Voncento 250 Iu Fviii//600 Iu Vwf
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Voncento 250 IU FVIII/ 600 IU VWF (5 ml rozpouštědlo) prášek a rozpouštědlo pro injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně:
- Factor VIII coagulationis humanus 250 IU1 (lidský koagulační faktor VIII2 (FVIII))
- Factor von Willebrand humanus 600 IU3 (lidský von Willebrandův faktor2 (VWF))
Po rekonstituci v 5 ml roztok obsahuje 50 IU/ml lidského koagulačního faktoru VIII a 120 IU/ml lidského von Willebrandova faktoru.
Pomocná látka se známým účinkem:
Sodík přibližně 128,2 mmol/l (2,95 mg/ml). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok.
Bílý prášek a čiré, bezbarvé rozpouštědlo pro injekční/infuzní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
von Willebrandova choroba (VWD)
Profylaxe a léčba hemoragie nebo krvácení během chirurgických zákroků u pacientů trpících VWD v případech, kdy je léčba samotným desmopressinem (DDAVP) neúčinná nebo kontraindikovaná.
Hemofilie A (vrozený deficit FVIII)
Profylaxe a léčba krvácení u pacientů trpících hemofilií A.
4.2 Dávkování a způsob podání
Léčba VWD a hemofilie A se má provádět pod dohledem lékaře, který má zkušenosti s léčbou poruch hemostázy.
Při rozhodování o využití přípravku u domácí léčby při krvácení u pacientů s VWD a s hemofilií A určí konkrétního pacienta ošetřující lékař, který zajistí odpovídající vyškolení v podávání přípravku a toto podávání kontroluje v pravidelných intervalech.
Poměr mezi FVIII:C a VWF:RCo v injekční lahvičce je přibližně 1:2,4.
Dávkování von Willebrandova choroba
Je důležité výpočem stanovit dávku na základě specifikovaného počtu IU VWF:RCo.
1 IU/kg VWF:RCo obvykle zvýší hladinu VWF:RCo v oběhu o 0,02 IU/ml (2 %).
Je třeba dosáhnout hladiny VWF:RCo > 0,6 IU/ml (60 %) a hladiny FVIII:C > 0,4 IU/ml (40 %).
Požadovaná léčba
K dosažení hemostázy se obyčejně doporučuje 40 - 80 IU/kg von Willebrandova faktoru (VWF:RCo), což odpovídá 20 - 40 IU FVIII:C/kg tělesné hmotnosti (BW).
Úvodní dávka 80 IU/kg VWF:RCo může být potřebná zejména u pacientů s VWD typu 3, kde k udržení adekvátních hladin mohou být nutné vyšší dávky než v případě jiných typů VWD.
Prevence krvácení během chirurgického zákroku
K prevenci masivního krvácení během nebo po chirurgickém zákroku se má přípravek začít podávat 1-2 hodiny před chirurgickým zákrokem.
Příslušná dávka se má podávat opakovaně každých 12 - 24 hodin. Dávka a trvání léčby závisí na klinickém stavu pacienta, na typu a závažnosti krvácení a na hladinách VWF:RCo a FVIII:C.
Při používání přípravku s VWF obsahujícího FVIII by si měl být ošetřující lékař vědomý, že pokračování v léčbě může zapříčinit nadměrný vzestup hladin FVIII:C. Po 24 - 48 hodinách léčby by se mělo zvážit snížení dávek a/nebo prodloužení intervalu mezi dávkami nebo použití přípravku s VWF obsahující nízkou hladinu FVIII, aby se zabránilo nadměrnému vzestupu hladiny FVIII:C (viz bod 5.2).
Profylaktická léčba
Při dlouhodobé profylaxi u pacientů s VWD má být zvažována dávka 25-40 IU VWF:RCo/kg tělesné hmotnosti při frekvenci 1-3 krát týdně. U pacientů s gastrointestinálním krvácení nebo menoragií mohou být nezbytné kratší intervaly nebo vyšší dávky. Dávka a trvání léčby bude záviset na klinickém stavu pacienta, jakož i plazmatických hladinách VWF:RCo a FVIII:C.
Pediatrická populace s VWD Léčba krvácení
U pediatrických pacientů se obvykle doporučuje pro léčbu krvácení 40 až 80 IU/kg von Willebrandova faktoru (VWF:RCo), odpovídající 20 až 40 IU FVIII:C/kg tělesné hmotnosti (BW).
Profylaktická léčba
Pacienti ve věku 12 až 18 roků: Dávkování je založeno na stejných pokynech jako pro dospělé.
Pacienti ve věku <12 let: Na základě výsledků z klinické studie s pediatrickými pacienty do 12 let, ve které bylo prokázáno, že mají nižší expozici VWF, by mělo být uvažováno profylaktické rozmezí dávek 40 - 80 IU VWF:RCo/kg tělesné hmotnosti 1 až 3x týdně. (viz bod 5.2).
Dávka a trvání léčby bude záviset na klinickém stavu pacienta, jakož i plazmatických hladinách VWF:RCo a FVffl:C.
Hemofilie A
Je důležité výpočem stanovit dávku na základě specifikovaného počtu IU FVIII:C.
Dávka a délka substituční terapie závisí na závažnosti deficitu FVIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek FVIII se vyjadřuje v mezinárodních jednotkách (International Units - IU), které jsou stanovené oproti současnému standardu WHO pro léčivé přípravky obsahující FVIII. Aktivita FVIII v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v IU (vzhledem k mezinárodnímu standardu pro FVIII v plazmě).
1 IU aktivity FVIII odpovídá množství FVIII v 1 ml normální lidské plazmy.
Požadovaná léčba
Výpočet požadované dávky FVIII vychází z empirického předpokladu, že podáníl IU FVIII na kg tělesné hmotnosti zvýší plazmatickou aktivitu FVIII v plazmě o přibližně 2 % normální aktivity (in vivo recovery 2 IU/dl). Požadovaná dávka se stanoví podle následujícího vzorce:
Požadovaný počet jednotek = tělesná hmotnost [kg] x požadovaný vzestup FVIII [% nebo IU/dl] x 0,5
Množství, které se má podat, a frekvence podávání mají vždy směřovat ke klinické účinnosti v individuálních případech.
V případě následujících hemoragických příhod nemá aktivita FVIII během daného období klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může posloužit jako návod pro stanovení dávky v případech krvácení a při chirurgických výkonech:
|
Stupeň krvácení/typ chirurgického výkonu |
Požadovaná hladina FVIII (% nebo IU/dl) |
Frekvence dávkování (hodiny)/délka trvání léčby (dny) |
|
Krvácení | ||
|
Časný hemartros, krvácení do svalstva nebo do ústní dutiny |
O 1 o <N |
Infuzi opakovat každých 12 - 24 hodin po dobu nejméně jednoho dne, dokud se krvácení nezastaví, což se projeví ústupem bolesti nebo zahojením. |
|
Rozsáhlejší hemartros, krvácení do svalstva nebo hematom |
O kO 1 o co |
Infuzi opakovat každých 12 - 24 hodin po dobu 3 - 4 dní nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
o o 1 o kO |
Infuzi opakovat každých 8 - 24 hodin, dokud nepomine ohrožení života. |
|
Chirurgické výkony | ||
|
Menší chirurgický výkon včetně extrakce zubů |
o kO 1 o co |
Infuzi opakovat každých 24 hodin po dobu nejméně jednoho dne až do zahojení. |
|
Velké chirurgické výkony |
80 -100 (před a po operaci) |
Infuzi opakovat každých 8 - 24 hodin, dokud nedojde k uspokojivému zahojení rány, potom pokračovat v léčbě nejméně dalších 7 dní a udržovat aktivitu FVIII mezi 30 % - 60 % (IU/dl). |
Během léčby se doporučuje náležitá kontrola hladin FVIII kvůli stanovení dávky, která má být podávaná, a frekvenci opakovaných infuzí. V případě velkých chirurgických zákroků je nevyhnutelný přesný monitoring substituční terapie prostřednictvím koagulační analýzy (aktivity plazmatického FVIII). U jednotlivých pacientů může být odpověď na léčbu faktorem VIII rozdílná, v závislosti na odlišných hladinách in vivo recovery a různém biologickém poločasu.
Profylaktická léčba
V případě dlouhodobé profylaxe u pacientů s těžkou formou hemofilie A se obyčejně doporučuje dávka 20 až 40 IU FVIII na kilogram tělesné hmotnosti v intervalech 2 až 3 dní. V některých případech, zejména u mladších pacientů, může být nutný kratší interval mezi dávkami nebo vyšší dávky.
Dříve neléčení pacienti
Bezpečnost a účinnost přípravku Voncento u dříve neléčených pacientů nebyla dosud stanovena.
Pediatrická populace s hemofilií A
Dávkování u hemofilie A u dospívajících ve věku od 12 do 18 let je založeno na tělesné hmotnosti, a proto pro něj platí stejná doporučení jako u dospělých. V některých případech je nezbytný kratší interval dávek nebo vyšší dávky. Frekvence podávání by měla být v každém jednotlivém případě vždy zaměřená na klinickou účinnost.
Bezpečnost a účinnost přípravku Voncento u dětí <12 let s hemofilií A nebyla stanovena. Žádné údaje nejsou k dispozici.
Starší osoby
U starších osob není potřebná žádná úprava dávek.
Způsob podání K intravenóznímu podání.
Přípravek rekonstituujte podle postupu v bodě 6.6. Rekonstituovaný přípravek se podává pomalu intravenózně injekcí/infuzí rychlostí příjemnou pro pacienta.
Rychlost podávání injekce nebo infuze nemá přesáhnout 6 ml za minutu. Pacienta je nutno sledovat, zda nemá bezprostřední reakci. Jestliže se vyskytne jakákoli reakce v souvislosti s podávaním přípravku Voncento, musí se podle klinického stavu pacienta rychlost podávání přípravku snížit nebo podávání zastavit (viz rovněž bod 4.4).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Jsou možné reakce z přecitlivělosti alergického typu. Pokud se objeví příznaky hypersenzitivity, pacienti by měli být poučeni, aby okamžitě přerušili používání léčivého přípravku a kontaktovali svého lékaře. Pacienti mají být informováni o prvních příznacích reakcí z přecitlivělosti jako je kopřivka, generalizovaná vyrážka, pocit tísně na hrudi, sípání, hypotenze a anafylaxe. V případě šoku je nutné dodržovat všeobecné lékařské postupy pro léčbu šoku.
Virová bezpečnost
Ke standardním opatřením k prevenci infekcí, které jsou následkem používání přípravků vyrobených z lidské krve nebo plazmy, patří výběr dárců, kontrola jednotlivých odběrů a plazmatických poolů zaměřená na specifické markery infekce a provedení efektivních výrobních kroků k inaktivaci/odstranění virů. Navzdory těmto opatřením není možné úplně vyloučit možnost přenosu infekčních agens, pokud podávané přípravky pocházejí z lidské krve nebo plazmy. Toto platí i pro neznámé nebo nové viry a další patogeny.
Tato opatření se považují za účinná pro obalené viry, jako jsou virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV) a neobalený virus hepatitidy A (HAV).
Provedená opatření mohou mít jen omezenou účinnost proti neobaleným virům, jako je parvovirus B19.
Infekce způsobená parvovirem B19 může být závažná u těhotných žen (infekce plodu) a jedinců s imunodeficiencí nebo zvýšenou erytropoézou (např. hemolytická anémie).
U pacientů, kteří pravidelně/opakovaně dostávají přípravky s faktorem Vin/VWF vyrobené z lidské plazmy, je třeba zvážit možnosti vhodného očkování (hepatitida A a B).
Důrazně se doporučuje, aby byl při každém podání přípravku Voncento pacientovi zaznamenaný název a číslo šarže přípravku, aby se udržovaly záznamy o propojení mezi pacientem a šarží přípravku.
von Willebrandova choroba
Existuje riziko vzniku trombóz zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Rizikoví pacienti musí být proto sledováni kvůli zachycení prvních příznaků trombózy. Měla by být provedena profylaktická opatření proti venózním trombózám v souladu s aktuálními doporučeními.
Při léčbě přípravkem s VWF obsahujícím FVIII má mít ošetřující lékař na paměti, že pokračování v léčbě může zapříčinit nadměrný vzestup FVIII:C. U pacientů, kteří dostávají přípravek s VWF obsahující FVIII, musí být sledovány hladiny FVIII:C v plazmě kvůli tomu, aby nebyly hladiny FVIII:C v plazmě dlouhodobě vysoké, což by mohlo zvýšit riziko výskytu trombóz, a současně je potřebné zvážit zavedení protitrombotických opatření (viz též bod 5.2).
U pacientů s VWD, zejména typu 3, se mohou vytvořit neutralizační protilátky (inhibitory) proti VWF. Jestliže se nedaří dosáhnout očekávanou hladinu aktivity VWF:RCo v plazmě nebo jestliže krvácení není kontrolované vhodnou dávkou, je potřebné uskutečnit odpovídající analýzu kvůli stanovení přítomnosti inhibitoru VWF. U pacientů s vysokými hladinami inhibitoru může být terapie nejen neúčinná, ale může vést i k anafylaktoidní reakci a je třeba uvažovat o jiných možnostech léčby.
Hemofilie A
Inhibitory
U pacientů s hemofilií A je známou komplikací tvorba neutralizačních protilátek (inhibitorů) proti FVIII. Tyto inhibitory jsou obyčejně IgG imunoglobuliny proti prokoagulační aktivitě FVIII, které jsou kvantifikovány v jednotkách Bethesda (BU) na ml plazmy, pomocí modifikovaného testu. Riziko vzniku inhibitorů koreluje s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dní expozice. Zřídka se mohou inhibitory vytvořit po prvních 100 dnech expozice.
Případy opakovaného výskytu inhibitoru (nízké titry) byly pozorovány po přechodu z jednoho přípravku, obsahujícího faktor VIII, na jiný, u pacientů, kteří už byli léčení předtím po dobu více než 100 dní a u kterých byla už předtím zaznamenaná tvorba inhibitoru. Proto se doporučuje důkladné monitorování všech pacientů, pokud se týká výskytu inhibitoru, po jakémkoli přechodu na jiný přípravek.
Obecně mají být všichni pacienti léčení lidským koagulačním faktorem FVIII pozorně monitorovaní kvůli tvorbě inhibitorů a to prostřednictvím klinického sledování a laboratorních testů.
Pokud nejsou dosaženy očekávané hladiny faktoru VIII v plazmě, nebo pokud není krvácení zvládnuto odpovídající dávkou, mělo být provedeno testování na přítomnost inhibitoru FVIII. U pacientů s vysokými hladinami inhibitoru může být léčba faktorem VIII neúčinná a měly by být zváženy jiné možnosti léčby. Léčba těchto pacientů by měla být vedena lékaři se zkušenostmi s péčí o pacienty s hemofilií A a s inhibitory faktoru VIII.
Komplikace související s katetrem
Jestliže je třeba použít zařízení pro centrální žilní přístup (CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katetru.
Obsah sodíku
Tento přípravek obsahuje až 14,75 mg (0,64 mmol) sodíku v jedné injekční lahvičce. Toto množství se musí zohlednit u pacientů s dietou s kontrolovaným příjmem sodíku.
Pediatrická populace
Uvedená upozornění a opatření se vztahují jak na dospělé tak na pediatrické pacienty.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly studovány žádné interakce VWF a FVIII s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S přípravkem Voncento se neprováděly reprodukční studie u zvířat. von Willebrandova choroba
Zkušenosti s léčbou těhotných nebo kojících žen nejsou k dispozici. Přípravek Voncento by měl být podáván těhotným nebo kojícím ženám s deficitem VWF pouze tehdy, pokud je jasně indikován, přičemž je nutno brát v úvahu, že porod představuje zvýšené riziko krvácivých příhod u těchto pacientek.
Hemofilie A
Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou dostupné zkušenosti, týkající se léčby během těhotenství a kojení. Proto by přípravek Voncento měl být používán během těhotenství a kojení pouze tehdy, pokud je jasně indikován.
Fertilita
Nejsou dostupné žádné údaje, týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje Voncento nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Následující nežádoucí účinky se mohou vyskytnout u dospělých a dospívajících během léčby přípravkem Voncento: hypersenzitivita nebo alergické reakce, tromboembolické příhody, horečka, bolest hlavy, poruchy chuti a abnormální hladiny testů jaterních funkcí. Navíc se mohou u pacientů vyvinout inhibitory FVIII a VWF.
Seznam nežádoucích účinků v tabulce
Tabulka uvedená níže je podle klasifikace orgánových systémů MedDRA.
Frekvence byly hodnoceny podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až <1/10), méně časté (> 1/1 000 až <1/100), vzácné (> 1 / 10000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinky |
Frekvence |
|
MedDRA |
|
Poruchy krve a lymfatického systému |
inhibice FVIII inhibice VWF |
velmi vzácné velmi vzácné |
|
Poruchy imunitního systému |
hypersenzitivita (zahrnující tachykardii, bolest na hrudi, nepříjemný pocit na hrudi a bolest v zádech) |
velmi vzácné |
|
Poruchy nervového systému |
porucha chuti |
velmi vzácné |
|
Cévní poruchy |
tromboembolická příhoda |
velmi vzácné |
|
Celkové poruchy a reakce |
velmi vzácné | |
|
v místě aplikace |
velmi vzácné | |
|
Vyšetření |
abnormální testy jaterních funkcí |
velmi vzácné |
Popis vybraných nežádoucích účinků
Hypersenzitivita (alergické reakce): přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, zimnici, zarudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargii, nauzeu, neklid, tachykardii, napětí na hrudi (včetně bolesti na hrudi a nepříjemného pocitu na hrudi), bolest zad, mravenčení, zvracení, sípání), byly pozorovány a mohou se v některých případech rozvinout do závažné anafylaxe (včetně šoku).
Inhibice FVIII: U pacientů s hemofilií A se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se jako nedostatečná klinická odpověď. V takových případech se doporučuje vyhledat specializované pracoviště k léčbě hemofilie. Nejsou žádné zkušenosti z klinických studií s podáváním přípravku Voncento u dříve neléčených pacientů (PUPs). Proto nejsou v současné době k dispozici žádné validní údaje o výskytu klinicky významných specifických inhibitorů.
Inhibice VWF: U pacientů s VWD, zejména typu 3, se mohou vytvořit neutralizační protilátky (inhibitory) proti VWF. Jestliže takové inhibitory vzniknou, tento stav se projeví jako nedostatečná klinická odpověď.Tyto protilátky jsou precipitační a může dojít současně k anafylaktické reakci. Proto pacienti, u kterých se vyskytla anafylaktická reakce, by měli být vyšetřeni na přítomnost inhibitorů.
Ve všech těchto případech se doporučuje vyhledat specializované pracoviště k léčbě hemofilie.
Tromboembolické příhody: U pacientů s VWD existuje riziko vzniku tromboembolických příhod, zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory.
Pacientům, kterým se podávají přípravky s VWF obsahující FVIII se může výrazně zvýšit hladina FVIII:C v plazmě, čímž se zvyšuje riziko tromboembolických příhod (viz též bod 4.4).
Informace o bezpečnosti s ohledem na přenosná agens viz též bod 4.4.
Pediatrická populace
Očekává se, že frekvence, typ a závažnost nežádoucích účinků u dětí budou stejné jako u dospělých. Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pět případů předávkování bylo hlášeno z klinických studií. Žádné nežádoucí účinky nebyly spojené s těmito hlášeními.
Riziko trombózy nelze v případě vysokého předávkování vyloučit, zejména u pacientů s VWD.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragikum: Koagulační faktory, von Willebrandův faktor
a koagulační faktor VIII v kombinaci.
Kód ATC: B02BD06
von Willebrandova choroba
Exogenně podávaný VWF vyrobený z lidské plazmy působí stejně jako endogenní VWF.
Podávání VWF umožňuje korekci hemostatických abnormalit, které se projevují u pacientů
s deficitem VWF (VWD) na dvou úrovních:
- VWF obnovuje adhezi krevních destiček na cévní subendotel v místě poranění cévy (protože váže cévní subendotel a membránu destiček) a zprostředkuje primární hemostázu, což se projeví zkrácením krvácení. Tento účinek nastupuje okamžitě a je známé, že ve velké míře závisí na úrovni polymerace proteinu.
- VWF způsobuje opožděnou korekci souvisejícího deficitu FVIII. Při intravenózním podání se VWF váže na endogenní FVIII (který je pacientem normálně produkován) a prostřednictvím stabilizace tohoto faktoru zabraňuje jeho rychlé degradaci.
Z tohoto důvodu podávání čistého VWF (přípravek s VWF s nízkou hladinou FVIII) sekundárně obnovuje hladinu FVIII:C na normální úroveň po první infuzi s mírným opožděním.
- Podávání přípravku s VWF obsahujícího FVIII:C obnovuje normální hladinu FVIII:C okamžitě po první infuzi.
Hemofilie A
Exogenně podávaný FVIII vyrobený z lidské plazmy se chová stejně jako endogenní FVIII.
Komplex FVIII/VWF se skládá ze dvou molekul (FVIII a VWF) s odlišnými fyziologickými funkcemi.
Po podání pacientovi s hemofilií se FVIII váže na VWF v krevním oběhu pacienta.
Aktivovaný FVIII působí jako kofaktor aktivovaného faktoru IX, který urychluje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin potom přeměňuje fibrinogen na fibrin a umožňuje tvorbu krevní sraženiny. Hemofilie A je dědičná porucha koagulace krve vázaná na pohlaví, která vzniká v důsledku snížené hladiny FVIII a jejím následkem je těžké krvácení do kloubů, svalů nebo vnitřních orgánů, či už spontánním nebo v důsledku náhodného poranění nebo chirurgického výkonu. Substituční terapií se hladina FVIII zvyšuje, což dočasně upravuje deficienci faktoru a také snižuje tendenci ke krvácení.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Voncento u pacientů s hemofilií A od narození do mladších než 12 let (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
von Willebrandova choroba
Farmakokinetické vlastnosti přípravku Voncento byly hodnoceny u pacientů s VWD ve stavu, kdy nedocházelo ke krvácení.
|
VWF:RCo |
VWF:Ag |
VWF:CB |
FYIII:C | |||||||||||||
|
Parametr |
N |
Střední hodnot a |
SD |
Rozsah |
N |
Střední hodnot a |
SD |
Rozsah |
N |
Střední hodnot a |
SD |
Rozsah |
N |
Střední hodnot a |
SD |
Rozsah |
|
Přírůstková recovery (kg/ml) |
12 |
0,017 |
0,0 02 |
0,01 0,02 |
12 |
0,018 |
0,0 02 |
0,01 0,02 |
12 |
0,020 |
0,0 04 |
0,02 0,02 |
12 |
0,025 |
0,0 06 |
0,02 0,04 |
|
Poločas (h) |
8 |
13,7 |
9,2 |
6,1 35,1 |
12 |
18,3 |
4,0 |
11,4 27,0 |
12 |
16,0 |
4,6 |
9,4 25,1 |
10 |
28,0 |
15, 7 |
7,7 57,5 |
|
AUCo-72 (h*IU/ml) |
12 |
17,7 |
9,7 |
12,7 22,7 |
12 |
37,8 |
13, 3 |
22,6 64,7 |
12 |
24,8 |
8,8 |
14,8 41,1 |
11 |
34,0 |
16, 2 |
13,2 66,8 |
|
MRT (h) |
8 |
14,0 |
5,0 |
8,6 25,5 |
12 |
23,6 |
5,0 |
15,3 33,6 |
12 |
20,0 |
4,4 |
11,6 28,6 |
10 |
43,1 |
22, 1 |
15,6 85,1 |
|
Cmax (IU/ml) |
12 |
1,65 |
0,6 3 |
10,93 3,36 |
12 |
2,29 |
0,5 9 |
1,52 3,66 |
12 |
1,68 |
0,5 0 |
1,04 2,66 |
12 |
0,96 |
0,2 5 |
0,57 1,32 |
|
Tmax (h) |
12 |
0,25a |
0,25 1,03 |
12 |
0,25a |
0,25 1,00 |
12 |
0,25a |
0,25 1,00 |
12 |
1,00a |
0,25 30,00 | ||||
|
Cmin (IU/ml) |
12 |
0,01 |
0,0 1 |
0,00 0,03 |
12 |
0,10 |
0,0 5 |
0,02 0,17 |
12 |
0,05 |
0,0 2 |
0,02 0,09 |
12 |
0,21 |
0,1 8 |
0,03 0,59 |
|
Celková clearance (ml/(h*kg) |
12 |
6,09 |
1,6 6 |
3,06 9,32 |
12 |
3,57 |
0,6 9 |
2,61 4,78 |
12 |
3,53 |
0,8 9 |
2,32 4,77 |
11 |
1,33 |
0,5 9 |
0,62 2,47 |
|
Vss (ml/kg) |
8 |
74,8 |
35, 3 |
44,7 158,0 |
12 |
82,8 |
18, 6 |
64,5 128,4 |
12 |
68,6 |
15, 7 |
47,5 93,7 |
10 |
48,1 |
15, 3 |
24,8 72,9 |
a medián
AUC = plocha pod křivkou, Cmax = maximální plazmatická koncentrace; Cmin = minimální koncentrace v plazmě; IU = mezinárodní jednotka; MRT = střední rezidenční čas, N = počet subjektů, SD = směrodatná odchylka; tmax = doba maximální koncentrace; Vss = distribuční objem v ustáleném stavu, VWF:Ag = von Willebrandův faktor: antigen, VWF:CB = von Willebrandův faktor: vazba kolagenu, VWF:RCo = von Willebrandův faktor: ristocetin kofaktor, FVIII:C = faktor VIII: koagulant.
Relativní obsah HMW (vysoké molekulové hmotnosti) VWF multimerů v přípravku Voncento je v průměru 86 % ve srovnání s normální lidskou plasmou (NHP).
Hemofilie A
Farmakokinetické vlastnosti přípravku Voncento byly hodnocené u pacientů s hemofilií A ve stavu, kdy nedocházelo ke krvácení.
|
FVIII:C | ||||
|
Parametr |
N |
Střední hodnota |
SD |
Rozsah |
|
Přírůstková recovery (kg/ml) |
16 |
0,021 |
0,006 |
0,011-0,032 |
|
Poločas (h) |
16 |
13,40 |
2,53 |
8,78-18,51 |
|
AUC0-48 (h*IU/ml) |
16 |
13,79 |
3,79 |
7,04-21,79 |
|
MRT (h) |
16 |
16,96 |
3,68 |
11,29-26,31 |
|
Cmax (IU/ml) |
16 |
1,07 |
0,28 |
0,57-1,57 |
|
Tmax (h) |
16 |
0,81 |
0,94 |
0,42-4,03 |
|
Cmin (IU/ml) |
16 |
0,060 |
0,028 |
0,021-0,111 |
|
Celková clearance (ml/(h*kg) |
16 |
3,92 |
1,22 |
2,30-7,11 |
|
Vss (ml/kg) |
16 |
65,33 |
20,65 |
35,07-113,06 |
AUC = plocha pod křivkou, Cmax = maximální plazmatická koncentrace; Cmin = minimální koncentrace v plazmě; IU = mezinárodní jednotka; MRT = střední rezidenční čas, N = počet subjektů, SD = směrodatná odchylka; tmax = doba maximální koncentrace; Vss = distribuční objem v ustáleném stavu; FVIII:C = faktor VIII:koagulant.
Pediatrická populace
von Willebrandova choroba
Farmakokinetické údaje u pacientů s von Willebrandovou chorobou jsou velmi podobné těm, které pozorovány u dospělé populace.
PK z jedné dávky 80 IU VWF: RCo/kg tělesné hmotnosti byla hodnocena u pediatrických pacientů mladších než 12 let s těžkým VWD (viz tabulka níže). Následující infoze, maximální koncentrace vWF markerů (VWF: RCo, VWF:Ag a VWF:CB) a FVIII:C bylo dosaženo okamžitě s průměrným IR 0,013 - 0,016 (IU/ml)/(IU/kg) pro VWF markery a 0,017-0,021 (IU/ml)/(IU/kg) pro FVIII:C. Eliminace t1/2 VWF markerů byla mezi 9,7 a 12,9 hodin vzhledem k tomu, že FVIII:C měl delší t1/2 přibližně 18 h vzhledem k plató efektu, který může představovat čistý vliv na snížení hladiny exogenního FVIII, v kombinaci s rostoucí endogenní úrovní FVIII. PK parametry z opakovaného PK vyhodnocení byly podobné těm z původního PK. Expozice přípravkem Voncento a dispozice byly srovnatelné mezi subjekty <6 let věku a 6-12 let.
Základní nastavení počátečních PK parametrů VWF a FVIII:C u subjektů <6 (n = 9) a 6-12 let starých (n = 5).
|
VWF:RCo |
VWF:Ag |
VWF:CB |
FVIII:C | |||||||||||||
|
Parametr |
n |
Mean |
N |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
|
(SD) |
(SD) |
(SD) |
(SD) |
(SD) |
(SD) |
(SD) |
(SD) | |||||||||
|
< 6 let |
6-12 let |
< 6 let |
6-12 let |
< 6 let |
6-12 let |
< 6 let |
6-12 let | |||||||||
|
Přírůstková |
9 |
0,013 |
5 |
0,015 |
9 |
0,013 |
5 |
0,016 |
9 |
0,013 |
5 |
0,014 |
8 |
0,021 |
5 |
0,017 |
|
recovery |
(0,003 |
(0,003 |
(0,003) |
(0,003) |
(0,002) |
(0,002) |
(0,012) |
(0,007) | ||||||||
|
(kg/ml) |
) |
) | ||||||||||||||
|
Poločas (h) |
5 |
12,9 |
3 |
10,4 |
8 |
12,0 |
5 |
10,8 |
8 |
10,6 |
5 |
9,7 (2,0) |
4 |
19,4 |
3 |
17,0 |
|
(8,1) |
(1,7) |
(4,6) |
(1,4) |
(2,7) |
(1,2) |
(13,7) | ||||||||||
|
AUC0-72 |
9 |
8,9 |
5 |
10,1 |
9 |
19,5 |
5 |
22,1 |
9 |
16,2 |
5 |
15,8 |
8 |
16,4 |
3 |
17,7 |
|
(h*IU/ml) |
(4,6) |
(4,7) |
(7,1) |
(3,6) |
(3,9) |
(2,8) |
(7,7) |
(14,5) | ||||||||
|
MRT (h) |
5 |
17,3 |
3 |
11,5 |
8 |
15,4 |
5 |
13,5 |
8 |
13,0 |
5 |
12,0 |
4 |
26,0 |
3 |
22,3 |
|
(12,3) |
(2,6) |
(7,0) |
(1,7) |
(4,0) |
(2,4) |
(1,9) |
(19,7) | |||||||||
|
Cmax(IU/ml) |
9 |
1,03 |
5 |
1,18 |
9 |
1,60 |
5 |
1,83 |
9 |
1,49 |
5 |
1,41 |
8 |
0,73 |
5 |
0,60 |
|
(0,23) |
(0,26) |
(0,22) |
(0,43) |
(0,27) |
(0,25) |
(0,31) |
(0,27) | |||||||||
|
Tmax (h) f |
9 |
0,55 |
5 |
0,56 |
9 |
0,55 |
5 |
0,56 |
9 |
0,55 |
5 |
0,56 |
8 |
4,25 |
5 |
0,56 |
|
(0,04) |
(0,04) |
(0,04) |
(0,04) |
(0,04) |
(0,04) |
(7,84) |
(0,04) | |||||||||
|
Celková |
5 |
8,91 |
3 |
7,33 |
8 |
6,84 |
5 |
4,83 |
8 |
7,27 |
5 |
6,28 |
4 |
2,52 |
3 |
10,6 |
|
clearance (ml/(h*kg) |
(5,79) |
(1,24) |
(3,60) |
(0,25) |
(2,42) |
(0,74) |
(1,13) |
(13,5) | ||||||||
|
Vss (ml/kg) |
5 |
102,8 |
3 |
82,3 |
8 |
89,0 |
5 |
64,8 |
8 |
87,4 |
5 |
74,9 |
4 |
65,2 |
3 |
97,4 |
|
(32,2) |
(10,5) |
(24,4) |
(6,6) |
(14,4) |
(15,9) |
(27,9) |
(42,6) | |||||||||
AUC = plocha pod křivkou; Cmax = maximální plazmatická koncentrace; IU = mezinárodní jednotka; MRT =
střední rezidenční čas; N = počet subjektů; SD = směrodatná odchylka; tmax = doba k dosažení maximální koncentrace; Vss = distribuční objem v ustáleném stavu; VWF:Ag = von Willebrandův faktor: antigen; VWF:CB = von Willebrandův faktor: vazba kolagenu; VWF:RCo = von Willebrandův faktor: ristocetin kofaktor, FVIIkC = faktor VIII: koagulant.
Hemofilie A
Žádné farmakokinetické údaje nejsou k dispozici u pacientů s hemofilií A mladších než 12 let.
5.3 Predklinické údaje vztahující se k bezpečnosti
Voncento obsahuje léčivé látky FVIII a VWF, které jsou získané z lidské plazmy, a působí jako endogenní složky plazmy. Předklinické studie s opakovaným podáváním dávek (chronická toxicita, kancerogenita a mutagenita) se z důvodu vzniku protilátek, které se tvoří po podání heterologní lidské bílkoviny, nemohly provést na konvenčních živočišných modelech.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Chlorid vápenatý, lidský albumin, chlorid sodný, natrium-citrát, sacharóza, trometamol
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky, ředidly a rozpouštědly s výjimkou těch, které jsou uvedeny v bodě 6.1.
6.3 Doba použitelnosti
3 roky.
Po rekonstituci byla chemická a fyzikální stabilita po otevření před použitím prokázána na dobu 8 hodin při pokojové teplotě (do 25 °C). Z mikrobiologického hlediska má být přípravek použít okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2 až 8 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Chraňte před mrazem. Uchovávejte lahvičku v krabičce, aby byl přípravek chráněn před světlem. Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení Vnitřní obal
Prášek (250 IU/600 IU) v injekční lahvičce (sklo typu I) se zátkou (pryž) diskem (plast) a víčkem (hliník).
5 ml rozpouštědla v injekční lahvičce (sklo typu I) se zátkou (pryž) diskem (plast) a víčkem (hliník). Velikost balení
Balení s 250 IU/600 IU:
1 injekční lahvička s práškem 1 injekční lahvička s rozpouštědlem
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Všeobecné pokyny
Roztok má být čirý nebo slabě opalizující. Po filtraci/natáhnutí (viz dále) se rekonstituovaný přípravek před aplikací vizuálně zkontroluje, zda neobsahuje částice nebo nezměnil zbarvení. Nepoužívejte viditelně zakalené roztoky nebo roztoky, které po filtraci ještě obsahují vločky nebo částice. Rekonstituce a natáhnutí roztoku se musí provádět za aseptických podmínek.
Rekonstituce
Ohřejte rozpouštědlo na pokojovou teplotu. Před otevřením balení Mix2Vial odstraňte ochranné víčko z injekční lahvičky s práškem a rozpouštědlem, zátky očistěte antiseptickým roztokem a nechte oschnout.

1. Otevřete balení Mix2Vial sloupnutím víčka. Nevytahujte Mix2Vial z blistru!
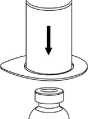
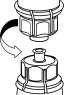
2. Postavte injekční lahvičku s rozpouštědlem na rovný a čistý povrch a pevně ji držte. Uchopte Mix2Vial společně s blistrem a zatlačte hrot konce modrého adaptéru přímo dolů přes zátku injekční lahvičky s rozpouštědlem.
1
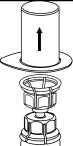
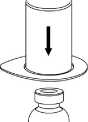
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle nahoru. Přesvědčte se, že jste vytáhli jen blistrový obal, nikoli soupravu Mix2Vial.
4. Postavte injekční lahvičku s přípravkem na rovný a pevný povrch. Otočte injekční lahvičku s rozpouštědlem a připojeným setem Mix2Vial dnem nahoru a zatlačte hrot průhledného konce adaptéru rovně dolů přes zátku injekční lahvičky s přípravkem. Rozpouštědlo automaticky přeteče do injekční lahvičky s přípravkem.



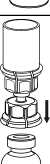
5. Uchopte jednou rukou část setu Mix2Vial uchycenou na injekční lahvičce s přípravkem. Druhou rukou uchopte část setu uchycenou na injekční lahvičce s rozpouštědlem a odšroubováním proti směru hodinových ručiček rozdělte opatrně set na dvě části, aby nedošlo k nadměrnému nahromadění pěny při rozpouštění přípravku.
Injekční lahvičku s rozpouštědlem a připojeným modrým adaptérem setu Mix2Vial odložte.
6. Injekční lahvičku s připojeným průhledným adaptérem jemně otáčejte, dokud se přípravek úplně nerozpustí. Injekční lahvičkou netřepejte.
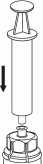
7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Injekční lahvičku s přípravkem nechejte postavenou dnem dolů a připojte na ni injekční stříkačku s nástavcem Mix2Vial Luer Lock šroubováním ve směru hodinových ručiček. Vstříkněte vzduch do injekční lahvičky s přípravkem.
Natáhnutí a aplikace
|
k |
8. Otočte systém dnem vzhůru a současně držte píst injekční stříkačky stlačený. Natáhněte roztok do injekční stříkačky pomalým vytahováním pístu. | ||
|
< |
s |
5 9 |
9. Po natáhnutí roztoku do injekční stříkačky uchopte pevně válec stříkačky (píst směřuje stále dolů) a odpojte průhledný adaptér setu Mix2Vial od injekční stříkačky odšroubováním proti směru hodinových ručiček. |
K injekci přípravku Voncento je nutné používat pouze dodávanou aplikační soupravu, protože v důsledku adsorpce lidského koagulačního faktoru VIII na vnitřní povrch některého injekčního/infuzního zařízení by mohlo dojít k selhání léčby.
V případě, že je požadováno velké množství přípravku Voncento, je možné spojit obsah několika lahviček přípravku Voncento pomocí komerčně dostupné infuzní sady (např. injekční pumpy pro intravenózní aplikaci léků). Nicméně v těchto případech se již rekonstituovaný roztok Voncento nesmí dále ředit.
Roztok podávejte pomalu intravenózně (viz bod 4.2). Ubezpečte se, že do naplněné injekční stříkačky nepronikla krev.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH Emil-von-Behring-Strasse 76 35041 Marburg Německo
8. REGISTRAČNÍ ČÍSLO
EU/1/13/857/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 12. srpna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Voncento 500 IU FVIII/1200 IU VWF (10 ml rozpouštědlo) prášek a rozpouštědlo pro injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně:
- Factor VIII coagulationis humanus 500 IU4 (lidský koagulační faktor VIII5 (FVIII))
- Factor von Willebrand humanus 1200 IU6 (lidský von Willebrandův faktor5 (VWF))
Po rekonstituci v 10 ml roztok obsahuje 50 IU/ml lidského koagulačního faktoru VIII a 120 IU/ml lidského von Willebrandova faktoru.
Pomocná látka se známým účinkem:
Sodík přibližně 128,2 mmol/l (2,95 mg/ml). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok.
Bílý prášek a čiré, bezbarvé rozpouštědlo pro injekční/infuzní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
von Willebrandova choroba (VWD)
Profylaxe a léčba hemoragie nebo krvácení během chirurgických zákroků u pacientů trpících VWD v případech, kdy je léčba samotným desmopressinem (DDAVP) neúčinná nebo kontraindikovaná.
Hemofilie A (vrozený deficit FVIII)
Profylaxe a léčba krvácení u pacientů trpících hemofilií A.
4.2 Dávkování a způsob podání
Léčba VWD a hemofilie A se má provádět pod dohledem lékaře, který má zkušenosti s léčbou poruch hemostázy.
Při rozhodování o využití přípravku u domácí léčby při krvácení u pacientů s VWD a s hemofilií A určí konkrétního pacienta ošetřující lékař, který zajistí odpovídající vyškolení v podávání přípravku a toto podávání kontroluje v pravidelných intervalech.
Poměr mezi FVIII:C a VWF:RCo v injekční lahvičce je přibližně 1:2,4.
Dávkování von Willebrandova choroba
Je důležité výpočem stanovit dávku na základě specifikovaného počtu IU VWF:RCo.
1 IU/kg VWF:RCo obvykle zvýší hladinu VWF:RCo v oběhu o 0,02 IU/ml (2 %).
Je třeba dosáhnout hladiny VWF:RCo > 0,6 IU/ml (60 %) a hladina FVIII:C > 0,4 IU/ml (40 %).
Požadovaná léčba
K dosažení hemostázy se obyčejně doporučuje 40 - 80 IU/kg von Willebrandova faktoru (VWF:RCo), což odpovídá 20 - 40 IU FVIII:C/kg tělesné hmotnosti (BW).
Úvodní dávka 80 IU/kg VWF:RCo může být potřebná zejména u pacientů s VWD typu 3, kde k udržení adekvátních hladin mohou být nutné vyšší dávky než v případě jiných typů VWD.
Prevence krvácení během chirurgického zákroku
K prevenci masivního krvácení během nebo po chirurgickém zákroku se má přípravek začít podávat 1-2 hodiny před chirurgickým zákrokem.
Příslušná dávka se má podávat opakovaně každých 12 - 24 hodin. Dávka a trvání léčby závisí na klinickém stavu pacienta, na typu a závažnosti krvácení a na hladinách VWF:RCo a FVIII:C.
Při používání přípravku s VWF obsahujícího FVIII by si měl být ošetřující lékař vědomý, že pokračování v léčbě může zapříčinit nadměrný vzestup hladin FVIII:C. Po 24 - 48 hodinách léčby by se mělo zvážit snížení dávek a/nebo prodloužení intervalu mezi dávkami nebo použití přípravku s VWF obsahující nízkou hladinu FVIII, aby se zabránilo nadměrnému vzestupu hladiny FVIII:C (viz bod 5.2).
Profylaktická léčba
Při dlouhodobé profylaxi u pacientů s VWD má být zvažována dávka 25-40 IU VWF:RCo/kg tělesné hmotnosti při frekvenci 1-3 krát týdně. U pacientů s gastrointestinálním krvácení nebo menoragií mohou být nezbytné kratší intervaly nebo vyšší dávky. Dávka a trvání léčby bude záviset na klinickém stavu pacienta, jakož i plazmatických hladinách VWF:RCo a FVIII:C.
Pediatrická populace s VWD Léčba krvácení
U pediatrických pacientů se obvykle doporučuje pro léčbu krvácení 40 až 80 IU/kg von Willebrandova faktoru (VWF:RCo), odpovídající 20 až 40 IU FVIII:C/kg tělesné hmotnosti (BW).
Profylaktická léčba
Pacienti ve věku 12 až 18 roků: Dávkování je založeno na stejných pokynech jako pro dospělé.
Pacienti ve věku <12 let: Na základě výsledků z klinické studie s pediatrickými pacienty do 12 let, ve které bylo prokázáno, že mají nižší expozici VWF, by mělo být uvažováno profylaktické rozmezí dávek 40 - 80 IU VWF:RCo/kg tělesné hmotnosti 1 až 3x týdně. (viz bod 5.2).
Dávka a trvání léčby bude záviset na klinickém stavu pacienta, jakož i plazmatických hladinách VWF:RCo a FVffl:C.
Hemofilie A
Je důležité výpočem stanovit dávku na základě specifikovaného počtu IU FVIII:C.
Dávka a délka substituční terapie závisí na závažnosti deficitu FVIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek FVIII se vyjadřuje v mezinárodních jednotkách (International Units - IU), které jsou stanovené oproti současnému standardu WHO pro léčivé přípravky obsahující FVIII. Aktivita FVIII v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v IU (vzhledem k mezinárodnímu standardu pro FVIII v plazmě).
1 IU aktivity FVIII odpovídá množství FVIII v 1 ml normální lidské plazmy.
Požadovaná léčba
Výpočet požadované dávky FVIII vychází z empirického předpokladu, že podáníl IU FVIII na kg tělesné hmotnosti zvýší plazmatickou aktivitu FVIII v plazmě o přibližně 2 % normální aktivity (in vivo recovery 2 IU/dl). Požadovaná dávka se stanoví podle následujícího vzorce:
Požadovaý počet jednotek = tělesná hmotnost [kg] x požadovaný vzestup FVIII [% nebo IU/dl] x 0,5
Množství, které se má podat a frekvence podávání mají vždy směřovat ke klinické účinnosti v individuálních případech.
V případě následujících hemoragických příhod nemá aktivita FVIII během daného období klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může posloužit jako návod pro stanovení dávky v případech krvácení a při chirurgických výkonech:
|
Stupeň krvácení/typ chirurgického výkonu |
Požadovaná hladina FVIII (% nebo IU/dl) |
Frekvence dávkování (hodiny)/délka trvání léčby (dny) |
|
Krvácení | ||
|
Časný hemartros, krvácení do svalstva nebo do ústní dutiny |
O 1 o <N |
Infuzi opakovat každých 12 - 24 hodin po dobu nejméně jednoho dne, dokud se krvácení nezastaví, což se projeví ústupem bolesti nebo zahojením. |
|
Rozsáhlejší hemartros, krvácení do svalstva nebo hematom |
O kO 1 o co |
Infuzi opakovat každých 12 - 24 hodin po dobu 3 - 4 dní nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
o o 1 o kO |
Infuzi opakovat každých 8 - 24 hodin, dokud nepomine ohrožení života. |
|
Chirurgické výkony | ||
|
Menší chirurgický výkon včetně extrakce zubů |
o kO 1 o co |
Infuzi opakovat každých 24 hodin po dobu nejméně jednoho dne až do zahojení. |
|
Velké chirurgické výkony |
80 -100 (před a po operaci) |
Infuzi opakovat každých 8 - 24 hodin, dokud nedojde k uspokojivému zahojení rány, potom pokračovat v léčbě nejméně dalších 7 dní a udržovat aktivitu FVIII mezi 30 % - 60 % (IU/dl). |
Během léčby se doporučuje náležitá kontrola hladin FVIII kvůli stanovení dávky, která má být podávaná, a frekvenci opakovaných infuzí. V případě velkých chirurgických zákroků je nevyhnutelný přesný monitoring substituční terapie prostřednictvím koagulační analýzy (aktivity plazmatického FVIII). U jednotlivých pacientů může být odpověď na léčbu faktorem VIII rozdílná, v závislosti na odlišných hladinách in vivo recovery a různém biologickém poločasu.
Profylaktická léčba
V případě dlouhodobé profylaxe u pacientů s těžkou formou hemofilie A se obyčejně doporučuje dávka 20 až 40 IU FVIII na kilogram tělesné hmotnosti v intervalech 2 až 3 dní. V některých případech, zejména u mladších pacientů, může být nutný kratší interval mezi dávkami nebo vyšší dávky.
Dříve neléčení pacienti
Bezpečnost a účinnost přípravku Voncento u dříve neléčených pacientů nebyla dosud stanovena.
Pediatrická populace s hemofilií A
Dávkování u hemofilie A u dospívajících ve věku od 12 do 18 let je založeno na tělesné hmotnosti, a proto pro něj platí stejná doporučení jako u dospělých. V některých případech je nezbytný kratší interval dávek nebo vyšší dávky. Frekvence podávání by měla být v každém jednotlivém případě vždy zaměřená na klinickou účinnost.
Bezpečnost a účinnost přípravku Voncento u dětí <12 let s hemofilií A nebyla stanovena. Žádné údaje nejsou k dispozici.
Starší osoby
U starších osob není potřebná žádná úprava dávek.
Způsob podání K intravenóznímu podání.
Přípravek rekonstituujte podle postupu v bodě 6.6. Rekonstituovaný přípravek se podává pomalu intravenózně injekcí/infuzí rychlostí příjemnou pro pacienta.
Rychlost podávání injekce nebo infuze nemá přesáhnout 6 ml za minutu. Pacienta je nutno sledovat, zda nemá bezprostřední reakci. Jestliže se vyskytne jakákoli reakce v souvislosti s podávaním přípravku Voncento, musí se podle klinického stavu pacienta rychlost podávání přípravku snížit nebo podávání zastavit (viz rovněž bod 4.4).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Jsou možné reakce z přecitlivělosti alergického typu. Pokud se objeví příznaky hypersenzitivity, pacienti by měli být poučeni, aby okamžitě přerušili používání léčivého přípravku a kontaktovali svého lékaře. Pacienti mají být informováni o prvních příznacích reakcí z přecitlivělosti jako je kopřivka, generalizovaná vyrážka, pocit tísně na hrudi, sípání, hypotenze a anafylaxe. V případě šoku je nutné dodržovat všeobecné lékařské postupy pro léčbu šoku.
Virová bezpečnost
Ke standardním opatřením k prevenci infekcí, které jsou následkem používání přípravků, vyrobených z lidské krve nebo plazmy, patří výběr dárců, kontrola jednotlivých odběrů a plazmatických poolů, zaměřená na specifické markery infekce a provedení efektivních výrobních kroků k inaktivaci/odstranění virů. Navzdory těmto opatřením není možné úplně vyloučit možnost přenosu infekčních agens, pokud podávané přípravky pocházejí z lidské krve nebo plazmy. Toto platí i pro neznámé nebo nové viry a další patogeny.
Tato opatření se považují za účinná pro obalené viry, jako jsou virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV) a neobalený virus hepatitidy A (HAV).
Provedená opatření mohou mít jen omezenou účinnost proti neobaleným virům, jako je parvovirus B19.
Infekce způsobená parvovirem B19 může být závažná u těhotných žen (infekce plodu) a jedinců s imunodeficiencí nebo zvýšenou erytropoézou (např. hemolytická anémie).
U pacientů, kteří pravidelně/opakovaně dostávají přípravky s faktorem Vin/VWF, vyrobené z lidské plazmy, je třeba zvážit možnosti vhodného očkování (hepatitida A a B).
Důrazně se doporučuje, aby byl při každém podání přípravku Voncento pacientovi zaznamenaný název a číslo šarže přípravku, aby se udržovaly záznamy o propojení mezi pacientem a šarží přípravku.
von Willebrandova choroba
Existuje riziko vzniku trombóz zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Rizikoví pacienti musí být proto sledováni kvůli zachycení prvních příznaků trombózy. Měla by být provedena profylaktická opatření proti venózním trombózám v souladu s aktuálními doporučeními.
Při léčbě přípravkem s VWF obsahujícím FVIII má mít ošetřující lékař na paměti, že pokračování v léčbě může zapříčinit nadměrný vzestup FVIII:C. U pacientů, kteří dostávají přípravek VWF obsahující FVIII, musí být sledovány hladiny FVIII:C v plazmě kvůli tomu, aby nebyly hladiny FVIII:C v plazmě dlouhodobě vysoké, což by mohlo zvýšit riziko výskytu trombóz, a současně je potřebné zvážit zavedení protitrombotických opatření (viz též bod 5.2).
U pacientů s VWD, zejména typu 3, se mohou vytvořit neutralizační protilátky (inhibitory) proti VWF. Jestliže se nedaří dosáhnout očekávanou hladinu aktivity VWF:RCo v plazmě nebo jestliže krvácení není kontrolované vhodnou dávkou, je potřebné uskutečnit odpovídající analýzu kvůli stanovení přítomnosti inhibitoru VWF. U pacientů s vysokými hladinami inhibitoru může být terapie nejen neúčinná, ale může vést i k anafylaktoidní reakci a je třeba uvažovat o jiných možnostech léčby.
Hemofilie A
Inhibitory
U pacientů s hemofilií A je známou komplikací tvorba neutralizačních protilátek (inhibitorů) proti FVIII. Tyto inhibitory jsou obyčejně IgG imunoglobuliny proti prokoagulační aktivitě FVIII, které jsou kvantifikovány v jednotkách Bethesda (BU) na ml plazmy, pomocí modifikovaného testu. Riziko vzniku inhibitorů koreluje s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dní expozice. Zřídka se mohou inhibitory vytvořit po prvních 100 dnech expozice.
Případy opakovaného výskytu inhibitoru (nízké titry) byly pozorovány po přechodu z jednoho přípravku, obsahujícího faktor VIII, na jiný, u pacientů, kteří už byli léčení předtím po dobu více než 100 dní a u kterých byla už předtím zaznamenaná tvorba inhibitoru. Proto se doporučuje důkladné monitorování všech pacientů, pokud se týká výskytu inhibitoru, po jakémkoli přechodu na jiný přípravek.
Obecně mají být všichni pacienti, léčení lidským koagulačním faktorem FVIII, pozorně monitorovaní kvůli tvorbě inhibitorů a to prostřednictvím klinického sledování a laboratorních testů.
Pokud nejsou dosaženy očekávané hladiny faktoru VIII v plazmě, nebo pokud není krvácení zvládnuto odpovídající dávkou, mělo být provedeno testování na přítomnost inhibitoru FVIII. U pacientů s vysokými hladinami inhibitoru může být léčba faktorem VIII neúčinná a měly by být zváženy jiné možnosti léčby. Léčba těchto pacientů by měla být vedena lékaři se zkušenostmi s péčí o pacienty s hemofilií A a s inhibitory faktoru VIII.
Komplikace související s katetrem
Jestliže je třeba použít zařízení pro centrální žilní přístup (CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katetru.
Obsah sodíku
Tento přípravek obsahuje až 29,50 mg (1,28 mmol) sodíku v jedné injekční lahvičce. Toto množství se musí zohlednit u pacientů s dietou s kontrolovaným příjmem sodíku.
Pediatrická populace
Uvedená upozornění a opatření se vztahují jak na dospělé tak na pediatrické pacienty.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly studovány žádné interakce VWF a FVIII s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S přípravkem Voncento se neprováděly reprodukční studie u zvířat. von Willebrandova choroba
Zkušenosti s léčbou těhotných nebo kojících žen nejsou k dispozici. Přípravek Voncento by měl být podáván těhotným nebo kojícím ženám s deficitem VWF pouze tehdy, pokud je jasně indikován, přičemž je nutno brát v úvahu, že porod představuje zvýšené riziko krvácivých příhod u těchto pacientek.
Hemofilie A
Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou dostupné zkušenosti, týkající se léčby během těhotenství a kojení. Proto by přípravek Voncento měl být používán během těhotenství a kojení pouze tehdy, pokud je jasně indikován.
Fertilita
Nejsou dostupné žádné údaje, týkající se fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje Voncento nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Následující nežádoucí účinky se mohou vyskytnout u dospělých a dospívajících během léčby přípravkem Voncento: hypersenzitivita nebo alergické reakce, tromboembolické příhody, horečka, bolesti hlavy, poruchy chuti a abnormální hladiny testů jaterních funkcí. Navíc se mohou u pacientů vyvinout inhibitory FVIII a VWF.
Seznam nežádoucích účinků v tabulce
Tabulka uvedená níže je podle klasifikace orgánových systémů MedDRA.
Frekvence byly hodnoceny podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až <1/10), méně časté (> 1/1 000 až <1/100), vzácné (> 1 / 10000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinky |
Frekvence |
|
MedDRA |
|
Poruchy krve a lymfatického systému |
inhibice FVIII inhibice VWF |
velmi vzácné velmi vzácné |
|
Poruchy imunitního systému |
hypersenzitivita (zahrnující tachykardii, bolest na hrudi, nepříjemný pocit na hrudi a bolest v zádech) |
velmi vzácné |
|
Poruchy nervového systému |
porucha chuti |
velmi vzácné |
|
Cévní poruchy |
tromboembolická příhoda |
velmi vzácné |
|
Celkové poruchy a reakce |
velmi vzácné | |
|
v místě aplikace |
velmi vzácné | |
|
Vyšetření |
abnormální testy jaterních funkcí |
velmi vzácné |
Popis vybraných nežádoucích účinků
Hypersenzitivita (alergické reakce): přecitlivělost nebo alergické reakce (které mohou zahrnovat angioedém, pálení a štípání v místě infuze, zimnici, zarudnutí, generalizovanou kopřivku, bolest hlavy, vyrážku, hypotenzi, letargie, nauzeu, neklid, tachykardii, napětí na hrudi (včetně bolesti na hrudi a nepříjemného pocitu na hrudi), bolest zad, mravenčení, zvracení, sípání), byly pozorovány a mohou se v některých případech rozvinout do závažné anafylaxe (včetně šoku).
Inhibice FVIII: U pacientů s hemofilií A se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se jako nedostatečná klinická odpověď. V takových případech se doporučuje vyhledat specializované pracoviště k léčbě hemofilie. Nejsou žádné zkušenosti z klinických studií s podáváním přípravku Voncento u dříve neléčených pacientů (PUPs). Proto nejsou v současné době k dispozici žádné validní údaje o výskytu klinicky významných specifických inhibitorů.
Inhibice VWF: U pacientů s VWD, zejména typu 3, se mohou vytvořit neutralizační protilátky (inhibitory) proti VWF. Jestliže takové inhibitory vzniknou, tento stav se projeví jako nedostatečná klinická odpověď.Tyto protilátky jsou precipitační a může dojít současně k anafylaktické reakci. Proto pacienti, u kterých se vyskytla anafylaktická reakce, by měli být vyšetřeni na přítomnost inhibitorů.
Ve všech těchto případech se doporučuje vyhledat specializované pracoviště k léčbě hemofilie.
Tromboembolické příhody: U pacientů s VWD existuje riziko vzniku tromboembolických příhod, zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory.
Pacientům, kterým se podávají přípravky s VWF obsahující FVIII se může výrazně zvýšit hladina FVIII:C v plazmě, čímž se zvyšuje riziko tromboembolických příhod (viz též bod 4.4).
Informace o bezpečnosti s ohledem na přenosná agens viz též bod 4.4.
Pediatrická populace
Očekává se, že frekvence, typ a závažnost nežádoucích účinků u dětí budou stejné jako u dospělých. Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pět případů předávkování bylo hlášeno z klinických studií. Žádné nežádoucí účinky nebyly spojené s těmito hlášeními.
Riziko trombózy nelze v případě vysokého předávkování vyloučit, zejména u pacientů s VWD.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragikum: Koagulační faktory, von Willebrandův faktor
a koagulační faktor VIII v kombinaci.
Kód ATC: B02BD06
von Willebrandova choroba
Exogenně podávaný VWF vyrobený z lidské plazmy působí stejně jako endogenní VWF.
Podávání VWF umožňuje korekci hemostatických abnormalit, které se projevují u pacientů
s deficitem VWF (VWD) na dvou úrovních:
- VWF obnovuje adhezi krevních destiček na cévní subendotel v místě poranění cévy (protože váže cévní subendotel a membránu destiček) a zprostředkuje primární hemostázi, což se projeví zkrácením krvácení. Tento účinek nastupuje okamžitě a je známé, že ve velké míře závisí na úrovni polymerace proteinu.
- VWF způsobuje opožděnou korekci souvisejícího deficitu FVIII. Při intravenózním podání se VWF váže na endogenní FVIII (který je pacientem normálně produkován) a prostřednictvím stabilizace tohoto faktoru zabraňuje jeho rychlé degradaci.
Z tohoto důvodu podávání čistého VWF (přípravek s VWF s nízkou hladinou FVIII) sekundárně obnovuje hladinu FVIII:C na normální úroveň po první infuzi s mírným opožděním.
- Podávání přípravku s VWF obsahujícího FVIII:C obnovuje normální hladinu FVIII:C okamžitě po první infuzi.
Hemofilie A
Exogenně podávaný FVIII vyrobený z lidské plazmy se chová stejně jako endogenní FVIII.
Komplex FVIII/VWF se skládá ze dvou molekul (FVIII a VWF) s odlišnými fyziologickými funkcemi.
Po podání pacientovi s hemofilií se FVIII váže na VWF v krevním oběhu pacienta.
Aktivovaný FVIII působí jako kofaktor aktivovaného faktoru IX, který urychluje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X přeměňuje protrombin na trombin. Trombin potom přeměňuje fibrinogen na fibrin a umožňuje tvorbu krevní sraženiny. Hemofilie A je dědičná porucha koagulace krve vázaná na pohlaví, která vzniká v důsledku snížené hladiny FVIII a jejím následkem je těžké krvácení do kloubů, svalů nebo vnitřních orgánů, či už spontánním nebo v důsledku náhodného poranění nebo chirurgického výkonu. Substituční terapií se hladina FVIII zvyšuje, což dočasně upravuje deficienci faktoru a také snižuje tendenci ke krvácení.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Voncento u pacientů s hemofilií A od narození do mladších než 12 let (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
von Willebrandova choroba
Farmakokinetické vlastnosti přípravku Voncento byly hodnoceny u pacientů s VWD ve stavu, kdy nedocházelo ke krvácení.
|
VWF:RCo |
VWF:Ag |
VWF:CB |
FVIII:C | |||||||||||||
|
Parametr |
N |
Středn í hodno ta |
SD |
Rozsah |
N |
Střední hodnot a |
SD |
Rozsah |
N |
Střední hodnot a |
SD |
Rozsah |
N |
Střední hodnot a |
SD |
Rozsah |
|
Přírůstková recovery (kg/ml) |
12 |
0,017 |
0,0 02 |
0,01 0,02 |
12 |
0,018 |
0,0 02 |
0,01 0,02 |
12 |
0,020 |
0,0 04 |
0,02 0,02 |
12 |
0,025 |
0,0 06 |
0,02 0,04 |
|
Poločas (h) |
8 |
13,7 |
9,2 |
6,1 35,1 |
12 |
18,3 |
4,0 |
11,4 27,0 |
12 |
16,0 |
4,6 |
9,4 25,1 |
10 |
28,0 |
15, 7 |
7,7 57,5 |
|
AUCo-72 (h*IU/ml) |
12 |
17,7 |
9,7 |
12,7 22,7 |
12 |
37,8 |
13, 3 |
22,6 64,7 |
12 |
24,8 |
8,8 |
14,8 41,1 |
11 |
34,0 |
16, 2 |
13,2 66,8 |
|
MRT (h) |
8 |
14,0 |
5,0 |
8,6 25,5 |
12 |
23,6 |
5,0 |
15,3 33,6 |
12 |
20,0 |
4,4 |
11,6 28,6 |
10 |
43,1 |
22, 1 |
15,6 85,1 |
|
C '--max (IU/ml) |
12 |
1,65 |
0,6 3 |
10,93 3,36 |
12 |
2,29 |
0,5 9 |
1,52 3,66 |
12 |
1,68 |
0,5 0 |
1,04 2,66 |
12 |
0,96 |
0,2 5 |
0,57 1,32 |
|
Tmax (h) |
12 |
0,25a |
0,25 1,03 |
12 |
0,25a |
0,25 1,00 |
12 |
0,25a |
0,25 1,00 |
12 |
1,00a |
0,25 30,00 | ||||
|
C5 O C7§. 3 |
12 |
0,01 |
0,0 1 |
0,00 0,03 |
12 |
0,10 |
0,0 5 |
0,02 0,17 |
12 |
0,05 |
0,0 2 |
0,02 0,09 |
12 |
0,21 |
0,1 8 |
0,03 0,59 |
|
Celková clearance (ml/(h*kg) |
12 |
6,09 |
1,6 6 |
3,06 9,32 |
12 |
3,57 |
0,6 9 |
2,61 4,78 |
12 |
3,53 |
0,8 9 |
2,32 4,77 |
11 |
1,33 |
0,5 9 |
0,62 2,47 |
|
Vss (ml/kg) |
8 |
74,8 |
35, 3 |
44,7 158,0 |
12 |
82,8 |
18, 6 |
64,5 128,4 |
12 |
68,6 |
15, 7 |
47,5 93,7 |
10 |
48,1 |
15, 3 |
24,8 72,9 |
a medián
AUC = plocha pod křivkou, Cmax = maximální plazmatická koncentrace; Cmin = minimální koncentrace v plazmě; IU = mezinárodní jednotka; MRT = střední rezidenční čas, N = počet subjektů, SD = směrodatná odchylka; tmax = dobak maximální koncentrace; Vss = distribuční objem v ustáleném stavu, VWF:Ag = von Willebrandův faktor: antigen, VWF:CB = von Willebrandův faktor: vazba kolagenu, VWF:RCo = von Willebrandův faktor: ristocetin kofaktor, FVIII:C = faktor VIII: koagulant.
Relativní obsah HMW (vysoké molekulové hmotnosti) VWF multimerů v přípravku Voncento je v průměru 86 % ve srovnání s normální lidskou plazmou (NHP).
Hemofilie A
Farmakokinetické vlastnosti přípravku Voncento byly hodnocené u pacientů s hemofilií A ve stavu, kdy nedocházelo ke krvácení.
|
FVIII:C | ||||
|
Parametr |
N |
Střední hodnota |
SD |
Rozsah |
|
Přírůstková recovery (kg/ml) |
16 |
0,021 |
0,006 |
0,011-0,032 |
|
Poločas (h) |
16 |
13,40 |
2,53 |
8,78-18,51 |
|
AUC0-48 (h*IU/ml) |
16 |
13,79 |
3,79 |
7,04-21,79 |
|
MRT (h) |
16 |
16,96 |
3,68 |
11,29-26,31 |
|
Cmax (IU/ml) |
16 |
1,07 |
0,28 |
0,57-1,57 |
|
Tmax (h) |
16 |
0,81 |
0,94 |
0,42-4,03 |
|
Cmin (IU/ml) |
16 |
0,060 |
0,028 |
0,021-0,111 |
|
Celková clearance (ml/(h*kg) |
16 |
3,92 |
1,22 |
2,30-7,11 |
|
Vss (ml/kg) |
16 |
65,33 |
20,65 |
35,07-113,06 |
AUC = plocha pod křivkou, Cmax = maximální plazmatická koncentrace; Cmin = minimální koncentrace v plazmě; IU = mezinárodní jednotka; MRT = střední rezidenční čas, N = počet subjektů, SD = směrodatná odchylka; tmax = doba maximální koncentrace; Vss = distribuční objem v ustáleném stavu; FVIII:C = faktor VIII:koagulant.
Pediatrická populace
von Willebrandova choroba
Farmakokinetické údaje u pacientů s von Willebrandovou chorobou jsou velmi podobné těm, které pozorovány u dospělé populace.
PK z jedné dávky 80 IU VWF: RCo/kg tělesné hmotnosti byla hodnocena u pediatrických pacientů mladších než 12 let s těžkým VWD (viz tabulka níže). Následující infoze, maximální koncentrace vWF markerů (VWF: RCo, VWF:Ag a VWF:CB) a FVIII:C bylo dosaženo okamžitě s průměrným IR 0,013 - 0,016 (IU/ml)/(IU/kg) pro VWF markery a 0,017-0,021 (IU/ml)/(IU/kg) pro FVIII:C. Eliminace t1/2 VWF markerů byla mezi 9,7 a 12,9 hodin vzhledem k tomu, že FVIII:C měl delší t1/2 přibližně 18 h vzhledem k plató efektu, který může představovat čistý vliv na snížení hladiny exogenního FVIII, v kombinaci s rostoucí endogenní úrovní FVIII. PK parametry z opakovaného PK vyhodnocení byly podobné těm z původního PK. Expozice přípravkem Voncento a dispozice byly srovnatelné mezi subjekty <6 let věku a 6-12 let.
Základní nastavení počátečních PK parametrů VWF a FVIII:C u subjektů <6 (n = 9) a 6-12 let starých (n = 5).
|
VWF:RCo |
VWF:Ag |
VWF:CB |
FVIII:C | |||||||||||||
|
Parametr |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
n |
Mean |
|
(SD) |
(SD) |
(SD) |
(SD) |
(SD) |
(SD) |
(SD) |
(SD) | |||||||||
|
< 6 let |
6-12 let |
< 6 let |
6-12 let |
< 6 let |
6-12 let |
< 6 let |
6-12 let | |||||||||
|
Přírůstková |
9 |
0,013 |
5 |
0,015 |
9 |
0,013 |
5 |
0,016 |
9 |
0,013 |
5 |
0,014 |
8 |
0,021 |
5 |
0,017 |
|
recovery |
(0,003 |
(0,003 |
(0,003) |
(0,003) |
(0,002) |
(0,002) |
(0,012) |
(0,007) | ||||||||
|
(kg/ml) |
) |
) | ||||||||||||||
|
Poločas (h) |
5 |
12,9 |
3 |
10,4 |
8 |
12,0 |
5 |
10,8 |
8 |
10,6 |
5 |
9,7 (2,0) |
4 |
19,4 |
3 |
17,0 |
|
(8,1) |
(1,7) |
(4,6) |
(1,4) |
(2,7) |
(1,2) |
(13,7) | ||||||||||
|
AUC0-72 |
9 |
8,9 |
5 |
10,1 |
9 |
19,5 |
5 |
22,1 |
9 |
16,2 |
5 |
15,8 |
8 |
16,4 |
3 |
17,7 |
|
(h*IU/ml) |
(4,6) |
(4,7) |
(7,1) |
(3,6) |
(3,9) |
(2,8) |
(7,7) |
(14,5) | ||||||||
|
MRT (h) |
5 |
17,3 |
3 |
11,5 |
8 |
15,4 |
5 |
13,5 |
8 |
13,0 |
5 |
12,0 |
4 |
26,0 |
3 |
22,3 |
|
(12,3) |
(2,6) |
(7,0) |
(1,7) |
(4,0) |
(2,4) |
(1,9) |
(19,7) | |||||||||
|
Cmax(IU/ml) |
9 |
1,03 |
5 |
1,18 |
9 |
1,60 |
5 |
1,83 |
9 |
1,49 |
5 |
1,41 |
8 |
0,73 |
5 |
0,60 |
|
(0,23) |
(0,26) |
(0,22) |
(0,43) |
(0,27) |
(0,25) |
(0,31) |
(0,27) | |||||||||
|
Tmax (h) f |
9 |
0,55 |
5 |
0,56 |
9 |
0,55 |
5 |
0,56 |
9 |
0,55 |
5 |
0,56 |
8 |
4,25 |
5 |
0,56 |
|
(0,04) |
(0,04) |
(0,04) |
(0,04) |
(0,04) |
(0,04) |
(7,84) |
(0,04) | |||||||||
|
Celková |
5 |
8,91 |
3 |
7,33 |
8 |
6,84 |
5 |
4,83 |
8 |
7,27 |
5 |
6,28 |
4 |
2,52 |
3 |
10,6 |
|
clearance (ml/(h*kg) |
(5,79) |
(1,24) |
(3,60) |
(0,25) |
(2,42) |
(0,74) |
(1,13) |
(13,5) | ||||||||
|
Vss (ml/kg) |
5 |
102,8 |
3 |
82,3 |
8 |
89,0 |
5 |
64,8 |
8 |
87,4 |
5 |
74,9 |
4 |
65,2 |
3 |
97,4 |
|
(32,2) |
(10,5) |
(24,4) |
(6,6) |
(14,4) |
(15,9) |
(27,9) |
(42,6) | |||||||||
AUC = plocha pod křivkou; Cmax = maximální plazmatická koncentrace; IU = mezinárodní jednotka; MRT =
střední rezidenční čas; N = počet subjektů; SD = směrodatná odchylka; tmax = doba k dosažení maximální koncentrace; Vss = distribuční objem v ustáleném stavu; VWF:Ag = von Willebrandův faktor: antigen; VWF:CB = von Willebrandův faktor: vazba kolagenu; VWF:RCo = von Willebrandův faktor: ristocetin kofaktor, FVIILC = faktor VIII: koagulant.
Hemofilie A
Žádné farmakokinetické údaje nejsou k dispozici u pacientů s hemofilií A mladších než 12 let.
5.3 Predklinické údaje vztahující se k bezpečnosti
Voncento obsahuje léčivé látky FVIII a VWF, které jsou získané z lidské plazmy, a působí jako endogenní složky plazmy. Předklinické studie s opakovaným podáváním dávek (chronická toxicita, kancerogenita a mutagenita) se z důvodu vzniku protilátek, které se tvoří po podání heterologní lidské bílkoviny, nemohly provést na konvenčních živočišných modelech.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Chlorid vápenatý, lidský albumin, chlorid sodný, natrium-citrát, sacharóza, trometamol
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky, ředidly a rozpouštědly s výjimkou těch, které jsou uvedeny v bodě 6.1.
6.3 Doba použitelnosti
3 roky.
Po rekonstituci byla prokázaná chemická a fyzikální stabilita po otevření před použitím prokázána na dobu 8 hodin při pokojové teplotě (do 25°C). Z mikrobiologického hlediska má být přípravek použít okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2 až 8 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Chraňte před mrazem. Uchovávejte lahvičku v krabičce, aby byl přípravek chráněn před světlem. Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení Vnitřní obal
Prášek (500 IU/1200 IU) v injekční lahvičce (sklo typu I) se zátkou (pryž) diskem (plast) a víčkem (hliník).
10 ml rozpouštědla v injekční lahvičce (sklo typu I) se zátkou (pryž) diskem (plast) a víčkem (hliník). Velikost balení
Balení s 500 IU/1200 IU:
1 injekční lahvička s práškem 1 injekční lahvička s rozpouštědlem
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Všeobecné pokyny
Roztok má být čirý nebo slabě opalizující. Po filtraci/natáhnutí (viz dále) se rekonstituovaný přípravek před aplikací vizuálně zkontroluje, zda neobsahuje částice nebo nezměnil zbarvení. Nepoužívejte viditelně zakalené roztoky nebo roztoky, které po filtraci ještě obsahují vločky nebo částice. Rekonstituce a natáhnutí roztoku se musí provádět za aseptických podmínek.
Rekonstituce
Ohřejte rozpouštědlo na pokojovou teplotu. Před otevřením balení Mix2Vial odstraňte ochranné víčko z injekční lahvičky s práškem a rozpouštědlem, zátky očistěte antiseptickým roztokem a nechte oschnout.

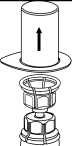
2. Postavte injekční lahvičku s rozpouštědlem na rovný a čistý povrch a pevně ji držte. Uchopte Mix2Vial společně s blistrem a zatlačte hrot konce modrého adaptéru přímo dolů přes zátku injekční lahvičky s rozpouštědlem.
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle nahoru. Přesvědčte se, že jste vytáhli jen blistrový obal, nikoli soupravu Mix2Vial.
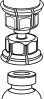
4. Postavte injekční lahvičku s přípravkem na rovný a pevný povrch. Otočte injekční lahvičku s rozpouštědlem a připojeným setem Mix2Vial dnem nahoru a zatlačte hrot průhledného konce adaptéru rovně dolů přes zátku injekční lahvičky s přípravkem. Rozpouštědlo automaticky přeteče do injekční lahvičky s přípravkem.
■CTT
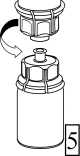


5. Uchopte jednou rukou část setu Mix2Vial uchycenou na injekční lahvičce s přípravkem. Druhou rukou uchopte část setu uchycenou na injekční lahvičce s rozpouštědlem a odšroubováním proti směru hodinových ručiček rozdělte opatrně set na dvě části, aby nedošlo k nadměrnému nahromadění pěny při rozpouštění přípravku.
Injekční lahvičku s rozpouštědlem a připojeným modrým adaptérem setu Mix2Vial odložte.
6. Injekční lahvičku s připojeným průhledným adaptérem jemně otáčejte, dokud se přípravek úplně nerozpustí. Injekční lahvičkou netřepejte.

7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Injekční lahvičku s přípravkem nechejte postavenou dnem dolů a připojte injekční stříkačku s nástavcem Mix2Vial Luer Lock šroubováním ve směru hodinových ručiček. Vstříkněte vzduch do injekční lahvičky s přípravkem.
Natáhnutí a aplikace
|
k |
8. Otočte systém dnem vzhůru a současně držte píst injekční stříkačky stlačený. Natáhněte roztok do injekční stříkačky pomalým vytahováním pístu. | ||
|
< |
A s |
9 |
9. Po natáhnutí roztoku do injekční stříkačky uchopte pevně válec stříkačky (píst směřuje stále dolů) a odpojte průhledný adaptér setu Mix2Vial od injekční stříkačky odšroubováním proti směru hodinových ručiček. |
K injekci přípravku Voncento je nutné používat pouze dodávanou aplikační soupravu, protože v důsledku adsorpce lidského koagulačního faktoru VIII na vnitřní povrch některého injekčního / infuzního zařízení by mohlo dojít k selhání léčby.
V případě, že je požadováno velké množství přípravku Voncento, je možné spojit obsah několika lahviček přípravku Voncento pomocí komerčně dostupné infuzní sady (např. injekční pumpy pro intravenózní aplikaci léků). Nicméně v těchto případech se již rekonstituovaný roztok Voncento nesmí dále ředit.
Roztok podávejte pomalu intravenózně (viz bod 4.2). Ubezpečte se, že do naplněné injekční stříkačky nepronikla krev.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH Emil-von-Behring-Strasse 76 35041 Marburg Německo
8. REGISTRAČNÍ ČÍSLO
EU/1/13/857/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 12. srpna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Voncento 500 IU FVIII/1200 IU VWF (5 ml rozpouštědlo) prášek a rozpouštědlo pro injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje nominálně:
- Factor VIII coagulationis humanus 500 IU7 (lidský koagulační faktor VIII8 (FVIII))
- Factor von Willebrand humanus 1200 IU9 (lidský von Willebrandův faktor8 (VWF))
Po rekonstituci v 5 ml roztok obsahuje 100 IU/ml lidského koagulačního faktoru VIII a 240 IU/ml lidského von Willebrandova faktoru.
Pomocná látka se známým účinkem:
Sodík přibližně 128,2 mmol/l (2,95 mg/ml). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok.
Bílý prášek a čiré, bezbarvé rozpouštědlo pro injekční/infuzní roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
von Willebrandova choroba (VWD)
Profylaxe a léčba hemoragie nebo krvácení během chirurgických zákroků u pacientů trpících VWD v případech, kdy je léčba samotným desmopressinem (DDAVP) neúčinná nebo kontraindikovaná.
Hemofilie A (vrozený deficit FVIII)
Profylaxe a léčba krvácení u pacientů trpících hemofilií A.
4.2 Dávkování a způsob podání
Léčba VWD a hemofilie A se má provádět pod dohledem lékaře, který má zkušenosti s léčbou poruch hemostázy.
Při rozhodování o využití přípravku u domácí léčby při krvácení u pacientů s VWD a s hemofilií A určí konkrétního pacienta ošetřující lékař, který zajistí odpovídající vyškolení v podávání přípravku a toto podávání kontroluje v pravidelných intervalech.
Poměr mezi FVIII:C a VWF:RCo v injekční lahvičce je přibližně 1:2,4.
Dávkování von Willebrandova choroba
Je důležité výpočem stanovit dávku na základě specifikovaného počtu IU VWF:RCo.
1 IU/kg VWF:RCo obvykle zvýší hladinu VWF:RCo v oběhu o 0,02 IU/ml (2 %).
Je třeba dosáhnout hladiny VWF:RCo > 0,6 IU/ml (60 %) a hladiny FVIII:C > 0,4 IU/ml (40 %).
Požadovaná léčba
K dosažení hemostázy se obyčejně doporučuje 40 - 80 IU/kg von Willebrandova faktoru (VWF:RCo), což odpovídá 20 - 40 IU FVIII:C/kg tělesné hmotnosti (BW).
Úvodní dávka 80 IU/kg VWF:RCo může být potřebná zejména u pacientů s VWD typu 3, kde k udržení adekvátních hladin mohou být nutné vyšší dávky než v případě jiných typů VWD.
Prevence krvácení během chirurgického zákroku
K prevenci masivního krvácení během nebo po chirurgickém zákroku se má přípravek začít podávat 1-2 hodiny před chirurgickým zákrokem.
Příslušná dávka se má podávat opakovaně každých 12 - 24 hodin. Dávka a trvání léčby závisí na klinickém stavu pacienta, na typu a závažnosti krvácení a na hladinách VWF:RCo a FVIII:C.
Při používání přípravku s VWF obsahujícího FVIII by si měl být ošetřující lékař vědomý, že pokračování v léčbě může zapříčinit nadměrný vzestup hladin FVIII:C. Po 24 - 48 hodinách léčby by se mělo zvážit snížení dávek a/nebo prodloužení intervalu mezi dávkami nebo použití přípravku s VWF obsahující nízkou hladinu FVIII, aby se zabránilo nadměrnému vzestupu hladiny FVIII:C (viz bod 5.2).
Profylaktická léčba
Při dlouhodobé profylaxi u pacientů s VWD má být zvažována dávka 25-40 IU VWF:RCo/kg tělesné hmotnosti při frekvenci 1-3 krát týdně. U pacientů s gastrointestinálním krvácení nebo menoragií mohou být nezbytné kratší intervaly nebo vyšší dávky. Dávka a trvání léčby bude záviset na klinickém stavu pacienta, jakož i plazmatických hladinách VWF:RCo a FVIII:C.
Pediatrická populace s VWD Léčba krvácení
U pediatrických pacientů se obvykle doporučuje pro léčbu krvácení 40 až 80 IU/kg von Willebrandova faktoru (VWF:RCo), odpovídající 20 až 40 IU FVIII:C/kg tělesné hmotnosti (BW).
Profylaktická léčba
Pacienti ve věku 12 až 18 roků: Dávkování je založeno na stejných pokynech jako pro dospělé.
Pacienti ve věku <12 let: Na základě výsledků z klinické studie s pediatrickými pacienty do 12 let, ve které bylo prokázáno, že mají nižší expozici VWF, by mělo být uvažováno profylaktické rozmezí dávek 40 - 80 IU VWF:RCo/kg tělesné hmotnosti 1 až 3x týdně. (viz bod 5.2).
Dávka a trvání léčby bude záviset na klinickém stavu pacienta, jakož i plazmatických hladinách VWF:RCo a FVffl:C.
Hemofilie A
Je důležité výpočem stanovit dávku na základě specifikovaného počtu IU FVIII:C.
Dávka a délka substituční terapie závisí na závažnosti deficitu FVIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek FVIII se vyjadřuje v mezinárodních jednotkách (International Units - IU), které jsou stanovené oproti současnému standardu WHO pro léčivé přípravky obsahující FVIII. Aktivita FVIII v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v IU (vzhledem k mezinárodnímu standardu pro FVIII v plazmě).
1 IU aktivity FVIII odpovídá množství FVIII v 1 ml normální lidské plazmy.
Požadovaná léčba
Výpočet požadované dávky FVIII vychází z empirického předpokladu, že podáníl IU FVIII na kg tělesné hmotnosti zvýší plazmatickou aktivitu FVIII v plazmě o přibližně 2 % normální aktivity (in vivo recovery 2 IU/dl). Požadovaná dávka se stanoví podle následujícího vzorce:
Požadovaný počet jednotek = tělesná hmotnost [kg] x požadovaný vzestup FVIII [% nebo IU/dl] x 0,5
Množství, které se má podat a frekvence podávání mají vždy směřovat ke klinické účinnosti v individuálních případech.
V případě následujících hemoragických příhod nemá aktivita FVIII během daného období klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může posloužit jako návod pro stanovení dávky v případech krvácení a při chirurgických výkonech:
|
Stupeň krvácení/typ chirurgického výkonu |
Požadovaná hladina FVIII (% nebo IU/dl) |
Frekvence dávkování (hodiny)/délka trvání léčby (dny) |
|
Krvácení | ||
|
Časný hemartros, krvácení do svalstva nebo do ústní dutiny |
O 1 o <N |
Infuzi opakovat každých 12 - 24 hodin po dobunejméně jednoho dne, dokud se krvácení nezastaví, což se projeví ústupem bolesti nebo zahojením. |
|
Rozsáhlejší hemartros, krvácení do svalstva nebo hematom |
O kO 1 o co |
Infuzi opakovat každých 12 - 24 hodin po dobu3 - 4 dní nebo déle, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
o o 1 o kO |
Infuzi opakovat každých 8 - 24 hodin, dokud nepomine ohrožení života. |
|
Chirurgické výkony | ||
|
Menší chirurgický výkon včetně extrakce zubů |
o kO 1 o co |
Infuzi opakovat každých 24 hodin po dobunejméně jednoho dne až do zahojení. |
|
Velké chirurgické výkony |
80 -100 (před a po operaci) |
Infuzi opakovat každých 8 - 24 hodin, dokud nedojde k uspokojivému zahojení rány, potom pokračovat v léčbě nejméně |
dalších 7 dní a udržovat aktivitu FVIII mezi 30 % - 60 % (IU/dl).
Během léčby se doporučuje náležitá kontrola hladin FVIII kvůli stanovení dávky, která má být podávaná, a frekvenci opakovaných infuzí. V případě velkých chirurgických zákroků je nevyhnutelný přesný monitoring substituční terapie prostřednictvím koagulační analýzy (aktivity plazmatického FVIII). U jednotlivých pacientů může být odpověď na léčbu faktorem VIII rozdílná, v závislosti na odlišných hladinách in vivo recovery a různém biologickém poločasu.
Profylaktická léčba
V případě dlouhodobé profylaxe u pacientů s těžkou formou hemofilie A se obyčejně doporučuje dávka 20 až 40 IU FVIII na kilogram tělesné hmotnosti v intervalech 2 až 3 dní. V některých případech, zejména u mladších pacientů, může být nutný kratší interval mezi dávkami nebo vyšší dávky.
Dříve neléčení pacienti
Bezpečnost a účinnost přípravku Voncento u dříve neléčených pacientů nebyla dosud stanovena.
Pediatrická populace s hemofilií A
Dávkování v u hemofilie A u dospívajících ve věku od 12 do 18 let je založeno na tělesné hmotnosti, a proto pro něj platí stejná doporučení jako u dospělých. V některých případech je nezbytný kratší interval dávek nebo vyšší dávky. Frekvence podávání by měla být v každém jednotlivém případě vždy zaměřená na klinickou účinnost.
Bezpečnost a účinnost přípravku Voncento u dětí <12 let s hemofilií A nebyla stanovena. Žádné údaje nejsou k dispozici.
Starší osoby
U starších osob není potřebná žádná úprava dávek.
Způsob podání K intravenóznímu podání.
Přípravek rekonstituujte podle postupu v bodě 6.6. Rekonstituovaný přípravek se podává pomalu intravenózně injekcí/infuzí rychlostí příjemnou pro pacienta.
Rychlost podávání injekce nebo infuze nemá přesáhnout 6 ml za minutu. Pacienta je nutno sledovat, zda nemá bezprostřední reakci. Jestliže se vyskytne jakákoli reakce v souvislosti s podávaním přípravku Voncento, musí se podle klinického stavu pacienta rychlost podávání přípravku snížit nebo podávání zastavit (viz rovněž bod 4.4).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodu 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Jsou možné reakce z přecitlivělosti alergického typu. Pokud se objeví příznaky hypersenzitivity, pacienti by měli být poučeni, aby přerušili používání léčivého přípravku a kontaktovali svého lékaře. Pacienti mají být informováni o prvních příznacích reakcí z přecitlivělosti jako je kopřivka, generalizovaná vyrážka, pocit tísně na hrudi, sípání, hypotenze a anafylaxe. V případě šoku je nutné dodržovat všeobecné lékařské postupy pro léčbu šoku.
Virová bezpečnost
Ke standardním opatřením k prevenci infekcí, které jsou následkem používání přípravků, vyrobených z lidské krve nebo plazmy, patří výběr dárců, kontrola jednotlivých odběrů a plazmatických poolů, zaměřená na specifické markery infekce a provedení efektivních výrobních kroků k inaktivaci/odstranění virů. Navzdory těmto opatřením není možné úplně vyloučit možnost přenosu infekčních agens, pokud podávané přípravky pocházejí z lidské krve nebo plazmy. Toto platí i pro neznámé nebo nové viry a další patogeny.
Tato opatření se považují za účinná pro obalené viry, jako jsou virus lidské imunodeficience (HIV), virus hepatitidy B (HBV) a virus hepatitidy C (HCV) a neobalený virus hepatitidy A (HAV).
Provedenáopatření mohou mít jen omezenou účinnost proti neobaleným virům, jako je parvovirus B19.
Infekce způsobená parvovirem B19 může být závažná u těhotných žen (infekce plodu) a jedinců s imunodeficiencí nebo zvýšenou erytropoézou (např. hemolytická anémie).
U pacientů, kteří pravidelně/opakovaně dostávají přípravky s faktorem Vin/VWF, vyrobené z lidské plazmy, je třeba zvážit možnosti vhodného očkování (hepatitida A a B).
Důrazně se doporučuje, aby byl při každém podání přípravku Voncento pacientovi zaznamenaný název a číslo šarže přípravku, aby se udržovaly záznamy o propojení mezi pacientem a šarží přípravku.
von Willebrandova choroba
Existuje riziko vzniku trombóz zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Rizikoví pacienti musí být proto sledováni kvůli zachycení prvních příznaků trombózy. Měla by být provedena profylaktická opatření proti venózním trombózám v souladu s aktuálními doporučeními.
Při léčbě přípravkem s VWF obsahujícím FVIII má mít ošetřující lékař na paměti, že pokračování v léčbě může zapříčinit nadměrný vzestup FVIII:C. U pacientů, kteří dostávají přípravek VWF obsahující FVIII, musí být sledovány hladiny FVIII:C v plazmě kvůli tomu, aby nebyly hladiny FVIII:C v plazmě dlouhodobě vysoké, což by mohlo zvýšit riziko výskytu trombóz, a současně je potřebné zvážit zavedení protitrombotických opatření (viz též bod 5.2).
U pacientů s VWD, zejména typu 3, se mohou vytvořit neutralizační protilátky (inhibitory) proti VWF. Jestliže se nedaří dosáhnout očekávanou hladinu aktivity VWF:RCo v plazmě nebo jestliže krvácení není kontrolované vhodnou dávkou, je potřebné uskutečnit odpovídající analýzu kvůli stanovení přítomnosti inhibitoru VWF. U pacientů s vysokými hladinami inhibitoru může být terapie nejen neúčinná, ale může vést i k anafylaktoidní reakci a je třeba uvažovat o jiných možnostech léčby.
Hemofilie A
Inhibitory
U pacientů s hemofilií A je známou komplikací tvorba neutralizačních protilátek (inhibitorů) proti FVIII. Tyto inhibitory jsou obyčejně IgG imunoglobuliny proti prokoagulační aktivitě FVIII, které jsou kvantifikovány v jednotkách Bethesda (BU) na ml plazmy, pomocí modifikovaného testu. Riziko vzniku inhibitorů koreluje s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dní expozice. Zřídka se mohou inhibitory vytvořit po prvních 100 dnech expozice.
Případy opakovaného výskytu inhibitoru (nízké titry) byly pozorovány po přechodu z jednoho přípravku, obsahujícího faktor VIII, na jiný, u pacientů, kteří už byli léčení předtím po dobu více než 100 dní a u kterých byla už předtím zaznamenaná tvorba inhibitoru. Proto se doporučuje důkladné monitorování všech pacientů, pokud se týká výskytu inhibitoru, po jakémkoli přechodu na jiný přípravek.
Obecně mají být všichni pacienti, léčení lidským koagulačním faktorem FVIII, pozorně monitorovaní kvůli tvorbě inhibitorů a to prostřednictvím klinického sledování a laboratorních testů.
Pokud nejsou dosaženy očekávané hladiny faktoru VIII v plazmě, nebo pokud není krvácení zvládnuto odpovídající dávkou, mělo být provedeno testování na přítomnost inhibitoru FVIII. U pacientů s vysokými hladinami inhibitoru může být léčba faktorem VIII neúčinná a měly by být zváženy jiné možnosti léčby. Léčba těchto pacientů by měla být vedena lékaři se zkušenostmi s péčí o pacienty s hemofilií A a s inhibitory faktoru VIII.
Komplikace související s katetrem
Jestliže je třeba použít zařízení pro centrální žilní přístup (CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katetru.
Obsah sodíku
Tento přípravek obsahuje až 14,75 mg (0,64 mmol) sodíku v jedné injekční lahvičce. Toto množství se musí zohlednit u pacientů s dietou s kontrolovaným příjmem sodíku.
Pediatrická populace
Uvedená upozornění a opatření se vztahují jak na dospělé tak na pediatrické pacienty.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly studovány žádné interakce VWF a FVIII s jinými léčivými přípravky.