Victoza 6 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Victoza 6 mg/ml injekční roztok v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
1 ml roztoku obsahuje liraglutidum 6 mg *. Jedno předplněné pero obsahuje liraglutidum 18 mg ve 3 ml.
* Analog lidského glukagonu podobného peptidu-1 (GLP-1) vyrobený rekombinantní DNA technologií v Saccharomyces cerevisiae.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok
Čirý bezbarvý či téměř bezbarvý izotonický roztok; pH = 8,15.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Victoza je určen k léčbě dospělých s diabetes mellitus 2. typu s cílem dosáhnout kontroly glykemie:
Monoterapie
Pokud samotná dieta a cvičení nezajistí dostatečnou kontrolu glykemie u pacientů, pro něž je léčba metforminem nevhodná z důvodu nesnášenlivosti nebo kontraindikací.
Kombinovaná terapie
V kombinaci s perorálními léčivými přípravky ke snížení hladiny glukózy v krvi a/nebo bazálními inzuliny, pokud tyto přípravky spolu s dietou a cvičením neposkytují dostatečnou kontrolu glykemie (dostupné údaje o různých kombinacích viz body 4.4 a 5.1).
4.2 Dávkování a způsob podání
Dávkování
Z důvodu zlepšení gastrointestinální snášenlivosti je počáteční dávka 0,6 mg liraglutidu denně. Minimálně po jednom týdnu má být dávka zvýšena na 1,2 mg. Na základě klinické odpovědi lze u některých pacientů předpokládat přínos při zvýšení dávky ze 1,2 mg na 1,8 mg. Proto pro další zlepšení kontroly glykemie se minimálně po jednom týdnu může zvýšit dávka na 1,8 mg. Denní dávky vyšší než 1,8 mg se nedoporučují.
Přípravek Victoza může být přidán ke stávající terapii metforminem nebo ke kombinované terapii metforminem a thiazolidindionem. Dosavadní dávka metforminu a thiazolidindionu může být ponechána nezměněná.
Přípravek Victoza může být přidán ke stávající terapii sulfonylureou nebo ke kombinované terapii metforminem a sulfonylureou nebo bazálním inzulinem. V případě, že je přípravek Victoza přidán k terapii sulfonylureou nebo bazálním inzulinem, je třeba zvážit snížení dávky sulfonylurey nebo bazálního inzulinu, aby se zmenšilo riziko hypoglykemie (viz bod 4.4).
Selfmonitoring glukózy v krvi pro nastavení dávky přípravku Victoza není nutný. Při zahájení léčby přípravkem Victoza v kombinaci se sulfonylureou nebo bazálním inzulínem však může být selfmomtoring glukózy v krvi nezbytný k nastavení dávky sulfonylurey nebo bazálního inzulinu.
Zvláštní skupiny _pacientů
Starší pacienti (věk > 65 let)
Z důvodu věku není nutná žádná úprava dávkování. Zkušenosti s léčbou pacientů ve věku > 75 let jsou omezené (viz bod 5.2).
Porucha funkce ledvin
U pacientů s lehkou či středně těžkou poruchou funkce ledvin (clearance kreatininu 60 - 90 ml/min, respektive 30 - 59 ml/min) není nutná žádná úprava dávkování. Nejsou žádné zkušenosti s léčbou pacientů s těžkou poruchou funkce ledvin (clearance kreatininu nižší než 30 ml/min). Přípravek Victoza v současnosti není doporučen pro použití u pacientů s těžkou poruchou funkce ledvin včetně pacientů v konečném stadiu selhání ledvin (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou nebo středně závažnou poruchou funkce jater není doporučena žádná úprava dávky. Použití přípravku Victoza se nedoporučuje u pacientů se závažnou poruchou funkce jater (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Victoza u dětí a dospívajících do 18 let nebyla stanovena (viz bod 5.1). Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Victoza nesmí být podán intravenózně nebo intramuskulárně.
Přípravek Victoza se podává jedenkrát denně kdykoli v průběhu dne, nezávisle na jídle, a může být aplikován subkutánně do břicha, stehna nebo horní části paže. Místo aplikace i doba aplikace injekce mohou být změněny bez úpravy dávkování. Je však výhodnější podávat přípravek Victoza přibližně ve stejnou denní dobu, která je pro pacienta nejvhodnější. Další pokyny o podávání viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Liraglutid nesmí být používán u pacientů s diabetes mellitus 1. typu nebo k léčbě diabetické ketoacidózy.
Liraglutid není náhrada za inzulin.
S podáváním pacientům s městnavým srdečním selháním třídy I-II podle New York Heart Association (NYHA) jsou pouze omezené zkušenosti. Liraglutid proto musí být používán s opatrností. Nejsou žádné zkušenosti s podáváním pacientům s městnavým srdečním selháním třídy III-IV podle NYHA. Liraglutid proto není u těchto pacientů doporučován.
S podáváním pacientům se zánětlivým onemocněním střev a diabetickou gastroparézou jsou pouze omezené zkušenosti. Používání liraglutidu není u těchto pacientů doporučeno, neboť je spojeno s přechodnými gastrointestinálními nežádoucími účinky, včetně nauzey, zvracení a průjmu.
Akutní pankreatitida
Použití agonistů GLP-1 receptoru bylo spojeno s rizikem vzniku akutní pankreatitidy. Bylo hlášeno několik případů akutní pankreatitidy. Pacienti musí být informováni o charakteristických příznacích akutní pankreatitidy. Je-li podezření na pankreatitidu, musí být přípravek Victoza vysazen. Pokud je akutní pankreatitida potvrzena, nesmí být léčba přípravkem Victoza znovu zahájena. U pacientů s prodělanou pankreatitidou je nutno dbát zvláštní opatrnosti.
Onemocnění štítné žlázy
V klinických studiích byly hlášeny nežádoucí účinky na štítnou žlázu včetně zvýšené hladiny kalcitoninu v krvi, zvětšení štítné žlázy a neoplazie štítné žlázy, a to zvláště u pacientů s již dříve existujícím onemocněním štítné žlázy. Liraglutid proto musí být používán s opatrností.
Hypoglykemie
Pacienti, kterým je podáván liraglutid v kombinaci se sulfonylureou nebo bazálním inzulinem, mohou mít zvýšené riziko hypoglykemie (viz bod 4.8). Riziko hypoglykemie se může zmenšit snížením dávky sulfonylurey nebo bazálního inzulinu.
Dehydratace
U pacientů léčených liraglutidem byly hlášeny známky a příznaky dehydratace včetně poruchy funkce ledvin a akutního selhání ledvin. Pacienti používající liraglutid musí být upozorněni na potenciální riziko dehydratace v případě gastrointestinálních nežádoucích účinků a musí být seznámeni s bezpečnostními opatřeními, která mají učinit, aby zabránili úbytku tekutin.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
In vitro je u liraglutidu prokázán velmi nízký potenciál pro farmakokinetické interakce s jinými léčivými látkami s vlivem na cytochrom P450 a vazbu na plasmatické proteiny.
Malé zpoždění ve vyprazdňování žaludku při používání liraglutidu může ovlivnit absorpci současně podávaných perorálních léčivých přípravků. Studie interakcí neprokázaly žádné klinicky významné zpoždění absorpce, a proto není vyžadována úprava dávky. Několik pacientů léčených liraglutidem hlásilo nejméně jednu epizodu těžkého průjmu. Průjem může ovlivnit absorpci současně podávaných perorálních léčivých přípravků.
Warfarin a další deriváty kumarinu
Nebyly provedeny žádné studie interakcí. Klinicky významné interakce s léčivými látkami se špatnou rozpustností nebo s úzkým terapeutickým indexem, jako je warfarin, nemohou být vyloučeny. Po zahájení léčby liraglutidem se u pacientů užívajících warfarin nebo další deriváty kumarinu doporučuje častější monitorování INR (International Normalised Ratio).
Paracetamol
Liraglutid neměnil celkovou expozici paracetamolu po podání 1 000 mg v jedné dávce. Hodnota Cmax paracetamolu byla snížena o 31 % a střední hodnota tmax byla zpožděna na 15 min. Při současném podávání paracetamolu není nutná žádná úprava dávkování.
Atorvastatin
Liraglutid neměnil celkovou expozici atorvastatinu v klinicky významné míře po podání 40 mg atorvastatinu v jedné dávce. Proto při podávání s liraglutidem není nutná žádná úprava dávky atorvastatinu. Při podávání s liraglutidem se hodnota Cmax atorvastatinu snížila o 38 % a střední hodnota tmax se zpozdila z 1 hod na 3 hod.
Griseofulvin
Liraglutid neměnil celkovou expozici griseofulvinu po podání 500 mg griseofulvinu v jedné dávce. Hodnota Cmax griseofulvinu byla zvýšena o 37 %, zatímco ke změně střední hodnoty tmax nedošlo. Úprava dávky griseofulvinu a jiných látek s nízkou rozpustností a vysokou permeabilitou není nutná.
Digoxin
Podání 1 mg digoxinu v jedné dávce spolu s liraglutidem vedlo ke snížení AUC digoxinu o 16 %, hodnota Cmax byla snížena o 31 %. Medián tmax digoxinu byl zpožděn z 1 hod na 1,5 hod. Na základě těchto výsledků není nutná úprava dávky digoxinu.
Lisinopril
Podání 20 mg lisinoprilu v jedné dávce spolu s liraglutidem vedlo ke snížení AUC lisinoprilu o 15 %, hodnota Cmax byla snížena o 27 %. Medián tmax lisinoprilu byl při podávání liraglutidu zpožděn z 6 hod na 8 hod. Na základě těchto výsledků není nutná úprava dávky lisinoprilu.
Perorální antikoncepční přípravky
Po podání jednorázové dávky perorálního antikoncepčního přípravku snižoval liraglutid Cmax ethinyloestradiolu a levonorgestrelu o 12 a 13 %. Hodnota tmax byla u obou látek při podání liraglutidu zpožděna o 1,5 hod. Nedošlo k žádnému klinicky významnému účinku na celkovou expozici ethinyloestradiolu ani levonorgestrelu. Předpokládá se proto, že antikoncepční účinek není při současném podávání liraglutidu ovlivněn.
Inzulín
Při podání jednorázové dávky inzulinu detemir 0,5 j./kg s liraglutidem v dávce 1,8 mg pacientům s diabetem 2. typu nebyly v rovnovážném stavu pozorovány žádné farmakokinetické nebo farmakodynamické interakce mezi liraglutidem a inzulinem detemir.
4.6 Fertilita, těhotenství a kojení
Adekvátní údaje o podávání liraglutidu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známé.
Liraglutid nemá být během těhotenství podáván, místo něj se doporučuje používání inzulinu. Pokud si pacientka přeje otěhotnět nebo otěhotní, musí být léčba přípravkem Victoza přerušena.
Kojení
Není známo, zda dochází k exkreci liraglutidu do lidského mateřského mléka. Studie na zvířatech ukázaly, že přenos liraglutidu a strukturně blízkých metabolitů do mléka je nízký. Neklinické studie ukázaly snížení neonatálního růstu u kojených potkaních mláďat spojené s léčbou (viz bod 5.3). Vzhledem k chybějícím zkušenostem nemá být přípravek Victoza používán během kojení.
Fertilita
S výjimkou lehkého snížení počtu živých nidovaných vajíček neprokázaly studie na zvířatech škodlivé účinky v souvislosti s fertilitou.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Victoza nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
Pacientům má být doporučeno provést opatření, aby během řízení a obsluhy strojů předešli hypoglykémii, zvláště pokud je přípravek Victoza používán v kombinaci se sulfonylureou nebo bazálním inzulinem.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V pěti velkých dlouhodobých klinických studiích se více než 2 500 pacientů léčilo samotným přípravkem Victoza nebo v kombinaci s metforminem, sulfonylureou (s metforminem nebo bez metforminu) nebo metforminem s rosiglitazonem.
Nejčastěji hlášené nežádoucí účinky byly v klinických studiích gastrointestinální poruchy: nauzea a průjem byly velmi časté, zatímco zvracení, zácpa, bolest břicha a dyspepsie byly časté. Na počátku léčby se mohou tyto gastrointestinální nežádoucí účinky vyskytovat častěji. Tyto reakce obvykle po několika dnech či týdnech trvalé léčby zeslábnou. Bolesti hlavy a nasofaryngitis byly také časté. Častá byla dále hypoglykemie, a pokud byl liraglutid použit v kombinaci se sulfonylureou, byla hypoglykemie velmi častá. Závažná hypoglykemie byla pozorována hlavně při kombinované léčbě se sulfonylureou.
Seznam nežádoucích účinků v tabulce
V tabulce 1 jsou uvedeny nežádoucí účinky hlášené během fáze 3 dlouhodobých kontrolovaných klinických hodnocení a ze spontánních (postmarketingových) hlášení. Frekvence výskytu související se spontánními (postmarketingovými) hlášeními byly vypočteny na základě jejich výskytu ve fázi 3 klinických hodnocení.
Frekvence výskytu jsou definovány takto: Velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti výskytu jsou nežádoucí účinky seřazeny v pořadí podle klesající závažnosti.
Tabulka 1 Nežádoucí účinky zjištěné ve fázi 3 dlouhodobých kontrolovaných hodnocení a ze spontánních (postmarketingových) hlášení
|
Třídy orgánových systémů podle databáze MedDRA |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
|
Infekce a infestace |
Nasofaryngitis Bronchitis | ||||
|
Poruchy imunitního systému |
Anafylaktické reakce | ||||
|
Poruchy metabolismu a výživy |
Hypoglykemie Snížená chuť k jídlu |
Dehydratace | |||
|
Poruchy nervového systému |
Bolest hlavy Závratě | ||||
|
Srdeční poruchy |
Zvýšená srdeční frekvence |
|
Gastrointestinální poruchy |
Zvracení Dyspepsie Bolest břicha v epigastriu Zácpa Gastritis Nadýmání Distenze břicha Gastroezofageální refluxní choroba Břišní diskomfort Bolest zubů |
Intestinální obstrukce |
Pankreatitida (včetně nekrotizující pankreatitidy) | ||
|
Poruchy kůže a podkožní tkáně |
Kopřivka | ||||
|
Poruchy ledvin a močových cest |
Porucha funkce ledvin Akutní selhání ledvin | ||||
|
Celkové poruchy a reakce v místě aplikace |
Únava Reakce v místě vpichu | ||||
|
Vyšetření |
Zvýšené hladiny lipázy* Zvýšené hladiny amylázy* |
* Pouze z kontrolovaných klinických studií fáze 3b a 4, kde byly měřeny.
Popis vybraných nežádoucích účinků
V klinických studiích s liraglutidem v monoterapii byl výskyt hypoglykémií při léčbě liraglutidem nižší než výskyt hlášený u pacientů léčených aktivním komparátorem (glimepiridem). Nejčastěji hlášené nežádoucí účinky byly gastrointestinální poruchy, infekce a infestace.
Hypoglykemie
Většina potvrzených hypoglykemických epizod v klinických hodnoceních byla nezávažná. Hypoglykemické epizody pozorované v klinickém hodnocení liraglutidu použitým v monoterapii nebyly závažné. Závažná hypoglykemie se může vyskytnout méně často a byla pozorována hlavně tehdy, když byl liraglutid kombinován se sulfonylureou (0,02 případů/pacientorok). Bylo pozorováno velmi málo epizod (0,001 případů/pacientorok) při podávání liraglutidu v kombinaci s jinými perorálními antidiabetiky, než je sulfonylurea. Riziko hypoglykemie při kombinované léčbě bazálním inzulinem a liraglutidem je nízké (1,0 příhoda na pacientorok, viz bod 5.1).
Gastrointestinální nežádoucí účinky
Při kombinaci liraglutidu s metforminem hlásilo 20,7 % pacientů nejméně jednu epizodu nauzey a 12,6 % pacientů hlásilo nejméně jednu epizodu průjmu. Při kombinaci liraglutidu se sulfonylureou hlásilo 9,1 % pacientů nejméně jednu epizodu nauzey a 7,9 % pacientů hlásilo nejméně jednu epizodu průjmu. Největší počet epizod byly lehké až středně závažné epizody a docházelo k nim v závislosti na dávce. S pokračující léčbou se frekvence a závažnost snižovala u většiny pacientů, kteří v počátku trpěli nauzeou.
U pacientů ve věku > 70 let se může při léčbě liraglutidem projevit více gastrointestinálních účinků.
U pacientů s lehkou a středně těžkou poruchou funkce ledvin (clearance kreatininu 60 - 90 ml/min, respektive 30 - 59 ml/min) se může při léčbě liraglutidem projevit více gastrointestinálních účinků.
Vysazení
Četnost vysazení z důvodu nežádoucích účinků byla v dlouhodobých kontrolovaných studiích (26 týdnů nebo delších) 7,8 % u pacientů léčených liraglutidem a 3,4 % u pacientů léčených komparátorem. Nejčastějšími nežádoucími účinky, které vedly k vysazení u nemocných léčených liraglutidem, byla nauzea (2,8 % pacientů) a zvracení (1,5 %).
Reakce v místě vpichu
Reakce v místě vpichu byly během dlouhodobých kontrolovaných klinických hodnocení (26 týdnů nebo delších) hlášeny přibližně u 2 % pacientů léčených přípravkem Victoza. Tyto reakce byly obvykle lehké.
Pankreatitis
Během dlouhodobých klinických studií s přípravkem Victoza bylo hlášeno několik případů akutní pankreatitidy (< 0,2 %). Pankreatitida byla rovněž hlášena v postmarketingovém sledování.
Alergické reakce
Alergické reakce včetně kopřivky, vyrážky a svědění byly hlášeny po uvedení přípravku Victoza na trh.
Po uvedení přípravku Victoza na trh bylo hlášeno několik případů anafylaktických reakcí s dalšími příznaky jako hypotenze, palpitace, dušnost a edém. Ve všech dlouhodobých klinických hodnoceních s přípravkem Victoza bylo hlášeno několik případů (0,05 %) angioedému.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Z klinických hodnocení a po uvedení přípravku na trh bylo hlášeno až 40násobné (72 mg) předávkování oproti doporučené udržovací dávce. Celkově byly pacienty hlášeny těžká nauzea, zvracení a průjem. Žádný z pacientů nehlásil těžkou hypoglykemii. Všichni pacienti se zotavili bez komplikací.
V případě předávkování má být zahájena vhodná podpůrná léčba podle klinických známek a příznaků, které se u pacienta vyskytnou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antidiabetika, jiná antidiabetika, kromě inzulinů. ATC kód: A10BX07 Mechanismus účinku
Liraglutid je analog GLP-1 s 97% sekvenční homologií s lidským GLP-1, který se váže na receptor GLP-1 a aktivuje jej. Receptor GLP-1 je vazebným místem přirozeného GLP-1, endogenního inkretinového hormonu, který potencuje sekreci inzulinu závislou na glukóze pankreatickými beta buňkami. Na rozdíl od přirozeného GLP-1 má liraglutid u lidí farmakokinetický a farmakodynamický profil vhodný pro podávání jednou denně. Po subkutánním podání je protrahovaný profil účinku založen na třech mechanismech: na shlukování, které má za následek pomalou absorpci, na vazbě na albumin a na vyšší enzymatické stabilitě vůči enzymům dipeptidylpeptidáze -4 (DPP-4) a neutrální endopeptidáze (NEP), což vede k delšímu plasmatickému poločasu.
Účinek liraglutidu je zprostředkován specifickou interakcí s receptory GLP-1 vedoucí ke zvýšení cyklického adenosinmonofosfátu (cAMP). Liraglutid stimuluje sekreci inzulinu v závislosti na koncentraci glukózy. Současně liraglutid snižuje nepřiměřeně vysokou sekreci glukagonu, rovněž v závislosti na koncentraci glukózy. Když je tedy koncentrace glukózy v krvi vysoká, je stimulována sekrece inzulinu a sekrece glukagonu je inhibována. A naopak, při hypoglykémii liraglutid snižuje sekreci inzulinu a nesnižuje sekreci glukagonu. Mechanismus snižování koncentrace glukózy v krvi zahrnuje rovněž mírné zpomalení vyprazdňování žaludku. Liraglutid snižuje tělesnou hmotnost a množství tělesného tuku mechanismem, který zahrnuje snížení hladu a snížení příjmu energie.
GLP-1 je fyziologický regulátor chuti k jídlu a příjmu potravy, ale přesný mechanismus účinku není zcela jasný. Ve studiích na zvířatech vedla periferní aplikace liraglutidu k vychytávání ve specifických oblastech mozku zapojených do regulace chuti k jídlu, kde liraglutid prostřednictvím specifické aktivace receptoru GLP-1 (GLP-1R) zvyšoval klíčové signály sytosti a snižoval klíčové signály hladu, čímž docházelo ke snižování tělesné hmotnosti.
Farmakodynamické účinky
Účinek liraglutidu trvá 24 hodin a zlepšuje u pacientů s diabetem 2. typu kontrolu glykemie snížením hladiny glukózy v krvi nalačno i postprandiálně.
Klinická účinnost a bezpečnost
K vyhodnocení účinku liraglutidu na kontrolu glykemie bylo provedeno pět dvojitě zaslepených randomizovaných kontrolovaných klinických hodnocení (tabulky 2 až 5). Léčba liraglutidem vedla ve srovnání s placebem ke klinicky i statisticky významnému zlepšení glykovaného hemoglobinu A1c (HbA1c), koncentraci plasmatické glukózy nalačno i postprandiálně.
Tato klinická hodnocení byla provedena u 3 978 pacientů s diabetem 2. typu (2 501 pacient byl léčen liraglutidem), 53,7 % tvořili muži a 46,3 % tvořily ženy, 797 pacientů (508 léčených liraglutidem) bylo ve věku > 65 let a 113 pacientů (66 léčených liraglutidem) bylo ve věku > 75 let.
Byly provedeny dodatečné studie s liraglutidem, zahrnující 1 901 pacienta ve čtyřech nezaslepených, randomizovaných kontrolovaných klinických studiích (464, 658, 323 a 177 pacientů na klinickou studii), a jedna dvojitě zaslepená, randomizovaná kontrolovaná klinická studie s pacienty s onemocněním diabetes mellitus 2. typu a středně těžkou poruchou funkce ledvin (279 pacientů).
• Kontrola glykemie
Monoterapie
U pacientů dříve léčených buď dietou a cvičením nebo monoterapií PAD (při nejvýše poloviční maximální dávce) vedla monoterapie liraglutidem po dobu 52 týdnů v porovnání s glimepiridem v dávce 8 mg ke statisticky významnému a přetrvávajícímu snížení HbA1c (-0,84 % pro dávku 1,2 mg a -1,14 % pro dávku 1,8 mg oproti -0,51% pro komparátor) (tabulka 2).
Kombinace s perorálními antidiabetiky
Liraglutid v kombinované terapii s metforminem, glimepiridem nebo metforminem a rosiglitazonem, trvající 26 týdnů, vedl ke statisticky významnému (p < 0,0001) a trvalému snížení HbA1c ve srovnání s pacienty dostávajícími placebo (tabulka 2).
Tabulka 2 Liraglutid v monoterapii (52 týdnů) a v kpmbinaci s PAD (26 týdnů)
|
N |
Průměrná |
Průměrná |
Pacienti (%), |
Průměrná |
Průměrná | |
|
výchozí |
změna |
kteří dosáhli |
výchozí |
změna | ||
|
hodnota |
HbA1c proti |
HbA1c<7% |
hodnota |
hmotnosti | ||
|
HbA1c (%) |
výchozí |
hmotnosti |
proti | |||
|
hodnotě (%) |
(kg) |
výchozí |
|
hodnotě (kg) | ||||||
|
Monoterapie | ||||||
|
Liraglutid 1,2 mg |
251 |
8,18 |
-0,84* |
42,8'; 58,33 |
92,1 |
-2,05** |
|
Liraglutid 1,8 mg |
246 |
8,19 |
-1,14** |
50,9'; 62,03 |
92,6 |
-2,45** |
|
Glimepirid 8 mg/den |
248 |
8,23 |
-0,51 |
27,8'; 30,83 |
93,3 |
1,12 |
|
Přidání k metforminu (2 000 mg/den) | ||||||
|
Liraglutid 1,2 mg |
240 |
8,3 |
-0,97+ |
35,3'; 52,82 |
88,5 |
-2,58** |
|
Liraglutid 1,8 mg |
242 |
8,4 |
-1,00+ |
42,4'; 66,32 |
88,0 |
-2,79** |
|
Placebo |
121 |
8,4 |
0,09 |
10,8'; 22,52 |
91,0 |
-1,51 |
|
Glimepirid 4 mg/den |
242 |
8,4 |
-0,98 |
36,3'; 56,02 |
89,0 |
0,95 |
|
Přidání ke glimepiridu (4 mg/den) | ||||||
|
Liraglutid 1,2 mg |
228 |
8,5 |
-1,08** |
34,5'; 57,42 |
80,0 |
0,32** |
|
Liraglutid 1,8 mg |
234 |
8,5 |
-1,13** |
41,6'; 55,92 |
83,0 |
-0,23** |
|
Placebo |
114 |
8,4 |
0,23 |
7,5'; 11,82 |
81,9 |
-0,10 |
|
Rosiglitazon 4 mg/den |
231 |
8,4 |
-0,44 |
21,9'; 36,12 |
80,6 |
2,11 |
|
Přidání k metforminu (2 000 mg/den) + rosiglitazonu (4 mg dvakrát denně) | ||||||
|
Liraglutid 1,2 mg |
177 |
8,48 |
-1,48** |
57,5' |
95,3 |
-1,02** |
|
Liraglutid 1,8 mg |
178 |
8,56 |
-1,48** |
53,7' |
94,9 |
-2,02** |
|
Placebo |
175 |
8,42 |
-0,54 |
28,1' |
98,5 |
0,60 |
|
Přidání k metforminu (2 000 mg/den) + glimepiridu (4 mg/den) | ||||||
|
Liraglutid 1,8 mg |
230 |
8,3 |
-1,33* |
53,1' |
85,8 |
-1,81** |
|
Placebo |
114 |
8,3 |
-0,24 |
15,3' |
85,4 |
-0,42 |
|
Inzulin glargin4 |
232 |
8,1 |
-1,09 |
45,8' |
85,2 |
1,62 |
Superiorita (p<0,01) vs. aktivní komparátor; **Superiorita (p<0,0001) vs. aktivní komparátor; +Non-inferiorita (p<0,0001) vs. aktivní komparátor
'všichni pacienti; 2předchozí monoterapie PAD; 3pacienti dříve léčení dietou
4dávkování inzulinu glargin bylo otevřené a bylo prováděno podle Pokynu na úpravu dávky inzulinu glargin. Titraci dávky inzulinu glargin prováděli pacienti po proškolení zkoušejícím:
Pokyn na úpravu dávky inzulinu glargin
|
Hodnota FPG změřená pacientem |
Zvýšení dávky inzulinu glargin (IU) |
|
<5,5 mmol/l (<100 mg/dl) cílová hodnota |
Žádná úprava |
|
>5,5 a <6,7 mmol/l (>100 a <120 mg/dl) |
0 - 2 IUa |
|
>6,7 mmol/l (>120 mg/dl) |
2 IU |
a podle individualizovaného doporučení zkoušejícího při předchozí návštěvě například v závislosti na výskytu hypoglykemie
Kombinace s inzulínem
V klinické studii trvající 104 týdny dosáhlo 57 % pacientů s diabetes mellitus 2. typu léčených inzulínem degludek v kombinaci s metforminem cílových hodnot HbAic < 7,0 %. Zbývající pacienti pokračovali ve 26týdenní otevřené klinické studii a byli randomizováni do větve s přidáním liraglutidu nebo do větve s přidáním jedné dávky inzulinu aspart (k hlavnímu jídlu). Ve větvi inzulin degludek + liraglutid byla dávka inzulinu snížena o 20 %, aby bylo minimalizováno riziko hypoglykemie. Přidání liraglutidu mělo za následek statisticky významně větší snížení HbAJc (-0,73 % u liraglutidu oproti -0,40% u komparátoru) a tělesné hmotnosti (-3,03 oproti 0,72 kg). Výskyt hypoglykemických příhod (na pacientorok léčby) byl statisticky významně nižší po přidání liraglutidu ve srovnání s přidáním jedné dávky inzulinu aspart (1,0 oproti 8,15; poměr: 0,13; 95% CI: 0,08 až 0,21).
V klinické studii trvající 52 týdnů a zahrnující pacienty, kteří nedosáhli cílových hodnot glykemie léčbou samotnými liraglutidem a metforminem, mělo přidání inzulinu detemir k liraglutidu (v dávce 1,8 mg) a metforminu za následek snížení HbA1c oproti výchozí hladině o 0,54 % ve srovnání s 0,20 % u kontrolní skupiny léčené liraglutidem v dávce 1,8 mg a metforminem. Úbytek tělesné hmotnosti zůstal zachován. Došlo k malému nárůstu ve výskytu lehkých hypoglykemických příhod (0,23 oproti 0,03 příhod na pacientoroky).
Použití u pacientů s poškozením funkce ledvin
Ve dvojitě zaslepené klinické studii srovnávající účinnost a bezpečnost liraglutidu oproti placebu (liraglutid podáván v dávce 1,8 mg jako přídatná léčba k inzulinu a/nebo PAD) u pacientů s onemocněním diabetes mellitus 2. typu a se středně těžkou poruchou funkce ledvin byl liraglutid superiorní ve srovnání s podáváním placeba, neboť po 26 týdnech snižoval HbA1c (-1,05 % oproti -0,38 %).
Ve srovnání s placebem dosáhl při léčbě liraglutidem signifikantně vyšší počet pacientů HbA1c pod 7 % (52,8 % oproti 19,5 %). V obou skupinách bylo pozorováno snížení tělesné hmotnosti: -2,4 kg u liraglutidu oproti -1,09 kg u placeba. V obou skupinách bylo srovnatelné riziko hypoglykemických příhod. Bezpečnostní profil liraglutidu byl obecně podobný jako v ostatních klinických studiích s liraglutidem.
• Podíl pacientů, kteří dosáhli snížení HbA1c
Léčba samotným liraglutidem vedla během 52 týdnů u statisticky významně vyššího podílu pacientů k dosažení HbA1c < 6,5 % v porovnání s pacienty, kteří byli léčení glimepiridem (37,6 % pro 1,8 mg a 28,0 % pro 1,2 mg oproti 16,2 % pro komparátor).
Léčba liraglutidem v kombinované terapii s metforminem, glimepiridem nebo metforminem a rosiglitazonem vedla ke statisticky významně vyššímu podílu pacientů, kteří dosáhli HbA1c < 6,5 % během 26 týdnů, ve srovnání s pacienty, kteří dostávali tyto léky samotné.
• Plasmatická hladina glukózy nalačno
Léčba samotným liraglutidem a v kombinaci s jedním či dvěma perorálními antidiabetiky vedla ke snížení plasmatické hladiny glukózy nalačno o 13 - 43,5 mg/dl (0,72 - 2,42 mmol/l). Toto snížení bylo pozorováno v prvních dvou týdnech léčby.
• Postprandiální glykemie
Liraglutid snižuje postprandiální glykémii po všech třech denních jídlech o 31 - 49 mg/dl (1,68 -2,71 mmol/l).
• Funkce beta buněk
Klinická hodnocení s liraglutidem ukazují zlepšenou funkci beta buněk na základě stanovení, jako je hodnocení modelu homeostázy pro beta buňky (HOMA-B) a poměru proinzulinu k inzulinu. Bylo prokázáno zlepšení první a druhé fáze sekrece inzulinu po 52 týdnech léčby liraglutidem na podskupině pacientů s diabetem 2. typu (N = 29).
• Tělesná hmotnost
Podávání liraglutidu samotného a v kombinaci s metforminem, metforminem a glimepiridem nebo metforminem a rosiglitazonem bylo spojeno s trvalým snížením tělesné hmotnosti v průběhu hodnocení v rozsahu 1,0 kg až 2,8 kg.
Významnější snížení tělesné hmotnosti bylo pozorováno se zvyšujícím se body mass indexem (BMI) na počátku.
• Kardiovaskulární zhodnocení
Krevní tlak
Po dobu trvání hodnocení snižoval liraglutid systolický krevní tlak v průměru o 2,3 až 6,7 mmHg oproti výchozímu a ve srovnání s aktivním komparátorem byl pokles 1,9 až 4,5 mmHg.
Post-hoc analýza závažných těžkých kardiovaskulárních nežádoucích příhod (úmrtí z kardiovaskulárních příčin, infarkt myokardu, cévní mozková příhoda) zahrnující data ze všech střednědobých a dlouhodobých studií (fáze 2 a 3, délka trvání od 26 do 100 týdnů) s 5 607 pacienty (3 651 z nich bylo léčeno liraglutidem) prokázala nezvýšené kardiovaskulární riziko (poměr výskytu 0,75 (95% CI 0,35; 1,63) u kombinovaného konečného výstupu pro liraglutid oproti všem komparátorům (metformin, glimepirid, rosiglitazon, inzulin glargin, placebo). Pacienti s vysokým kardiovaskulárním rizikem byli ze studií vyloučeni a četnost výskytu závažných těžkých kardiovaskulárních nežádoucích příhod ve studiích byla nízká (6,02 na 1 000 pacientoroků u pacientů léčených liraglutidem a 10,45 u pacientů léčených všemi komparátory), znemožňující vytvoření spolehlivých závěrů.
• Imunogenicita
V souvislosti s potenciálně imunogenními vlastnostmi léčivých přípravků obsahujících bílkoviny nebo peptidy se mohou u pacientů léčených liraglutidem tvořit protilátky proti liraglutidu. V průměru se protilátky vytvořily u 8,6 % pacientů. Tvorba protilátek nebyla spojena se sníženou účinností liraglutidu.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Victoza u jedné nebo více podskupin pediatrické populace s diabetes mellitus 2. typu (informace o použití u dětí viz bod 4.2).
Další klinické údaje:
V otevřeném klinickém hodnocení, prováděném u pacientů nedostatečně kontrolovaných léčbou metforminem (průměrná hodnota HbA1c 8,5%), která srovnávala účinnost a bezpečnost liraglutidu (v dávce 1,2 mg a 1,8 mg) a sitagliptinu (DPP-4 inhibitor, v dávce 100 mg), byla léčba liraglutidem v obou dávkováních po 26 týdnech statisticky úspěšnější než léčba sitagliptinem co se týče snížení HbA1c (-1,24 % resp. -1,50 % oproti -0,90 %, p<0,0001). Pacienti léčení liraglutidem dosáhli významného úbytku tělesné hmotnosti ve srovnání s pacienty léčenými sitagliptinem (-2,9 kg a -3,4 kg oproti -1,0 kg, p<0,0001). Přechodnou nauzeu pociťoval větší podíl pacientů léčených liraglutidem ve srovnání s pacienty léčenými sitagliptinem (20,8 % a 27,1 % u liraglutidu oproti 4,6 % v případě sitagliptinu). Redukce HbA1c a lepší výsledky oproti sitagliptinu pozorované po 26 týdnech léčby liraglutidem (v dávce 1,2 mg a 1,8 mg) přetrvávaly i po 52 týdnech léčby (-1,29 % a -1,51 % oproti -0,88 %, p<0,0001). Převedení pacientů ze sitagliptinu na liraglutid po 52 týdnech léčby vedlo
k dalšímu a statisticky významnému snížení HbAJc (-0,24 % a -0,45 %, 95% CI: -0,41 až -0,07 a -0,67 až -0,23) v 78. týdnu. Formální kontrolní skupina však nebyla k dispozici.
V otevřeném klinickém hodnocení, prováděném u pacientů nedostatečně kontrolovaných léčbou metforminem a/nebo sulfonylureou (průměrná hodnota HbAJc 8,3%), která srovnávala účinnost a bezpečnost liraglutidu v dávce 1,8 mg jednou denně a exenatidu v dávce 10 ^.g dvakrát denně, byla léčba liraglutidem po 26 týdnech statisticky úspěšnější než léčba exenatidem, co se týče snížení HbA1c (-1,12 % oproti -0,79 %, odhadovaný léčebný rozdíl: -0,33; 95% CI: -0,47 až -0,18). Významně vyšší počet pacientů dosáhl při užívání liraglutidu HbAic pod 7 % ve srovnání s exenatidem (54,2 % oproti 43,4 %, p=0,0015). Při léčbě oběma přípravky bylo dosaženo průměrného úbytku tělesné hmotnosti přibližně 3 kg. Převedení pacientů z exenatidu na liraglutid po 26 týdnech léčby vedlo k dalšímu a statisticky významnému snížení HbAJc (-0,32 %, 95% CI: -0,41 až -0,24) ve 40. týdnu. Formální kontrolní skupina však nebyla k dispozici. Během 26 týdnů léčby došlo u 235 pacientů užívajících liraglutid k 12 závažným nežádoucím příhodám (5,1%), zatímco u 232 pacientů užívajících exenatid to bylo 6 závažných nežádoucích příhod (2,6 %). Ve vztahu ke třídám orgánových systémů nebyly údaje o nežádoucích účincích konzistentní.
V otevřeném klinickém hodnocení srovnávajícím účinnost a bezpečnost liraglutidu v dávce 1,8 mg s lixisenatidem v dávce 20 ^g u 404 pacientů nedostatečně kontrolovaných léčbou metforminem (průměrná hodnota HbAJc 8,4 %) prokázal liraglutid superioritu k lixisenatidu snížením HbAJc po
26 týdnech léčby (-1,83 % oproti -1,21 %, p<0,0001). Významně vyšší počet pacientů dosáhl hodnoty HbA1c pod 7 % při léčbě liraglutidem v porovnání s lixisenatidem (74,2 % oproti 45,5 %, p<0,0001) a také cílové hodnoty HbA1c <6,5 % (54,6 % oproti 26,2 %, p<0,0001). Snížení tělesné hmotnosti bylo pozorováno v obou větvích (-4,3 kg u liraglutidu a -3,7 kg u lixisenatidu). Gastrointestinální nežádoucí účinky byly hlášeny častěji při léčbě liraglutidem (43,6 % oproti 37,1 %).
5.2 Farmakokinetické vlastnosti
Absorpce
Absorpce liraglutidu po subkutánním podání je pomalá a dosahuje maximální koncentrace za 8 -12 hodin po podání. Zjištěná maximální koncentrace liraglutidu byla 9,4 nmol/l u subkutánní jednorázové dávky liraglutidu 0,6 mg. Po dávce 1,8 mg liraglutidu dosáhla průměrná koncentrace liraglutidu v ustáleném stavu (AUCt/24) přibližně 34 nmol/l. Expozice liraglutidem se zvyšovala úměrně dávce. Variační koeficient u jednoho subjektu pro AUC liraglutidu byl po podání jednorázové dávky 11 %.
Absolutní biologická dostupnost liraglutidu po subkutánním podání je přibližně 55 %.
Distribuce
Zdánlivý distribuční objem po subkutánním podání je 11 - 17 l. Střední distribuční objem po intravenózním podání liraglutidu je 0,07 l/kg. Liraglutid se značně váže na plasmatické proteiny (> 98 %).
Biotransformace
Během 24 hodin po podání jednorázové dávky radioaktivně značeného liraglutidu s [3H] zdravým subjektům byla největší složka v plasmě intaktní liraglutid. Byly zjištěny dva méně významné metabolity v plasmě (< 9 % a < 5 % celkové expozice plasmatické radioaktivity). Liraglutid je metabolizován podobným způsobem jako velké bílkoviny, aniž by byl zjištěn určitý orgán jako hlavní cesta eliminace.
Eliminace
Po podání dávky liraglutidu značeného [3H] nebyl intaktní liraglutid zjištěn v moči ani ve stolici. Pouze menší část podané radioaktivity byla vyloučena jako metabolity liraglutidu močí nebo stolicí (6 % a 5 %). Radioaktivita v moči a stolici byla vylučována hlavně během prvních 6 - 8 dnů a odpovídala třem méně významným metabolitům liraglutidu.
Průměrná clearance po subkutánním podání jednorázové dávky liraglutidu je přibližně 1,2 l/h s poločasem eliminace přibližně 13 hodin.
Zvláštní skupiny pacientů
Starší pacienti:
Podle výsledků farmakokinetické studie na zdravých subjektech a analýzy populačních farmakokinetických dat u pacientů (18 až 80 let) nemá věk žádný klinicky významný vliv na farmakokinetiku liraglutidu.
Pohlaví:
Podle výsledků analýzy populačních farmakokinetických dat u mužských a ženských pacientů a farmakokinetické studie na zdravých subjektech nemá pohlaví žádný klinicky významný vliv na farmakokinetiku liraglutidu.
Etnický původ:
Podle výsledků populační farmakokinetické analýzy, která zahrnovala pacienty z bělošských, černošských, asijských a hispánských skupin, nemá etnický původ žádný klinicky významný vliv na farmakokinetiku liraglutidu.
Obezita:
Populační farmakokinetické analýzy ukazují, že body mass index (BMI) nemá významný vliv na farmakokinetiku liraglutidu.
Porucha funkce jater:
Farmakokinetika liraglutidu byla hodnocena u pacientů s různým stupněm poruchy funkce jater ve studii s jednorázovou dávkou. U pacientů s lehkou až středně těžkou poruchou funkce jater byla expozice liraglutidem snížena ve srovnání se zdravými pacienty o 13 - 23 %.
Expozice byla významně nižší (44 %) u pacientů s těžkou poruchou funkce jater (skóre Child Pugh > 9).
Porucha funkce ledvin:
U pacientů s poruchou funkce ledvin byla expozice liraglutidem ve srovnání s jedinci s normální funkcí ledvin snížena. U pacientů s lehkou (clearance kreatininu CrCl 50 - 80 ml/min), středně těžkou (CrCl 30 - 50 ml/min) a těžkou (CrCl < 30 ml/min) poruchou funkce ledvin a u terminálního selhání ledvin vyžadujícího dialýzu byla expozice liraglutidem snížena o 33 %, 14 %, 27 % a 26 %.
Podobně ve 26týdenní klinické studii s pacienty s onemocněním diabetes mellitus 2. typu a se středně těžkou poruchou funkce ledvin (CrCl 30 - 59 ml/min, viz bod 5.1) byla expozice liraglutidu o 26 % nižší v porovnání se samostatnou klinickou studií, v níž byli zahrnuti pacienti s onemocněním diabetes mellitus 2. typu s normální funkcí ledvin či s lehkou poruchou funkce ledvin.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Ve dvouletých studiích karcinogenicity na potkanech a myších byly pozorovány nonletální tumory C-buněk štítné žlázy. U potkanů nebyl pozorován NOAEL (No observed adverse effect level). Tyto tumory nebyly pozorovány u opic léčených po 20 měsíců. Tyto nálezy u hlodavců jsou způsobeny nongenotoxickým a receptory zprostředkovaným mechanismem specifickým pro GLP-1, na který jsou hlodavci zvláště citliví. Význam pro člověka je pravděpodobně nízký, ale nemůže být zcela vyloučen. Žádné jiné tumory spojené s léčbou nebyly zjištěny.
Studie na zvířatech neprokázaly přímý škodlivý vliv týkající se fertility, ale lehce zvýšenou míru časných úmrtí embryí po nejvyšší dávce. Podávání přípravku Victoza ve střední fázi těhotenství vedlo ke snížení tělesné hmotnosti matky a zpomalení růstu plodu s neprůkazným vlivem na žebra u potkanů a kosterní změny u králíků. Růst novorozenců byl u potkanů během expozice přípravkem Victoza snížen a efekt přetrvával u skupiny s vysokou dávkou i po odstavení. Není známo, zda je redukovaný růst mláďat způsoben sníženým příjmem mléka v důsledku přímého vlivu GLP-1, nebo sníženou tvorbou mateřského mléka při sníženém kalorickém příjmu.
Po intraarteriální injekci liraglutidu králíkům byly pozorovány lehká až střední hemoragie, erytém a otoky v místě vpichu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát hydrogenfosforečnanu sodného
Propylenglykol
Fenol
Voda na injekci
6.2 Inkompatibility
Látky přidané k přípravku Victoza mohou způsobit degradaci liraglutidu. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
30 měsíců
Po prvním použití: 1 měsíc
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Neuchovávejte v blízkosti mrazicího oddílu.
Po prvním použití: Uchovávejte při teplotě do 30 °C nebo v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Ponechávejte uzávěr na peru, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Zásobní vložka (sklo typu 1) s pístem (bromobutyl) a zátkou (bromobutyl/polyisopren) obsažená v jednorázovém multidávkovém předplněném peru zhotoveném z polyolefinu a polyacetalu.
Jedno pero obsahuje 3 ml roztoku, postačující pro 30 dávek po 0,6 mg, 15 dávek po 1,2 mg nebo 10 dávek po 1,8 mg.
Velikost balení po 1, 2, 3, 5 nebo 10 předplněných perech.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Victoza nesmí být použit v případě, že není čirý a bezbarvý či téměř bezbarvý.
Přípravek Victoza nesmí být použit v případě, že byl zmražen.
Přípravek Victoza může být podáván jehlami o délce do 8 mm a o síle do 32G. Pero je navrženo k použití s jednorázovými jehlami NovoFine nebo NovoTwist.
Jehly nejsou součástí balení.
Pacient musí být poučen, že má injekční jehlu po každé injekci zlikvidovat v souladu s místními požadavky a pero má uchovávat bez nasazené jehly. Tím se zabrání kontaminaci, infekci a vytékání přípravku. Také se tím zajistí přesnost dávkování.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
8. REGISTRAČNÍ ČÍSLA
EU/1/09/529/001 - 005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. června 2009
Datum posledního prodloužení registrace: 11. dubna 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Novo Nordisk A/S Hallas Allé DK-4400 Kalundborg Dánsko
Název a adresa výrobce odpovědného za propouštění šarží
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
B PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Victoza 6 mg/ml injekční roztok v předplněném peru Liraglutidum
2. OBSAH LÉČIVÉ LÁTKY
1 ml obsahuje liraglutidum 6 mg. Jedno předplněné pero obsahuje liraglutidum 18 mg,
3. SEZNAM POMOCNÝCH LÁTEK
dihydrát hydrogenfosforečnanu sodného, propylenglykol, fenol, voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 pero
2 pera
3 pera 5 per 10 per
Jedno pero obsahuje 3 ml roztoku postačující na 30 dávek po 0,6 mg, 15 dávek po 1,2 mg nebo 10 dávek po 1,8 mg.
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Pero Victoza je určeno k použití s jednorázovými jehlami NovoFine nebo NovoTwist. Jehly nejsou součástí balení.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Neuchovávejte pero s nasazenou jehlou. Určeno pouze pro jednu osobu.
8. POUŽITELNOST
Použitelné do
1 měsíc po prvním použití pero zlikvidujte.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce. Chraňte před mrazem.
Po prvním použití pera uchovávejte při teplotě do 30 °C nebo v chladničce. Chraňte před mrazem. Ponechávejte uzávěr na peru, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
12. REGISTRAČNÍ ČÍSLA
EU/1/09/529/001 1 x 3 ml EU/1/09/529/002 2 x 3 ml EU/1/09/529/003 3 x 3 ml EU/1/09/529/004 5 x 3 ml EU/1/09/529/005 10 x 3 ml
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Victoza
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK NA PŘEDPLNĚNÉ PERO
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Victoza 6 mg/ml injekce Liraglutidum Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
c.s.
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
Novo Nordisk A/S
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Victoza 6 mg/ml injekční roztok v předplněném peru
Liraglutidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek Victoza a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Victoza používat
3. Jak se přípravek Victoza používá
4. Možné nežádoucí účinky
5. Jak přípravek V ictoza uchovávat
6. Obsah balení a další informace
1. Co je přípravek Victoza a k čemu se používá
Přípravek Victoza obsahuje léčivou látku liraglutid. Pomáhá při snižování hladiny krevního cukru pouze tehdy, je-li cukr v krvi příliš vysoký. Také zpomaluje průchod jídla žaludkem.
Přípravek Victoza se používá samostatně, pokud dieta a cvičení samotné nedostatečně kontrolují hladinu cukru v krvi a pokud nemůžete používat metformin (další lék k léčbě cukrovky).
Přípravek Victoza se používá s dalšími léky k léčbě diabetu, pokud tyto léky nedostatečně kontrolují hladinu cukru v krvi. Tyto přípravky mohou být:
• perorální antidiabetika (jako například metformin, pioglitazon, přípravky obsahující sulfonylureu) a/nebo bazální inzulin (typ inzulinu, jehož účinek trvá celý den).
2. Čemu musíte věnovat pozornost, než začnete přípravek Victoza používat Nepoužívejte přípravek Victoza
- jestliže jste alergický(á) na liraglutid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou:
• před použitím přípravku Victoza
• pokud trpíte onemocněním slinivky břišní nebo jste je někdy prodělal(a).
Tento přípravek nesmí být používán, pokud máte diabetes 1. typu (vaše tělo neprodukuje vůbec žádný inzulin) nebo diabetickou ketoacidózu (komplikaci diabetu doprovázenou vysokou hladinou cukru v krvi a hlubokým zrychleným dýcháním). Tento přípravek není inzulin, a nesmí být proto používán jako náhrada inzulinu.
Pokud máte závažné onemocnění ledvin či pokud podstupujete dialýzu, použití přípravku Victoza se nedoporučuje.
Použití přípravku Victoza se nedoporučuje, pokud máte závažné onemocnění jater.
S použitím tohoto přípravku u pacientů se srdečním selháním jsou jen malé či žádné zkušenosti. Trpíte-li závažným srdečním selháním, není používání tohoto přípravku doporučeno.
Použití tohoto přípravku není doporučeno, pokud máte závažné žaludeční nebo střevní potíže, které mají za následek zpožděné vyprazdňování žaludku (tzv. gastroparéza), či zánětlivé onemocnění střev.
Pokud máte příznaky akutní pankreatitidy, jako je trvalá intenzivní bolest břicha, měl(a) byste se okamžitě obrátit na svého lékaře (viz bod 4).
Pokud trpíte onemocněním štítné žlázy včetně uzlovitého zbytnění štítné žlázy a zvětšení štítné žlázy, obraťte se na lékaře.
Na počátku léčby přípravkem Victoza můžete někdy pociťovat úbytek tekutin/dehydrataci, například v případě zvracení, pocitu na zvracení a průjmu. Je důležité, abyste zabránil(a) dehydrataci pitím dostatečného množství tekutin. Pokud máte nějaké otázky či obavy, obraťte se na ošetřujícího lékaře.
Děti a dospívající
Vzhledem k tomu, že bezpečnost a účinnost přípravku Victoza u dětí a dospívajících do 18 let zatím nebyly stanoveny, nedoporučuje se v této věkové skupině přípravek Victoza používat.
Další léčivé přípravky a přípravek Victoza
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Zvláště byste měl(a) svému lékaři, lékárníkovi nebo zdravotní sestře oznámit, jestli používáte léky k léčbě cukrovky obsahující některou z následujících léčivých látek:
• sulfonylureu (jako je např. glimepirid nebo glibenklamid). Při používání přípravku Victoza spolu se sulfonylureou vás může postihnout hypoglykemie (nízká hladina cukru v krvi), protože sulfonylurey zvyšují riziko hypoglykemie. Když poprvé začínáte používat tyto léky společně, lékař vám může doporučit snížit dávku přípravku obsahujícího sulfonylureu. Pročtěte si prosím varovné příznaky nízké hladiny cukru v krvi uvedené v bodě 4. Pokud rovněž užíváte sulfonylureu (například glimepirid nebo glibenklamid), může vám lékař říci, abyste si měřil(a) hladinu cukru v krvi. To pomůže lékaři při rozhodování, zda je nutno dávku sulfonylurey změnit.
• warfarin či další perorální antikoagulační léčivé přípravky. Lékař může požadovat častější provádění krevních testů k určení schopnosti vytvářet krevní sraženinu.
Těhotenství a kojení
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem. Přípravek Victoza se nemá používat během těhotenství, protože není známo, zda nemůže poškodit nenarozené dítě.
Není známo, zda přípravek Victoza přechází do mateřského mléka. Proto během období kojení tento přípravek nepoužívejte.
Řízení dopravních prostředků a obsluha strojů
Nízká hladina cukru v krvi (hypoglykemie) může snižovat vaši schopnost koncentrovat se. Neřiďte ani neobsluhujte stroje, pokud pociťujete příznaky hypoglykemie. Varovné příznaky nízké hladiny cukru v krvi najdete v bodě 4. Další informace týkající se tohoto problému získáte u ošetřujícího lékaře.
3. Jak se přípravek Victoza používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
• Počáteční dávka j e 0,6 mg j edenkrát denně, po dobu minimálně j ednoho týdne.
• Váš lékař vám řekne, kdy dávku zvýšit na 1,2 mg jedenkrát denně.
• Váš lékař vám může říci, abyste dále zvýšil(a) dávku na 1,8 mg jedenkrát denně, pokud při dávce 1,2 mg nebude hladina cukru v krvi dostatečně uspokojivá.
Neměňte dávku, pokud vám to váš lékař nedoporučí.
Přípravek Victoza je určen k injekční aplikaci do podkoží (subkutánní). Neaplikujte si injekci do žíly nebo do svalu. Nejvhodnější místo k aplikaci je přední část stehen, přední část pasu (břicho) nebo horní část paže.
Injekci si můžete aplikovat kdykoli v průběhu dne, bez ohledu na jídlo. Je však výhodnější podávat přípravek Victoza vždy přibližně ve stejnou denní dobu, která je pro vás nejvhodnější.
Než použijete pero poprvé, ukáže vám váš lékař nebo zdravotní sestra, jak se pero používá.
Podrobné pokyny k použití jsou uvedeny na druhé straně této příbalové informace.
Jestliže jste použil(a) více přípravku Victoza, než jste měl(a)
Pokud použijete více přípravku Victoza, než jste měl(a), sdělte to okamžitě svému lékaři, neboť byste mohl(a) vyžadovat léčbu. Můžete trpět pocitem na zvracení, zvracet nebo mít průjem.
Jestliže jste zapomněl(a) použít přípravek Victoza
Pokud zapomenete jednu dávku, použijte přípravek Victoza hned, jak si na to vzpomenete.
Pokud je to však za více než 12 hodin od doby, kdy jste měl(a) přípravek Victoza použít, vynechejte zapomenutou dávku. Pak si vezměte další dávku následující den jako obvykle.
Neberte si dávku navíc ani nezvyšujte dávku následující den, abyste vynechanou dávku nahradil(a).
Jestliže jste přestal(a) používat přípravek Victoza
Nepřestávejte používat přípravek Victoza, aniž byste se poradil(a) se svým lékařem. Pokud přestanete používat přípravek Victoza, může vám stoupnout hladina cukru v krvi.
Máte-li jakékoli další otázky, týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
rw r V r Vil r r v» 1
Závažné nežádoucí účinky
Časté: mohou se projevit až u 1 z 10 pacientů
• Hypoglykemie (nízká hladina cukru v krvi). Varovné příznaky nízké hladiny cukru v krvi se mohou objevit náhle a mohou zahrnovat: chladný pot, chladnou bledou pokožku, bolest hlavy, rychlý srdeční rytmus, nevolnost, pocit velkého hladu, změny vidění, ospalost, slabost, nervozitu, pocit úzkosti, zmatenost, potíže s koncentrací, třes. Váš lékař vám řekne, jak nízkou hladinu cukru v krvi léčit a co dělat, když zaznamenáte tyto varovné příznaky. Hypoglykemie u vás může nastat s větší pravděpodobností, pokud také používáte sulfonylureu nebo bazální inzulin. Před tím, než začnete používat přípravek Victoza, může vám lékař snížit dávku těchto léků.
Vzácné: mohou se projevit až u 1 pacienta z 1 000
• Závažná forma alergické reakce (anafylaktická reakce) s dalšími příznaky jako dýchací obtíže, otoky hrdla a obličeje, rychlý srdeční tep atd. Pokud se u vás tyto příznaky projeví, musíte okamžitě vyhledat lékařskou pomoc a informovat ošetřujícího lékaře, jakmile to bude možné.
• Střevní obstrukce. Závažná forma zácpy spojená s dalšími příznaky jako jsou bolest břicha,
nadýmání, zvracení apod.
Velmi vzácné: mohou se projevit až u 1 pacienta z 10 000
• Případy zánětu slinivky (pankreatitida). Pankreatitida může být závažný, potencionálně život ohrožující stav. Pocítíte-li následující závažné příznaky, ukončete používání přípravku Victoza a okamžitě kontaktujte lékaře:
Silná a přetrvávající bolest břicha (v oblasti žaludku), která může zasahovat až do zad, rovněž nevolnost a zvracení. Mohou to být příznaky zánětu slinivky břišní (pankreatitida).
Další nežádoucí účinky
Velmi časté: mohou se projevit u více než 1 pacienta z 10
• Nevolnost (pocit na zvracení). Ta obvykle časem přejde.
• Průjem. Ten obvykle časem přejde.
Časté:
• Zvracení
Při zahájení léčby přípravkem Victoza můžete v některých případech pociťovat ztrátu tekutin/dehydrataci (například pokud zvracíte, máte pocit na zvracení nebo průjem). Je důležité zabránit dehydrataci pitím velkého množství tekutin.
• Bolest hlavy
• Zažívací potíže
• Zánět žaludku (gastritida). Příznaky zahrnují bolest břicha, pocit na zvracení a zvracení.
• Gastroezofageální refluxní choroba (GERD). Příznaky zahrnují pálení žáhy.
• Bolest nebo nadmutí břicha
• Nepříj emný pocit v oblasti břicha
• Zácpa
• Nadýmání (větry)
• Snížená chuť k j ídlu
• Bronchitida (zánět průdušek)
• Nachlazení
• Závratě
• Zrychlený srdeční tep
• Únava
• Bolest zubů
• Reakce v místě vpichu (jako podlitiny, bolest, podráždění, svědění a vyrážka)
• Zvýšení hladin pankreatických enzymů (např. lipázy a amylázy)
Méně časté: mohou se projevit až u 1 ze 100 pacientů
• Alergické reakce jako svědění a kopřivka (typ kožní vyrážky)
• Dehydratace, někdy spojená se sníženou funkcí ledvin
• Malátnost (slabost)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Victoza uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku pera a na krabičce za „Použitelné do“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Před otevřením:
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Neuchovávejte v blízkosti mrazicího oddílu.
Během používání:
Pero můžete uchovávat při teplotě do 30 °C nebo v chladničce (2 °C - 8 °C) mimo mrazicí oddíl po dobu 1 měsíce. Chraňte před mrazem.
Pokud pero nepoužíváte, ponechejte uzávěr na peru, aby byl přípravek chráněn před světlem.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý či téměř bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Victoza obsahuje
- Léčivou látkou je liraglutidum. 1 ml injekčního roztoku obsahuje liraglutidum 6 mg. Jedno předplněné pero obsahuje liraglutidum 18 mg.
- Pomocnými látkami j sou dihydrát hydrogenfosforečnanu sodného, propylenglykol, fenol a voda na injekci.
Jak přípravek Victoza vypadá a co obsahuje toto balení
Přípravek Victoza je dodáván jako čirý bezbarvý či téměř bezbarvý injekční roztok v předplněném peru. Jedno pero obsahuje 3 ml roztoku postačující na 30 dávek po 0,6 mg, 15 dávek po 1,2 mg nebo 10 dávek po 1,8 mg.
Přípravek Victoza je dostupný v baleních obsahujících 1, 2, 3, 5 nebo 10 per. Na trhu nemusí být k dispozici všechny velikosti balení.
Jehly nejsou součástí balení.
Držitel rozhodnutí o registraci a výrobce
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dánsko
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
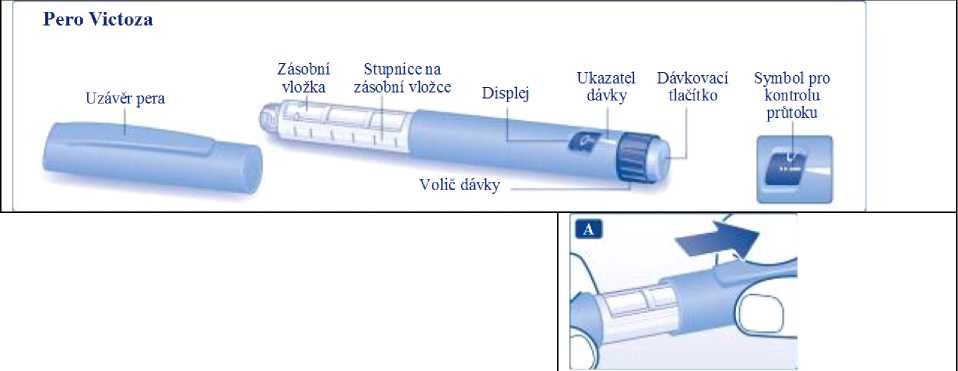
POKYNY PRO POUŽITÍ PERA VICTOZA Před použitím pera si pečlivě přečtěte tyto pokyny.
Pero obsahuje 18 mg liraglutidu. Můžete zvolit dávku 0,6 mg, 1,2 mg a 1,8 mg.
Pero je určeno k použití s jednorázovými injekčními jehlami NovoFine nebo NovoTwist o délce do 8 mm a síle do 32G (0,25/0,23 mm).
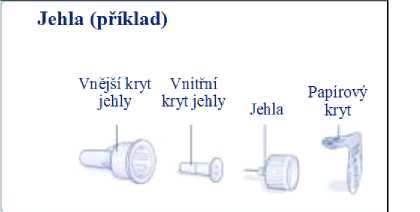
Příprava pera
Sejměte uzávěr pera.
Zkontrolujte název a barvu na štítku pera, abyste se ujistil(a), že obsahuje liraglutid. Použití nesprávného léku by mohlo způsobit vážné poškození zdraví.
Sejměte papírový kryt z nové jednorázové jehly. Našroubujte jehlu rovně a pevně na pero.
Sejměte vnější kryt jehly a ponechejte si jej pro pozdější potřebu.
Sejměte vnitřní kryt jehly a zahoďte jej.
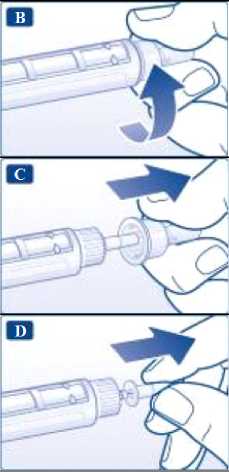
A Pro každou injekci vždy použijte novou jehlu. Tím snížíte riziko kontaminace, infekce, úniku liraglutidu, ucpání jehly a nepřesného dávkování.
A Dbejte na to, aby se jehla neohnula nebo nepoškodila.
A Nikdy nezkoušejte nasadit vnitřní kryt jehly zpět. Mohl(a) byste se jehlou poranit.
Péče o pero
• Nepokoušejte se pero opravovat ani je rozebírat.
• Chraňte pero před prachem, špínou a tekutinami všeho druhu.
• Pero čistěte hadříkem navlhčeným slabým saponátem.
• Pero se nepokoušejte omývat, namáčet či mazat - pero by se tím mohlo poškodit.
Pero ani jehly nepůjčujte nikomu dalšímu. Uchovávejte pero z dosahu jiných osob, zvláště dětí.
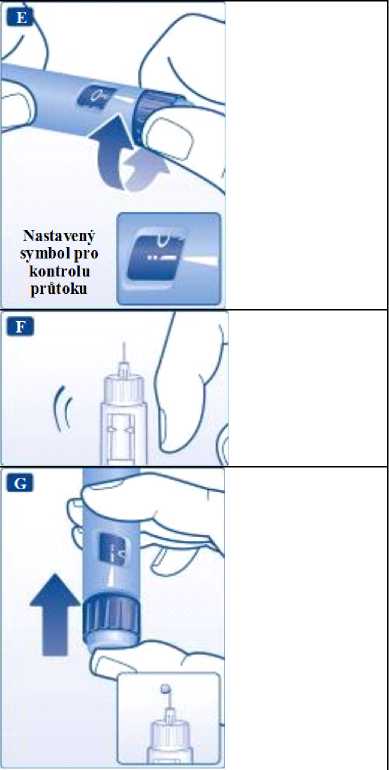
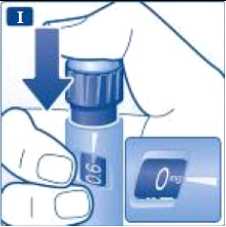
Pokaždé, když začínáte používat nové pero, zkontrolujte průtok
Než použijete nové pero, vždy zkontrolujte průtok. Pokud již pero používáte, přejděte ke kroku H, „Nastavení dávky“.
Otáčejte voličem dávky tak, aby symbol pro kontrolu průtoku byl proti ukazateli dávky.
Držte pero s jehlou směrem vzhůru. Jemně několikrát prstem poklepejte na zásobní vložku. Tím se vzduchové bublinky shromáždí v horní části zásobní vložky.
Držte jehlu směrem vzhůru a tiskněte dávkovací tlačítko, dokud nebude proti ukazateli dávky 0.
Na hrotu jehly se má objevit kapka liraglutidu. Pokud se kapka neobjeví, opakujte kroky E až G a to až čtyřikrát.
Pokud se stále kapka liraglutidu neobjeví, vyměňte jehlu a opakujte kroky E až G ještě jednou.
Pokud se opět neobjeví kapka liraglutidu, pero nepoužívejte. Znamená to totiž, že pero je vadné a musíte použít nové._
A Pokud vám pero upadlo na tvrdý povrch nebo máte podezření, že je poškozené, vždy použijte novou jednorázovou jehlu a před injekcí zkontrolujte průtok.
Nastavení dávky
Vždy zkontrolujte, že je ukazatel dávky na nule
Otáčejte voličem dávky, dokud potřebná dávka nebude proti ukazateli dávky (0,6 mg, 1,2 mg nebo 1,8 mg).
Pokud jste omylem zvolil(a) špatnou dávku, jednoduše ji změňte otáčením voliče dávky zpět nebo dopředu, dokud proti ukazateli dávky nebude správná dávka.
Při otáčení voliče dávky směrem zpět postupujte opatrně, abyste nezmáčkl (a) dávkovací tlačítko. Mohlo by totiž dojít k úniku liraglutidu.
Pokud se volič dávky zastaví dříve, než proti ukazateli dávky, kterou potřebujete, není již v peru dostatek liraglutidu pro celou dávku. Pak můžete:
Rozdělit dávku do dvou injekcí:
Otáčejte voličem dávky jedním nebo druhým směrem,

dokud proti ukazateli dávky nebude 0,6 mg nebo 1,2 mg. Aplikujte si dávku. Poté si připravte nové injekční pero a aplikujte zbývající počet miligramů, abyste doplnil(a) svou dávku na úplnou.
Rozdělit dávku mezi staré a nové pero můžete, pouze pokud jste proškolen(a) či pokud Vám to zdravotnický personál doporučí. K propočtu dávky použijte kalkulačku. Pokud byste dávku rozdělil(a) nesprávně, mohl(a) byste si aplikovat liraglutidu příliš mnoho či příliš málo.
Aplikovat pomocí nového pera celou dávku:
Pokud se volič dávky zastaví dříve, než se proti ukazateli dávky objeví 0,6 mg, připravte si nové pero a aplikujte celou dávku novým perem.
A Nepokoušejte se zvolit jiné dávky než 0,6 mg, 1,2 mg nebo 1,8 mg. Aby bylo zajištěno, že dostanete správnou dávku, musí být čísla na displeji přesně proti ukazateli dávky.
Při otáčení voličem dávky se ozývá cvakání. Toto cvakání nepoužívejte pro nastavení vaší dávky.
Nepoužívejte stupnici na zásobní vložce ke změření množství liraglutidu k aplikaci, není dostatečně přesná.
Aplikace dávky
Vpíchněte jehlu do kůže. Použijte injekční techniku, kterou vám doporučil Váš lékař nebo zdravotní sestra. Pak postupujte podle níže uvedených pokynů:
Tiskněte dávkovací tlačítko, dokud proti ukazateli dávky nebude nula. Dbejte, abyste se při aplikaci injekce nedotkl(a) displeje ostatními prsty nebo abyste nestiskl(a) volič dávky šikmo, protože byste tím mohl(a) injekci zablokovat. Přidržte dávkovací tlačítko stisknuté a ponechejte jehlu pod kůží po dobu nejméně šesti sekund, aby bylo zajištěno, že aplikujete celou dávku.
Vytáhněte jehlu.
Poté můžete na hrotu jehly vidět kapku liraglutidu. To je běžné a nemá to žádný vliv na dávku.
Zasuňte hrot jehly do vnějšího krytu jehly, aniž byste se dotkl(a) jehly či vnějšího krytu jehly.
Jakmile je jehla uvnitř, opatrně velký vnější kryt jehly zcela dotlačte. Pak jehlu odšroubujte. Opatrně jehlu zlikvidujte a vložte uzávěr pera zpět na pero.
Je-li pero prázdné, opatrně jej zlikvidujte bez nasazené jehly. Pero a jehlu zlikvidujte v souladu s místními
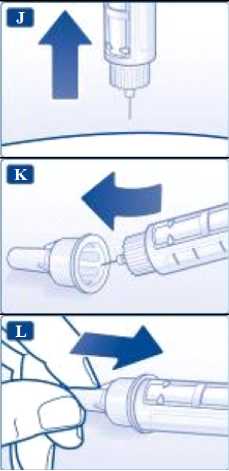
požadavky.
A Injekční jehlu po každé injekci sejměte a pero uchovávejte bez nasazené jehly.
A Tím se snižuje riziko kontaminace, infekce, úniku liraglutidu, ucpání jehly a nepřesného dávkování.
A Ošetřující osoby musí být velmi opatrné pn manipulaci s použitými jehlami, aby zabránily _poranění jehlou a přenosu infekce._
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) pro liraglutid dospěl výbor CHMP k těmto vědeckým závěrům:
Na základě signálu zvýšené hladiny lipázy a případů zvýšení průměrných hodnot lipázy získaných z klinických studií a z poregistračních zdrojů výbor PRAC zastává stanovisko, že informace o přípravku mají být aktualizovány. Do bodu 4.8 souhrnu údajů o přípravku mají být přidány nežádoucí účinky „zvýšené hladiny lipázy“ a „zvýšené hladiny amylázy“ s častou četností výskytu. Příbalová informace má být aktualizována odpovídajícím způsobem.
Proto s ohledem na údaje uvedené v hodnocené zprávě PSUR výbor PRAC dospěl k závěru, že změny informací o přípravku u léčivých přípravků obsahujících liraglutid jsou opodstatněné.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se liraglutidu výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících liraglutid zůstává nezměněný, a to pod podmínkou, že v informacích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
35