Vedrop 50 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Vedrop 50 mg/ml perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje tocoferolum alfa RRR 50 mg, ve formě tocofersolanum, což odpovídá až 74,5 IU tocoferolum.
Pomocné látky:
Jeden ml obsahuje 6 mg sodné soli methylparabenu (E219), 4 mg sodné soli ethylparahydroxybenzoátu (E215) a 0,18 mM (4,1 mg) sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok
Mírně viskózní, nažloutlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Vedrop je indikován při deficitu vitamínu E způsobenému malabsorpcí v zažívacím traktu u pediatrických pacientů s vrozenou nebo dědičnou chronickou cholestázou, ve věku od narození (donošení novorozenci) až do 18 let.
4.2 Dávkování a způsob podání
Léčbu Vedropem musí zahájit a sledovat lékař se zkušenostmi s léčbou pacientů trpících vrozenou nebo dědičnou chronickou cholestázou.
Biologická dostupnost vitamínu E z Vedropu se liší od hodnot jiných léčivých přípravků. Dávku je třeba předepsat v mg tokoferolu-alfa-RRR (ve formě tokofersolanu). Plazmatické hladiny vitamínu E je nutné kontrolovat jednou měsíčně přinejmenším několik prvních měsíců léčby, později pak v pravidelných intervalech, a v případě potřeby dávky přiměřeně upravovat.
Dávkování
Doporučená celková denní dávka u dětí trpících vrozenou nebo dědičnou chronickou cholestázou je 0,34 ml/kg/den (17 mg/kg tokoferolu-alfa-RRR ve formě tokofersolanu). Dávka by měla být předepsána v ml.
Dávka by měla být upravena podle plazmatické hladiny vitamínu E.
K výpočtu vhodné dávky Vedropu je třeba vydělit předepsanou dávku tokoferolu alfa (v mg) číslem 50. Výsledkem je objem Vedropu v ml:
Dávka Vedropu (v ml) = dávka d-alfa-tokoferolu (v mg)
50
Následující tabulka uvádí vhodný objem perorálního roztoku podle tělesné hmotnosti pacienta.
|
Tělesná hmotnost (kg) |
Objem perorálního roztoku (ml) |
|
3 |
1,0 |
|
4 |
1,4 |
|
5 |
1,7 |
|
6 |
2,0 |
|
7 |
2,4 |
|
8 |
2,7 |
|
9 |
3,1 |
|
10 |
3,4 |
|
15 |
5.1 |
Zvláštní populace
Porucha funkce jater nebo ledvin
Zkušenosti s léčbou tokofersolanem u pacientů s poruchou funkce ledvin nebo primární poruchou funkce jater neprokázaly nutnost úpravy dávkovacího schématu přípravku Vedrop (viz bod 4.4).
Způsob podání
Vedrop je podáván perorálně s vodou nebo bez ní. 1ml nebo 2ml perorální stříkačky přiložené v balení usnadňují odměření přesné předepsané dávky.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Vedrop se nesmí podávat nedonošeným novorozencům.
4.4 Zvláštní upozornění a opatření pro použití
Velké dávky vitamínu E podle literatury zvyšují tendenci ke krvácení u pacientů, kteří mají deficit vitaminu K nebo perorálně užívají jeho antagonisty. Doporučujeme proto monitorovat protrombinový čas a INR. Možná bude třeba upravit dávku perorálně užívaného antikoagulancia během léčby Vedropem a po ní.
Protože údaje týkající se pacientů s poruchou funkce ledvin jsou omezené, je třeba Vedrop podávat pacientům s poruchou funkce ledvin (například i dehydratovaným) opatrně a s pečlivým sledováním funkce ledvin (viz bod 4.2).
Vedrop je třeba podávat pacientům s primární poruchou funkce jater opatrně a s pečlivým sledováním jaterní funkce (viz bod 4.2).
Vedrop obsahuje sodnou sůl methylparabenu (E219) a sodnou sůl ethylparahydroxybenzoátu (E215), které mohou způsobit alergickou reakci (pravděpodobně zpožděnou).
Tento léčivý přípravek obsahuje sodík. To je třeba vzít v úvahu u pacientů na dietě s řízeným obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Při podávání s antagonisty vitamínu K se doporučuje sledovat koagulační funkce (viz bod 4.4).
Tokofersolan (vzhledem k inhibičním účinkům na transporter P-glykoproteinu) může také posilovat střevní absorpci jiných současně podávaných vitamínů rozpustných v tucích (A, D, E, K) nebo vysoce lipofilních léčivých přípravků (například steroidů, antibiotik, antihistaminik, cyklosporinu a takrolimu). Proto musí být zajištěno sledování účinku, a pokud je to třeba tak i úprava dávek.
4.6 Fertilita, těhotenství a kojení
Nejsou k dispozici klinické údaje o podávání tokofersolanu během těhotenství. Studie na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Při předepisování těhotným ženám nutno postupovat opatrně.
Kojení
Není známo, zda se tokofersolan vylučuje do lidského mateřského mléka. Jeho vylučování do mléka nebylo u zvířat ověřováno. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby tokofersolanem pro matku je nutno rozhodnout, zda pokračovat v kojení nebo jej přerušit nebo zda pokračovat v podávání přípravku Vedrop nebo jeho podávání přerušit.
Fertilita
Nejsou dostupné žádné údaje.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vedrop nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejčastěji hlášeným nežádoucím účinkem během léčby je průjem.
Tabulkový přehled nežádoucích reakcí
Hlášené nežádoucí účinky jsou uvedeny dále a setříděny podle tříd orgánových systémů a četnosti.
Četnosti jsou definovány takto: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třída orgánových systémů |
Nežádoucí účinky |
|
Gastrointestinální poruchy |
Časté: průjem Není známo: bolest břicha |
|
Poruchy kůže a podkožní tkáně | |
|
Celkové poruchy a reakce v místě aplikace |
Méně časté: tělesná slabost, bolest hlavy |
|
Vyšetření |
Méně časté: abnormální hladina sodíku v séru, abnormální hladina draslíku v séru, zvýšení hladiny transamináz |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Velké dávky vitamínu E mohou způsobit průjem, bolest břicha a jiné gastrointestinální poruchy.
Při předávkování je třeba navrhnout symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Vitaminy, jiné nekombinované vitaminové přípravky, ATC kód: A11HA08
Vitamín E je hlavním antioxidantem rozpustným v tucích v organismu. Působí jako molekula eliminující volné radikálové řetězce, blokuje peroxidaci mastných kyselin a podporuje stabilitu a neporušenost buněčných membrán.
Tento léčivý přípravek byl registrován za „výjimečných okolností”.
Znamená to, že vzhledem k vzácné povaze onemocnění, pro které je indikován, nebylo možné získat úplné informace o prospěšnosti a rizicích tohoto léčivého přípravku.
Evropská agentura pro léčivé přípravky každoročně vyhodnotí jakékoli nově dostupné informace a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Léčivá látka d-alfa-tokoferol-polyetylenglykol 1000 sukcinát (tokofersolan) je prodrug; aktivním metabolitem je d-alfa-tokoferol. Tokofersolan v nízkých koncentracích vytváří micely stimulující absorpci nepolárních lipidů, například vitamínů rozpustných v tucích. Kritická micelární koncentrace je nízká (0,04 až 0,06 mmol/l).
K hydrolýze tokofersolanu dochází v lumen střeva. Po vstřebání buňkami se alfa-tokoferolová složka objevuje v chylomikronech v míze a vypadá stejně jako vitamín E přijímaný z potravy. Příjem buňkami nevyžaduje žádné receptory, vazebné proteiny ani zvláštní metabolické procesy a neprobíhá formou pinocytózy. Absorpce izotopicky značeného tokofersolanu ukázala normální spektrum a dynamiku u lipoproteinů: hladina alfa-tokoferolu nejprve dosáhla maxima v chylomikronech, poté v lipoproteinech s velmi nízkou hustotou (VLDL), a nakonec v lipoproteinech s nízkou hustotou (LDL) a lipoproteidech s vysokou hustotou (HDL), a stejně tak i klesala podobně jako u kontrolních osob.
Při studii u 12 zdravých dobrovolníků byly hodnoty tokofersolanu porovnávány s referenčním vitamínem E mísitelným s vodou, a to po jediné perorální nasycovací dávce 1200 IU. Relativní biologická dostupnost tokofersolanu měla tendenci k vyšším hodnotám (Frel 1,01 ± 1,74) s AUC0-t 0,383 ± 0,203 pM.h/mg, Cmax 0,013 ± 0,006, tmax 6,0 h (6,0 - 24,0), a tm 29,7 h (16,0 - 59,5).
V jiné podobné studii tokofersolan vykazoval vyšší biologickou dostupnost než referenční vitamin E mísitelný s vodou u pediatrických pacientů s chronickou cholestázou (n=6); absorpce byla významně vyšší, jak ukazuje maximální nárůst plazmatické koncentrace (p=0,008) a AUC (p=0,0026).
Distribuce
Vitamín E se nachází hlavně na buněčných membránách, v mitochondriích a mikrozomech, a je široce distribuován (erytrocyty, mozek, svaly, játra, trombocyty), přičemž hlavním rezervoárem jsou tukové tkáně.
Eliminace
Vitamín E je vylučován hlavně ve žluči (75 %) a stolici, buď jako volný tokoferol, nebo jako jeho oxidované formy. Moč je pouze vedlejší cestou eliminace vitamínu E (vylučován jako glukuronidový konjugát).
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje v literatuře získané na základě konvenčních studií toxicity po opakovaném podávání, genotoxicity a reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Kalium-sorbát
Sodná sůl methylparabenu (E219)
Sodná sůl ethylparahydroxybenzoátu (E215)
Glycerol
Dodekahydrát hydrogenfosforečnanu sodného Kyselina chlorovodíková 35%
Čištěná voda
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
Po prvním otevření lahvičky: 1 měsíc.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Lahvička z hnědého skla typu III se šroubovacím uzávěrem a těsněním z polyetylénu (HDPE resp. LDPE). Perorální stříkačky s pouzdrem z polyetylénu (LDPE) a pístem z polystyrolu. Každá lahvička obsahuje 10 ml, 20 ml nebo 60 ml perorálního roztoku.
Krabičky obsahují:
■ jednu 10ml lahvičku a jednu 1ml stříkačku pro perorální podání
■ jednu 20ml lahvičku a jednu 1ml stříkačku pro perorální podání
■ jednu 60ml lahvičku a jednu 2ml stříkačku pro perorální podání
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Podávanou dávku je třeba z lahvičky odsát do perorální stříkačky, která je součástí balení.
1ml stříkačka pro perorální podání je kalibrována v rozsahu 0,05 až 1 ml po 0,05ml stupních. Jeden dílek 1ml stříkačky pro perorální podání odpovídá 2,5 mg d-alfa-tokoferolu ve formě tokofersolanu.
2ml stříkačka pro perorální podání j e kalibrována v rozsahu 0,1 až 2 ml po 0,1ml stupních. Jeden dílek 2ml stříkačky pro perorální podání odpovídá 5 mg d-alfa-tokoferolu ve formě tokofersolanu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Orphan Europe SARL Immeuble „le Wilson“
70, avenue du Général de Gaulle
92800 Puteaux
Francie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/09/533/001 10ml lahvička EU/1/09/533/002 20ml lahvička EU/1/09/533/003 60ml lahvička
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 24. července 2009
Datum posledního prodloužení registrace: 23. dubna 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury
pro léčivé přípravky
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Jméno a adresa výrobce odpovědného za propouštění šarží
Orphan Europe SARL Immeuble „Le Wilson“
70, avenue du Général de Gaulle
92800 Puteaux
Francie
nebo
Orphan Europe SARL
Parc d’Activités des Peupliers
39, rue des Peupliers, Batiment K
F-92000 Nanterre
Francie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
Tato registrace byla schválena za „výjimečných okolností“, a proto podle článku 14(8) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
V rámci každoročního přehodnocení dodá držitel rozhodnutí o registraci jednou za rok průběžnou zprávu z ustaveného registru pacientů s vrozenou chronickou cholestázou nebo s dědičnou cholestázou. |
Průběžné zprávy budou předkládány jednou za rok v rámci ročního přehodnocení. |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vedrop 50 mg/ml perorální roztok Tocofersolanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje tocoferolum alfa RRR 50 mg ve formě tocofersolanum, což odpovídá až 74,5 IU tocoferolum.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: sodná sůl methylparabenu (E219), sodná sůl ethylparahydroxybenzoátu (E215). Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální roztok
10ml lahvička a 1ml stříkačka pro perorální podání. 20ml lahvička a 1ml stříkačka pro perorální podání. 60ml lahvička a 2ml stříkačka pro perorální podání.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Zlikvidujte jeden měsíc po prvním otevření.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Orphan Europe SARL Immeuble „Le Wilson“
70, avenue du Général de Gaulle
92800 Puteaux
Francie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/09/533/001 10ml lahvička EU/1/09/533/002 20ml lahvička EU/1/09/533/003 60ml lahvička
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vedrop 50 mg/ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Vedrop 50 mg/ml perorální roztok Tocofersolanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje tocoferolum alfa RRR 50 mg ve formě tocofersolanum, což odpovídá až 74,5 IU tocoferolum.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: sodná sůl methylparabenu (E219), sodná sůl ethylparahydroxybenzoátu (E215). Další informace najdete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální roztok 10ml 20ml 60ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DETÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Zlikvidujte jeden měsíc po otevření
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Orphan Europe SARL Immeuble „Le Wilson“
70, avenue du Général de Gaulle
92800 Puteaux
Francie
13. ČÍSLO ŠARŽE
EU/1/09/533/001 10ml lahvička EU/1/09/533/002 20ml lahvička EU/1/09/533/003 60ml lahvička
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku je vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Vedrop 50 mg/ml
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Vedrop 50 mg/ml perorální roztok
Tokofersolanum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to
i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Vedrop a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Vedrop užívat
3. Jak se Vedrop užívá
4. Možné nežádoucí účinky
5. Jak Vedrop uchovávat
6. Obsah balení a další informace
1. Co je Vedrop a k čemu se používá
Vedrop obsahuje vitamín E (ve formě tokofersolanu). Používá se k léčbě nedostatku vitamínu E způsobenému malabsorpcí v zažívacím traktu (kdy jsou živiny z jídla v zažívacím traktu špatně vstřebávány) u pacientů od narození (donošených novorozenců) až do věku 18 let trpících chronickou cholestázou (dědičné nebo vrozené onemocnění, kde se žluč nedostane z jater do střev).
2. Čemu musíte věnovat pozornost, než začnete Vedrop užívat Neužívejte přípravek Vedrop
- jestliže jste alergický(á) na vitamín E (d-alfa-tokoferol) nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- Vedrop se nesmí používat u nedonošených novorozenců.
Upozornění a opatření
Před užitím přípravku Vedrop se poraďte se svým lékařem, pokud máte:
■ Potíže s ledvinami nebo jste dehydratovaný(á). Vedrop by měl být užíván se zvýšenou opatrností a funkci ledvin je nutno pečlivě sledovat, protože polyetylenglykol, součást léčivé látky tokofersolanu, může ledviny poškozovat.
■ Potíže s játry. Vedrop užívejte se zvýšenou opatrností a funkci jater je nutno pečlivě sledovat.
Další léčivé přípravky a Vedrop
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Informujte lékaře nebo lékárníka, jestliže užíváte:
■ Některé léky určené k ředění krve (perorální antikoagulační přípravky, například warfarin). Lékař vás požádá, abyste pravidelně docházel(a) na krevní testy, a případně upraví dávky, aby předešel vyššímu riziku krvácení.
■ Vitamíny rozpustné v tucích (například vitamín A, D, E nebo K) nebo léky vysoce rozpustné v tucích (například kortikoidy, cyklosporin, tacrolimus a antihistaminika). Vedrop může zvyšovat jejich vstřebávání v zažívacím traktu, a lékař proto bude sledovat účinky léčby a v případě potřeby upraví dávky.
Těhotenství a kojení
O expozici tomuto přípravku během těhotenství nejsou k dispozici žádné klinické údaje. Informujte svého
lékaře, jestliže jste těhotná, lékař rozhodne, zda můžete lék užívat.
Neexistují údaje o tom, zda je přípravek přítomen v mateřském mléce. Informujte svého lékaře, jestliže
hodláte kojit. Lékař vám poradí, co je pro Vás a Vaše dítě nejlepší.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv lék.
Řízení dopravních prostředků a obsluha strojů
Vedrop pravděpodobně neovlivní Vaši schopnost řídit a obsluhovat stroje.
Vedrop obsahuje sodnou sůl methylparabenu (E219) a sodnou sůl ethylparahydroxybenzoátu (E215),
které mohou způsobit alergickou reakci (okamžitou i zpožděnou).
Vedrop obsahuje 0,18 mmol (4,1 mg) sodíku na 1 ml. Poraďte se s lékařem, jestliže dodržujete dietu
s kontrolovaným obsahem sodíku..
3. Jak se Vedrop užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Obvyklá dávka přípravku je 0,34 mg/kg tělesné hmotnosti denně.
Lékař vám dávku předepíše v ml.
Dávka bude upravena lékařem podle hladiny vitamínu E v krvi.
Způsob podání
Roztok polykejte s vodou nebo bez ní. K podání používejte vždy pouze stříkačku pro perorální podání (pro podání ústy) přiloženou v krabičce.
Vedrop můžete užívat před jídlem nebo během jídla, s vodou nebo bez ní.
Odměření dávky:
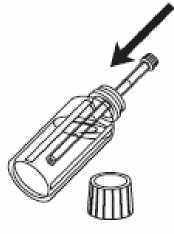
1- Otevřete lahvičku.
2- Vsuňte do ní stříkačku přiloženou v balení.
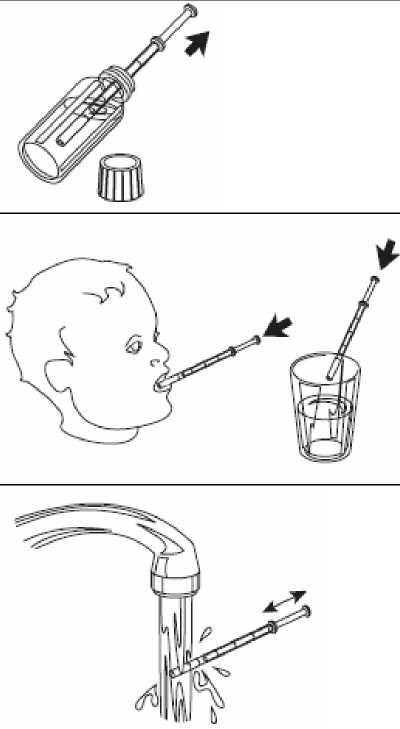
3- Naplňte stříkačku tekutinou vytažením pístu až po značku, která odpovídá množství v mililitrech (ml) předepsanému lékařem.
4- Vytáhněte stříkačku z lahvičky.
5- Vyprázdněte obsah stříkačky zatlačením pístu až nadoraz, a to buď:
- přímo do úst, nebo
- do sklenice vody, a tu poté celou vypijte.
6- Zavřete lahvičku.
7- Vypláchněte stříkačku vodou.
Jestliže jste užil(a) více Vedropu, než jste měl(a):
Jestliže užijete velkou dávku vitamínu E, může se projevit přechodný průjem a bolesti břicha. Jestliže příznaky přetrvávají déle než dva dny, poraďte se se svým lékařem nebo lékárníkem.
Jestliže jste zapomněl(a) Vedrop užít
Vynechte zapomenutou dávku a pokračujte v pravidelném podávání příště. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat Vedrop
Nepřestávejte užívat léčbu bez předchozí domluvy se svým lékařem, neboť nedostatek vitamínu E se může znovu objevit a odrazit se na Vašem zdraví. Před ukončením léčby se obraťte na svého lékaře nebo lékárníka.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Uváděny jsou následující nežádoucí účinky:
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
• průjem
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
• astenie (pocit slabosti)
• bolest hlavy
• ztráta vlasů a ochlupení
• svědění
• vyrážka (výsev na kůži)
• abnormální hladina sodíku v krvi
• abnormální hladina draslíku v krvi
• zvýšení hladiny transamináz (jaterních enzymů)
Není známo (četnost nelze z dostupných údajů určit)
• bolesti břicha
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Vedrop uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za slovem EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
• Lahvičku zlikvidujte jeden měsíc po prvním otevření, i když v ní zbývá roztok.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Vedrop obsahuje
- Léčivou látkou je tocofersolanum. Jeden ml roztoku obsahuje tocoferolum alfa RRR 50 mg ve formě tocofersolanum, což odpovídá až 74,5 IU tocoferolum.
- Dalšími složkami jsou: kalium-sorbát, sodná sůl methylparabenu (E219) a sodná sůl ethylparahydroxybenzoátu (E215) (více informací o těchto dvou složkách naleznete v závěru bodu 2), glycerol, dodekahydrát hydrogenfosforečnanu sodného, kyselina chlorovodíková 35%, čištěná voda.
Jak Vedrop vypadá a co obsahuje toto balení
Vedrop je mírně viskózní, nažloutlý roztok k perorálnímu podání, dodávaný v hnědé skleněné lahvičce. Lahvička obsahuje 10 ml, 20 ml nebo 60 ml perorálního roztoku. Každá krabička obsahuje jednu lahvičku a jednu stříkačku pro perorální podání, a to 1ml stříkačku u 10ml nebo 20ml lahviček, nebo 2ml stříkačku u 60ml lahviček.
Držitel rozhodnutí o registraci
Orphan Europe SARL Immeuble „Le Wilson“
70, avenue du General de Gaulle
92800 Puteaux
Francie
Výrobce
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Francie
nebo
Orphan Europe SARL
Parc d’Activités des Peupliers
39, rue des Peupliers, Batiment K
F-92000 Nanterre
Francie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgique/Belgie/Belgien Lietuva
Orphan Europe Benelux Orphan Europe AB
Koning Albert I laan 48 bus 3 Isafjordsgatan 30C, plan 3
BE-1780 Wemmel (Brussels) S-164 40 Kista
Tél/Tel: +32 2 46101 36 Švedija
Tel: + 46 8 545 80 230
Btarapna
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm
TepMaHHH
Tel: +49 731 140 554 0
Česká republika
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Německo
Tel: +49 731 140 554 0
Danmark
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sverige
Tlf : +46 8 545 80 230
Deutschland
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Tel: +49 731 140 554 0
Luxembourg/Luxemburg
Orphan Europe Benelux Koning Albert I laan 48 bus 3 BE-1780 Wemmel(Brussels) Belgique/Belgien Tél/Tel: +32 2 46101 36
Magyarország
Orphan Europe (Germany) GmbH
Eberhard-Finckh-StraBe 55
D-89075 Ulm
Németország
Tel: +49 731 140 554 0
Malta
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Franza
Tel: +33 1 47 73 64 58 Nederland
Orphan Europe Benelux Koning Albert I Iaan 48 bus 3 BE-1780 Wemmel (Brussels) Belgie
Tel: +32 2 46101 36
Eesti
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Rootsi
Tel: + 46 8 545 80 230 EXXáSa
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
rakkía
Tpk: +33 1 47 73 64 58 Espaňa
Orphan Europe, S.L.
C/ Isla de la Palma, 37, 2a planta E-28700 San Sebastián de los Reyes, Madrid Tel: + 34 91 659 28 90
France
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Tél: +33 (0)1 47 73 64 58
Hrvatska
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Francuska
Tél: +33 (0)1 47 73 64 58 Ireland
Orphan Europe (UK) Ltd.
Isis House, 43 Station road Henley-on-Thames Oxfordshire RG9 1AT - UK United Kingdom Tel: +44 1491 414333
Island
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sví^jóó
Simi:+46 8 545 80 230
Norge
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sverige
Tlf : +46 8 545 80 230 Osterreich
Orphan Europe (Germany) GmbH
Eberhard-Finckh-StraBe 55
D-89075 Ulm
Deutschland
Tel: +49 731 140 554 0
Polska
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Niemcy
Tel: +49 731 140 554 0 Portugal
Orphan Europe, S.L.
C/ Isla de la Palma, 37, 2a planta
E-28700 San Sebastián de los Reyes, Madrid
Espanha
Tel: +34 91 659 28 90 Románia
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Germania
Tel: +49 731 140 554 0
Slovenija
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Nemčija
Tel: +49 731 140 554 0
Slovenská republika
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Nemecko
Tel: +49 731 140 554 0
Italia
Orphan Europe (Italy) Srl Via Marostica, 1 I-20146 Milano Tel: +39 02 487 87 173
Kúrcpog
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F - 92800 Puteaux
Ta^Ma
Tr(k : +33 1 47 73 64 58 Latvija
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Zviedrija
Tel: + 46 8 545 80 230
Suomi/Finland
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sverige
Puh/Tel : +46 8 545 80 230 Sverige
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Tel : +46 8 545 80 230
United Kingdom
Orphan Europe (UK) Ltd. Isis House, 43 Station road Henley-on-Thames Oxfordshire RG9 1AT - UK Tel: +44 (0)1491 414333
Tato příbalová informace byla naposledy revidována
Tento léčivý přípravek byl registrován za „výjimečných okolností”. Znamená to, že vzhledem ke vzácné povaze tohoto onemocnění nebylo možné získat o tomto léčivém přípravku úplné informace.
Evropská agentura pro léčivé přípravky každoročně vyhodnotí jakékoli nové informace týkající se tohoto léčivého přípravku a tato příbalová informace bude podle potřeby aktualizována.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro
léčivé přípravky:
24