat antikoncepci. Vzhledem k tomu, že Wakix může snižovat účinnost hormonální antikoncepce, je nutné používat alternativní metodu účinné antikoncepce.
Kojení
U zvířat přípravek Wakix přecházel do mateřského mléka. Pacientky užívající přípravek Wakix musí přestat kojit.
Řízení dopravních prostředků a obsluha strojů
Je nutné dbát zvýšené opatrnosti v případě činností, které vyžadují pozornost, jako například řízení auta nebo práce se stroji. Pokud si nejste jistý(á), zda Váš stav nemá negativní dopad na Vaši schopnost řídit, obraťte se na svého lékaře.
3. Jak se přípravek Wakix užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Léčba se za normálních okolností zahajuje dávkou 9 mg jedenkrát denně a postupně se zvyšuje v průběhu tří týdnů na nejvhodnější dávku. Váš lékař může kdykoli dávku zvýšit nebo snížit v závislosti na tom, jak dobře u Vás přípravek zabírá a jak dobře ho snášíte.
Než pocítíte účinek přípravku, může to trvat několik dní. Maximální účinek se obvykle projevuje po několika týdnech.
Neprovádějte změny dávkování přípravku Wakix na základě vlastního rozhodnutí. Veškeré změny dávkování musí být předepsány a monitorovány Vaším lékařem.
V případě dávky 4,5 mg užijte jednu 4,5 mg tabletu.
V případě dávky 9 mg užijte dvě 4,5 mg tablety.
V případě dávky 18 mg užijte jednu 18 mg tabletu.
V případě dávky 36 mg užijte dvě 18 mg tablety.
Přípravek Wakix užívejte perorálně jednou denně ráno se snídaní.
Neužívejte dávku přípravku Wakix odpoledne, mohlo by pak pro Vás být obtížné usnout.
Jestliže jste užil(a) více přípravku Wakix, než jste měl(a)
Jestliže jste užil(a) příliš mnoho tablet přípravku Wakix, okamžitě kontaktujte pohotovost v nejbližší nemocnici nebo informujte svého lékaře či lékárníka. Může se u Vás vyskytnout bolest hlavy, bolest břicha, nevolnost nebo podráždění. Můžete mít také potíže s usínáním. Tuto příbalovou informaci a případně zbylé tablety vezměte s sebou.
Jestliže jste zapomněl(a) užít přípravek Wakix
Jestliže jste zapomněl(a) užít přípravek Wakix, užijte další dávku v obvyklý čas, nezdvojnásobujte dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Wakix
Přípravek Wakix byste měl(a) dále užívat po dobu určenou Vaším lékařem. S užíváním přípravku Wakix nepřestávejte náhle na základě vlastního rozhodnutí.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Možné nežádoucí účinky
4.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 pacientů):
- Potíže při usínání, pocity úzkosti, pocity podrážděnosti, depresivní pocity, potíže se spánkem
- Bolesti hlavy, pocit točení hlavy (závrať), ztráta rovnováhy, třes
- Nevolnost, zvracení, zažívací potíže
- Únava
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 pacientů):
- Pocení
- Snížená nebo zvýšená chuť k j ídlu
- Otok
- Nervozita, vidění nebo slyšení věcí, které neexistují
- Změny emocí
- Abnormální sny
- Napětí
- Obtíže při usínání na začátku noci, uprostřed noci, nebo na konci noci, obtíže udržet souvislý spánek, nadměrná spavost, ospalost
- Stav lhostejnosti s nedostatkem emocí
- Noční můry
- Pocity neklidu a neschopnost udržet se v klidu
- Panické reakce
- Pozměněný nebo zvýšený sexuální zájem
- Náhlé nebo přechodné epizody svalové slabosti, nekontrolovatelných svalových křečí nebo pohybů jedné nohy
- Narušení pozornosti
- Migréna
- Epilepsie
- Slabost
- Narušení pohybů, pomalé pohyby těla
- Pocit brnění, lechtání, píchání nebo pálení kůže
- Náhlé a nepředvídatelné fáze hybnosti a nehybnosti
- Pocit vrávorání
- Snížená ostrost vidění, abnormální kontrakce nebo tiky očního víčka
- Slyšení zvuků bez přítomnosti vnějšího zdroje zvuku
- Abnormální tlukot srdce, pomalý nebo rychlý tep, zvýšení nebo snížení krevního tlaku, návaly
horka
- Zívání
- Sucho v ústech
- Průjem, bolest břicha, nepříjemný pocit nebo bolest v břišní oblasti, zácpa, pálení žáhy, bolest a nepříjemné pocity v žaludku, gastritida, nadměrná kyselost zažívacího ústrojí
- Svědění, neobvyklé zarudnutí kůže v oblasti nosu a tváří, nadměrné pocení
- Bolest kloubů, bolest zad, svalová ztuhlost, svalová slabost, bolest svalů a kostí, bolest prstů na nohou a na rukou
- Abnormální močení
- Nepravidelné krvácení z dělohy
- Ztráta síly nebo extrémní únava, bolest na hrudi, malátnost, otok
- Nárůst tělesné hmotnosti, úbytek tělesné hmotnosti, abnormální srdeční činnost (EKG), abnormální krevní obraz s ohledem na funkci jater.
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1000 pacientů):
- Ztráta chuti k j ídlu, zvýšená chuť k j ídlu
- Abnormální chování, zmatený stav, depresivní nálada, podrážděnost, pocity emoční a duševní nepohody, pocity slyšení nebo vidění věcí, které neexistují během spánku
- Ztráta vědomí, tenzní bolesti hlavy, potíže s pamětí, nízká kvalita spánku
- Mírná bolest břicha, obtížné nebo bolestivé polykání, plynatost, zánět zažívacího ústrojí
- Kožní infekce, abnormálně vysoká citlivost na sluneční světlo
- Bolest krku, hrudníku
- Spontánní potrat
- Bolest, noční pocení, pocit útlaku
- Vysoká koncentrace enzymu kreatinin fosfokinázy v krvi, abnormální celkový fyzický stav, změna elektrických záznamů srdce (EKG)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku_č. V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Wakix uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za zkratkou EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádná zvláštní opatření pro uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Wakix obsahuje
Léčivou látkou je pitolisantum.
Wakix 4,5 mg tablety
Jedna tableta obsahuje pitolisanti hydrochloridum 5 mg, což odpovídá pitolisantum 4,45 mg.
Wakix 18 mg tablety
Jedna tableta obsahuje pitolisanti hydrochloridum 20 mg, což odpovídá pitolisantum 17,8 mg.
Dalšími složkami jsou mikrokrystalická celulóza, krospovidon, mastek, magnesium-stearát, koloidní bezvodý oxid křemičitý, polyvinylalkohol, oxid titaničitý, makrogol 3350.
Jak přípravek Wakix vypadá a co obsahuje toto balení
Wakix 4,5 mg se prodává ve formě bílé, kulaté, potahované tablety o průměru 3,7 mm, bikonvexního tvaru s označením „5“ na jedné straně.
Wakix 18 mg se prodává ve formě bílé, kulaté, potahované tablety o průměru 7,5 mm, bikonvexního tvaru s označením „20“ na jedné straně.
Wakix se dodává v lahvičce s 30 tabletami.
Držitel rozhodnutí o registraci
Bioprojet Pharma 9, rue Rameau 75002 Paris Francie
Výrobce
Rottendorf
ZI N°2 de Prouvy
Rouvignies
1 rue de Nungesser
59121 Prouvy
Francie
Patheon
40 Boulevard de Champaret 38300 Bourgoin-Jallieu Francie
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http: //www .ema.europa.eu.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP. QR kód bude doplněn URL {webová stránka EMA a související informace o přípravku nejsou zatím známy} Informace naleznete také při použití QR kódu níže.
33